

Abstract
Inositol-requiring enzyme 1 (IRE1) is an orchestrator of the unfolded protein response (UPR), the cellular response to endoplasmic reticulum (ER) stress that plays a crucial role in tumor development. IRE1 signaling is the most evolutionary conserved branch of the UPR. Under ER stress, the IRE1 luminal domain undergoes a conformational change to multimerize, resulting in trans-autophosphorylation and activation of the cytosolic kinase and endoribonuclease domain. Adenosine triphosphate-competitive inhibitors that bind to the IRE1 kinase site can modulate the activity of the RNase domain through an allosteric relationship between the IRE1 kinase and RNase domains. The current study aims at the investigation of available structural data of the IRE1 kinase domain and provides insights into the design of novel kinase inhibitors. To this end, a detailed analysis of IRE1 kinase active site and investigation of suitable structures for virtual screening studies were performed. The results indicate in silico target fishing as an appropriate strategy for the identification of novel IRE1 kinase binders, further validating the robustness of the in silico protocol. Importantly, the study highlights the kinase-inhibiting RNase attenuator (KIRA)-bound protein data bank 4U6R structure as the best protein structure to perform virtual screening to develop diverse and more potent KIRA-like IRE1 kinase inhibitors that are capable of allosterically affecting the RNase activity.
1. Introduction
The failure of cells to appropriately fold and modify secretory and transmembrane proteins leads to the accumulation of misfolded proteins in the endoplasmic reticulum (ER).1 Under these conditions of “ER stress”, the unfolded protein response (UPR) is initiated by the activation of three sensor proteins on the ER membrane: inositol-requiring protein 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6).2 Among the three branches, the IRE1 pathway is the most evolutionarily conserved and represents the sole branch of the UPR in yeast.3 This pathway plays a critical role in a variety of physiological and disease conditions, including B cell and adipocyte differentiation, secretory capacity of pancreatic beta cells and salivary organs, neurodegeneration, obesity, and insulin resistance.2 Hence, a detailed understanding of the regulatory mechanisms underlying mammalian IRE1 activation is essential to the development of therapeutics.4
IRE1 is an ER-resident type 1 transmembrane protein that contains an N-terminal luminal domain, a transmembrane domain, and cytoplasmic C-terminal kinase and endoribonuclease (RNase) effector domains.5 Mammalian IRE1 is present in two distinct isoforms, α and β. IRE1α (hereafter called IRE1) is ubiquitously expressed and plays an important role in how cells and organisms respond to ER stress, whereas IRE1β is expressed primarily in the epithelial cells of the gastrointestinal tract and the lung but is absent in the liver and pancreas and participates in mucosal secretion and lipid transport in the gut.6
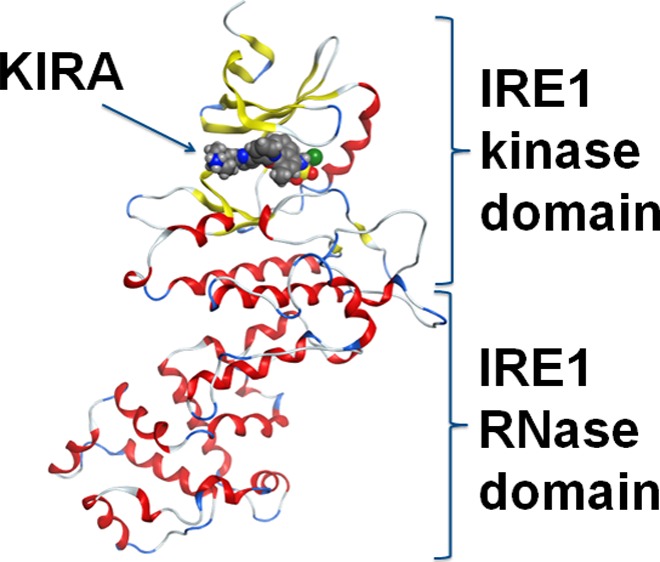
Several X-ray crystal structures of the IRE1 cytosolic domain in complex with different exogenous and endogenous ligands are available in the protein data bank (PDB) (Table 1). The first crystallographic structure of the IRE1 cytosolic domain (PDB code: 2RIO) was arranged in a back-to-back conformation (dimer interface area: 3800 Å2), with adenosine diphosphate (ADP) and Mg2+ in the adenosine triphosphate (ATP)-binding site.7 The IRE1 kinase domain has a β-stranded N-terminal lobe and an α-helical C-terminal lobe1 (Figure 1) with Lys599 crucial for kinase activity.8 On the basis of structural and biochemical analyses, the kinase domain was found to display similarity to several protein kinases, including proto-oncogene tyrosine-protein kinase Src (c-Src), epidermal growth factor receptor, PKR, general control nonderepressible 2 (GCN2), cyclin-dependent kinase 2 (CDK2), and mitogen-activated protein kinase kinase 1 (MAP2K1).9 The RNase domain, on the other hand, is unique to IRE1 and was originally named as kinase extension ribonuclease.7 It is composed of eight α helices connected via short loops and with His1061 and Tyr1043 reported to be crucial for catalyzing the cleavage of RNA.7 Crystallographic data (PDB code: 3FBV, PDB code: 3SDM), in vivo and in vitro data,10 and studies of the kinetics of RNA cleavage9 have demonstrated the formation of high-order supramolecular IRE1 multimers. Although different arrangements of dimeric complexes are crystallized, both back-to-back (PDB codes: 3SDJ, 3SDM, 3LJ0, 3LJ1, 3LJ2, 3FBV, and 2RIO) and face-to-face (PDB code: 3P23), the only form compatible with an oligomerization state is the back-to-back dimer.9
Table 1. Crystal Structures Available for IRE1 in Different Organisms.
| PDB code | ligand IDa | resolution (Å) | organism | assembly | # of mutation (s) | references |
|---|---|---|---|---|---|---|
| 3SDJ | APJ (2) | 3.65 | Saccharomyces cerevisiae | oligomer | 29 | (18) |
| 3SDM | (Apo) | 6.6 | S. cerevisiae | oligomer | 28 | (19) |
| 3LJ0 | ADP (1) | 3.2 | S. cerevisiae | dimer | 24 | (20) |
| 3LJ1 | DKI (3) | 3.33 | S. cerevisiae | dimer | 24 | (21) |
| 3LJ2 | IZA (4) | 3.33 | S. cerevisiae | dimer | 24 | (21) |
| 3FBV | APJ (2) | 3.2 | S. cerevisiae | oligomer | 28 | (22) |
| 2RIO | ADP (1) | 2.4 | S. cerevisiae | dimer | wild-type | (7) |
| 2BE1 | (Apo) | 2.98 | S. cerevisiae | dimer | wild-type | (23) |
| 4PL3 | ADP (1) | 2.9 | Mus musculus | dimer | 1 | (12) |
| 4PL4 | ADP (1) | 3.0 | M. musculus | tetramer | 1 | (12) |
| 4PL5 | ADP (1) | 3.4 | M. musculus | tetramer | 1 | (12) |
| 5HGI | (Apo) | 2.58 | Homo sapiens | dimer | wild-type | (13) |
| 4YZ9 | 4K7 (5) | 2.46 | H. sapiens | dimer | wild-type | (24) |
| 4YZC | STU (8) | 2.49 | H. sapiens | monomer | wild-type | (25) |
| 4YZD | ADP (1) | 3.1 | H. sapiens | monomer | wild-type | (25) |
| 4Z7G | (Apo) | 2.6 | H. sapiens | monomer | wild-type | (26) |
| 4Z7H | 4L5 (6) | 2.9 | H. sapiens | monomer | wild-type | (26) |
| 4U6R | 3E4 (7) | 2.5 | H. sapiens | monomer | 1 | (11) |
| 3P23 | ADP (1) | 2.7 | H. sapiens | dimer | 1 | (27) |
| 2HZ6 | (Apo) | 3.1 | H. sapiens | dimer | 2 | (28) |
Figure 1.

(A) Ribbon diagram representing the structure of the IRE1 kinase and RNase domains (PDB code: 4U6R). β-strands are shown in blue and α-helices in red. Binding mode of (B) exogenous ligand kinase-inhibiting RNase attenuators (KIRAs) (PDB code: 4U6R) and (C) ADP (endogenous ligand) (PDB code: 3P23).
Interest in targeting the IRE1 pathway as a therapeutic strategy has prompted the search for highly selective IRE1 modulators.11 Because the RNase activity of IRE1 is essential for the resolution of ER stress and proper functioning of the UPR, modulation of this activity is targeted. Several of these efforts have focused on modulating the RNase domain directly.12 In addition, characterization of the allosteric relationship between the kinase and RNase domains has allowed for the modulation of IRE1α with small organic molecules targeting the ATP-binding site.11,13 The RNase activity can be either enhanced or reduced by targeting this site.13 The first characterized compounds inhibiting the kinase domain (type I IRE1 kinase inhibitors) were found to activate the RNase domain.13 Available IRE1 crystal structures reveal the conformational changes induced by these inhibitors in the kinase domain that drive the monomeric form to the active dimeric state, resulting in RNase activation.13 Recently, an imidazopyrazine scaffold was discovered to provide a new series of ATP-competitive inhibitors, called KIRAs, that block the RNase activity through binding to the kinase domain.13 Whereas type I IRE1 kinase inhibitors increase the dimerization or oligomerization state of IRE1 and thus activate the RNase, the KIRAs stabilize the IRE1 monomeric form and thereby inhibit the RNase activity.1,13 Whereas an active kinase conformation is characterized by a DFG-in motif and helix-αC-in conformation, KIRA compounds stabilize the helix-αC-out conformation, thereby hindering dimer formation.13 Interestingly, this chemical scaffold shows a promising selectivity profile against a series of 100 other kinases.11
In this study, we elucidate atomistic details of differences in ligand recognition between exogenous and endogenous ligands in the IRE1 kinase active site utilizing a range of in silico approaches. Molecular docking and cross-docking (inverse docking) studies were performed with available IRE1 protein structures and reported inhibitors. In general, cross-docking involves docking ligands of a protein to different crystal structures or different proteins.14 Because crystallographic structures of IRE1 are increasingly deposited in PDB, the application of docking methods is essential for both virtual screening (VS) method development and calibrating scoring functions. These studies could aid structure-based drug design in the identification of new potent and selective IRE1 kinase modulators. The analysis can also provide valuable insights for creating focused small-molecule libraries, thereby increasing the hit rates in drug discovery screening campaigns toward the IRE1 kinase active site.
2. Methods
2.1. Selection and Preparation of IRE1 Crystal Structures
At the time of the current study, six X-ray structures of yeast IRE1 cocrystallized with small organic molecules in the kinase active site (exogenous or endogenous compounds), and one apo structure, were present in PDB15 (Table 1). Three X-ray structures of IRE1 cocrystallized with the endogenous ligand ADP in the kinase active site were available for mouse and six X-ray structures cocrystallized with ADP or exogenous ligands in the kinase active site for human IRE1, and two apo structures were present (Table 1).
The crystal structures obtained from PDB were categorized according to holo or apo, that is, whether or not they have a ligand molecule in their active site. Each crystal structure was prepared using the Schrödinger protein preparation wizard.16 Hydrogen atoms were added, and possible metal binding states were generated. The protonation and tautomeric states of Asp, Glu, Arg, Lys, and His were adjusted to match a pH of 7.4, and possible orientations of Asn and Gln residues were generated. Hydrogen bond sampling with the adjustment of active site water molecule orientations was performed using PROPKA at pH 7.4. Water molecules with fewer than two hydrogen bonds to nonwaters were deleted. Finally, the protein–ligand complexes were subjected to geometry refinement using the OPLS2005 force field17 in restrained minimizations.
2.2. Ligand Preparation
The cocrystallized ligands (Table 1) and KIRA analogues13 (Table S1) were extracted and used for cross-docking studies. The ligands are displayed in Figure 2 and Table S1. The ligands were prepared using Ligprep29 from the Schrödinger suite.30 The OPLS2005 force field17 was used in all ligand preparation steps. Possible protonation and ionization states were enumerated for each ligand using an ionizer at pH 7.4. Possible stereoisomers, tautomeric states, and metal-binding states were generated.
Figure 2.
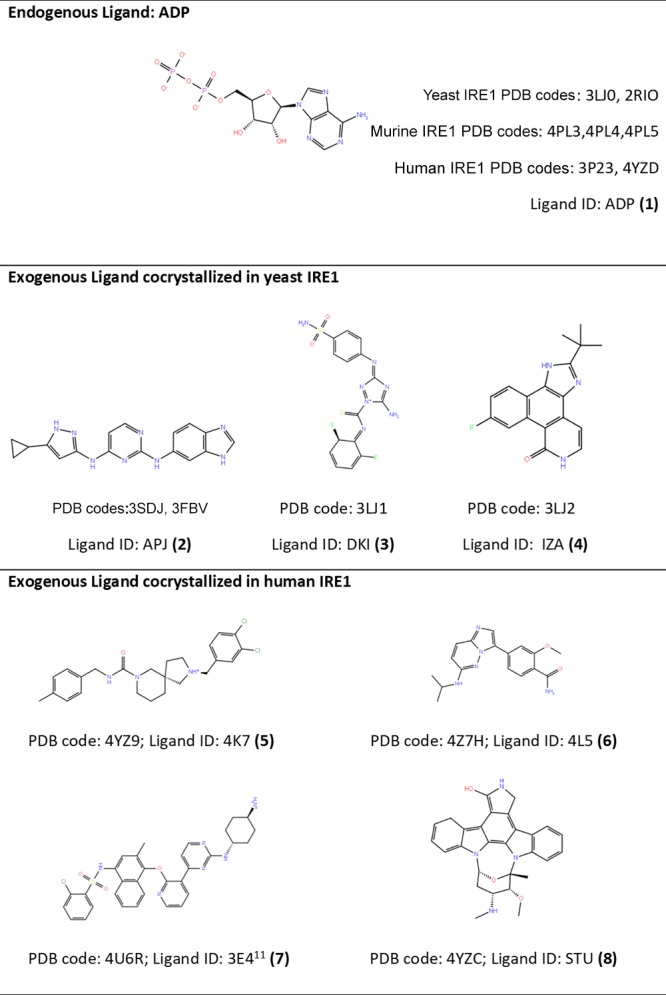
Ligands cocrystallized in the IRE1 kinase active site.
2.3. Key Interaction Points
To investigate the key ligand interactions in a more quantitative manner, we evaluated the individual electrostatic and hydrophobic contributions to the interaction energy of each amino acid residue involved in binding to the corresponding ligands. The electrostatic contribution was calculated on the basis of the nonbonded electrostatic interaction energy term of the force field, whereas the hydrophobic contributions were computed using the directional hydrophobic interaction term based on contact surfaces as implemented in the molecular operating environment scoring function.31 As an outcome, an energy (in kcal/mol) is associated with the electrostatic contribution, whereas a score (the higher the better) is related to the hydrophobic contribution. Finally, the interaction energy patterns are displayed as heat maps. The identification of key residues involved in the interactions highlight possible patterns in the binding modes of the compounds.
In this study, the kinase active sites of IRE1 from all three organisms (S. cerevisiae, M. musculus, and H. sapiens) were characterized and assessed for their ability to bind druglike molecules. By investigating the interaction between all ligands in all organisms, selectivity is addressed from both a ligand and a protein (organism) perspective.
2.4. Molecular Docking
Three sets of docking studies were performed, each based on the same docking methodology. In order to validate the suitability of the docking approach, benchmark studies were first performed in which each ligand was removed and redocked into the respective protein crystal structure. Following this, cross-docking studies against the full pool of IRE1 cytosolic structures were performed. In addition, a set of 25 KIRA analogues were docked and compared with the available experimental inhibition data (Table S1). Docking was done using the Glide program32 in Schrödinger,30 with the receptor grid prepared using the OPLS2005 force field.17 The grid center was set at the centroid of the cocrystallized ligand, and the cubic grid had a side length of 20 Å. For the apo structures, the centroid of Lys599, a residue crucial for the kinase activity, was set to be the center of the grid. All structures were superposed by the protein alignment of the kinase domain (i.e., sequence numbers 571–832 of the cytosolic domain; numbering in hIRE1), and the cocrystallized ligands were docked against each target. No constraints were used in any of the receptor grids. Flexible ligand sampling was considered in the docking procedure using the XP (extra precision) docking mode. All other parameters were set to defaults according to the Glide docking process. We ranked the compounds by performing a postdocking analysis in the form of a normalization based on the glide docking score. The normalization approach suggested by Vigers and Rizzi33 is shown by the following equations:
| 1 |
| 2 |
| 3 |
where Sij is the calculated glide docking score for the ith compound and jth pocket (in kcal mol–1) and Sij′ is the modified score for compound i in the active site j. μi and σi are the average values and standard deviations of the scores for compound i across all pockets j. Sij is also termed the multiple active site correction (MASC) score.33
3. Results and Discussion
3.1. IRE1 Structural Analysis
In order to uncover IRE1 kinase active site-specific sequence motifs, multiple sequence alignments were performed. Sequence similarity and sequence identity analysis of IRE1 in different species revealed that the primary sequence of the cytosolic domain of yeast IRE1 has ∼39% sequence identity compared with that of human IRE1 (hIRE1), whereas murine IRE1 displays a sequence identity of more than 85% compared to human IRE1 (Figure S1). The sequence similarity to hIRE1 is also higher for the murine protein, ∼55% for yeast IRE1 and >89% for murine IRE1. Furthermore, residue conservation within the kinase active site was examined. Identifying residues expected to be involved in functional specificity and targeting these specific residues with appropriate small organic molecules could be an attractive approach to design novel IRE1 modulators. Amino-acid residues at a distance of 5.0 Å from the cocrystallized ligands in human IRE1 are highly conserved through the species and are highlighted in Figures S1 and S2. The primary structure of the yeast kinase active site domain of IRE1 shows ∼73% sequence identity (85% sequence similarity) compared with that of human IRE1, whereas murine and human IRE1 displays a sequence identity of 100%. The available structural data were also examined (Figures S4 and S5). On the basis of the superposition of available structures, the Cα root-mean-square deviation (rmsd) comparison reveals highly similar conformations among all structures and between all species (S3, S4, and S5). The three-dimensional (3D) structures of the cytosolic domain of yeast IRE1 show 1–3 Å displacement compared with that of human IRE1, whereas murine IRE1 structures display values between 0.5 and 1.5 Å. Structures 4YZ9 and 3P23 can be considered as outliers (Figure S4) with slightly higher rmsd values. In these two structures, the loops display small reorientations explaining the Cα rmsd deviation. The rmsd deviation of ∼2 Å for 4U6R PDB is due to the ligand-induced displacement of the helix-αC13 (Figure S4).
Also, the amino-acid residues in the IRE1 kinase active site are highly conserved in terms of 3D conformation among all species (Figure S5). The 3D structure of the yeast kinase active site domain of IRE1 shows values between 1.78 and 2.79 Å compared with that of human IRE1, whereas murine IRE1 displays values between 0.48 and 1.64 Å compared to hIRE1 (Figure S5). Again, 4YZ9 displays the largest deviations in rmsd values.
3.2. Dissecting IRE1 Kinase Active Site Small Organic Molecule Interactions
One of the aims of the present study is to investigate and characterize key interactions between bound ligands and the IRE1 kinase active site residues. From all the crystallographic data, three heat maps were inferred based on the different organisms (yeast, murine, and human), and the nature of compounds was analyzed (exogenous or endogenous). To dissect the ligand-enzyme recognition system in a more quantitative manner, we calculated the individual electrostatic and hydrophobic contributions to the interaction energy of each amino-acid residue within a distance of 5.0 Å from the cocrystallized ligands.
With the calculated per residue electrostatic and hydrophobic energy interaction contribution values, the heat maps depicted in Figure 3 (human) and Supporting Information Figures S6 and S7 (yeast and murine, respectively) were obtained.
Figure 3.

Per amino-acid interaction energy map for the cocrystallized compounds in the human IRE1 kinase-binding site. (A) Electrostatic energy values (kcal mol–1); (B) hydrophobic score (arbitrary units). PDB IDs 3P23 and 4YZD have ADP as a cocrystalized ligand and PDB IDs 4YZ9, 4YZC, 4Z7H, and 4U6R have exogenous ligands cocrystallized. *Dephosphorylated IRE1α cocrystallized with KIRA.13
These analyses provide information regarding structure–activity relationships. First of all, the different binding modes of endogenous ligands (ADP) and exogenous ligands are highlighted (cf. Figures 1 and S8). The key interaction points (KIPs) (Figure 3A) indicate several fundamental groups with favorable electrostatic interaction energy with the exogenous ligands (colored blue), namely, Lys599, Glu643, Leu644, Cys645, Ala646, Glu651, and Asp711. On the other hand, the endogenous ligands cocrystallized in hIRE1 establish a favorable interaction with Gly580, Thr584, Val586, Ala597, Lys599, Glu643, Leu644, Cys645, Lys690, and Ser710, whereas repulsive ones are mediated by Glu651, Asn693, and Asp711. This can be rationalized in terms of the negatively charged phosphate groups of ADP, absent in the exogenous ligands (Figure 2). In particular, we note that for the residues on the right half of the electrostatic interaction map (Figure 3A; residue Ala646 onward), the interaction between endogenous versus exogenous ligands is essentially reversed. As seen in Figure 1, these residues form the lower part of the active site cavity, which hence is a more selective/sensitive region for discriminating between different ligands.
In addition, from the analysis we see that the hydrophobic interaction contributes poorly to stabilize the endogenous ligand compared to exogenous ligands (Figure 3B). For exogenous ligands, hydrophobic interaction is established with Leu577, Val586, Ile642, and Leu695, whereas only minor hydrophobic interaction is displayed between ADP and residues Val586 and Leu695.
Comparison of the apo structure (PDB code: 5HGI) with holo structures cocrystallized with ADP (PDB code: 3P23) or KIRA (PDB code: 4U6R), respectively, reveals the significant impact on the electrostatic surface and size of the active site, in the absence or presence of endogenous or exogenous ligands (Figure 4).
Figure 4.

Electrostatic surface representation of the hIRE1 kinase active site in (A) apo form (PDB code: 5HGI) (B) with the KIRA molecule cocrystallized (PDB code: 4U6R) and (C) with ADP cocrystallized (PDB code: 3P23). Standard coloring scheme was used: red for negative, white for neutral, and blue for positive electrostatic potential.
Analysis of IRE1–ligand interaction in the active sites of different species can be useful in validating residues expected to be involved in ligand binding (Figures S6 and S7). For ADP, the interacting residues crucial for binding are highly conserved between the different species. Electrostatic interaction is seen between ADP and Gly580, Lys599, and Lys690 (numbered according to the human PDB code) in all three organisms (Figures S6 and S7). This confirms the importance of these interactions for the correct accommodation of the endogenous ligand in the kinase pocket. Repulsive interaction with Asn693 and Asp711 is also conserved through the species, as are the hydrophobic interactions with Val586 and Leu695.
For the exogenous compounds, comparisons of the binding mode were only possible with yeast because no exogenous ligands have been cocrystallized with murine IRE1. Despite the difference between these two organisms, some useful insights can also be obtained from yeast IRE1. Hydrophobic interactions between the ligand and Leu577 (Leu680 in yeast), Val586 (Val689 in yeast), and Leu695 (Leu804 in yeast) are very well conserved. Electrostatic interaction with Lys599 (Lys702 in yeast) and Glu651 (Asp754 in yeast) is not conserved, whereas electrostatic interaction with Glu643 (Glu746 in yeast), Cys645 (Cys748), and Asn693 (Asn802) is conserved, underlining the importance of those particular residues for the correct binding to the target protein.
Hence, albeit crystal structures are highly similar (low Cα rmsd), the slight difference in sequence between human and yeast is clearly reflected in the interaction fingerprints.
3.3. Docking Studies
An essential prerequisite when performing a docking study is the accessibility of a target structure. The success of a docking simulation relies heavily on the use of appropriate and accurate protein structures, with holo structures in general giving the best performance.34 To evaluate the performance of different IRE1 structures to host various ligands, benchmark docking, cross-docking (inverse docking), and evaluation toward a set of RNase effectors were performed. A protein structure able to better accommodate the higher number of small organic molecules with a conformation similar to the cocrystallized one could be considered a good candidate for VS studies. Throughout this work, the entire pool of IRE1 crystallographic structures was used (Table 1), and all cocrystallized ligands (Figure 2) available from the experimental studies. The pipeline can be summarized in the following.
3.3.1. Self-Docking
To evaluate the ability of the docking protocol to successfully replicate the crystallographic binding mode, a self-docking benchmark was first performed.
3.3.2. Total Cross-Docking
Cross-docking studies (inverse docking) were executed among the whole pool of IRE1 cytosolic structures available, to discriminate between the IRE1 structures able to bind the majority number of the cocrystallized ligands and those structures clearly unsuitable for virtual high-throughput screening campaigns.
3.3.3. Normalized Values of Docking Score as the Ranking Method
To differentiate between the IRE1 structures, a postdocking analysis in the form of a normalization or standardization was performed.33 This allows us to measure the specificity or promiscuity of each IRE1 structure with respect to accommodation of the ligands in the active site pocket. Evaluating the ability of the overall IRE1 crystallographic protein structure to host compounds could be valuable information to ameliorate future VS campaigns.
3.3.4. Self- and Cross-Docking: Rmsd Analysis
Reproducing the bound conformation of a ligand in its crystallographic structure is one of the initial challenges in molecular docking studies. Rmsd values between the best-scoring pose and the cocrystallized conformation of the ligand were calculated and are visualized as a heat map in Figure 5A. Each column highlights different IRE1 PDB structures, and each row represents the different cocrystallized ligands.
Figure 5.

(A) Self-docking and cross-docking results for cocrystallized ligands (Figure 2) with available cocrystal and apo structures (Table 1; listed by PDB ID). Values shown are the rmsd (in angstrom) of the best-scoring docked pose to the crystal structure pose. The rmsds are highlighted using a colorimetric scale from blue to red for values from 0 to 10 Å. The diagonal (in squares) represents self-docking results. (B) Superposition of the predicted best-scoring poses of IRE1 cocrystallized compounds (blue) on the experimental ones (tan). For each pose, rmsd is reported. *Dephosphorylated IRE1α cocrystallized with KIRA.13 **KIRA cocrystallized in the 4U6R PDB structure.
Out of the eight small organic molecules cocrystallized in the different IRE1 structures, seven were used in these studies, as STU(8) cocrystallized in the 4YZC PDB structure was not able to attain good poses that fit in the active site of 4Z7G, 4YZ9, and 4Z7H without clashes in structures. For the 4YZ9 PDB structure, no valid poses were obtained after minimization, indicating poor fit for the ligand in the active site. For the 4Z7H and 4Z7G structures, all poses were rejected during postdocking minimization.
The results are excellent, with rmsd values <1.5 Å, validating the accuracy of the software in reproducing the pose of the cocrystallized ligands (Figure 5A,B). Understandably, there are three exceptions; ADP cocrystallized in 2RIO, 4PL4, and 4PL5 all of which show higher rmsd values. For the 4PL4 and 4PL5 structures, cocrystallized ADP is characterized by having high B-factor values, whereas the result for 2RIO can be ascribed to the general flexibility of the ADP molecule. Our computational workflow was designed to identify a unique pose (conformational state) per compound. Evaluating a larger number of poses by visual inspection may be a good strategy to cherry-pick the pose closest to the experimental one but does not allow for automatization and predictability.
Moreover, that different ligand conformations and orientations are possible in the kinase active site is clearly evident from the ligands 3E4 (7) and 4K7 (5) cocrystallized in the 4U6R and 4YZ9 PDB structures, respectively (Figure S9). As expected, reproduction of these two conformations using a protein structure from a complex containing a different ligand could be challenging (Figure 5A). This is clearly evident by the high rmsd values when these two ligands were docked in different IRE1 structures (Figure 5A).
In addition, using the apo structures generated high rmsd values in the docked ligand poses compared to the cocrystallized ligand conformations (Figure 5A). The data support the general findings that reproducing correct ligand native conformation using apo structures, where no native ligand has been cocrystallized, is generally worse compared to using holo structures.34
3.3.5. MASC Score Analysis
The second step was to assess the performance of the docking program in ranking the series of cocrystallized ligands toward the full pool of IRE1 crystal structures. Docking score analysis can be problematic if several cocrystallized ligands score well against the full pool of protein structures, as is the case herein (Figure S10). Except for the KIRA ligand, the ranking of the other cocrystallized ligands proved difficult as highlighted by essentially the same docking score value throughout the different IRE1 crystallographic structures (Figure S10). In order to resolve this problem, we used MASC scoring.33 The MASC score is a useful tool, as it incorporates signed information describing how far a value is from the average and in which direction.35 In our studies, the more negative the MASC score, the more unique is the accommodation of a specific ligand in a specific structure, whereas a large set with similar (near-zero) values indicates promiscuity (either among ligands or among possible binding partners for a specific ligand).
Despite the fact that no direct binding experiments are available to compare the cross-docking results (only IC50 values are reported11,13), the data obtained are very encouraging in ranking these series of ligands cocrystallized in different IRE1 structures. Ligands docked into their respective cocrystallized structures were correctly ranked with better scores compared to other ligands docked in that same structure (Figure 6A). For ADP, the best binding targets stand out to be 2RIO, 3LJ0, 3P23, 4PL5, and 4YZD, which all represent structures with ADP cocrystallized. This is clearly relevant and encouraging information for successful VS studies.
Figure 6.
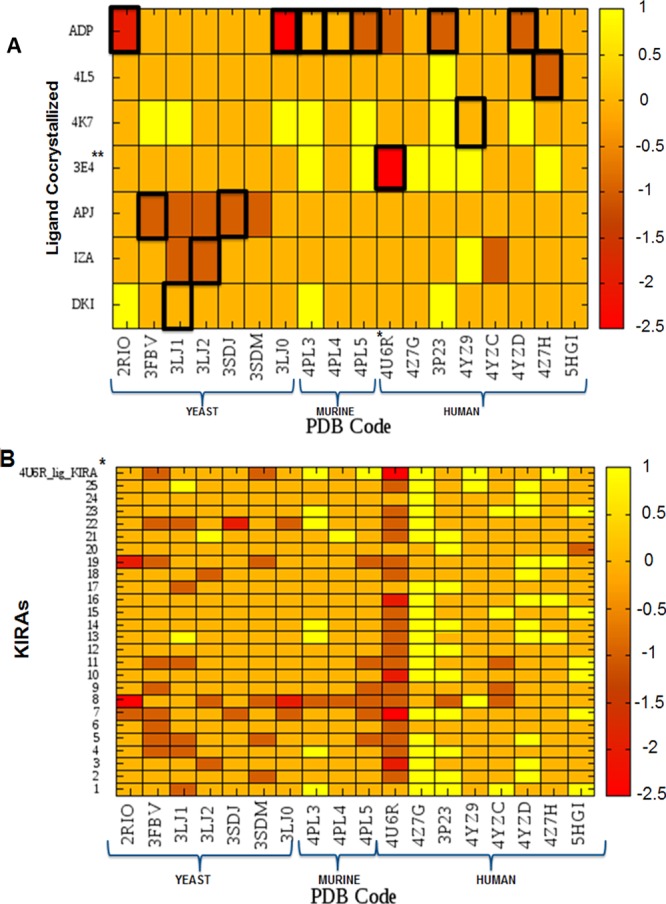
(A) MASC score values for each ligand (y-values) in each IRE1 PDB structure (x-values), represented by a colorimetric scale going from red (−2.5) to yellow (1). Cocrystallized ligands are highlighted with boxes for each IRE1 structure. *Dephosphorylated IRE1 cocrystallized with KIRA.13 **KIRA cocrystallized in the 4U6R PDB structure. (B) MASC score values of KIRA analogues (1–25) for each IRE1 PDB structure represented by a colorimetric scale, going from red (−2.5) to yellow (1). *KIRA cocrystallized in the PDB structure 4U6R.
In addition, multiple IRE1 crystallographic structures score well for a couple of compounds. For instance, the ligand APJ (2) cocrystallized in 3SDJ and 3FBV scores very well against the yeast IRE1 structures that did not contain ADP in the kinase active site, whereas their binding in the murine/human IRE1 is worse. An explanation for the poor score against the IRE1 structures of other organisms can be seen in the noted differences in sequence and in residues contributing to the favorable interaction of ligands in the yeast versus human and murine IRE1. A screening campaign using yeast IRE1 will hence not render the compounds best suited for targeting the human analogue, despite the highly conserved nature of the protein. Other interesting information was obtained from investigating the heat map of the apo structures 5HGI and 4Z7G (Figure 6A). Both of them show MASC values close to zero for all ligands, indicating promiscuity. In a VS campaign, this could generate misleading results because these two structures could enrich the population of false positives. Finally, it is important to note that these docking simulations characterize the PDB structure 4U6R as the better one to accommodate a subset of ATP-competitive ligands (KIRAs13) that allosterically inactivate the RNase domain through binding to the kinase domain.
In order to confirm the PDB structure 4U6R as the best model to accommodate this type of KIRA allosteric inactivators, 25 KIRA compounds13 shown in Table S1 were docked into each of the 18 IRE1 crystallographic active sites. The MASC score and docking score results are shown in Figures 6B and S10, respectively.
All KIRAs were found to score well in the 4U6R structure (Figure 6B). This can be understood from the fact that the allosteric action of the KIRAs involves a structural distortion in the active site, moving helix-αC to in turn affect dimer formation. Understandably, the docking score and MASC score identified the ligand cocrystallized in 4U6R as the best ligand for its cognate active site (i.e., higher docking score and larger negative MASC score).
Unexpectedly, the 3FBV PDB structure revealed a negative MASC score for several of the KIRA compounds studied. The results could be rationalized by visual inspection of the docked poses, showing that this structure establishes favorable interactions from amino acids Leu680, Val689, Lys702, and Asp754 (corresponding to Leu577, Val586, Lys599, and Asp651 in human), relevant for the correct accommodation of KIRA-like compounds.
Furthermore, the PDB structures 4Z7G, 3P23, and 4YZD perform worse against a large set of KIRA compounds. From visual inspection of the docking poses, several steric clashes were identified explaining the low docking scores. 4Z7G is the apo structure of hIRE1 and again highlights the importance of using a holo structure for docking. 3P23 and 4YZD were both cocrystallized with ADP, emphasizing the plasticity of the active site pocket (Figure 4). Interestingly, one KIRA compound (KIRA analogue 20) binds very selectively to the apo structure 5HGI. Only in the 5HGI structure, the compound is able to attain a binding pose such that it can establish a favorable interaction with Leu577, Val586, Lys599, and Leu695, validating that these amino acids are crucial for the correct accommodation of KIRA-like compounds inside the IRE1 kinase active site.
Comparing the docking scores and MASC scores to the reported IC50 values for kinase and RNase activity (Table S1), we conclude that there is no specific pattern identified that singles out strong binders as strong effectors in the current work. This can be explained by several factors: the small range of experimental values (i.e., 0.1–20 μM), absence of explicit dissociation constant (Kd) values, and the inherent limitations and approximations of docking methods.36,37
4. Conclusions and Perspective
In the present study, we have tried to address fundamental questions related to docking, accuracy in reproducing ligand poses in the crystal structures, and importance of appropriate protein conformations in reproducing ligand poses. We have investigated a series of small organic molecules cocrystallized in the IRE1 kinase active site and validated the importance of molecular docking approaches for identifying novel IRE1 modulators. The performance of the docking algorithm and MASC score to rank compounds in a VS campaign was evaluated, and we conclude that the application of a combined procedure that takes into consideration self-docking and cross-docking studies will raise the chance of successful VS studies.
The Glide docking program performed well. The poses of cocrystallized ligands were reproduced in almost all cases. In some cross-docking cases, we saw high variability in docking results to a target receptor structure (i.e., lack of consistency between the MASC score and rmsd analysis). When non-native ligands are docked in these crystal structures, there is a high chance of retrieving false positives. Cross-docking the ligand set to the apo structures (PDB codes: 5HGI and 4Z7G) resulted in high rmsd values. The apo structures are thus not suitable for VS studies.
The current study identified the holo receptor conformation of the PDB code 4U6R as highly suitable for docking-based VS aiming to identify novel KIRA-like compounds capable of allosterically inhibiting the RNase activity. KIRA induces a conformational change upon binding,13 which explains the difference in selectivity for this particular structure.
In addition, we have compared and determined the most important interactions of the ligands cocrystallized into the kinase active site of hIRE1, opening the possibility for a rational VS approach. Key residues involved in the binding of small organic molecules in the IRE1 kinase active site were identified. In agreement with experimental results,8 the sequence similarity and 3D comparison between different species and interaction energy fingerprint analysis highlight the importance of Lys599 for correct ligand binding. In addition, from the cocrystallized structures numbered as per hIRE1 PDB codes (Table 1), Leu577, Val586, and Leu695 were predicted to stabilize ligand binding by hydrophobic interactions. These key residues, involved in strong electrostatic and hydrophobic interactions with almost all cocrystallized ligands, could be exploited as filtering tools during a VS campaign. Postprocessing the VS results by filtering toward the presence or absence of these favorable interactions could further enhance the detection of putative binders. The methodology employed in this study can be adapted to any protein for studying selectivity and choosing the right starting structure for VS. All information acquired from this study is currently being applied by our research groups to understand the detailed molecular interactions required for selective IRE1α inhibition. This knowledge coupled with relevant biochemical assays can be directly applicable for developing new modulators of hIRE1α.
Acknowledgments
This research was funded by the EU’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant 675448 (TRAINERS). The Faculty of Science at the University of Gothenburg and the Swedish Science Research Council (VR; grant number 2014-3914) are gratefully acknowledged for financial support (L.A.E.), and the Swedish National Infrastructure for Computing is also acknowledged for allocations of computing time at the C3SE Supercomputing Center at Chalmers (Göteborg).
Glossary
Abbreviations
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- ER
endoplasmic reticulum
- IRE1
inositol-requiring enzyme 1
- hIRE1
human inositol-requiring enzyme 1
- KIRAs
kinase-inhibiting RNase attenuators
- rmsd
root-mean-square deviation
- UPR
unfolded protein response
- VS
virtual screening
- SAR
structure–activity relationship
- MASC
multiple active site correction
- KIP
key interaction point
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- ATF6
activating transcription factor 6
- Kd
dissociation constant
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01404.
Molecular images of the series of KIRA compounds and their IC50 values; table with experimental activities; sequence alignment of the IRE1 cytosolic domain in different organisms; multiple sequence alignment of the IRE1 cytosolic domain in eight different organisms using clustal; superposition of the 3D structures of IRE1 in different organisms; rmsd matrix values in angstrom of the positions of the Cα atoms for each pair of the IRE1 cytosolic domain structures; rmsd matrix values in angstrom of the positions of the Cα atoms for each pair of IRE1 kinase active site domain structures; per amino-acid interaction energy map for cocrystallized compounds inside the yeast IRE1 kinase-binding site; per amino-acid interaction energy map for cocrystallized compounds inside the murine IRE1 kinase-binding site; schematic representation of the ligand interactions; superposition of all available 3D structures of IRE1 in different organisms; docking score values returned by each cocrystallized ligand (y-values) for each IRE1 PDB structure (x-values) represented by a colorimetric scale; and docking score of each KIRA analogue (y-values 1–25) for each IRE1 PDB structure (x-values) represented by a colorimetric scale (PDF)
Author Contributions
All authors formulated the project. A.C. and C.C. performed the calculations and initial analyses and wrote the first version of the manuscript. All authors contributed to the production of subsequent versions of the manuscript and additional analyses.
The authors declare the following competing financial interest(s): A.M.G., A.S., and L.A.E. are directors and shareholders of Cell Stress Discoveries, Ltd.
Supplementary Material
References
- Maly D. J.; Papa F. R. Druggable Sensors of the Unfolded Protein Response. Nat. Chem. Biol. 2014, 10, 892–901. 10.1038/nchembio.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C.; Chevet E.; Harding H. P. Targeting the Unfolded Protein Response in Disease. Nat. Rev. Drug Discovery 2013, 12, 703–719. 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Xue Z.; He Y.; Ye K.; Gu Z.; Mao Y.; Qi L. A Conserved Structural Determinant Located at the Interdomain Region of Mammalian Inositol-requiring Enzyme 1α. J. Biol. Chem. 2011, 286, 30859–30866. 10.1074/jbc.m111.273714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S. S.; Kaufman R. J. Targeting Endoplasmic Reticulum Stress in Metabolic Disease. Expert Opin. Ther. Targets 2013, 17, 437–448. 10.1517/14728222.2013.756471. [DOI] [PubMed] [Google Scholar]
- Feldman H. C.; Tong M.; Wang L.; Meza-Acevedo R.; Gobillot T. A.; Lebedev I.; Gliedt M. J.; Hari S. B.; Mitra A. K.; Backes B. J.; et al. Structural and Functional Analysis of the Allosteric Inhibition of IRE1alpha with ATP-Competitive Ligands. ACS Chem. Biol. 2016, 11, 2195–2205. 10.1021/acschembio.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkaew D.; Chattopadhyay A.; King M. D.; Chunhacha P.; Liu Z.; Stevenson H. L.; Chen Y.; Sinthujaroen P.; McDougal O. M.; Fujise K. Fortilin Binds IRE1α and Prevents ER Stress from Signaling Apoptotic Cell Death. Nat. Commun. 2017, 8, 18. 10.1038/s41467-017-00029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. P. K.; Dey M.; Neculai D.; Cao C.; Dever T. E.; Sicheri F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W.; Welihinda A. A.; Kaufman R. J. A Stress Response Pathway from the Endoplasmic Reticulum to the Nucleus Requires a Novel Bifunctional Protein Kinase/Endoribonuclease (Ire1p) in Mammalian Cells. Genes Dev. 1998, 12, 1812–1824. 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A.; Walter P. Structural Basis of the Unfolded Protein Response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- Korennykh A. V.; Egea P. F.; Korostelev A. A.; Finer-Moore J.; Zhang C.; Shokat K. M.; Stroud R. M.; Walter P. The Unfolded Protein Response Signals through High-Order Assembly of Ire1. Nature 2009, 457, 687–693. 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington P. E.; Biswas K.; Malwitz D.; Tasker A. S.; Mohr C.; Andrews K. L.; Dellamaggiore K.; Kendall R.; Beckmann H.; Jaeckel P.; et al. Unfolded Protein Response in Cancer: IRE1α Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med. Chem. Lett. 2015, 6, 68–72. 10.1021/ml500315b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches M.; Duffy N. M.; Talukdar M.; Thevakumaran N.; Chiovitti D.; Canny M. D.; Lee K.; Kurinov I.; Uehling D.; Al-Awar R.; et al. Structure and Mechanism of Action of the Hydroxy–aryl–aldehyde Class of IRE1 Endoribonuclease Inhibitors. Nat. Commun. 2014, 5, 4202. 10.1038/ncomms5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman H. C.; Tong M.; Wang L.; Meza-Acevedo R.; Gobillot T. A.; Lebedev I.; Gliedt M. J.; Hari S. B.; Mitra A. K.; Backes B. J.; et al. Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands. ACS Chem. Biol. 2016, 11, 2195–2205. 10.1021/acschembio.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso V.; Sturlese M.; Cuzzolin A.; Moro S. Combining Self- and Cross-Docking as Benchmark Tools: The Performance of DockBench in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 251–264. 10.1007/s10822-017-0051-4. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Banks J. L.; Beard H. S.; Cao Y.; Cho A. E.; Damm W.; Farid R.; Felts A. K.; Halgren T. A.; Mainz D. T.; Maple J. R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2009, 26, 1752–1780. 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A. V.; Korostelev A. A.; Egea P. F.; Finer-Moore J.; Stroud R. M.; Zhang C.; Shokat K. M.; Walter P. Structural and Functional Basis for RNA Cleavage by Ire1. BMC Biol. 2011, 9, 47. 10.1186/1741-7007-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A. V.; Egea P. F.; Korostelev A. A.; Finer-moore J.; Stroud R. M.; Zhang C.; Shokat K. M.; Walter P. Cofactor-Mediated Conformational Control in the Bifunctional Kinase/RNase Ire1. BMC Biol. 2011, 9, 48. 10.1186/1741-7007-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R. L.; Zhang Y.; Lee K. P. K.; Harding H. P.; Haynes C. M.; Price J.; Sicheri F.; Ron D. Flavonol Activation Defines an Unanticipated Ligand-Binding Site in the Kinase-RNase Domain of IRE1. Mol. Cell 2010, 38, 291–304. 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R. L.; Zhang Y.; Lee K. P. K.; Harding H. P.; Haynes C. M.; Price J.; Sicheri F.; Ron D. Flavonol Activation Defines an Unanticipated Ligand-Binding Site in the Kinase-RNase Domain of IRE1. Mol. Cell 2010, 38, 291–304. 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A. V.; Egea P. F.; Korostelev A. A.; Finer-Moore J.; Zhang C.; Shokat K. M.; Stroud R. M.; Walter P. The Unfolded Protein Response Signals through High-Order Assembly of Ire1. Nature 2009, 457, 687–693. 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle J. J.; Finer-Moore J. S.; Papa F. R.; Stroud R. M.; Walter P. On the Mechanism of Sensing Unfolded Protein in the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18773–18784. 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concha N. O.; Smallwood A.; Bonnette W.; Totoritis R.; Zhang G.; Federowicz K.; Yang J.; Qi H.; Chen S.; Campobasso N.; et al. Long-Range Inhibitor-Induced Conformational Regulation of Human IRE1 Endoribonuclease Activity. Mol. Pharmacol. 2015, 88, 1011–1023. 10.1124/mol.115.100917. [DOI] [PubMed] [Google Scholar]
- Concha N. O.; Smallwood A.; Bonnette W.; Totoritis R.; Zhang G.; Federowicz K.; Yang J.; Qi H.; Chen S.; Campobasso N.; et al. Long-Range Inhibitor-Induced Conformational Regulation of Human IRE1 Endoribonuclease Activity. Mol. Pharmacol. 2015, 88, 1011–1023. 10.1124/mol.115.100917. [DOI] [PubMed] [Google Scholar]
- Joshi A.; Newbatt Y.; McAndrew P. C.; Stubbs M.; Burke R.; Richards M. W.; Bhatia C.; Caldwell J. J.; McHardy T.; Collins I.; et al. Molecular Mechanisms of Human IRE1 Activation through Dimerization and Ligand Binding. OncoTarget 2015, 6, 13019–13035. 10.18632/oncotarget.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M. M. U.; Bagratuni T.; Davenport E. L.; Nowak P. R.; Silva-Santisteban M. C.; Hardcastle A.; McAndrews C.; Rowlands M. G.; Morgan G. J.; Aherne W.; et al. Structure of the Ire1 Autophosphorylation Complex and Implications for the Unfolded Protein Response. EMBO J. 2011, 30, 894–905. 10.1038/emboj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Liu C. Y.; Back S. H.; Clark R. L.; Peisach D.; Xu Z.; Kaufman R. J. The Crystal Structure of Human IRE1 Luminal Domain Reveals a Conserved Dimerization Interface Required for Activation of the Unfolded Protein Response. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14343–14348. 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2015-4: LigPrep, Schrödinger, LLC, New York, NY, 2015.
- Schrödinger Release 2015-4: Maestro, Schrödinger, LLC, New York, NY, 2015.
- Molecular Operating Environment (MOE), 2015.10: Chemical Computing Group ULC; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015. [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Vigers G. P. A.; Rizzi J. P. Multiple Active Site Corrections for Docking and Virtual Screening. J. Med. Chem. 2004, 47, 80–89. 10.1021/jm030161o. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Shoichet B. K. Information Decay in Molecular Docking Screens against Holo , Apo , and Modeled Conformations of Enzymes. J. Med. Chem. 2003, 46, 2895–2907. 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- Saenz-Méndez P.; Eriksson M.; Eriksson L. A. Ligand Selectivity between the ADP-Ribosylating Toxins: An Inverse- Docking Study for Multitarget Drug Discovery. ACS Omega 2017, 2, 1710–1719. 10.1021/acsomega.7b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-C. Beware of Docking!. Trends Pharmacol. Sci. 2015, 36, 78–95. 10.1016/j.tips.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. Binding of small-molecule ligands to proteins: ″what you see″ is not always ″what you get″. Struct. 2009, 17, 489–498. 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.