

Abstract
Fundamental research on Parkinson's disease (PD) most often focuses on the ability of α‐synuclein (aS) to form oligomers and amyloids, and how such species promote brain cell death. However, there are indications that aS also plays a gene‐regulatory role in the cell nucleus. Here, the interaction between monomeric aS and DNA in vitro has been investigated with single‐molecule techniques. Using a nanofluidic channel system, it was discovered that aS binds to DNA and by studying the DNA–protein complexes at different confinements we determined that aS binding increases the persistence length of DNA from 70 to 90 nm at high coverage. By atomic force microscopy it was revealed that at low protein‐to‐DNA ratio, the aS binding occurs as small protein clusters scattered along the DNA; at high protein‐to‐DNA ratio, the DNA is fully covered by protein. As DNA‐aS interactions may play roles in PD, it is of importance to characterize biophysical properties of such complexes in detail.
Keywords: biochemistry, DNA, nanofluidics, proteins, synuclein
Introduction
Parkinson's disease (PD) is a neurodegenerative disease that is estimated to affect 2 % of the population older than 60 years.1 α‐synuclein (aS) is a 140‐residue protein whose assembly process into amyloid fibers is directly related to PD.2 The consequences of amyloid fiber formation are associated with several human neurodegenerative diseases in addition to PD, such as Alzheimer's, ALS, type 2 diabetes and Huntington's disease.3 Despite the fact that amyloid fibers are formed by a unique protein in each disease, they all have a common cross‐β conformation; that is, β‐sheets that are packed perpendicular to the fiber axis. In PD, the aS fibers accumulate in cytosolic inclusions, called Lewy bodies, that are found in dopamine neurons in the brains of patients along with loss of such neurons in the substantia nigra.4 The function of aS in cells has been suggested to involve synaptic vesicle release and trafficking5 and, indeed, aS is present at presynaptic nerve terminals6 near synaptic vesicles.7 Although intrinsically disordered in solution, the N‐terminus of aS forms an amphipathic α‐helix upon membrane binding.8
Mostly overlooked, aS has been reported to be present in the cell nucleus, in addition to the cytoplasm, and it appears to travel in and out in a dynamic fashion.9 Notably, the fraction of aS in the nucleus increases upon chemically stimulated oxidative stress10 and it was reported to modulate transcription of the master mitochondrial transcription activator, PGC1alpha.10 aS‐induced changes in transcription of PGC1alpha negatively affected mitochondrial morphology and function. Moreover, chromatin‐bound oxidized aS was found to cause double‐strand breaks in neuronal DNA that resulted in cell death11 and DNA has been reported to stimulate aS amyloid formation in vitro.12 These studies clearly emphasize that aS may have additional roles in PD that are related to nuclear gene regulation. However, to date, there is only limited biophysical knowledge of the interaction between monomeric aS and DNA.
Here we used nanofluidics, in combination with other biophysical techniques, to characterize the interactions between monomeric aS and DNA on the single DNA molecule level. Nanofluidic channels have emerged as a novel method to study DNA–protein interactions on the single DNA molecule level.13 When DNA is confined in nanofluidic channels it has to stretch to accommodate the mismatch between the relaxed size of the DNA and the small channel size. The degree of stretching is sensitive to the surrounding environment, and to molecules interacting with DNA. Therefore, this method is well suited for studying proteins that cause conformational changes in the DNA, and protein‐induced DNA stiffening14 as well as compaction15 have been reported.
Results and Discussion
To address putative structural changes in aS and DNA when they interact, we first employed bulk solution experiments. We found that the far‐UV circular dichroism (CD) signal of the protein did not change significantly upon mixing monomeric aS with DNA (even upon adding large excess of DNA to ensure all protein is bound). Thus, the disordered structure of aS remains also in the presence of DNA (Figure S1A, Supporting Information). Our CD results are in agreement with previous data on aS interactions with genomic DNA, although larger structural changes in aS were found when the protein is bound to super‐coiled or single‐stranded DNA.12a, 16 To probe how DNA is affected upon aS binding, we turned to flow linear dichroism (LD) which is a bulk method to probe polarized absorption of aligned chromophores.17 DNA molecules align with the flow in a Couette cell, resulting in negative LD where the bases absorb as the base pairs are arranged perpendicular to the flow orientation. LD data for DNA alone and for DNA mixed with aS (using excess protein to ensure all DNA will be bound to protein) show that although there are indications of aS binding in the 210 nm absorption range (where the protein absorbs), there is no effect on the LD of the DNA bases (260 nm range) (Figure S1B). This implies that aS binding to DNA does not alter the ability of the DNA to align with the flow which in turn means that the physical properties of the DNA base pairs are not significantly affected. To obtain deeper insights on the DNA–aS interaction we turned to single DNA molecule methods.
We assessed the physical properties of single DNA molecules upon interactions with monomeric aS in nanofluidic channels. For this we mixed 48.5 kbp λ‐DNA (5 μm base pairs) with increasing concentrations of aS (up to 40 μm; reaching 8:1 aS‐to‐base pair ratio) and determined the extension of individual DNA–protein complexes in a channel with a 100×150 nm2 geometry. Since aS is not fluorescently labeled, we used the dye YOYO‐1, a well characterized fluorophore that binds to DNA by bis‐intercalating its aromatic subunits between DNA base pairs, to visualize the DNA.18 From the extension data, it is clear that the DNA molecule is stretched in the presence of aS, and that the effect increases with increasing aS concentration (Figure 1). The increase in DNA extension is about 30 % at the highest protein concentration used (40 μm). The highest aS concentration is eight times higher than the base‐pair concentration, suggesting that the binding of aS to DNA is weak. When performed at higher ionic strength (1×TE buffer with 30 mm NaCl instead of only 1×TE buffer, Figure 2 A) in the narrow channels, there is very little increase in extension of the DNA even at the highest protein concentration, suggesting that binding of aS to DNA is to a large extent driven by electrostatic interactions. The N‐terminal part of aS is positively charged and this part of aS (which also is the part that interacts with lipid vesicles) probably interacts with the negative DNA backbone. It is however worth noting that purely multivalent cationic interactions (aS carries 15 lysine residues)19 would lead to compaction of DNA, which would be directly visible in the channels. This is not the case for aS, at least at the concentrations investigated here, implying that aS binding is also governed by other forces, such as hydrophobic and/or specific interactions.
Figure 1.
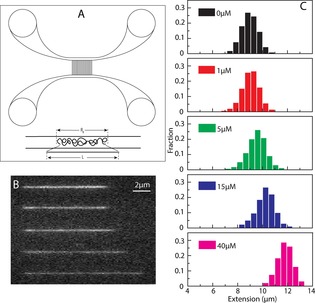
(A) Schematic illustration of the nanofluidic chip design (left). The channel system comprises pairs of micro‐channels, spanned by an array of straight nanochannels. The cartoon shows DNA polymers confined inside a nanochannel. DNA will be partially stretched along the nanochannel, with an extension R∥, shorter than its contour length L. (B) Montage of fluorescence images of λ‐DNA with aS. From up to bottom: 0, 1, 5, 15 and 40 μm aS. The YOYO:bp ratio is 1:50. (C) Extension distribution of λ‐DNA molecules in 150×100 nm2 nanochannels at different concentrations of aS.
Figure 2.
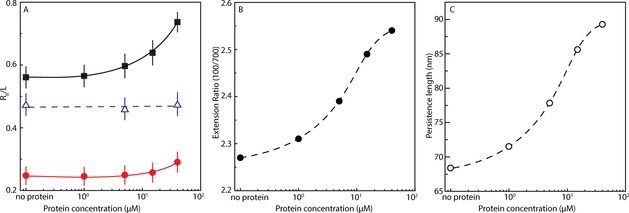
(A) Relative extension R∥ L−1 of λ‐DNA in 1×TE buffer in 150×700 nm2 channels (•, red) and 150×100 nm2 channels (▪, black), and λ‐DNA in 1×TE buffer with 30 mm NaCl in 150×100 nm2 channels (Δ, blue), versus the α‐synuclein concentration. (B) The ratio between the extension of λ‐DNA molecules in 150×100 nm2 and 150×700 nm2 channels versus aS concentration. (C) Fitted data for DNA persistence length versus aS concentration. The dashed and solid curves are drawn as an aid to the eye.
The observed homogeneous YOYO‐1 emission along the DNA upon aS interaction suggests that there is no large difference of aS preference between AT‐ and GC‐rich regions of the DNA,20 since λ‐DNA has one half of the molecule rich in AT and the other rich in GC basepairs.
We also performed the experiments in a wider channel (700×150 nm2), where the DNA is not as stretched as in the narrower channel. The idea was to compare the increase in extension when confinement changes and to use this parameter to determine the physical properties of the protein‐bound DNA.15a We found the effect on DNA upon aS binding to follow the same trend in the wider channel, that is, the DNA is extended with increasing concentration of aS, but the magnitude of the increase in DNA extension is smaller (Figure 2). This is reflected by an increase in the ratio of the extension at the two confinements as protein is added. Werner et al. developed a one‐parameter theoretical prediction for estimating the physical properties of DNA for square nanochannels in this size range.21 In the Supporting Information, we demonstrate how this prediction can be used to estimate the persistence length of DNA, with and without aS bound, by approximating the rectangular channel size by its geometric mean (Supporting Information data analysis, Figures S2 and S3). The conclusion from this analysis is that the persistence length gradually increases from around 70 nm for naked DNA to around 90 nm for DNA with 40 μm aS added (Figure 2). Thus, the DNA is stiffened by aS binding. However, the effect is slight when compared to, for example, the filament‐forming protein RecA that stiffens the DNA dramatically14 or the bottle‐brush diblock polypeptide used by Zhang et al. that resulted in an approximately four fold increase in persistence length.22
To complement the observations made using the nanofluidic channels, we turned to atomic force microscopy (AFM) imaging of DNA (1 kbp fragments; 5 μm base pairs) in the presence of aS (Figure 3). The AFM data demonstrate that aS interacts with DNA in small ‘clusters’ scattered on the DNA at low protein concentration (0.1 μm). Thus, AFM clearly demonstrates protein binding at low aS concentrations where little effect is found on DNA extension in the nanochannels. On the much larger length scale visualized in the nanochannel experiments, the AFM‐detected small protein clusters cannot be distinguished. Instead, the even YOYO‐1 emission along the 48.5 kbp DNA indicates homogeneous protein coverage at all protein concentrations when analyzed with this spatial resolution (Figure 1). AFM measurements at higher concentrations of aS (40 μm) show that the DNA becomes fully coated with protein. The height of the DNA‐protein complex is the same at high aS concentration as at low aS concentration (approx. 1.5 nm; Figure 3). The estimated height of the protein when bound to DNA is roughly 1 nm (1.5 nm for the complex minus 0.5 nm for bare DNA), which is much smaller (and may correspond to a single polypeptide) than the cross‐section of aS amyloid fibers, which is around 7–9 nm when probed by AFM.23 Thus, aS binding to DNA takes place next to already‐bound aS but not on top of pre‐bound aS.
Figure 3.
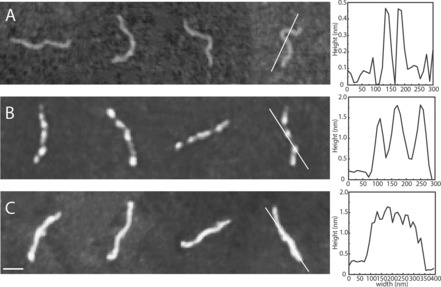
Tapping mode atomic force microscopy images of DNA (1 kb, 5 μm base pairs) with aS at a concentration of (A) 0 μm, (B) 0.1 μm and (C) 40 μm. The scale bar is 100 nm. The line in each panel corresponds to the height profile of the molecule shown to the right. Mica (A) and silica (B and C) surfaces were used (see Materials and Methods for details).
Notably, the AFM data also demonstrate that aS does not form oligomers or amyloid fibers under these conditions as those would have been directly visible in the images. Without the presence of beads and agitation (typical conditions for aS aggregation experiments), aS samples can be incubated for days without amyloid formation; the addition of DNA to such samples did not change this behavior. Thus, aS remains monomeric in our interaction experiments although smaller assemblies cannot be excluded. As the protein remains unstructured upon DNA binding, according to far‐UV CD, amyloid‐like β‐strand interactions between nearby aS polypeptides when bound to the DNA are excluded.
To ensure that YOYO‐1 does not affect aS binding to DNA or vice versa, we compared the YOYO‐1 fluorescence in the presence of aS alone to that in the presence of DNA alone as well as when in a mixture of aS and DNA. It appears that YOYO‐1 can interact weakly with aS alone as the YOYO‐1 fluorescence was somewhat increased in the presence of aS (about 10 % of the emission found upon DNA binding). For the mixture of YOYO‐1, aS and DNA, the fluorescence corresponds to the sum of the emission for YOYO‐1 with DNA alone and with aS alone (Figure S4); thus, there is no competition for YOYO‐1 between protein and DNA nor a competition for DNA between YOYO‐1 and aS at the conditions studied.
Conclusion
Using nanofluidic channels, we have discovered that aS binds to DNA, and upon protein binding, the DNA persistence length increases by 30 %. Upon complex formation, there is no structural change in aS and the DNA base pairing is not distorted. The latter is also evident from the fact that YOYO‐1 can intercalate between the base pairs also when aS is bound. Using an analogous approach, thorough single molecule experiments of DNA binding of the bacterial nucleoid proteins H‐NS, Hfq, and HU have been reported.15b, 22, 24 These proteins play pivotal roles in compaction, transcription, and replication of the bacterial genome. As observed here for aS‐DNA, H‐NS binding to DNA resulted in elongated DNA molecules with increased (doubled) persistence length.24b In contrast, HU and Hfq interactions induced contraction of the DNA that was proposed to involve protein‐mediated bridging of distal DNA segments.15b, 24a We observed no compaction of DNA by aS at the protein concentrations studied, although AFM show complete coverage of DNA with aS at the highest aS concentration. This may be surprising considering the high positive charge of aS at pH 7.4, but agrees with a similar study on a designed cationic polypeptide.22
Characterization of physical properties of aS‐DNA complexes are of importance as aS has been reported to act as a transcription modulator10 and inducer of double‐stranded DNA breaks upon oxidative stress11 in the nucleus of cells, but little is known about underlying molecular principles. Notably, in addition to aS, proteins and peptides involved in promoting other amyloid disorders, such Alzheimer's and prion diseases, seem to also have DNA‐binding properties.25 For example, the amyloid‐β peptide that forms amyloids in Alzheimer's was proposed to also act as a repressor of transcription from certain promoters.26 This suggests that DNA interactions may be a common (but mostly unnoticed) property of proteins associated with neurodegenerative disorders such that, in addition to toxic amyloid formation, these proteins may also alter expression profiles of disease‐modifying genes. Single DNA molecule techniques, such as the approach presented here, may be useful to characterize putative DNA binding of many other proteins involved in neurodegeneration.
Experimental Section
aS Preparation
Monomeric aS was prepared by transforming the wild‐type plasmid into BL21 (DE3) (Novagen) cells. Cells were grown in LB medium with 100 μg mL−1 carbenicillin at 37 0C. IPTG (isopropyl b‐d‐1‐thiogalactopyranoside, 1 mm) was added when OD600 reached 0.6 and the cells were harvested after overnight incubation at 25 °C. Cells were lysed by sonication in ice cold 20 mm Tris‐HCl buffer pH 8.0 with protease inhibitor cocktail (Roche), followed by treatment with universal nuclease (Pierce) for 15 minutes at room temperature. The lysate was then heated at 90 °C for 10 minutes and centrifuged for 30 minutes at 15 000 g. The supernatant was filtered, loaded onto pre‐equilibrated 5 mL HiTrap Q FF anion exchange column (GE Healthcare) and eluted using a linear NaCl gradient from 0–1 m in 20 mm Tris‐HCl buffer pH 8.0. Fractions containing α‐synuclein were pooled and concentrated with Ultra‐15 Ultracel 10 K centrifugal filter devices (Millipore). The concentrate was loaded onto Hiload 16/600 Superdex 75 pg column (GE Healthcare) and eluted with 20 mm Tris‐sulphate buffer pH 7.4. The sample purity was confirmed by a single band on SDS‐PAGE gel and a single elution peak in size exclusion chromatography. Prior to the experiments, the protein was loaded on a gel filtration column (Superdex 75 10/300 GL, GE Healthcare) and eluted with Tris‐EDTA buffer (10 mm Tris‐HCl; 1 mm EDTA, pH 7.4, Sigma–Aldrich). The protein concentration was determined using ϵ 280=5960 m −1 cm−1. In all experiments performed, aS was monomeric at the start and in no case were aS oligomers or amyloids observed throughout the measurements.
Circular dichroism
Far‐UV CD spectra of aS in Tris‐EDTA buffer (10 mm Tris‐HCl; 1 mm EDTA, pH 7.4) were recorded in the absence and presence of DNA using a Jasco J‐810 spectropolarimeter in 1 mm quartz cuvette with 1 nm steps, a bandwidth of 1 nm, a scanning speed of 50 nm min−1 and a response of 1 s. The molar ratios of protein:DNA base pair were: 1:0; 1:1; 1:2; 1:8; 1:25; 1:50. The spectra were averaged eight times and baseline subtracted. The CD data sets are reported as mean residue molar ellipticity (degrees M−1 m−1).
Linear dichroism
LD measurements were performed on a Chirascan CD spectropolarimeter. All spectra were recorded between 200 and 400 nm in 1 nm increments at a scan speed of 85 nm min−1 and a bandwidth of 1 nm. The alignment of the DNA was achieved by a custom‐made outer‐cylinder‐rotation Couette flow cell with a path length of 1 mm. The shear rate was 3100 s−1. At least three data accumulations were made to generate an average for each measurement. Baselines at zero shear gradient were collected and subtracted from all spectra.
Nanofluidics
DNA from phage lambda (λ‐DNA, Roche) was mixed with aS and then stained with YOYO‐1 (Invitrogen) at a ratio of 1 dye molecule per 25 base pairs. The mixture was incubated at 4 °C for at least 4 hours. The complexes were introduced into the nanofluidic system and equilibrated for 60 s before image capture. The DNA concentration was 5 μM (basepairs) in all samples. 3 % (v/v) β‐mercaptoethanol (Sigma–Aldrich) was added as an oxygen scavenger to suppress oxygen radical induced photo‐damage of the DNA. The buffer used was 1×TE (10 mm Tris and 1 mm EDTA) and 1×TE with 30 mm NaCl (pH 7.5).
The single DNA molecule experiments were performed in nanochannels with dimensions of 150×100 nm2 and 150×700 nm2. The devices were fabricated using advanced nanofabrication described elsewhere.27 The channel system consists of a pair of feeding channels (micro‐size), spanned by a set of parallel nanochannels. Figure 1 A shows a schematic illustration of the nanofluidic chip. The sample is loaded into the channel system from one of the four reservoirs that are connected to the feeding channels and moved into the nanochannels by pressure driven (N2) flow. To avoid non‐specific binding of protein to the negatively charged channel walls, the channels were coated with a lipid bilayer comprising 99 % 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC, Avanti) and 1 % N‐(fluorescein‐5‐thiocarbamoyl)‐1,2‐dihexadecanoyl‐sn‐Glycero‐3‐phosphoethanolamine, triethyl ammonium salt (fluorescein‐DHPE, Invitrogen) prior to the experiment. The coating procedure is described elsewhere.28
The DNA and DNA‐protein complexes were imaged using an epifluorescence microscope (Zeiss AxioObserver.Z1) equipped with a Photometrics Evolve EMCCD camera, a 63X oil immersion TIRF objective (NA=1.46) and a 1.6X optovar from Zeiss. Using the microscopy imaging software ZEN, 100 subsequent images were recorded with an exposure time of 100 ms. Data analysis was performed using a custom‐written MATLAB‐based software. Microscopy image stacks were used as input to the program. Images were first binarized by thresholding with a global average plus one‐fold of standard deviation. Taking advantage of the high contrast of the YOYO‐stained DNA fluorescence images, regions with higher brightness are directly considered as DNA objects without additional image filtering. Finally, the lengths of the DNA molecules were extracted by identifying the longest axis of the objects.
Atomic force microscopy
For the AFM studies, NoLimits 1000 bp DNA (ThermoFisher Scientific) was used. DNA was mixed with aS at different ratios and incubated at 4 °C for at least 4 hours. 10 μl of each aS‐DNA sample was deposited onto silicon substrates that were prepared freshly as follows. Silicon wafers were initially cleaned with acetone, isopropanol and ultrapure water, dried using a N2 gun and then exposed to O2 plasma at 350 W power for 3 minutes. The sample was left on the silicon surface for 10 min, then flushed with Milli‐Q water followed by drying under a stream of N2 gas. For bare DNA analysis, DNA samples with the addition of 0.2 mm MgCl2 were deposited on mica freshly cleaved in a similar procedure as for the silicon wafers (10 minutes adsorption time, washing with Milli‐Q and drying using N2 gas). The addition of multivalent salts, such as MgCl2, mediates the interaction between DNA and mica but because this interaction is weak, DNA chain statistics are not affected.29 The AFM images were recorded in air, with a Dimension ICON scanning probe microscope (Bruker) operating in peak force tapping mode. Images were acquired in the tapping mode with silicon (Si) probes (force constant of 1.45–15.1 N m−1, resonant frequency 87–230 kHz). The scanning rate was 1 Hz.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Kai Jiang holds a personal grant from the Wenner‐Gren Foundation. PWS and FW both acknowledge funding from the Olle Engqvist Foundation and the Swedish Research Council (PWS no 2015‐3881, FW no. 2015‐5062), and KDD acknowledges funding from the National Institutes of Health (R01‐HG006851). PWS also acknowledges funding from the Knut and Alice Wallenberg Foundation. We thank Ranjeet Kumar (Chalmers) for help with protein production and Yii‐Lih Lin (Chalmers) for the software for analyzing the nanochannel data.
K. Jiang, S. Rocha, A. Westling, S. Kesarimangalam, K. D. Dorfman, P. Wittung-Stafshede, F. Westerlund, Chem. Eur. J. 2018, 24, 15685.
Contributor Information
Prof. Pernilla Wittung‐Stafshede, Email: pernilla.wittung@chalmers.se.
Prof. Fredrik Westerlund, Email: Fredrik.westerlund@chalmers.se
References
- 1. Pringsheim T., Jette N., Frolkis A., Steeves T. D., Mov. Disord. 2014, 29, 1583–1590. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R., Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Gallegos S., Pacheco C., Peters C., Opazo C. M., Aguayo L. G., Frontiers in neuroscience 2015, 9, 59; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Roberts H. L., Brown D. R., Biomolecules 2015, 5, 282–305; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Anandhan A., Rodriguez-Rocha H., Bohovych I., Griggs A. M., Zavala-Flores L., Reyes-Reyes E. M., Seravalli J., Stanciu L. A., Lee J., Rochet J. C., Khalimonchuk O., Franco R., Neurobiol. Dis. 2015, 81, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fink A. L., Acc. Chem. Res. 2006, 39, 628–634. [DOI] [PubMed] [Google Scholar]
- 4. Galvin J. E., Lee V. M., Schmidt M. L., Tu P. H., Iwatsubo T., Trojanowski J. Q., Adv. Neurol. 1999, 80, 313–324. [PubMed] [Google Scholar]
- 5.
- 5a. Dev K. K., Hofele K., Barbieri S., Buchman V. L., van der Putten H., Neuropharmacology 2003, 45, 14–44; [DOI] [PubMed] [Google Scholar]
- 5b. Lassen L. B., Reimer L., Ferreira N., Betzer C., Jensen P. H., Brain Pathol. 2016, 26, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Maroteaux L., Campanelli J. T., Scheller R. H., J. Neurosci. 1988, 8, 2804–2815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H. A., Kittel A., Saitoh T., Neuron 1995, 14, 467–475. [DOI] [PubMed] [Google Scholar]
- 7. Cheng F., Vivacqua G., Yu S., J. Chem. Neuroanat. 2011, 42, 242–248. [DOI] [PubMed] [Google Scholar]
- 8. Eliezer D., Kutluay E., R. Bussell, Jr. , Browne G., J. Mol. Biol. 2001, 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Goers J., Manning-Bog A. B., McCormack A. L., Millett I. S., Doniach S., Di Monte D. A., Uversky V. N., Fink A. L., Biochemistry 2003, 42, 8465–8471; [DOI] [PubMed] [Google Scholar]
- 9b. Gonçalves S., Outeiro T. F., Mol. neurobiol. 2013, 47, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siddiqui A., Chinta S. J., Mallajosyula J. K., Rajagopolan S., Hanson I., Rane A., Melov S., Andersen J. K., Free Radical Biol. Med. 2012, 53, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vasquez V., Mitra J., Hegde P. M., Pandey A., Sengupta S., Mitra S., Rao K. S., Hegde M. L., J. Alzheimer's disease: JAD 2017, 60(s1), S133–S150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Hegde M. L., Rao K. S., Arch. Biochem. Biophys. 2007, 464, 57–69; [DOI] [PubMed] [Google Scholar]
- 12b. Cherny D., Hoyer W., Subramaniam V., Jovin T. M., J. mol. biol. 2004, 344, 929–938. [DOI] [PubMed] [Google Scholar]
- 13. Frykholm K., Nyberg L. K., Westerlund F., Integr. Biol. 2017, 9, 650–661. [DOI] [PubMed] [Google Scholar]
- 14. Frykholm K., Alizadehheidari M., Fritzsche J., Wigenius J., Modesti M., Persson F., Westerlund F., Small 2014, 10, 884–887. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Frykholm K., Berntsson R. P., Claesson M., de Battice L., Odegrip R., Stenmark P., Westerlund F., Nucleic Acids Res. 2016, 44, 7219–7227; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Jiang K., Zhang C., Guttula D., Liu F., van Kan J. A., Lavelle C., Kubiak K., Malabirade A., Lapp A., Arluison V., van der Maarel J. R., Nucleic acids res. 2015, 43, 4332–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma K. L., Song L. K., Yuan Y. H., Zhang Y., Yang J. L., Zhu P., Chen N. H., Cell. Mol. Neurobiol. 2014, 34, 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodger A., Norden B., Circular Dichroism & Linear Dichroism, Oxford University Press, Oxford, UK, 1997. [Google Scholar]
- 18. Nyberg L., Persson F., Akerman B., Westerlund F., Nucleic acids res. 2013, 41, e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plotegher N., Bubacco L., Ageing Res. Rev. 2016, 26, 62–71. [DOI] [PubMed] [Google Scholar]
- 20. Müller V., Westerlund F., Lab. Chip 2017, 17, 579–590. [DOI] [PubMed] [Google Scholar]
- 21. Werner E., Cheong G. K., Gupta D., Dorfman K. D., Mehlig B., Phys. Rev. Lett. 2017, 119, 268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C., Hernandez-Garcia A., Jiang K., Gong Z., Guttula D., Ng S. Y., Malar P. P., van Kan J. A., Dai L., Doyle P. S., Vries R., van der Maarel J. R., Nucleic acids res. 2013, 41, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Kiskis J., Horvath I., Wittung-Stafshede P., Rocha S., Q. Rev. Biophys. 2017, 50, e3; [DOI] [PubMed] [Google Scholar]
- 23b. Horvath I., Wittung-Stafshede P., Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Guttula D., Liu F., van Kan J. A., Arluison V., van der Maarel J. R. C., Soft Matter 2018, 14, 2322–2328; [DOI] [PubMed] [Google Scholar]
- 24b. Zhang C., Guttula D., Liu F., Malar P. P., Ng S. Y., Dai L., Doyle P. S., van Kan J. A., van der Maarel J. R., Soft Matter 2013, 9, 9593–9601. [DOI] [PubMed] [Google Scholar]
- 25. Camero S., Benitez M. J., Jimenez J. S., Adv. Protein Chem. Struct. Biol. 2013, 91, 37–63. [DOI] [PubMed] [Google Scholar]
- 26. Barucker C., Harmeier A., Weiske J., Fauler B., Albring K. F., Prokop S., Hildebrand P., Lurz R., Heppner F. L., Huber O., Multhaup G., J. Biol. Chem. 2014, 289, 20182–20191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Persson F., Tegenfeldt J. O., Chem. Soc. rev. 2010, 39, 985–999. [DOI] [PubMed] [Google Scholar]
- 28. Persson F., Fritzsche J., Mir K. U., Modesti M., Westerlund F., Tegenfeldt J. O., Nano Lett. 2012, 12, 2260–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Rivetti C., Guthold M., Bustamante C., J. mol. biol. 1996, 264, 919–932; [DOI] [PubMed] [Google Scholar]
- 29b. Wiggins P. A., van der Heijden T., Moreno-Herrero F., Spakowitz A., Phillips R., Widom J., Dekker C., Nelson P. C., Nat. Nanotechnol. 2006, 1, 137–141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary