

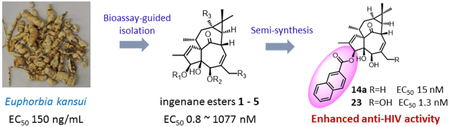
Abstract
Euphorbia kansui showed potent anti-HIV-1 activity during screening of a library composed of plant extracts from Euphorbiaceae and Thymelaeaceae families. Bioassay-guided isolation led to identification of ingenane esters as the active compounds. Further chemical modification resulted in 3-(2-naphthoyl)ingenol (23), which exhibited the most potent anti-HIV-1 activity. Compound 23 also acted as an HIV-1-latency-reversing agent on activation of HIV-1 replication in a latently infected U1 cell model and a T cell latent HIV-1 model JLat-A2.
Keywords: Euphorbia kansui, anti-HIV, latency-reversing agent, ingenane, 3-(2-naphthoyl)ingenol
Graphic Abstract
1. Introduction
Since the first recognized cases emerged in 1981, acquired immunodeficiency syndrome (AIDS) has caused more than 35 million deaths and currently more than 37 million individuals are infected with human immunodeficiency virus (HIV) worldwide [1]. Combination antiretroviral therapy (cART) can effectively control plasma viremia in many patients, although the virus is suppressed rather than truly eradicated, and requires life-long administration to prevent relapse [2-4]. Additionally, cART can be compromised by the unwanted side effects of current medications and by emergence of drug-resistant viruses. Thus, a major goal of current HIV/AIDS therapy continues to be the development of new anti-HIV compounds as well as drug regimens for eradication of the HIV virus.
One of the current strategies for HIV-1 eradication requires pharmacological reactivation of latent viruses, which is believed to make the virus and infected cells susceptible to immune clearance and cytopathic effects of the virus [5]. Recently, diterpenoids from Euphorbiaceae and Thymelaeaceae families have attracted much interest as natural drug candidates for reactivation of a latent virus. Prostratin, a non-tumor promoting phorbol ester from Euphorbiaceae plants, can inhibit HIV-1 infection and induce HIV-1 reactivation in latent infection cell models [6]. As we previously reported, gnidimacrin, a daphnane diterpene from a Thymelaeaceae plant, exhibits an dichotomous activity by reactivating latent HIV-1 and inhibiting nascent HIV-1 infection through selective activation of protein kinase C βI and βII at low picomolar concentrations [7,8].
In continuation of our biological screening program on Euphorbiaceae and Thymelaeaceae plant extracts for discovery of anti-HIV natural products, methanol extracts from the two plant families were evaluated for selective inhibition of HIV replication. The methanolic extract of Euphobia kansui roots showed the strongest anti-HIV activity (EC50 = 150 ng/mL) compared with other species, such as Daphne genkwa, Euphorbia fischeriana, Daphne giraldii, Wikstroemia indica, Euphorbia lathyris, Daphne odora (EC50 = 220 ~ 4500 ng/mL), hence confirming the great potential of natural extracts as a source of anti-HIV agents. In the present study, we report the screening, bioassay-guided isolation, and semi-synthesis of ingenane esters as potent anti-HIV agents. The most promising ingenane ester derivative was selected to be further investigated for its potential as an anti-HIV latency drug candidate.
2. Results and discussion
2.1. Bioassay-guided isolation and identification of ingenane esters as potent anti-HIV agents
The HIV inhibitory MeOH extract of E. kansui roots was subjected to liquid–liquid partitioning to give an active EtOAc fraction with an EC50 value of 110 ng/mL. The EtOAc fraction was fractionated by Diaion HP-20 chromatography to afford four major sub-fractions (E1–E4), among which, the E3-fraction exhibited the lowest IC50 value (28 ng/mL). The E3-fraction was subsequently purified by RP-HPLC to afford five anti-HIV active ingenane diterpenes (EC50 = 0.8 ~ 1076.9 nM). Their chemical structures were identified as 5-O-benzoyl-20-deoxyingenol (1) [9], 3-O-benzoyl-20-deoxyingenol (2) [9], kansuiphorin C (3) [10], ingenol monoacetate (4) [11], and 3-O-(2,3-dimethylbutanoyl)-13-O-dodecanoylingenol (5) [12], by detailed spectroscopic analyses (Figure 1). Meanwhile, three jatrophane diterpenoids, identified as kansuinin B (6) [13], kansuinin C (7) [13], and esulone A (8) [14], which were also isolated from the E3-fraction, showed no anti-HIV activity. Comparison of the anti-HIV data of1–5 indicated that a long-chain ester unit at C-13 led to remarkably increased activity, and acylation at C-3 produced stronger anti-HIV activity than that at C-5 or C-3,5. Furthermore, deacylated compounds (9 and 11), which were obtained by removal of the C-3 ester groups of 1 and 5 by alkaline hydrolysis, showed dramatically weaker anti-HIV activities than 2 and 5, suggesting that the 3-ester group was important for anti-HIV activity. Although 5 showed potent anti-HIV-1 activity [15], the structural similarity to phorbol 12-myristate 13-acetate (PMA), which is a potent tumor promoter and T-cell activator, reduced its impact for further anti-HIV drug development. Thus, 5 was excluded during further investigation.
Figure 1.

Compounds obtained from E. kansui by bioactivity-guided isolation.
2.2. Preparation of ingenane alcohols as starting materials for chemical synthesis of ingenane derivatives
To clarify the importance of the acyl group and discover more potent anti-HIV agents, a library of ingenane ester derivatives was synthesized from ingenane alcohols as starting materials. The total synthesis of ingenane alcohol has been achieved, but the process is complex (over 14 steps) and produces low yields (ca. 1%) [16-18]. In the present study, a simple and direct method was established to obtain ingenane alcohols from the E. kansui extract via a one-step deacylation as depicted in Scheme 1. Since the E. kansui extract contains numerous esters of dodecatrienoyl, benzoyl, and acetyl moieties, which are attached at polyhydroxyl groups on the ingenane skeleton, an LC-MS analysis was applied initially to monitor the simultaneous deacylation process. The ESI-MS fragmentation patterns of 1–5 indicated that 20-deoxyingenol, ingenol, and 13-oxyingenol esters readily produced fragmentation ions at m/z 297, 313, and 329, respectively, by the loss of organic acids (RCOOH) and/or one molecule of H2O (Supplementary data). On the basis of the summarized characteristic fragmentation ions, an LC-MS analysis in positive and negative-full scan modes combined with SIM channels (m/z 297, 313 and 329) was then applied to monitor the deacylation process (Figure 2). Consequently, methanolysis of E. kansui extract with K2CO3 in MeOH at room temperature for 4 h produced 20-deoxyingenol (9, 4%), ingenol (10, 2.5%), and 13-oxyingenol-13-dodecanoate (11, 0.8%) from the E3-fraction.
Scheme 1.

Preparation of starting materials.
Figure 2.

LC-MS monitoring of preparation of starting materials from E3-fraction of E. kansui extract. (a) TIC spectrum of E3-fraction in positive mode; (b) TIC spectrum of E3-fraction after deacylation in positive mode;(c) SIM chromatogram (m/z 297) of E3-fraction in positive mode; (d) SIM chromatograms (m/z 297) of E3-fraction after deacylation in positive mode; (e) SIM chromatogram (m/z 313) of E3-fraction in positive mode; (f) SIM chromatogram (m/z 313) of E3-fraction after deacylation in positive mode; (g) SIM chromatogram (m/z 329) of E3-fraction in positive mode; (h) SIM chromatogram (m/z 329) of E3-fraction after deacylation in positive mode.
2.3. Semi-synthesis of ingenane esters
The accessibility of these semi-synthetic ingenane derivatives increased their appeal as lead compounds for potential anti-HIV drug development. Compounds 9 and 10, which were obtained in higher amounts compared to 11, were selected as starting materials for further modification. Because no systematic anti-HIV studies on 20-deoxyingenane esters have been reported, we focused first on these compounds. 20-Deoxyingenol derivatives with different aliphatic and aromatic esters were synthesized as shown in Scheme 2. Derivatives 12a–14a, 12b–14b, and 18 (19–38% yields) were obtained by acylation of 9 with corresponding acids and DMAP/EDCI at room temperature for 4-48 h in a mixed solvent of super dehydrated DCM and DMF. Next, compounds 15–17 were prepared from 9 by reaction with the respective anhydride in the presence of DMAP and dehydrated pyridine. Due to the low reactivity of angelic anhydride, angeloylation of 9 was accelerated at 40 °C. Angelic anhydride easily undergoes isomerization to tiglic anhydride, which led to a pair of isomers, 3-tigloyl-20-deoxyingenol (16) and 3-angeloyl-20-deoxyingenol (17). The synthesis of ingenol ester derivatives 22 and 23 is depicted in Scheme 3. The acylation reactivity of hydroxyl groups in 10 is in the order of 20-OH > 3-OH ≈ 5-OH. Therefore, to selectively modify the C-3 hydroxyl group of 10, the hydroxyl groups at C-5 and C-20 were firstly protected as the monoacetonide (12) in the presence of p-TsOH.H2O and acetone. Compounds 20 and 21 were produced by selective esterification at the C-3 position of 19 with angelic anhydride and 2-naphthoic acid, respectively. Subsequently, deprotection was easily achieved using 2 M HCl in MeOH at room temperature to give the expected esters (22 and 23) in good yields. All reactions were monitored by LC-MS analyses, and the target compounds were purified by semi-preparative RP-HPLC.
Scheme 2.

Synthesis of 20-deoxyingenol ester derivatives.a
a Reagents and conditions: (i) respective acid, DMAP, EDCI, DMF/DCM, 0 °C→rt, 4-48 h; (ii) respective anhydride, DMAP, pyridine, rt or 40 °C, 24 h.
Scheme 3.

Synthesis of ingenol ester derivatives.a
a Reagents and conditions: (i) p-TsOH.H2O, acetone, rt, 2 h; (ii) angelic anhydride, CS2CO3, MeCN, rt, 4 h; (iii) 2-naphthoic acid, DMAP, EDCI, DMF/DCM, 0 °C→rt, 4 h; (iv) 2M HCl, MeOH, rt, 4 h
2.4. Anti-HIV replication activities
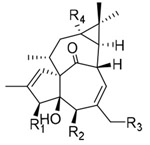
All synthesized ingenane analogues were evaluated for anti-HIV-1 replication activity. As shown in Table 1, 20-deoxyingenol (12a–18) and ingenol (22 and 23) analogues exhibited potent activity with IC50 values ranging from 1.3 to 630 nM. The potencies of the target compounds 14a and 23 were significantly improved by 800- and 8000-fold, as compared to those of the respective starting materials 9 and 10 (IC50 > 1000 nM). With the 20-deoxyingenol analogues, 3-esters exhibited more potent or comparable inhibitory activity against HIV-1 replication than the corresponding 5-esters. In a comparison of compound 2 with 15– 18, benzoate derivative 2 was more potent than 15–18 with elongated aliphatic ester and α,β-unsaturated alkyl ester groups, indicating that an aromatic moiety might enhance the activity. To further investigate the influence of the aromatic moiety, 20-deoxyingenol esters (12a–14b) containing different aromatic moieties were synthesized. The monosubstituted p-methoxybenzoyl esters (12a and 12b) and the trans-cinnamoyl esters (13a and 13b) exhibited decreased HIV-1 inhibitory activity in comparison to 1 and 2. The replacement of the phenyl group by a naphthyl group (14a and 14b) enhanced the inhibitory activity, suggesting that a conjugated aromatic system contributes positively to anti-HIV activity. Likewise, 3-(2-naphthoyl)ingenol (23) exhibited highly increased anti-HIV-1 replication activity with an IC50 value of 1.3 nM, approximately three-fold more potent than the 3-angeloyl ester (22). Derivative 23 was also greater than 10-fold more potent than AZT in the antiviral assays.
Table 1.
Anti-HIV replication activities of compounds in HIV-1 infected MT-4 cell lines.
 |
|||||||
|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 | anti-HIV EC50 (nM) |
cytotoxicity CC50 (μM) |
SI |
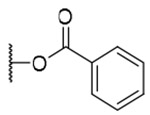
| 1 | OH | 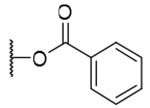 |
H | H | 42.6±14.0 | >9.2 | >216 |
| 2 | 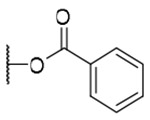 |
OH | H | H | 30.5±10.4 | >9.2 | >302 |
| 3 | 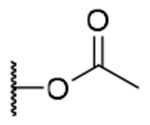 |
 |
H | H | 367.0±94.1 | >8.4 | >23 |
| 4 | OH | OH | 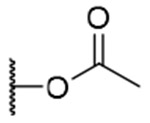 |
H | 1076.9±282.1 | >10.3 | >10 |
| 5 | 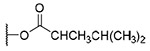 |
OH | OH | 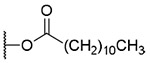 |
0.8±0.3 | >6.2 | >7500 |
| 9 | OH | OH | H | H | >12000 | >12.0 | >10 |
| 10 | OH | OH | OH | H | >11490 | >11.5 | >10 |
| 11 | OH | OH | OH | 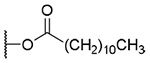 |
33.7±13.9 | >9.2 | >273 |
| 12a | 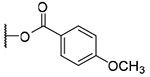 |
OH | H | H | 134.1±37.8 | >8.6 | >64 |
| 12b | OH | 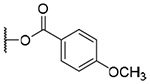 |
H | H | 433.5±144.0 | >8.6 | >20 |
| 13a | 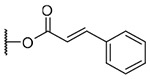 |
OH | H | H | 187.7±50.6 | >8.7 | >45 |
| 13b | OH | 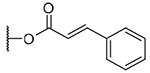 |
H | H | 396.1±105.6 | >8.7 | >22 |
| 14a | 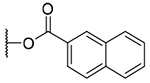 |
OH | H | H | 15.1±4.6 | >8.2 | >543 |
| 14b | OH | 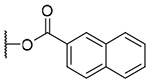 |
H | H | 15.5±5.5 | >8.2 | >179 |
| 15a | 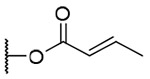 |
OH | H | H | 600±158.3 | >10.0 | >17 |
| 15b | OH | 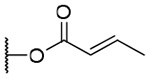 |
H | H | 630±144.0 | >10.0 | >16 |
| 16 | 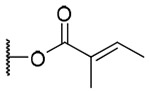 |
OH | H | H | 209.4±56.5 | >9.7 | >46 |
| 17 | 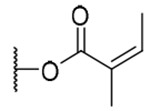 |
OH | H | H | 425.6±117.9 | >9.7 | >22.8 |
| 18 | 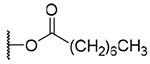 |
OH | H | H | 145.2±51.3 | >8.7 | >59.9 |
| 22 | 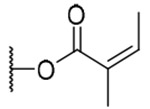 |
OH | OH | H | 4.5±1.9 | >9.3 | >2067 |
| 23 | 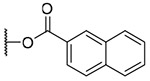 |
OH | OH | H | 1.3±0.5 | >8.0 | >6153 |
| AZT | 19.1±8.6 | >0.4 | >21 | ||||
SI, selectivity index, calculated as CC50/EC50.
2.5. Effects of compounds 22 and 23 on latent HIV-1 activation
Because compounds 22 and 23 both showed potent anti-HIV activity, we next evaluated whether they could reactivate latent HIV. For this purpose, the latently infected U1 cell line was used as an in vitro cell model for HIV-1 latent infection. The cells were treated with 22 or 23 at various concentrations for three days. In agreement with previous reports [19,20], compound 22 effectively reactivated latent HIV in vitro with an EC50 value 4.2 nM. Meanwhile, compound 23 caused marked reactivation of latent HIV-1 with relatively low cellular toxicity (EC50 2.4 nM, CC50 > 2.0 μM), approximately 2-fold and 100-fold higher than those of 22 and prostratin [8].
In addition to the U1 monocytic cell model, compounds 22 and 23 were also tested in a T cell latent HIV-1 model JLat-A2 [21]. Compounds 22 and 23 exhibited comparable potency in activation of green fluorescent protein (GFP) expression at both 10 and 50 nM. This result supports the notion that the ingenol derivatives can potently activate latent HIV-1 and may have potential to be developed as promising agents against latent HIV-1 infection.
3. Conclusion
Ingenane esters were obtained as the major anti-HIV constituents from the methanolic extract of Euphobia kansui roots by means of bioassay-guided isolation. A preliminary structure-activity relationship (SAR) comparison indicated that the ester group plays an important role in the inhibitory activity against HIV-1. Based on this SAR result, various ingenane ester derivatives were subsequently prepared from 20-deoxyingenol (9) and ingenol (10), which were obtained by a one-step deacylation from E. kansui extract. Among these analogues, derivative 23, 3-(2-naphthoyl)ingenol, was identified as the most promising candidate for development of ingenane diterpenoids as new effective anti-HIV agents that can inhibit HIV-1 infection and reactivate latent HIV-1.
4. Experimental section
4.1. Instrumentation and reagents
Optical rotations were measured on a JASCO P-2200 polarimeter in a 0.5-dm cell. The UV spectra were obtained with a Shimadzu UV-160 spectrophotometer. The IR spectra were measured on a JASCO FT/IR-4100 Fourier transform infrared spectrometer by the KBr disk method. The 1H and 13C NMR spectra were measured on a JEOL ECA-500 spectrometer with the deuterated solvent as the internal reference, and the chemical shifts are expressed in δ (ppm). HRFABMS and HRESITOFMS were conducted using a JEOL JMS-700 MStation and a JEOL JMST100LP AccuTOF LC-plus mass spectrometer, respectively. Diaion HP-20 (Mitsubishi Chemical Corporation, Tokyo, Japan) was used for column chromatography. RP-HPLC was performed on a Waters 515 HPLC pump, equipped with a Shodex RI-101 Differential Refractometer detector and a JASCO UV-970 intelligent UV/VIS detector. An RP-C18 silica gel column (YMC-Pack Pro C18, 150×20 mm) was used at a flow rate of 5.0 mL/min. Sep-Pak C18 and Sep-Pak silica cartridges were purchased from Waters (Milford, MA, USA). The purity of the tested compounds (>95%) was confirmed by HPLC-PDA analysis and 1H-NMR spectroscopic analysis. LC-MS analysis was conducted on a Shimadzu LCMS-8040 Triple Quadrupole LC/MS/MS Mass Spectrometer. Solvents for LC-MS analysis, and super dehydrated solvent (water in Max. 0.001%) and dehydrated solvent (water in Max. 0.005%) for synthesis were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
4.2. Plant material
The roots of E. Kansui, originated in Shanxi Province, P.R. China, were purchased in April 2015 and identified by Anhua Wang (Shenyang Pharmaceutical University). The voucher specimens (TE-E04) were deposited at the Department of Pharmacognosy, Faculty of Pharmaceutical Sciences, Toho University, Japan.
4.3. Extraction and isolation
Roots of E. kansui (950.0 g) were cut into small pieces and pulverized into a coarse powder, then the dried powders were extracted ultrasonically with MeOH (4 L × 3 h, 5 times). The methanolic extract was concentrated (28.5 g), suspended in H2O, and then partitioned with EtOAc. The EtOAc layer (17.6 g), which exhibited strong anti-HIV activity (EC50 = 150 ng/mL), was applied to an Diaion HP-20 column and eluted with a step gradient of MeOH-H2O (from 5:5 to 10:0, v/v), finally with acetone to afford four fractions (E1– E4). Fraction E3 showed the most potent anti-HIV activity (EC50 = 28 ng/mL). A part of fraction E3 (2.0 g / 4.0 g) was chromatographed by RP-HPLC with a gradient of MeOH–H2O to yield eleven subfractions (A1– A11). Fraction A10 (123 mg) was purified by RP-HPLC (100% CH3CN) to give 5 (9 mg). Fraction A5 (207 mg) was fractionated by RP-HPLC (75% CH3CN) to give 1 (19 mg), 2 (4 mg), and 3 (24 mg). Compounds 4 (2 mg), 6 (13 mg), 7 (4 mg) and 8 (4 mg) were isolated by RP-HPLC (65% CH3CN) from fraction A2 (137 mg).
4.4. Preparation of starting materials
A part of E3-fraction (0.8 g) from E. kansui extract was mixed with a solution of K2CO3 (160 mg in MeOH, 160 mL), and stirred for 4 h at room temperature. An LC-MS analysis in positive and negative full scan modes combined with SIM channels (m/z 329, 313, 297) was applied to monitor the deacylation process. The resulted deacylated extract was then subjected to Diaion HP-20 column chromatography, eluting with H2O, followed MeOH. The MeOH fraction was separated by RP-HPLC (MeCN-H2O, 50:50, 40:60 and 90:10) to yield 9 (32 mg), 10 (20 mg), and 11 (7 mg), respectively.
4.5. General procedure for preparation of 12a–14a, 12b–14b and 18
To a stirred solution of 9 (15 μmol) in a mixed solvent of super dehydrated DMF (0.5 mL) and DCM (2.5 mL) was added an appropriate acid and DMAP, and then chilled to 0 °C. A solution of EDCI (5 mg, 25 μmol) dissolved in super dehydrated DCM (0.5 mL) was added dropwise, and the reaction mixture was stirred at room temperature for 4–48 h under argon protection. After removal of solvent in vacuo, the residue was purified by RP-HPLC to give desired compounds.
3-(p-Methoxybenzoyl)-20-deoxyingenol (12a) and 5-(p-methoxybenzoyl)-20-deoxyingenol (12b). Compounds 12a and 12b were obtained from reaction with p-anisic acid (11 mg, 71 μmol) and DMAP (10 mg, 82 μmol) for 48 h. The residue was purified by RP-HPLC (MeCN-H2O, 75:25, 5 mL/min) to give 12a (2.1 mg, 30.0%, tR = 27.6 min) and 12b (1.7 mg, 24.4%, tR = 24.0 min). 12a: colorless oil; [α]D25 +90.0 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (4.80), 257 (4.50) nm; IR (KBr) max: 2928, 1719, 1606, 1444, 1510, 1460, 1259, 1168, 1100, 1027, 759 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.10 (1H, q, J = 1.4 Hz, H-1), 5.61 (1H, s, H-3), 3.72 (1H, brs, H-5), 5.75 (1H, dq, J = 4.8, 1.5 Hz, H-7), 4.01 (1H, ddq, J = 11.7, 4.8, 1.7 Hz, H-8), 2.51 (1H, m, H-11), 2.24 (1H, ddd, J = 15.4, 8.9, 2.8 Hz, H2-12a), 1.76 (1H, ddd, J = 15.4, 6.6, 5.2 Hz, H2-12b), 0.67 (1H, ddd, J = 8.9, 8.3, 6.6 Hz, H-13), 0.92 (1H, dd, J = 11.7, 8.3 Hz, H-14), 1.03 (3H, s, H3-16), 1.05 (3H, s, H3-17), 1.02 (3H, d, J = 6.6 Hz, H3-18), 1.81 (3H, d, J = 1.4 Hz, H3-19), 1.78 (3H, brs, H3-20). 7.98 (2H, dt, J = 8.9, 2.0 Hz, H-3',7'), 6.94 (2H, dt, J = 8.9, 2.0 Hz, H-4',6'), 3.85 (3H, s, OCH3-5'); 13C NMR (CDCl3, 125 MHz) δ 132.8 (C-1), 135.5 (C-2), 83.8 (C-3), 85.1 (C-4), 77.6 (C-5), 137.2 (C-6), 124.2 (C-7), 43.4 (C-8), 206.7 (C-9), 72.2 (C-10), 39.0 (C-11), 31.3 (C-12), 23.2 (C-13), 23.4 (C-14), 24.0 (C-15), 28.6 (C-16), 15.5 (C-17), 17.3 (C-18), 15.6 (C-19), 22.0 (C-20), 167.0 (C-1'), 121.8 (C-2'), 131.9 (C-3',7'), 113.9 (C-4',6'), 163.9 (C-5'), 55.5 (OCH3-5'); positive-ion HRFABMS m/z 489.2246 [M+Na]+, (calcd for C28H34O6Na, 489.2253). 12b: colorless oil; [α]D25-26.9 (c 0.15, CHCl3) ; UV (MeCN) λmax (log ε): 200 (4.79), 258 (4.45) nm; IR (KBr) max: 2925, 1714, 1605, 1511, 1457, 1257, 1169, 1098, 1030, 756 cm−1; 1H NMR (CDCl3, 500 MHz) δ 5.98 (1H, q, J = 1.2 Hz, H-1), 3.84 (1H, brs, H-3), 5.34 (1H, brs, H-5), 5.88 (1H, dq, J = 4.9, 1.7 Hz, H-7), 4.26 (1H, ddq, J = 11.8, 4.9, 1.7 Hz, H-8), 2.40 (1H, m, H-11), 2.33 (1H, ddd, J = 15.8, 8.9, 3.2 Hz, H2-12a), 1.77 (1H, ddd, J = 15.8, 6.3, 5.2 Hz, H212b), 0.70 (1H, ddd, J = 8.9, 8.3, 6.6 Hz, H-13), 0.96 (1H, dd, J = 11.8, 8.3 Hz, H-14), 1.05 (3H, s, H3-16), 1.15 (3H, s, H317), 0.98 (3H, d, J = 7.1 Hz, H3-18), 1.81 (3H, d, J = 1.2 Hz, H3-19), 1.58 (3H, brs, H3-20). 8.05 (2H, dt, J = 8.9, 2.0 Hz, H-3',7'), 6.92 (2H, dt, J = 8.9, 2.0 Hz, H-4',6'), 3.85 (3H, s, OCH3-5'); 13C NMR (CDCl3, 125 MHz) δ 130.1 (C-1), 135.0 (C-2), 80.4 (C-3), 85.3 (C-4), 77.2 (C-5), 139.2 (C-6), 125.9 (C-7), 44.1 (C-8), 207.1 (C-9), 73.0 (C-10), 39.5 (C-11), 31.2 (C-12), 23.3 (C-13), 23.4 (C-14), 24.0 (C-15), 28.5 (C-16), 15.7 (C-17), 17.6 (C-18), 15.4 (C-19), 21.6 (C-20), 165.1 (C-1'), 121.6 (C-2'), 132.2 (C-3',7'), 113.9 (C-4',6'), 163.9 (C-5'), 55.5 (OCH3-5'); positive-ion HRFABMS m/z 489.2282 [M+Na]+, (calcd for C28H34O6Na, 489.2253).
3-(trans-Cinnamoyl)-20-deoxyingenol (13a) and 5-(trans-cinnamoyl)-20-deoxyingenol (13b). Compounds 13a and 13b were obtained from reaction with trans-cinnamic acid (6 mg, 36 μmol) and DMAP (5 mg, 41 μmol) for 4 h. The residue was purified by RP-HPLC (MeCN-H2O, 80:20, 5 mL/min) to give 13a (1.3 mg, 18.8%, tR = 25.5 min) and 13b (1.8 mg, 26.0%, tR = 21.0 min). 13a: colorless oil; [α]25 D+76.6 (c 0.10, CHCl3); UV (MeCN) λ***max (log ε): 200 (4.74), 279 (4.67) nm; IR (KBr) max: 2923, 1711, 1638, 1450, 1381, 1310, 1168, 1023 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.09 (1H, brq, J = 1.2 Hz, H-1), 5.53 (1H, s, H-3), 3.69 (1H, brs, H-5), 5.75 (1H, dq, J = 4.6, 1.2 Hz, H-7), 4.02 (1H, ddq, J = 11.7, 4.6, 1.4 Hz, H-8), 2.47 (1H, m, H-11), 2.26 (1H, ddd, J = 15.7, 8.9, 3.2 Hz, H2-12a), 1.75 (1H, ddd, J = 15.7, 6.3, 4.8 Hz, H2-12b), 0.67 (1H, ddd, J = 8.9, 8.6, 6.3 Hz, H-13), 0.92 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.03 (3H, s, H3-16), 1.06 (3H, s, H3-17), 0.99 (3H, d, J = 7.2 Hz, H3-18), 1.80 (3H, d, J = 1.2 Hz, H3-19), 1.78 (3H, brs, H3-20), 6.49 (1H, d,J = 16.0 Hz, H-2'), 7.74 (1H, d, J = 16.0 Hz, H-3'), 7.54 (2H, dd, J = 6.0, 2.0 Hz, H-5',9'), 7.38 (2H, dd, J = 6.0, 1.2 Hz, H-6',8'), 7.39 (2H, dd, J = 6.0, 2.0 Hz, H-7'); 13C NMR (CDCl3, 125 MHz) δ 132.129 (C-1), 135.4 (C-2), 83.6 (C-3), 85.1 (C-4), 77.5 (C-5), 137.2 (C-6), 124.2 (C-7), 43.4 (C-8), 206.6 (C-9), 72.1 (C-10), 39.0 (C-11), 31.3 (C-12), 23.4 (C-13), 23.0 (C-14), 24.0 (C-15), 28.5 (C-16), 15.6 (C-17), 17.2 (C-18), 15.6 (C-19), 22.0 (C-20), 167.6 (C-1'), 117.2 (C-2'), 146.4 (C-3'), 134.1 (C-4') 129.0 (C-5',9'), 128.2 (C-6',8'), 130.7 (C-7'); positive-ion HRFABMS m/z 485.2331 [M+Na]+, (calcd for C29H34O5Na, 485.2304). 13b: colorless oil; [α]25 D-61.0 (c 0.15, CHCl3); UV (MeCN) λmax (log ε): 200 (4.68), 279 (4.65) nm; IR (KBr) max:3445, 2925, 1712, 1635, 1450, 1380, 1335, 1157, 988 cm−1; 1H NMR (CDCl3, 500 MHz) δ 5.97 (1H, q, J = 1.7 Hz, H-1), 3.82 (1H, brd, J = 4.9 Hz, H-3), 5.29 (1H, brs, H-5), 5.89 (1H, dq, J = 4.6, 1.7 Hz, H-7), 4.24 (1H, ddq, J = 11.7, 4.6, 2.3 Hz, H-8), 2.40 (1H, m, H-11), 2.32 (1H, ddd, J = 15.5, 8.6, 3.1 Hz, H2–12a), 1.76 (1H, ddd, J = 15.5, 6.3, 5.5 Hz, H2-12b), 0.69 (1H, ddd, J = 8.6, 8.3, 6.3 Hz, H-13), 0.96 (1H, dd, J = 11.7, 8.3 Hz, H-14), 1.05 (3H, s, H3-16), 1.15 (3H, s, H3-17), 0.99 (3H, d, J = 7.2 Hz, H3-18), 1.83 (3H, d, J = 1.7 Hz, H3-19), 1.61 (3H, brs, H3-20), 6.52 (1H, d, J = 16.0 Hz, H-2'), 7.78 (1H, d, J = 16.0 Hz, H-3'), 7.52 (2H, dd, J = 6.0, 2.3 Hz, H-5',9'), 7.37 (2H, dd, J = 6.0, 1.1 Hz, H-6',8'), 7.39 (1H, dd, J = 6.0, 2.3 Hz, H-7'); 13C NMR (CDCl3, 125 MHz) δ 132.8 (C-1), 134.8 (C-2), 80.3 (C-3), 85.3 (C-4), 77.6 (C-5), 139.1 (C-6), 124.2 (C-7), 43.4 (C-8), 206.7 (C-9), 73.0 (C-10), 39.0 (C-11), 31.2 (C-12), 23.2 (C-13), 23.3 (C-14), 24.0 (C-15), 28.5 (C-16), 15.5 (C-17), 17.6 (C-18), 15.3 (C-19), 21.6 (C-20), 166.7 (C-1'), 116.9 (C-2'), 146.8 (C-3'), 134.1 (C-4'), 129.0 (C-5',9'), 128.2 (C-6',8'), 130.7 (C-7'); positive-ion HRFABMS m/z 485.2326 [M+Na]+, (calcd for C29H34O5Na, 485.2304).
3-(2-Naphthoyl)-20-deoxyingenol (14a) and 5-(2-naphthoyl)-20-deoxyingenol (14b). Compounds 14a and 14b were obtained from reaction with 2-naphthoic acid (5 mg, 29 μmol) and DMAP (5 mg, 41 μmol) for 8 h. The residue was purified by RP-HPLC (MeCN-H2O, 80:20, 5 mL/min) to give 14a (2.3 mg, 31.6%, tR = 32.8 min) and 14b (2.2 mg, 30.2%, tR = 27.0 min). 14a: colorless oil; [α]25D +139.6 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (5.08), 238 (5.01), 281 (4.07) nm; IR (KBr) max: 3496, 2922, 1712, 1465, 1444, 1381, 1356, 1282, 1228, 1196, 1095 cm−1 1H NMR (CDCl3, 500 MHz) δ 6.16 (1H, brq, J = 1.7 Hz, H-1), 5.70 (1H, s, H-3), 3.77 (1H, brs, H-5), 5.79 (1H, dq, J = 4.8, 1.4 Hz, H-7), 4.04 (1H, ddq, J = 11.8, 4.8, 1.8 Hz, H-8), 2.58 (1H, m, H-11), 2.26 (1H, ddd, J = 15.8, 8.9, 3.2 Hz, H2-12a), 1.75 (1H, ddd, J = 15.8, 6.0, 5.1 Hz, H2-12b), 0.67 (1H, ddd, J = 8.9, 8.3, 6.3 Hz, H-13), 0.92 (1H, dd, J = 11.8, 8.3 Hz, H-14), 1.03 (3H, s, H3-16), 1.04 (3H, s, H3-17), 1.07 (3H, d, J = 6.9 Hz, H3-18), 1.80 (3H, d, J = 1.2 Hz, H3-19), 1.78 (3H, brs, H3-20), 8.02 (1H, dd, J = 8.6, 1.7 Hz, H-3'), 7.90 (1H, d, J = 8.6 Hz, H-4'), 7.89 (1H, brd, J = 8.0 Hz, H-5'), 7.60 (1H, ddd, J = 8.0, 6.9, 1.4 Hz, H-6'), 7.55 (1H, ddd, J = 8.0, 6.9, 1.4 Hz, H-7'), 7.96 (1H, brd, J = 8.0 Hz, H-8'), 8.59 (1H, s, H-9'); 13C NMR (CDCl3, 125 MHz) δ 133.3 (C-1), 135.5 (C-2), 84.2 (C-3), 85.2 (C-4), 77.6 (C-5), 137.3 (C-6), 124.2 (C-7), 43.3 (C-8), 207.2 (C-9), 72.2 (C-10), 39.0 (C-11), 31.1 (C-12), 23.0 (C-13), 23.2 (C-14), 23.8 (C-15), 28.4 (C-16), 15.4 (C-17), 17.1 (C-18), 15.2 (C-19), 21.8 (C-20), 167.9 (C-1'), 126.9 (C-2'), 125.3 (C-3'), 128.4 (C-4'), 128.0 (C-5'), 128.8 (C-6'), 126.8 (C-7'), 129.6 (C-8'), 131.7 (C-9'), 132.7 (C-10'), 136.0 (C-11'); positive-ion HRFABMS m/z 509.2314 [M+Na]+, (calcd for C31H34O5Na, 509.2304). 14b: colorless oil; [α]25D -53.6 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (4.98), 238 (5.12), 280 (4.20) nm; IR (KBr) max: 3450, 2924, 1715, 1465, 1444, 1381, 1356, 1277, 1227, 1195, 1091 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.00 (1H, brq, J = 1.5 Hz, H-1), 3.89 (1H, s, H-3), 5.47 (1H, brs, H-5), 5.92 (1H, dq, J = 4.5, 1.7 Hz, H-7), 4.30 (1H, ddq, J = 11.7, 4.5, 1.5 Hz, H-8), 2.42 (1H, m, H-11), 2.35 (1H, ddd, J = 15.8, 8.6, 2.9 Hz, H2-12a), 1.79 (1H, ddd, J = 15.8, 6.3, 5.1 Hz, H2-12b), 0.69 (1H, ddd, J = 8.6, 8.6, 6.3 Hz, H-13), 0.96 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.06 (3H, s, H3-16), 1.19 (3H, s, H3-17), 0.99 (3H, d, J = 6.9 Hz, H3-18), 1.83 (3H, d, J = 1.5 Hz, H3-19), 1.63 (3H, brs, H3-20), 8.09 (1H, dd, J = 8.6, 1.8 Hz, H-3'), 7.88 (1H, d, J = 8.6 Hz, H-4'), 7.88 (1H, brd, J = 8.0 Hz, H-5'), 7.60 (1H, ddd, J = 8.0, 6.9, 1.4 Hz, H-6'), 7.54 (1H, ddd, J = 8.0, 6.9, 1.4 Hz, H-7'), 7.95 (1H, brd, J = 8.0 Hz, H-8'), 8.68 (1H, s, H-9'); 13C NMR (CDCl3, 125 MHz) δ 132.8 (C-1), 139.1 (C-2), 80.4 (C-3), 85.3 (C-4), 78.0 (C-5), 134.9 (C-6), 124.2 (C-7), 43.4 (C-8), 207.1 (C-9), 73.1 (C-10), 39.6 (C-11), 31.2 (C-12), 23.3 (C-13), 23.4 (C-14), 24.0 (C-15), 28.5 (C-16), 15.6 (C-17), 17.6 (C-18), 15.4 (C-19), 21.7 (C-20), 166.5 (C-1'), 126.5 (C-2'), 125.4 (C-3'), 128.4 (C-4'), 127.8 (C-5'), 128.6 (C-6'), 126.9 (C-7'), 129.4 (C-8'), 131.7 (C-9'), 132.5 (C-10'), 135.8 (C-11'); positive-ion HRFABMS m/z 509.2324 [M +Na]+, (calcd for C31H34O5Na, 509.2304).
3-Octanoyl-20-deoxyingenol (18). Compound 18 was obtained from reaction with n-Octanoic acid (6 μL, 36 μmol) and DMAP (5 mg, 41 μmol) for 4 h. The residue was purified by RP-HPLC (MeCN-H2O, 90:10, 5 mL/min) to give 18 (1.4 mg, 20.4%, tR = 24.0 min). 18: colorless oil; [α]D25 +13.0 (c 0.10, CHCl3); UV (MeCN) λmax (log ε): 200 (4.45) nm; IR (KBr) max: 3484, 2926, 1723, 1458, 1380, 1260, 1160, 1024 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.04 (1H, brq, J = 1.5 Hz, H-1), 5.37 (1H, s, H-3), 3.66 (1H, brd, J = 5.5, H-5), 5.74 (1H, dq, J = 4.8, 1.5 Hz, H-7), 4.00 (1H, ddq, J = 11.8, 4.8, 1.7 Hz, H-8), 2.41 (1H, m, H-11), 2.24 (1H, ddd, J = 15.7, 8.8, 3.1 Hz, H2-12a), 1.75 (1H, ddq, J = 15.7, 6.3, 5.7 Hz, H2-12b), 0.66 (1H, ddd, J = 8.9, 8.6, 6.3 Hz, H-13), 0.90 (1H, dd, J = 11.8, 8.6 Hz, H-14), 1.03 (3H, s, H3-16), 1.06 (3H, s, H3-17), 0.97 (3H, d, J = 6.9 Hz, H3-18), 1.76 (3H, brs, H3-19), 1.76 (3H, brs, H3-20), 2.40 (2H, ddd, J = 7.4, 7.4, 2.6 Hz, H2-2'), 1.64 (2H, qui, J = 7.4 Hz, H2-;3'), 1.34-1.27 (2H, m, H2-4'), 1.34-1.27 (2H, m, H2-5'), 1.29-1.24 (2H, m, H2-6'), 1.32-1.24 (2H, m, H2-7'), 0.87 (3H, t, J = 7.2 Hz, H3-8'); 13C NMR (CDCl3, 125 MHz) δ 132.7 (C-1), 135.3 (C-2), 83.3 (C-3), 84.9 (C-4), 77.5 (C-5), 137.1 (C-6), 124.3 (C-7), 43.3 (C-8), 206.6 (C-9), 72.0 (C-10), 38.9 (C-11), 31.2 (C-12), 23.1 (C-13), 23.4 (C-14), 24.0 (C-15), 28.5 (C-16), 15.5 (C-17), 17.2 (C-18), 15.5 (C-19), 21.9 (C-20), 174.6 (C-1'), 34.5 (C-2'), 25.1 (C-3'), 29.0 (C-4'), 28.9 (C-5'), 31.7 (C-6'), 22.6 (C-7'), 14.0 (C-8'); positive-ion HRFABMS m/z 481.2957 [M+Na]+, (calcd for C25H34O5Na, 481.2930).
4.6. General procedure for preparation of 15a, 15b, 16 and 17.
To a stirred solution of 9 (15 μmol) in dehydrated pyridine (3 mL) was added corresponding acidic anhydride and DMAP (8 μmol). The mixture was stirred at room temperature or 40 °C for 24 h under argon protection. After removal of pyridine in vacuo, the residue was purified by RP-HPLC to give desired compounds.
3-Crotonoyl-20-deoxyingenol (15a) and 5-crotonoyl-20-deoxyingenol (15b). Compounds 15a and 15b were obtained from reaction with crotonic anhydride (5 μl, 33 μmol) at room temperature. The residue was purified by RP-HPLC (MeCN-H2O, 70:30, 5 ml/min) to give 15a (1.5 mg, 25.0%, tR = 27.0 min) and 15b (1.6 mg, 26.7%, tR = 21.0 min). 15a: colorless oil; [α]25 D +37.7 (c 0.15, CHCl3); UV (MeCN) λmax (log ε): 200 (4.71) nm; IR (KBr) max: 2919, 1718, 1656, 1444, 1379, 1190, 1009, 760 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.04 (1H, brq, J = 1.4 Hz, H-1), 5.46 (1H, s, H-3), 3.66 (1H, brd, J = 5.5 Hz, H-5), 5.74 (1H, dq, J = 4.6, 1.7 Hz, H-7), 4.00 (1H, ddq, J = 11.7, 4.6, 1.7 Hz, H-8), 2.41 (1H, m, H-11), 2.24 (1H, ddd, J = 15.7, 8.8, 3.1 Hz, H2-12a), 1.75 (1H, ddd, J = 15.7, 5.7, 5.7 Hz, H2-12b), 0.66 (1H, ddd, J = 8.7, 8.6, 6.6 Hz, H-13), 0.91 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.02 (3H, s, H3-16), 1.06 (3H, s, H3-17), 0.97 (3H, d, J = 6.9 Hz, H3-18), 1.76 (3H, brs, H3-19), 1.76 (3H, brs, H3-20), 5.92 (1H, dq, J = 15.5, 1.7 Hz, H-2'), 7.04 (1H, dq, J = 15.5, 6.9 Hz, H-3'), 1.91 (3H, dd, J = 6.9, 1.5 Hz, H3-4'); 13C NMR (CDCl3, 125 MHz) δ 129.5 (C-1), 135.5 (C-2), 83.2 (C-3), 85.0 (C-4), 77.5 (C-5), 137.2 (C-6), 124.1 (C-7), 43.4 (C-8), 206.7 (C-9), 72.0 (C-10), 38.9 (C-11), 31.2 (C-12), 23.2 (C-13), 23.4 (C-14), 24.0 (C-15), 28.5 (C-16), 15.5 (C-17), 17.2 (C-18), 15.5 (C-19), 21.9 (C-20), 167.0 (C-1'), 122.1 (C-2'), 146.5 (C-3'), 18.2 (C-4'); positive-ion HRFABMS m/z 423.2178 [M+Na]+, (calcd for C24H32O5Na, 423.2147). 15b: colorless oil; [α]25 D -18.1 (c 0.10, CHCl3); UV (MeCN) λmax (log ε): 200 (4.62) nm; IR (KBr) max: 2922, 1720, 1656, 1444, 1379, 1172, 1006, 757 cm−1; 1H NMR (CDCl3, 500 MHz) δ 5.94 (1H, brq, J = 1.4 Hz, H-1), 3.78 (1H, s, H-3), 5.20 (1H, brs, H-5), 5.86 (1H, dq, J = 4.6, 1.5 Hz, H-7), 4.20 (1H, ddq, J = 11.7, 4.6, 1.7 Hz, H-8), 2.38 (1H, m, H-11), 2.29 (1H, ddd, J = 15.7, 8.9, 3.2 Hz, H2-12a), 1.74 (1H, ddd, J = 15.7, 5.8, 5.8 Hz, H2-12b), 0.68 (1H, ddd, J = 8.9, 8.3, 6.3 Hz, H-13), 0.91 (1H, dd, J = 11.7, 8.3 Hz, H-14), 1.04 (3H, s, H3-16), 1.13 (3H, s, H3-17), 0.97 (3H, d, J = 6.9 Hz, H3-18), 1.81 (3H, brs, H3-19), 1.56 (3H, brs, H3-;20), 5.91 (1H, dq, J = 15.5, 1.7 Hz, H-2'), 7.09 (1H, dq, J = 15.5, 6.9 Hz, H-3'), 1.90 (3H, dd, J = 6.9, 1.5 Hz, H3-4'); 13C NMR (CDCl3, 125 MHz) δ 129.9 (C-1), 134.8 (C-2), 80.3 (C-3), 85.3 (C-4), 77.0 (C-5), 139.2 (C-6), 126.1 (C-7), 44.1 (C-8), 206.8 (C-9), 73.1 (C-10), 39.5 (C-11), 31.2 (C-12), 23.3 (C-13), 23.3 (C-14), 24.0 (C-15), 28.5 (C-16), 15.6 (C-17), 17.6 (C-18), 15.3 (C-19), 21.6 (C-20), 166.1 (C-1'), 121.7 (C-2'), 147.1 (C-3'), 18.1 (C-4'); positive-ion HRFABMS m/z 423.2174 [M+Na]+, (calcd for C24H32O5Na, 423.2147).
3-Tigloyl-20-deoxyingenol (16) and 3-angeloyl-20-deoxyingenol (17). Compounds 16 and 17 were obtained from reaction with angelic anhydride (16 μL, 88 μmol) at 40 °C. The residue was purified by RP-HPLC (MeCN-H2O, 75:25, 5 mL/min) to give 16 (2.1 mg, 33.8%, tR = 22.5 min) and 17 (2.2 mg, 35.4%, tR = 25.0 min). 16: colorless oil; [α]25D +39.1 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (4.54) nm; IR (KBr) max: 3424, 2927, 1710, 1651, 1456, 1380, 1267, 1153, 1072 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.04 (1H, brq, J = 1.4 Hz, H-1), 5.43 (1H, s, H-3), 3.67 (1H, brs, H-5), 5.73 (1H, dq, J = 4.9, 1.4 Hz, H-7), 4.20 (1H, ddq, J = 11.7, 4.9, 1.7 Hz, H-8), 2.42 (1H, m, H-11), 2.24 (1H, ddd, J = 15.8, 8.6, 2.9 Hz, H2-12a), 1.75 (1H, ddd, J = 15.8, 6.4, 5.2 Hz, H2-12b), 0.68 (1H, ddd, J = 8.6, 8.6, 6.4 Hz, H-13), 0.90 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.03 (3H, s, H3-16), 1.06 (3H, s, H3-17), 0.97 (3H, d, J = 6.8 Hz, H3-18), 1.76 (3H, brs, H3-19), 1.74 (3H, brs, H3-20), 6.89 (1H, qq, J = 7.2, 1.2 Hz, H-2'), 1.82 (1H, dq, J = 7.2, 1.2 Hz, H-3'), 1.85 (3H, qui, J = 1.2 Hz, H3-4'); 13C NMR (CDCl3, 125 MHz) δ 132.5 (C-1), 135.5 (C-2), 83.5 (C-3), 85.0 (C-4), 77.6 (C-5), 137.3 (C-6), 124.2 (C-7), 43.4 (C-8), 206.8 (C-9), 72.1 (C-10), 38.9 (C-11), 31.3 (C-12), 23.3 (C-13), 23.4 (C-14), 23.9 (C-15), 28.6 (C-16), 15.6 (C-17), 17.3 (C-18), 15.5 (C-19), 22.0 (C-20), 168.7 (C-1'), 128.2 (C-2'), 138.8 (C-3'), 14.6 (C-4'), 12.2 (C-5'); positive-ion HRFABMS m/z 437.2335 [M+Na]+, (calcd for C25H34O5Na, 437.2304). 17: colorless oil; [α]25D +12.3 (c 0.20, CHCl3); UV (MeCN) λmax (log μ): 200 (4.52) nm; IR (KBr) max: 3445, 2925, 1714, 1650, 1456, 1381, 1261, 1156, 1032 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.04 (1H, brq, J = 1.4 Hz, H-1), 5.45 (1H, s, H-3), 3.69 (1H, brs, H-5), 5.74 (1H, dq, J = 4.6, 1.7 Hz, H-7), 4.00 (1H, ddq, J = 11.8, 4.6, 1.7 Hz, H-8), 2.44 (1H, m, H-11), 2.24 (1H, ddd, J = 15.8, 8.9, 3.0 Hz, H2-12a), 1.72 (1H, ddd, J = 15.8, 6.4, 5.2 Hz, H2-12b), 0.66 (1H, ddd, J = 8.9, 8.3, 6.4 Hz, H-13), 0.91 (1H, dd, J = 11.8, 8.3 Hz, H-14), 1.03 (3H, s, H3-16), 1.06 (3H, s, H3-17), 0.95 (3H, d, J = 7.1 Hz, H3-18), 1.78 (3H, d, J = 1.4, H3-19), 1.77 (3H, brs, H3-20), 6.15 (1H, qq, J = 7.2, 1.4 Hz, H-2'), 2.00 (1H, dq, J = 7.2, 1.4 Hz, H-3'), 1.91 (3H, qui, J = 1.4 Hz, H3-4'); 13C NMR (CDCl3, 125 MHz) δ 132.6 (C-1), 135.4 (C-2), 83.5 (C-3), 84.9 (C-4), 77.6 (C-5), 137.2 (C-6), 124.2 (C-7), 43.4 (C-8), 206.7 (C-9), 72.1 (C-10), 38.8 (C-11), 31.2 (C-12), 23.2 (C-13), 23.4 (C-14), 24.0 (C-15), 28.6 (C-16), 15.5 (C-17), 17.2 (C-18), 15.6 (C-19), 21.9 (C-20), 168.5 (C-1'), 127.2 (C-2'), 139.9 (C-3'), 15.9 (C-4'), 20.7 (C-5'); positive-ion HRFABMS m/z 437.2329 [M+Na]+, (calcd for C25H34O5Na, 437.2304).
4.7. General procedure for preparation of 22 and 23
To a solution of 10 (32 μmol) in acetone (6 mL), p-TsOH.H2O (3 μmol) was added and stirred at room temperature for 2 h. After removal of solvent in vacuo, the residue was passed through a Sep-Pak C18 cartridge, and further purified by RP-HPLC (YMC Pack Pro C18, MeCN-H2O, 60:40, 5 mL/min) to give 19 (8.3 mg, 66.8%). Derivative 20 (3 mg, 95.7%) was prepared by 19 (7 μmol) reacted with angelic anhydride (28 μmol) and Cs2CO3 (28 μmol) in super dehydrated MeCN (2 mL) at room temperature for 4 h, and subsequent purification was achieved by chromatography using a Sep-Pak C18 cartridge (Waters). To a stirred solution of diol 19 (7 μmol) in super dehydrated DCM (3 mL), DMAP (25 μmol) and 2-naphthoic acid (28 μmol) were added, and then chilled to 0 °C. After a solution of EDCI (5 mg, 25 μmol) dissolved in super dehydrated DCM (0.5 mL) was added dropwise, the reaction mixture was stirred at room temperature for 4 h under argon protection. After removal of the solvent in vacuo, the residue was purified by RP-HPLC (YMC Pack Pro C18, MeCN-H2O, 80:20, 5.0 mL/min) to give 21 (2.6 mg, 68.6%). Then, to a solution of 20 (6 μmol) or 21 (5 μmol) in MeOH (2 mL), 2 M HCl (10 μL) was added and stirred at room temperature for 4 h. After removal of the solvent in vacuo, the residue was chromatographed on a Sep-Pak silica cartridge using Hexane-EtOAc (100:0 and 50:50, each 10 ml) to obtain 22 (2.5 mg, 91.1%) or 23 (2.1 mg, 87.5%).
3-Angeloylingenol (22): colorless oil; [α]25D +1.3 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (4.82) nm; IR (KBr) max: 3485, 2927, 1711, 1459, 1382, 1352, 1231, 1157, 1041 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.01 (1H, brq, J = 1.5 Hz, H-1), 5.53 (1H, s, H-3), 3.48 (1H, brs, H-5), 6.04 (1H, brd, J = 4.0, H-7), 4.11 (1H, m, H-8), 2.52 (1H, m, H-11), 2.25 (1H, ddd, J = 15.7, 8.9, 3.2 Hz, H2-12a), 1.75 (1H, ddd, J = 15.7, 6.0, 5.8 Hz, H2-12b), 0.67 (1H, ddd, J = 8.9, 8.6, 6.0 Hz, H-13), 0.92 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.03 (3H, s, H3-16), 1.07 (3H, s, H3-17), 0.99 (3H, d, J = 7.1 Hz, H3-18), 1.80 (3H, brs, H3-19), 4.13 (2H, brs, H2-20), 6.15 (1H, qq, J = 7.1, 1.5 Hz, H-3'), 2.00 (1H, dq, J = 7.2, 1.5 Hz, H-4'), 1.91 (3H, qui, J = 1.5 Hz, H3-5'); 13CNMR (CDCl3, 125 MHz) δ 132.9 (C-1), 135.4 (C-2), 82.6 (C-3), 84.7 (C-4), 77.5 (C-5), 139.2 (C-6), 128.5 (C-7), 43.5 (C-8), 206.6 (C-9), 72.1 (C-10), 38.3 (C-11), 31.1 (C-12), 23.1 (C-13), 23.4 (C-14), 23.9 (C-15), 28.5 (C-16), 15.5 (C-17), 17.3 (C-18), 15.5 (C-19), 67.4 (C-20), 168.6 (C-1'), 127.3 (C-2'), 139.9 (C-3'), 15.5 (C-4'), 20.7 (C-5'); positive-ion HRFABMS m/z 453.2279 [M+Ma]+, (calcd for C25H34O6, 453.2253).
3-(2-Naphthoyl)ingenol (23): colorless oil; [α]25 D +156.6 (c 0.20, CHCl3); UV (MeCN) λmax (log ε): 200 (4.981), 236 (5.06), 280 (4.15) nm; IR (KBr) max: 3416, 2925, 1719, 1467, 1381, 1355, 1283, 1229, 1197, 1097 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.13 (1H, brq, J = 1.7 Hz, H-1), 5.79 (1H, s, H-3), 4.13 (1H, brs, H-5), 6.06 (1H, dq, J = 4.6, 1.4 Hz, H-7), 4.15 (1H, m, H-8), 2.47 (1H, m, H-11), 2.25 (1H, ddd, J = 15.7, 8.9, 3.2 Hz, H2-12a), 1.77 (1H, ddd, J = 15.7, 6.0, 5.7 Hz, H2-12b), 0.69 (1H, ddd, J = 8.9, 8.6, 6.0 Hz, H-13), 0.93 (1H, dd, J = 11.7, 8.6 Hz, H-14), 1.01 (3H, s, H3-16), 1.02 (3H, s, H3-17), 0.99 (3H, d, J = 7.2 Hz, H3-18), 1.80 (3H, brd, J = 1.2 Hz, H3-19), 4.18 (2H, brs, H2-20), 8.03 (1H, dd, J = 8.6, 1.7 Hz, H-3'), 7.90 (1H, d, J = 8.6 Hz, H-4'), 7.88 (1H, brd, J = 8.0 Hz, H-5'), 7.60 (1H, ddd, J = 8.0, 6.9, 1.2 Hz, H-6'), 7.55 (1H, ddd, J = 8.0, 6.9, 1.2 Hz, H-7'), 7.95 (1H, brd, J = 8.0 Hz, H-8'), 8.59 (1H, s, H-9'); 13C NMR (CDCl3, 125 MHz) δ 132.6 (C-1), 135.7 (C-2), 83.6 (C-3), 85.0 (C-4), 77.2 (C-5), 138.9 (C-6), 124.2 (C-7), 43.6 (C-8), 206.5 (C-9), 72.1 (C-10), 38.6 (C-11), 31.2 (C-12), 23.4 (C-13), 23.1 (C-14), 24.0 (C-15), 28.5 (C-16), 15.5 (C-17), 17.4 (C-18), 15.6 (C-19), 67.5 (C-20), 167.6 (C-1'), 126.9 (C-2'), 125.2 (C-3'), 128.4 (C-4'), 127.8 (C-5'), 128.5 (C-6'), 126.8 (C-7'), 129.5 (C-8'), 131.5 (C-9'), 132.5 (C-10'), 135.7 (C-11'); positive-ion HRFABMS m/z 525.2282 [M+Ma]+, (calcd for C31H34O6Na, 525.2253).
4.8. Multi-cycle viral replication in MT4 cell assay
HIV-1 NL4-3 Nanoluc-sec at a dose of 50 TCID50/well was used to infect MT4 cells (1 × 105 cells/mL) in the presence of compounds at various concentrations in 96-well plates. On day 3 post-infection, supernatant samples were harvested and assayed for luciferase activity using the Promega Nano-Glo® Luciferase Assay System. The antiviral potency is defined as the drug concentration that reduces the luciferase activity by 50% (EC50).
4.9. Cytotoxicity Assay
A CytoTox-Glo™ cytotoxicity assay (Promega) was used to determine the cytotoxicity of the tested compounds. MT4 cells were cultured in the presence of various concentrations of the compounds for 3 days. Cytotoxicity of the compounds was determined by following the protocol provided by the manufacturer. The 50% cytotoxic concentration (CC50) was defined as the concentration that caused a 50% reduction of cell viability.
4.10. U1 cells model of HIV latency
U1 cells were used as an HIV-1 latency model. U1 cells (2×105 cells/mL) were incubated in the presence of various concentrations of compounds 22 and 23 at 37 ºC for 72 h. The culture supernatant was assayed for p24 with an HIV p24 ELISA kit (ZeptoMetrix) following the manufacture’s protocol. Drug concentration that activated HIV-1 p24 production by 50% is defined as the EC50 of the compounds. EC50 was determined with a non-linear regression analysis using the Biosoft software. The cytotoxicity against U937 cell was measured by the same method as that for MT4 cells.
4.10. Fluorescence-activated cell sorting (FACS) analysis of GFP-expressing J-Lat cells
J-Lat (A2) cells (1×106 cells/well) were incubated in the presence of various concentrations of HIV-1 latency reversing agents, compounds 22 and 23, at 37 °C for 72 h. The GFP-expressing cells were analyzed by using a BD LSRII/Fortessa cell analyzer (Becton-Dickinson).
Supplementary Material
Figure 3.

Fluorescence-activated cell sorting (FACS) analysis of GFP-expressing J-Lat cells in the presence of the compounds 22 and 23.
Highlights.
MeOH extract of Euphorbia kansui showed potent anti-HIV-1 activity.
Ingenane esters from Euphorbia kansui are potent anti-HIV-1 agents.
Naphthoyl group at C-3 of 20-deoxyingenol and ingenol enhanced anti-HIV-1 activity.
3-(2-Naphthoyl)ingenol is a potent HIV-1 latency reversing agent.
Acknowledgements
This investigation was supported by JSPS KAKENHI 17K08348 and 26460133 (W. Li). Further support was supplied by NIH Grants AI33066 (K.-H. Lee) and P30064518 (L. Huang) from the National Institute of Allergy and Infectious Diseases.
ABBREVITATIONS USED
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- p-TsOH.H2O
p-toluenesulfonic acid monohydrate
- RP-HPLC
reverse-phase high performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.eimech.xxxx. Deacylation of 1 and 5, LC-MS analysis, spectroscopic data of 10 and preparation of 19–21 in Supplementary data (PDF).
Notes
The authors declare no competing financial interest.
References
- [1].Organization, W. H., World Health Statistics 2017. Global Health Observatory (GHO) data 2016. [Google Scholar]
- [2].Dau B; Holodniy M Novel targets for antiretroviral therapy: clinical progress to date. Drugs 2009, 69, 31–50. [DOI] [PubMed] [Google Scholar]
- [3].Pereira CF; Paridaen JT Anti-HIV drug development–an overview. Curr. Pharm. Des 2004, 10, 4005–4037. [DOI] [PubMed] [Google Scholar]
- [4].Finzi D; Hermankova M; Pierson T; Carruth LM; Buck C; Chaisson RE; Quinn TC; Chadwick K; Margolick J; Brookmeyer R; Gallant J; Markowitz M; Ho DD; Richman DD Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [DOI] [PubMed] [Google Scholar]
- [5].Archin NM; Liberty AL; Kashuba AD; Choudhary SK; Kuruc JD; Crooks AM; Parker DC; Anderson EM; Kearney MF; Strain MC; Richman DD; Hudgens MG; Bosch RJ: Coffin JM; Eron JJ; Hazuda DJ; Margolis DM Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kulkosky J; Culnan DM; Roman J; Dornadula G; Schnell M; Boyd MR; Pomerantz RJ Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [DOI] [PubMed] [Google Scholar]
- [7].Lai W, Huang L, Zhu L, Ferrari G, Chan C, Li W; Lee KH; Chen CH Gnidimacrin, a potent anti-HIV diterpene, can eliminate latent HIV-1 ex vivo by activation of protein kinase Cβ. J. Med. Chem 2015, 58, 8638–8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang L; Ho P; Yu J; Zhu L; Lee KH; Chen CH Picomolar dichotomous activity of gnidimacrin against HIV-1. PLoS One 2011, 6, e26677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang L; Gao L; Li Z; Yan X; Yang Y; Tang Y; Gao Y; Ding A Bio-guided isolation of the cytotoxic terpenoids from the roots of Euphorbia kansui against human normal cell lines L-O2 and GES-1. Int. J. Mol. Sci 2012, 13, 11247–11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pan DJ; Hu CQ; Chang JJ; Lee TY; Chen YP; Hsu HY; Mcphail DR; Lee KH Kansuiphorin-C and-D, cytotoxic diterpenes from Euphorbia kansui. Phytochemistry 1991, 30, 1018–1020. [Google Scholar]
- [11].Jakupovic J; Jeske F; Morgenstern T; Tsichritzis F; Marco JA; Berendsohn W Diterpenes from Euphorbia segetalis. Phytochemistry 1998, 47, 1583–1600. [Google Scholar]
- [12].Matsumoto T; Cyong JC; Yamada H Stimulatory effects of ingenols from Euphorbia kansui on the expression of macrophage Fc receptor. Planta Med 1992, 58, 255–258. [DOI] [PubMed] [Google Scholar]
- [13].Wang LY; Wang NL; Yao XS; Miyata S; Kitanaka S Diterpenes from the roots of Euphorbia kansui and their in vitro effects on the cell division of Xenopus. J. Nat. Prod 2002, 65, 1246–1251. [DOI] [PubMed] [Google Scholar]
- [14].Manners GD; Wong RY The absolute stereochemical characterization of two new jatrophane diterpenes from Euphorbia esula. J. Chem. Soc. -Perkin Trans 1 1985, 2075–2081. [Google Scholar]
- [15].Wang P; Lu P; Qu X; Shen Y; Zeng H; Zhu X; Zhu Y; Li X; Wu H; Xu J; Lu H; Ma Z; Zhu H Reactivation of HIV-1 from latency by an ingenol derivative from Euphorbia kansui. Sci. Rep 2017, 7, 9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Winkler JD; Rouse MB; Greaney MF; Harrison SJ; Jean YT The first total synthesis of (+/−)-ingenol. J. Am. Chem. Soc 2002, 124, 9726–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McKerrall SJ; Jørgensen L; Kuttruff CA; Ungeheuer F; Baran PS Development of a concise synthesis of (+)-ingenol. J. Am. Chem. Soc 2014, 136, 5799–5810. [DOI] [PubMed] [Google Scholar]
- [18].Ohyoshi T; Funakubo S; Miyazawa Y; Niida K; Hayakawa I; Kigoshi H Total synthesis of (−)-13-oxyingenol and its natural derivative. Angew. Chem., Int. Ed 2012, 51, 4972–4975. [DOI] [PubMed] [Google Scholar]
- [19].Jiang G; Mendes EA; Kaiser P; Wong DP; Tang Y; Cai I; Fenton A; Melcher GP; Hildreth JE; Thompson GR; Wong JK; Dandekar S Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-kB signaling in combination with JQ1 induced p-TEFb activation. PLoS Pathog 2015, 11, e1005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brogdon J; Ziani W; Wang X; Veazey RS; Xu H In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep 2016, 6, 39032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jordan A; Defechereux P; Verdin E The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J 2001, 20, 1726–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.