

Abstract
Reactions that directly install nitrogen into C–H bonds of complex molecules are significant because of their potential to change the chemical and biological properties of a given compound. Although selective intramolecular C–H amination reactions are known, achieving high levels of reactivity, while maintaining excellent site-selectivity and functional-group tolerance, remains a challenge for intermolecular C–H amination. Herein, we report a manganese perchlorophthalocyanine catalyst [MnIII(ClPc)] for intermolecular benzylic C–H amination of bioactive molecules and natural products that proceeds with unprecedented levels of reactivity and site-selectivity. In the presence of Brønsted or Lewis acid, the [MnIII(ClPc)]-catalyzed C–H amination demonstrates unique tolerance for tertiary amine, pyridine and benzimidazole functionalities. Mechanistic studies suggest that C–H amination likely proceeds through an electrophilic metallonitrene intermediate via a stepwise pathway where C–H cleavage is the rate-determining step of the reaction. Collectively these mechanistic features contrast previous base-metal catalyzed C–H aminations and provide new opportunities for tunable selectivities.
Introduction of nitrogen into natural products and biologically active small molecules has the potential to drastically change the physical and biological properties of a given molecule.1 For example, ampicillin, the first broad spectrum penicillin derivative, differs from penicillin G by the presence of a benzylic amine (Figure 1a).2 In fact, the importance of nitrogen in the benzylic position of pharmacophores can be gleaned by its appearance in top selling FDA approved drugs such as imatinib, meclizine, clopidogrel, sertraline, rivastigmine, donepezil, and many others.3 In nature, such nitrogen functionality is installed through oxygenated intermediates.4 For example, in the biosynthesis of L-p-hydroxyphenylglycine, a crucial component of several peptidic natural products, the benzylic amine is installed from the benzylic ketone.5 Analogously, standard synthetic methods to install nitrogen rely on functional group transformations from pre-oxidized carbon-heteroatom (C–O, C–X, X = halogens) precursors.6–8 This approach limits direct installation of nitrogen into topologically and functionally complex molecules, often necessitating de novo syntheses. The ability to effect late-stage functionalization via direct and selective installation of nitrogen in complex molecules may expedite discovery processes for biologically active small molecules and re-invigorate exploration of natural product derived drug candidates.9,10
Figure 1. Converting C–H bonds to C–N bonds.
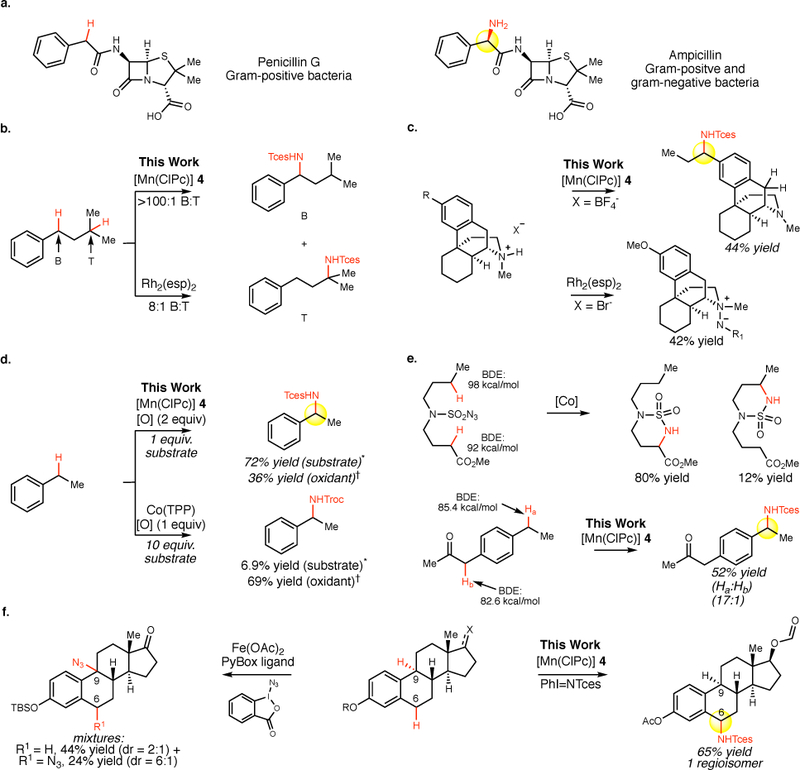
a. Ampicillin, a broad spectrum antibacterial drug differs from penicillin by an atomistic change of a benzylic C–H to C–N. b. Comparison of site-selectivities for benzylic versus tertiary C–H amination of Mn versus Rh catalyzed C–H aminations. Tces = 2,2,2-trichloroethoxysulfonamide. c. Chemoselectivity in a tertiary amine containing natural product of Mn versus Rh catalyzed C–H aminations. d. Comparison of reactivity of Mn versus Co catalyzed benzylic C–H aminations. *Yield based on equivalents of substrate used. †Yield based on equivalents of oxidant used. Troc = 2,2,2-trichloroethoxycarbonyl. e. Observed site-selectivity of Mn versus Co catalyzed C–H aminations. f. Comparison of site-selectivities among sterically differentiated benzylic C–H bonds on estrone/estradiol derivatives of a Mn-catalyzed reaction proceeding via a metallonitrene with previously reported Fe-catalyzed C–H azidation proceeding via a free radical intermediate.
Significant reactivity and selectivity challenges exist in the late-stage C–H amination of natural products and pharmaceuticals as many similar C–H bond types may be present in a molecule. Noble metal rhodium catalysis via metallonitrene intermediates is well-established for the intramolecular C–H amination of a wide range of C–H bond types.11–13 One advantage of proceeding via metallonitrene intermediates is that both C–H cleavage and functionalization are tightly regulated at the metal and therefore are amenable to tuning via ligand/metal modifications. Intermolecular rhodium catalyzed C–H aminations are emerging for benzylic, tertiary and allylic C–H aminations;14–17 however, chemo-and site-selectivity challenges exist in molecules having multiple reactive functionalities or C–H bonds. For example, in molecules having both tertiary and benzylic C–H bonds, inseparable product mixtures are formed (Figure 1b).14,15 Moreover, the ability to effect remote C–H amination in molecules having basic amines or heterocyclic functionality has not been demonstrated for this type of C–H amination (Figure 1c). Tertiary amine-containing natural products undergo α-amination/oxidation sequences to give amidines or direct N-amination to furnish hydrazine sulfamate zwitterions, with protection of nitrogen as an amine salt not shown to be tolerated.18
Recently, base-metal catalysts have emerged for highly selective intramolecular C–H aminations;19–21 however, analogous intermolecular processes are scarce.22,23 For example, cobalt-catalyzed intermolecular benzylic C–H aminations are not suitable for late-stage applications as they have formidable reactivity challenges requiring solvent quantities of substrates (Figure 1d), and intramolecular processes do not appear to discriminate C–H bonds based on electronic properties (Figure 1e).24,25 Copper-catalyzed processes can have significant site-and chemoselectivity issues, furnishing mixtures of aminated products with substrates as simple as ethylbenzene.26,27 Iron and manganese catalyzed intermolecular C–H azidations have been reported having tolerance for nitrogen heterocyclic functionality, but these proceed via free radical pathways that result in poor site-selectivities and long-lived substrate radicals which scramble stereochemistry and may lead to skeletal rearrangements (Figure 1f).28,29 Herein, we report a manganese perchlorophthalocyanine [MnIII(ClPc)] catalyst for a highly site-selective and functional group tolerant intermolecular benzylic C(sp3)–H amination. This methodology enables preparative base-metal nitrene mediated late-stage amination on a broad range of natural products and pharmaceuticals, including ones containing multiple benzylic sites and basic nitrogen (tertiary amines, pyridines, and benzimidazole) functionality.
Results and discussion
Reaction development.
Reaction development commenced by examining commercially available first row transition metal catalysts previously demonstrated to be effective in intramolecular C–H amination via metallonitrene intermediates.19,20,30 The combination of 2,2,2-trichloroethyl sulfamate (TcesNH2) and PhI(OPiv)2 oxidant in the presence of iron phthalocyanine gave no product whereas manganese phthalocyanines gave trace product (Table 1, entries 1–4). In the presence of cobalt (II) tetraphenylporphyrin (CoII(TPP)), no reactivity was observed under in situ iminoiodinane conditions or previously reported TrocN3 conditions (entry 5).24 Mechanistic studies for intramolecular C–H amination processes promoted by manganese phthalocyanines suggested that in situ formation of iminoiodinane, an equilibrium process that favors the sulfamate ester, may be partially rate-determining.20 Examining if elimination of this step could enhance reactivity by using preformed iminoiodinane (PhI=NTces) gave improved yields of benzylic amination across all the phthalocyanine catalysts examined, with [MnIII(Pc)]SbF6 giving the best results (entries 6–8). The use of non-coordinating hexafluoroantimonate (SbF6–) counterion is thought to increase reactivity in these systems by furnishing a cationic metal complex with enhanced electrophilicity.19,20 We further hypothesized that electron withdrawing ligand modifications may promote formation of an electrophilic metallonitrene that is more reactive towards intermolecular C–H amination. Gratifyingly, a novel perchlorinated manganese phthalocyanine catalyst [MnIII(ClPc)] 4 [[MnIII(ClPc)] 4 = [MnIII(Cl16Pc)]SbF6], readily accessed in one step from commercial reagents, significantly improved the yield of desired amination to 53% (Table 1 entry 9, Fig. 2a).31 Increasing the reaction temperature from 23 °C to 40 °C further increased the yield of aminated product to 68% (entry 10). Lowering the catalyst loading to 5 mol% or cutting the PhI=NTces to 1.0 equivalent led to a 47% and 42% yield, respectively (entries 11–12). Replacing the benzene solvent with 1,2-dichloroethane, a polar aprotic solvent compatible with polar substrates and complex molecules (vide infra), gave a synthetically useful 60% yield (entry 13). In the absence of AgSbF6, a synthetically useful 50% yield is still obtained (entry 14); whereas elimination of [MnIII(ClPc)] 4 resulted in no product formation (entry 15). Re-examination of CoII(TPP) with iminoiodinane under the optimal [MnIII(ClPc)] 4 conditions did not lead to a productive reaction (entry 16). In situ iminoiodinane formation under these optimal conditions was unproductive (entry 17). It is important to note however that iminoiodinane (PhI=NTces) can be readily synthesized from commercial starting materials and upon isolation can be stored for months (see Supplementary Information section II).32 Interestingly, using preformed iminoiodinane for rhodium-catalyzed intermolecular benzylic C–H amination was reported to result in a significant decrease in yield relative to the in situ method.14
Table 1.
Development of [MnIII(ClPc)]-catalyzed intermolecular benzylic C–H amination.
 | |||||
|---|---|---|---|---|---|
| Entry* | Sulfamate Ester | Oxidant | Catalyst | Temp | % yield (% rsm) |
| 1 | NH2Tces | Phl(OPiv)2 | [FeIII(Pc)]SbF6 | 23 °c | 0% (100%) |
| 2 | NH2Tces | Phl(OPiv)2 | [MnIII(Pc)]SbF6 | 23 °c | trace (96%) |
| 3† | NH2Tces | Phl(OPiv)2 | [MnIII(Pc)]SbF6 | 23 °c | trace (95%) |
| 4 | NH2Tces | Phl(OPiv)2 | [MnIII(BuPc)]SbF6 | 23 °c | trace (94%) |
| 5‡ | NH2Tces | Phl(OPiv)2 | CoII(TPP) | 23 °c | 0%(99%) |
| 6 | Phl=NTces | [FeIII(Pc)]SbF6 | 23 °c | 9%(85%) | |
| 7 | Phl=NTces | [MnIII(tBuPc)]SbF6 | 23 °c | 12%(88%) | |
| 8 | Phl=NTces | [MnIII(Pc)]SbF6 | 23 °c | 17%(83%) | |
| 9 | Phl=NTces | [MnIII(ClPc)]SbF6 | 23 °c | 53%(41%) | |
| 10 | Phl=NTces | [MnIII(ClPc)]SbF6 | 40 °c | 68%(32%) | |
| 11§ | Phl=NTces | [MnIII(ClPc)]SbF6 | 40 °c | 47%(49%) | |
| 12‖ | Phl=NTces | [MnIII(ClPc)]SbF6 | 40 °c | 42%(58%) | |
| 13¶ | Phl=NTces | [MnIII(ClPc)]SbF6 | 40 °c | 60%(24%) | |
| 14 | Phl=NTces | [MnIII(ClPc)]Cl | 40 °c | 0%(100%) | |
| 15 | Phl=NTces | AgSbF6 | 40 °c | trace(100%) | |
| 16 | Phl=NTces | CoII(TPP) | 40 °c | trace(95%) | |
| 17 | NH2 Tces | Phl(OPiv)2 | [MnIII(ClPc)]SbF6 | 40 °c | trace(87%) |
Reaction conditions: 1 (0.2 mmol, 1 equiv), catalyst (10 mol%), AgSbF6 (10 mol%), oxidant (2 equiv), C6H6 (0.5M), 5Å molecular sieves (40 mg), 8 h. Yields are of isolated products. Trace yield (<5%) and recovered starting material (rsm) are reported based on 1H NMR analysis of the crude reaction using 1,3,5-trimethylbenzene as an internal standard.
2.2 equiv of MgO used as an additive in the reaction.
A separate reaction with 1 (1 equiv), TrocN3 (2 equiv), CoII(TPP) (10 mol%), C6H6 (0.5M), 40 ˚C, 5Å molecular sieves (40 mg), 8 h resulted in 0% yield (88% rsm). TPP = tetraphenylporphyrin.
5 mol% catalyst used.
1 equiv of PhI=NTces used.
3Å molecular sieves used. 1,2-DCE used as solvent.
Figure 2. Intermolecular benzylic C–H amination substrate scope.
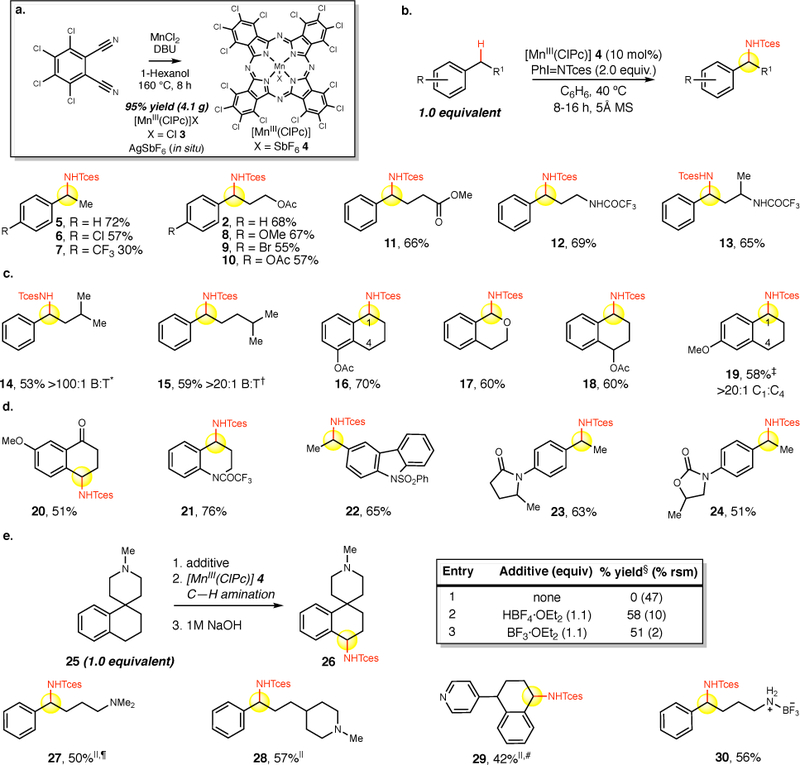
a. One-step, gram-scale synthesis of [MnIII(ClPc)]Cl from commercial materials. b. General reaction conditions and evaluation of aryl and alkyl electronic effects. Reaction conditions: substrate (0.2 mmol, 1 equiv), [MnIII(ClPc)]Cl 3 (10 mol%), AgSbF6 (10 mol%), PhI=NTces (2 equiv), C6H6 (0.5M), 5Å molecular sieves (40 mg), 40 ˚C, 8–16 h. Isolated yields are average of three runs. Diastereomeric ratios are 1:1 unless otherwise noted c. Evaluation of site-selectivity: benzylic versus tertiary and sterically and electronically differentiated benzylic C–H bonds. d. Nitrogen-containing heterocyclic substrates with electron-withdrawing groups on nitrogen. e. Brønsted and Lewis acid protection strategy for basic tertiary amine and pyridine-containing substrates. *No tertiary product detected by HPLC (detection limit 0.0002 mg/mL using authentic product). †No tertiary product detected in 1H NMR crude or after purification. ‡No monoamination product detected at C4 by HPLC (detection limit 0.00015 mg/mL using an authentic product); trace diamination detected using authentic product by HPLC, not seen by 1H NMR. An imine product at C1 was isolated in 12% yield. §Overall three step yield (1,2-DCE (0.5M): acid complexation, amination and decomplexation. ||3 equivalents of PhI=NTces. ¶15 mol% catalyst used. #3Å molecular sieves.
Reaction scope.
We next evaluated the substrate scope of this transformation under preparative conditions (all examples are with 1 equiv. of substrate) using the novel and readily accessible [MnIII(ClPc)] 4 catalyst, synthesized in one step as the chloride [MnIII(ClPc)]Cl 3 from commercial starting materials (95% yield, multi-gram scale, Figure 2a) followed by an in situ metathesis with equimolar AgSbF6. In contrast to results with previously reported base-metal catalysts, ethyl benzene furnished good yields and selectivities of aminated product (Figure 2b, 5, 72%).24,27 Significantly substituting the para position with electron-withdrawing functionality (Cl, σpara= 0.23, CF3, σpara= 0.54) led to diminished yields as the sigma value of the substituent increased (6, 57%; 7, 30%). A similar trend is observed when varying the aromatic ring electronics on screening substrate 1 where a bias for electron-neutral and electron-rich aromatic systems can be observed (2, 8–10, 55–68% yields). Remote electron-withdrawing aliphatic functionality is well tolerated in this reaction as seen with ester and trifluoroacetamide containing compounds undergoing [MnIII(ClPc)] 4 catalyzed aminations in good yields (11–13, 65–69% yields).
We explored the selectivity of the [MnIII(ClPc)] 4-catalyzed benzylic C–H amination in substrates containing more than one potential site for functionalization. In substrates containing both a benzylic and a tertiary site, we observed selective amination at the benzylic position with no tertiary amination observed by LC-MS and/or 1H NMR analysis (Figure 2c, 14, 53% yield, >100:1 benzylic:tertiary (B:T) amination; 15, 59% yield, >20:1 B:T). These results are orthogonal to noble metal catalyzed methods where mixtures (often inseparable) of benzylic and tertiary amination products are routinely observed (Figure 1b).14,15,17 In substrates containing multiple benzylic sites, we found an amination preference for the least sterically encumbered and most electron-rich site. For example, evaluation of 5-acetoxytetralin bearing two methylene benzylic sites (C1 and C4) revealed preferential amination at the C1 site over the C4 site, likely because of the 1,3 strain present at C4 (16, 70% yield). Oxygen substitution in cyclic ethers activates adjacent C–H bonds towards amination via hyperconjugation (17, 60% yield). In contrast oxygen with an electron withdrawing substituent like acyl deactivates the proximal C–H bond toward amination (18, 60% yield, 1:1 d.r.), likely due to a combination of electronic and steric deactivation. Electronic substitution on the aromatic ring can have substantial effects on site-selectivity and reactivity in this reaction. For example, 6-methoxytetralin contains two potential sites for oxidation, differentiated by the electron-donating para-methoxy group on the phenyl ring. We isolate amination product at C1 (19, 58%), the more electronically favored site, with only trace diaminated product detected in the crude reaction mixture (determined by HPLC). In contrast, under rhodium catalysis an inseparable 7:1 mixture of C1:C4 amination products is reported alongside a 15% yield of diamination product.14 The methoxy substitution on 7-methoxytetralone provides electronic activation to the benzylic methylene C–H bonds to achieve a synthetically useful 51% yield of aminated tetralone 20 (Figure 2d). It is significant to note that an unsubstituted α-tetralone affords only trace amounts of aminated product.
Evaluation of nitrogen containing heterocycles commenced using strongly electron-withdrawing protecting groups for secondary amine functionality. Tetrahydroquinoline is a viable substrate for C–H amination when the nitrogen is protected as a trifluoroacetamide (Figure 2d, 21, 76% yield). Carbazole, a key heterocycle in many alkaloids, was aminated at the pendent benzylic site in good yield (22, 65%) when the secondary amine was protected with a phenylsulfonyl group. Moderately basic amide-containing heterocycles were also examined under [MnIII(ClPc)] 4 catalysis: a N-aryl lactam derived substrate underwent benzylic C–H amination in good yield (23, 63% yield, 1:1 d.r.) and N-aryl oxazolidinone, a structural motif found in pharmaceuticals like linezolid, was aminated to form 24 in 51% yield (1:1 d.r.). The lack of diastereoselectivity observed with these substrates is likely attributed to the substrate’s stereogenic center being distal to the site of C–H amination.
Despite the high prevalence of basic tertiary amines and basic heterocycles in pharmaceuticals and natural products, previous methods for metal-catalyzed C–H amination proceeding via metallonitrene intermediates have not demonstrated tolerance for this functionality in remote C–H aminations. Indeed, examining this amination protocol in a substrate containing a tertiary piperidine yielded no benzylic amination product with 47% recovered starting material (Figure 2e, entry 1). We hypothesized that the basic amine may bind strongly to the metal, inhibiting catalysis and/or undergo α-amination or direct N-amination to furnish polar products that are not easily isolated.18 We examined Brønsted acid and Lewis acid complexation strategies that have been demonstrated to circumvent such issues in the context of aliphatic C–H hydroxylations.33–35 Both protection methods produced the desired product in good yield (26, 58% and 51% yields, entries 2–3). Analogous to previous reports, HBF4 has proven to be more effective for tertiary amines having steric bulk. However, the BF3 protecting method enhances substrate solubility and is beneficial with more polar substrates facing isolation challenges (vide infra).
This strategy was evaluated with four basic amine-containing substrates containing pharmaceutically common motifs. The linear 3° amine and N-methylpiperidine containing substrates were aminated using HBF4/[MnIII(ClPc)] 4 to afford 27 (Figure 2e, 50% yield) and 28 (57% yield), respectively. Pyridine, the most prevalent heteroaromatic in pharmaceuticals,36 was also tolerated using the HBF4 protonation strategy. A useful yield was observed for 2° benzylic amination in a pyridyl substituted tetrahydronaphthalene with no 3° benzylic C–H amination observed likely due to sterics and the strong electron-deactivation of the pyridyl ring upon nitrogen protonation (29, 42% yield, 1:1 d.r.). A primary amine-containing substrate can also be conveniently protected as a BF3 salt and aminated using the [MnIII(ClPc)] 4 catalyst to afford a product that can be readily isolated via silica gel chromatography (30, 56% yield), stored as a bench stable solid, and easily deprotected using a hydroxide base or fluoride anion.34
Late-stage benzylic C–H amination of bioactive molecules.
Site-selective intermolecular C–H functionalization processes that may be used at a late-stage constitute powerful methods because of their capacity to make atomistic changes that may profoundly impact a molecule’s biological and physical properties.37,38 We sought to evaluate the [MnIII(ClPc)] 4-catalyzed benzylic C–H amination in such pharmaceutical settings where late-stage amination may be beneficial (Figure 3a). FKGK11, a group VIA calcium-independent phospholipase A2, with a pentafluoroethyl carbonyl underwent benzylic amination to afford 31 in 51% yield. A retinoic acid receptor agonist analog39 having a biaryl moiety underwent benzylic amination to afford 32 in 76% yield, which could be readily deprotected using a Zn/Cu couple to afford the primary amine (33, 88% yield).14 Analogous to previous substrates, electron deficient nitrogen is well-tolerated: a P-glycoprotein inhibitor containing an imide functionality and a spiropiperidine σ receptor agonist precursor containing a 2° amine protected as a trifluoroacetamide were efficiently aminated in excellent yields to afford 34 (84% yield) and 35 (85% yield), respectively.40,41 Citalopram, a widely used antidepressant containing a linear 3° amine, was evaluated for late-stage C—H amination with HBF4/[MnIII(ClPc)] 4. Benzylic amination product 36 was obtained as a single diastereomer in 71% yield. It is significant to note that no intermolecular C–H amination methods have thus far demonstrated the capacity to preparatively aminate such a broad range of medicinally relevant substrates.
Figure 3. Late-stage benzylic C–H amination of bioactive molecules.
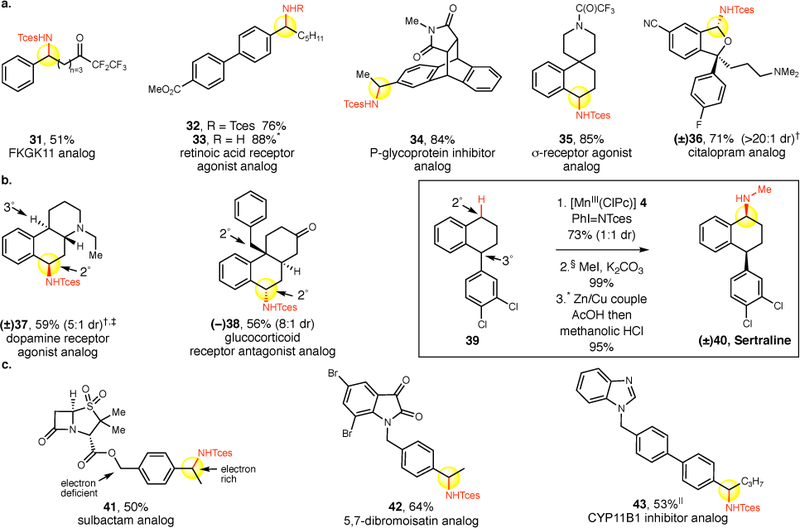
a. [MnIII(ClPc)] 4 catalyzed benzylic C–H amination (reaction conditions in Figure 2, isolated yields are average of three runs, diastereomeric ratios are 1:1 unless otherwise noted) selectively installs Tces-protected amines into bioactive molecules that can be converted to primary amines (32→33) b. Bioactive molecules with multiple reactive benzylic C–H bonds are selectively aminated at the most sterically accessible site. c. Bioactive molecules are selectively aminated at the most electron rich site. *Tces deprotection performed using Zn/Cu couple (10 equiv), MeOH:AcOH (1:1). After filtration through celite plug the concentrated white solid was stirred in methanolic HCl at 40 ˚C for 12 h to yield the corresponding amine. †3Å molecular sieves (40 mg) used. Substrate protonated with HBF4•OEt2 (1.1 equiv) in CH2Cl2 prior to amination then deprotonated with 1M NaOH in CH2Cl2 after amination. ‡3 equiv of iminoiodinane used. §Only the cis-isomer used for alkylation, deprotection. ||3Å molecular sieves (40 mg) used. Substrate complexed with BF3•OEt2 (1.1 equiv) in CH2Cl2 prior to amination and decomplexed with tetramethylethylenediamine (TMEDA) in CH2Cl2.
We next evaluated bioactive molecules with multiple benzylic C–H bond sites that differ primarily in their steric environments. In a dopamine receptor agonist analog having both 2° and 3° benzylic sites, the HBF4/[MnIII(ClPc)] 4 catalyzed amination protocol was effective at selectively functionalizing the more exposed 2° benzylic position to afford 37 (Figure 3b, 59% yield, 5:1 d.r.).42 Remarkably, even a glucocorticoid receptor agonist analog having two sterically differentiated 2° benzylic sites was selectively aminated at the less hindered site flanked by a methylene, rather than the one adjacent to a quaternary center, to furnish 38 as one regioisomer in 56% yield (8:1 d.r.).43 Anti-depressant sertraline 40 was accessed from 39 via a high yielding 3-step synthetic sequence. Site-selective amination of a 2° benzylic C–H bond in the presence of a sterically encumbered but highly activated 3° dibenzylic C–H bond proceeded in 73% yield (1:1 d.r. of an easily separable mixture of cis and trans diastereomers). N-methylation of the cis-diastereomer proceeded quantitatively followed by Tces removal with Zn/Cu couple (95% yield) to afford sertraline 40. These examples serve to highlight the high sensitivity of this C—H amination method to the steric environments of benzylic C–H bonds.
Bioactive molecules with electronically differentiated 2° benzylic C–H sites, proximal and distal to electron-withdrawing functionality were evaluated (Figure 3c). In an alkylated analog of the antibiotic sulbactam, a 50% yield (1:1 d.r.) of aminated product 41 was isolated at the more electron rich benzylic site distal from the ester linkage. A 5,7-dibromoisatin analog, shown to have anti colon cancer activity,44 was preferentially aminated at the benzylic site remote from the electron deficient nitrogen moiety to furnish 42 in 64% yield. We evaluated our C–H amination protocol in a CYP11B1 inhibitor analog45 having a benzimidazole functionality. Use of the BF3 complexation strategy led to an increase in substrate solubility, and we found amination occurred at the benzylic site remote from the electron deficient heteroaromatic to furnish 43 in 53% yield. These examples further underscore the [MnIII(ClPc)] 4-catalyzed benzylic C–H amination’s sensitivity to electronics with preparatively meaningful site-selectivities being achieved for amination at electron rich benzylic C–H bonds relative to sites proximal to electron withdrawing functionality. Such sensitivity to electronics is consistent with an electrophilic metallonitrene intermediate and inconsistent with a nonelectrophilic metalloradical intermediate, evoked in Co-mediated intramolecular C–H aminations that demonstrate poor sensitivity to substrate electronics (Figure 1e, vide infra).25
We explored the capacity for the [MnIII(ClPc)] catalyst to effect late-stage benzylic C–H amination in complex natural product settings (Figure 4). The COCF3-leelamine analog, a diterpene amine with two sterically differentiated 2° and 3° benzylic sites, was selectively aminated at the 2° benzylic site to afford 44 (62% yield, 6:1 dr) as a single regioisomer. The Zn/Cu couple Tces deprotection is chemoselective for the Tces amine in the presence of the COCF3-amine and furnished 45 in 70% yield. The 2,8-dioxabicyclo[3.3.1]nonane skeleton is contained in biflavonoid natural product motifs that exhibit intriguing medicinal properties which include antiviral, anti-inflammatory and antitumor properties.46 Benzylic amination in the presence of this biflavonoid natural product core was achieved to give 46 in a 73% yield (1:1 d.r.). Further diversification of the biflavonoid natural product core with a pyridine was performed, and using the HBF4/amination protocol, a 57% yield (1:1 d.r.) of benzylic amination product 47 was ultimately realized. Significantly, the acid sensitive ketal functionality remains intact under the HBF4 complexation protocol.
Figure 4. Late-stage benzylic C–H amination of natural products.
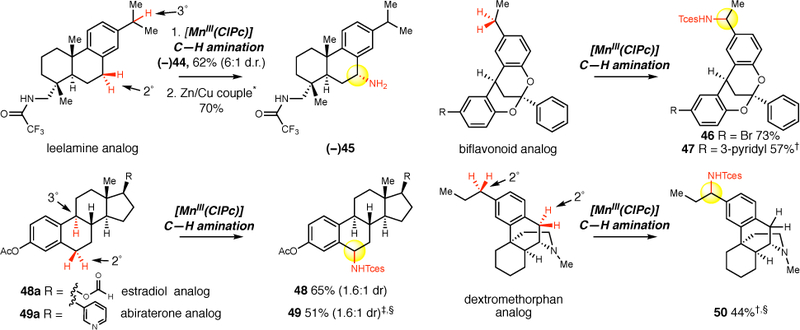
The late-stage functionalization of six natural product analogs is demonstrated with [MnIII(ClPc)] 4 catalysis (reaction conditions in Figure 2, isolated yields are average of three runs, diastereomeric ratios are 1:1 unless otherwise noted) to give preparative yields and excellent levels of site-selectivity. *Tces deprotection: Zn/Cu couple protocol (Figure 3). †3Å molecular sieves (40 mg) used. HBF4•OEt2 (1.1 equiv, CH2Cl2) amine protection followed by 1M NaOH (CH2Cl2) deprotection after amination. ‡3Å molecular sieves (40 mg) used. BF3•OEt2 (1.1 equiv, CH2Cl2) pyridine protection prior to amination, then deprotection with tetramethylethylenediamine in CH2Cl2 after amination. §3 equiv of iminoiodinane used.
Previously, low yields or highly complex mixtures of products were furnished from the C—H azidation of estrone analogs (Figure 1f).28,29 We evaluated a formylated OAc-estradiol analog and a structurally analogous pyridyl containing abiraterone analog, both having two potential amination sites (2° and 3° benzylic C(sp3) –H bonds) (Figure 4). In both cases, we observed highly site-selective 2° benzylic amination (3° benzylic amination was not observed) in synthetically useful yields (65% yield (1.6:1 d.r.) 48; 51% yield (1.6:1 d.r.) 49). In the case of the pyridyl containing steroid, complexation with BF3 was found to be beneficial in generating a BF3-pyridine complex soluble in 1,2-dichloroethane. These examples further underscore the ability of the [MnIII(ClPc)] 4 catalyst to discriminate between small differences in the steric environments of benzylic sites.
Previous efforts to diversify the 3° amine-containing alkaloid dextromethorphan using a rhodium nitrene provided amination at the 3° amine and the methyl group adjacent to the 3° amine (Figure 1c).18 We postulated that base-metal nitrene catalyzed amination at a benzylic site remote from the 3° amine would be possible after irreversible protonation of dextromethorphan with HBF4, as the protonated 3° amine renders the nearby C–H bonds electron deficient. Gratifyingly, our protonated dextromethorphan analog was aminated in 44% yield (1:1 d.r.) at the benzylic site most remote from the 3° amine (Figure 4).
Mechanistic studies.
We hypothesized the [MnIII(ClPc)] 4-catalyzed intermolecular benzylic C–H amination reaction proceeds via formation of an electrophilic metallonitrene followed by a rate-determining C–H cleavage step to afford a benzylic radical that undergoes rebound from the Mn-imido intermediate to form the benzylic aminated product (Figure 5a). Whereas in the intramolecular [MnIII(tBuPc)]-catalyzed C–H amination, a step prior to C–H cleavage, such as iminoiodinane formation, was found to be partially rate-determining,20 we envisioned that preforming the iminoiodinane would now render C–H cleavage as the rate-determining step. To assess this hypothesis, the intermolecular kinetic isotope effect (KIE) was measured to evaluate the effect of C–H cleavage on the overall reaction rate and compared to the intramolecular KIE that directly probes the C–H cleavage step (Figure 5b). Initial rates measured on parallel reactions with substrates 1 and 1-d2 gave a primary KIE of 2.5 ± 0.2. This value was closely matched with the intramolecular KIE of 3.0 ± 0.1 measured on monodeuterated substrate 51, suggesting that C–H cleavage is likely the rate-determining step of this intermolecular benzylic C–H amination. The intramolecular KIE is similar to those obtained with other base metal phthalocyanine catalysts (Mn and Fe) that catalyze intramolecular benzylic C–H aminations (KIE = 4.2 and 4.8, respectively),20 however is appreciably lower than that reported for cobalt porphyrins that catalyze intramolecular allylic C–H aminations (KIE = 6.2).47 Therefore, while this data supports our hypothesis that [MnIII(ClPc)] 4 proceeds through a stepwise mechanism, C–H bond breakage appears to occur to a much lesser extent in the transition structure than with Co porphyrin catalysis where such a stepwise mechanism has also been evoked.25,47
Figure 5. Mechanism of [MnIII(ClPc)] catalyzed benzylic C–H amination.
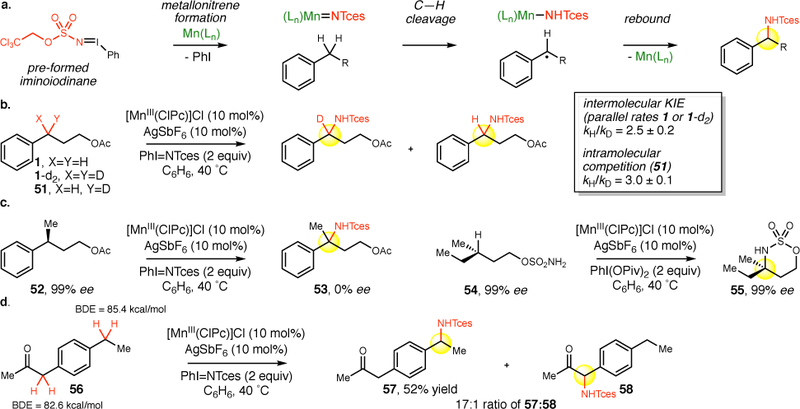
a. Proposed stepwise mechanism for the [MnIII(ClPc)] catalyzed benzylic C–H amination. b. C–H cleavage step is rate-determining step. Intermolecular kinetic isotope effect (KIE), evaluating the effect of C–H cleavage on overall reaction rates, closely matches the intramolecular KIE that directly probes the C–H cleavage step. c. The intermolecular rebound of the Mn-imido with a stabilized benzylic radical proceeds with racemization of stereogenic centers. Intramolecular rebound with an aliphatic carbon-centered radical proceeds with complete retention of stereochemistry. d. Site-selectivity of [MnIII(ClPc)] catalyzed C–H amination is probed in a substrate containing two 2˚ benzylic C(sp3)–H sites that differ electronically and in bond dissociation energies (BDE). The high site-selectivity for the most electron rich site having a higher BDE suggests that the reaction proceeds via an electrophilic metallonitrene intermediate. The 17:1 ratio of 57:58 was measured by HPLC.
We next evaluated the proposed rebound step of the Mn-imido with the substrate benzylic radical intermediate (Figure 5c). C–H amination at a defined benzylic stereocenter in 52 proceeds with complete erosion in stereochemistry with [MnIII(ClPc)] 4, supporting the intermediacy of a stabilized carbon-centered radical. Importantly, intramolecular C–H amination with [MnIII(ClPc)] 4 at a defined aliphatic stereocenter in 54 proceeds with complete stereoretention, as had been previously reported with [MnIII(tBuPc)].20 These results support a step-wise mechanism and suggest that the rate of the rebound step is dependent on the stability of the carbon-centered radical intermediate.
We sought to probe the nature of the aminating species to distinguish between an electrophilic metallonitrene that has been suggested for rhodium, manganese, and iron catalysis versus a nonelectrophilic metalloradical intermediate evoked in cobalt catalysis (Figure 5d). In intramolecular C–H amination studies, rhodium, manganese, and iron mediated C–H aminations preferred the more electron rich C–H site whereas cobalt mediated C–H aminations were relatively insensitive to electronics and exhibited site-selectivity based primarily on differences in bond dissociation energies (BDE, Figure 1e).11,20,25 We directly probed this by examining [MnIII(ClPc)] 4-catalyzed C–H amination in substrate 56, having two benzylic sites that are electronically distinct and have different BDEs (Figure 5d).48 [MnIII(ClPc)] 4 demonstrates a strong preference for amination at the more electron rich benzylic site having the higher BDE versus the electron deficient benzylic site α to a carbonyl having a lower BDE, furnishing 57 in 52% isolated yield with 17:1 site-selectivity (57:58). This outcome contrasts site-selectivity trends reported with nonelectrophilic metalloradical mediated cobalt reactions that preferentially aminate even highly electron deficient C–H bonds α to carbonyls because of their low BDEs (Figure 1e).25 Collectively, this data suggests that Mn phthalocyanine catalyzed benzylic C–H aminations proceed through a stepwise mechanism via electrophilic metallonitrene intermediates where C–H bond cleavage is tightly regulated at the metal center and occurs preferentially at the most electron rich and sterically accessible site.
Conclusion
A base-metal catalyzed intermolecular benzylic C–H amination is reported that proceeds with unprecedented levels of reactivity, site-selectivity and functional group tolerance. The [MnIII(ClPc)] 4 catalyst is synthesized in a one step, scalable reaction from abundant and sustainable commercial starting materials.49 The [MnIII(ClPc)] 4 catalyzed C–H amination demonstrates high site-selectivity in molecules containing multiple reactive C–H bonds based on steric and electronic differentiation.50 Moreover, the catalyst is compatible with both Brønsted and Lewis acid complexed basic nitrogen functionality, enabling remote C–H amination in molecules containing tertiary amines and heterocycles that cannot effectively be masked via alternative methods. Collectively, the combination of reactivity and selectivity makes this [MnIII(ClPc)] 4-catalyzed intermolecular benzylic C(sp3)–H amination uniquely effective at late-stage functionalization of bioactive molecules and natural products. Mechanistic studies suggest that this method proceeds via a stepwise C–H amination pathway with C–H cleavage as the rate-determining step. A competition study and empirical data on the impact of electronic effects on site-selectivities suggest that the nature of the key intermediate is that of an electrophilic metallonitrene. Collectively, these features significantly contrast with other base-metal catalyzed C–H aminations that are poorly reactive intermolecularly and generally proceed via intermediates with more free radical character that exhibit less control on site-selectivity. We envision that the high reactivity and selectivity of this reaction will make it uniquely effective for late-stage installation of valuable nitrogen functionality in complex molecule settings. Moreover, we anticipate that the mechanistic features will enable tunable control of site-selectivity when the reactivity is increased for intermolecular amination of other C(sp3)–H bond types.
Methods
A general procedure for the manganese-catalyzed benzylic amination is as follows. In a 10 mL round-bottom flask, 5Å powdered molecular sieves (40 mg) and a Teflon stir bar were added. The flask was sealed with a Suba Seal rubber septum, placed under vacuum, flame-dried for 45 seconds to activate the molecular sieves, cooled under a purged and completely air-free argon balloon and wrapped in foil to exclude light. Once cooled, solvent (0.40 mL, 0.5 M to substrate) and substrate (0.20 mmol, 1 equiv) were added and stirred for 10 minutes. Manganese (III) perchlorophthalocyanine chloride 3 (23.1 mg, 0.020 mmol, 0.1 equiv) and silver hexafluoroantimonate (6.9 mg, 0.020 mmol, 0.1 equiv) were weighed in a foil-wrapped 1 dram vial in the glove box and sealed with a Teflon cap. The vial was removed from the glove box and the contents added directly to the round bottom flask while maintaining an argon atmosphere, then stirred for 10 minutes at room temperature. In a 1 dram vial open to air, 2,2,2-trichloroethyl (phenyl-λ3-iodanylidene)sulfamate (172.2 mg, 0.40 mmol, 2 equiv) was weighed and added directly to the round bottom flask while maintaining an argon atmosphere. The Suba Seal rubber septum was replaced by a polyethylene cap, sealed tightly and placed in a 40 °C oil bath for 8 hours with stirring. Upon reaction completion, the reaction was filtered through a 1 inch silica gel plug using diethyl ether or ethyl acetate as the eluent. The crude material was concentrated and dry-loaded directly onto a silica gel column.
A general procedure for the manganese-catalyzed benzylic amination of basic nitrogen-containing substrates is as follows. In a 1-dram vial equipped with a stir bar were added the nitrogen-containing substrate (0.20 mmol, 1 equiv) and methylene chloride (DCM) (0.8 mL). Tetrafluoroboric acid diethyl ether complex (HBF4•OEt2) (30.2 μL, 35.6 mg, 0.22 mmol, 1.1 equiv) or boron trifluoride diethyl etherate (BF3•OEt2) (27.2 μL, 31.2 mg, 0.22 mmol, 1.1 equiv) was added dropwise while stirring at room temperature. The reaction mixture was stirred for 1 h. Upon reaction completion, the stir bar was removed, and the mixture was concentrated in vacuo and placed on vacuum overnight. A 10 mL round-bottom flask containing 3Å powdered molecular sieves (40 mg) and a Teflon stir bar was set up and flame-dried according to the general procedure above. In the 1-dram vial carrying the protonated substrate was added 0.2 mL of anhydrous 1,2-dichloroethane (DCE). The resulting solution or suspension was added into the round-bottom flask containing the 3Å molecular sieves. This process was repeated 2x with 0.1 mL DCE each time to ensure complete transfer. The amination reaction was then set up according to the general procedure above. The reaction was then placed in a 40 °C oil bath for 15 hours with stirring. Upon completion, the flask was removed from the oil bath. Sodium hydroxide solution (1M, 3 mL) and CH2Cl2 (3 mL) were added and the reaction mixture was vigorously stirred for 15 min (for HBF4 deprotection) or 4 h (for BF3 deprotection of amines), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3×5 mL). The organic layers were combined, dried over anhydrous potassium carbonate, filtered and concentrated via rotary evaporation. The crude material was purified by flash chromatography to afford the aminated product. For deprotection of BF3 in pyridine and benzimidazole containing substrates, tetramethylethylenediamine (TMEDA) (150 μL, 116 mg, 1.0 mmol, 5 equiv) was added instead of NaOH, followed by CH2Cl2 (1 mL). The resulting mixture was stirred for 4 h and loaded directly onto a silica column.
Supplementary Material
Acknowledgements
Financial support for this work was provided by the NIGMS Maximizing Investigators’ Research Award MIRA (R35 GM122525). J.R.C is an NIH Ruth Kirschstein Postdoctoral Fellow (1 F32GM112501–01A1). The authors thank Dr. Danielle L. Gray and Dr. Toby J. Woods for crystallographic analysis of compounds 36, 37, 38 and 45. We thank Dr. L. Zhu for assistance with nuclear magnetic resonance spectroscopy and acknowledge Dr. Takeshi Nanjo, Rulin Ma, Wei Liu, and Dr. Jennifer Griffin for checking our experimental procedure.
Footnotes
Data availability. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1587014 ((±)−36), 1587015 ((±)−37), 1587016 ((–)−38), 1587017 ((–)−44). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
Additional information
Supplementary information and chemical compound information are available in the online version of the paper. Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to M.C.W.
Competing financial interests
The University of Illinois has filed a patent application on the [Mn(ClPc)] catalyst for intermolecular C–H functionalization. The [Mn(ClPc)] catalyst (product # 901425) will be offered by MilliporeSigma through a license from the University of Illinois.
References
- 1.Richter MF et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acred P, Brown DM, Turner DH & Wilson MJ Pharmacology and Chemotherapy of ampicillin -A new broad-spectrum penicillin. Brit. J. Pharmacol 18, 356–369 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGrath NA, Brichacek M & Njardarson JT A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ 87, 1348–1349 (2010). [Google Scholar]
- 4.Hili R & Yudin AK Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol 2, 284–287 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Hubbard BK, Thomas MG & Walsh CT Biosynthesis of L-p-hydroxyphenylglycine, a non-proteinogenic amino acid constituent of peptide antibiotics. Cell Chem. Biol 7, 931–942 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Carey JS, Laffan D, Thomson C & Williams MT Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem 4, 2337–2347 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Dugger RW, Ragan JA & Ripin DHB Survey of GMP bulk reactions run in a research facility between 1985 and 2002. Org. Process Res. Dev 9, 253–258 (2005). [Google Scholar]
- 8.Roughley SD & Jordan AM The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem 54, 3451–3479 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Li JWH & Vederas JC Drug discovery and natural products: end of an era or an endless frontier? Science 325, 161–165 (2009). [DOI] [PubMed] [Google Scholar]
- 10.DeCorte BL Underexplored opportunities for natural products in drug discovery. J. Med. Chem 59, 9295–9304 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Roizen JL, Harvey ME & Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C—H bonds. Acc. Chem. Res 45, 911–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dequirez G, Pons V & Dauban P Nitrene chemistry in organic synthesis: still in its infancy? Angew. Chem. Int. Ed 51, 7384–7395 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Huard K & Lebel H N-tosyloxycarbamates as reagents in rhodium-catalyzed C–H amination reactions. Chem. Eur. J 14, 6222–6230 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Fiori KW & Du Bois J Catalytic intermolecular amination of C–H bonds: method development and mechanistic insights. J. Am. Chem. Soc 129, 562–568 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Roizen JL, Zalatan DN & Du Bois, J. Selective intermolecular amination of C–H bonds at tertiary carbon centers. Angew. Chem. Int. Ed 52, 11343–11346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang C et al. Efficient diastereoselective intermolecular rhodium-catalyzed C–H amination. Angew. Chem. Int. Ed 45, 4641–4644 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Bess EN et al. Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C–H amination reactions. J. Am. Chem. Soc 136, 5783–5789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J et al. Simultaneous structure–activity studies and arming of natural products by C–H amination reveal cellular targets of eupalmerin acetate. Nat. Chem 5, 510–517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paradine SM & White MC Iron-catalyzed intramolecular allylic C–H amination. J. Am. Chem. Soc 134, 2036–2039 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Paradine SM et al. A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp3) –H amination. Nat. Chem 7, 987–994 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hennessy ET & Betley TA Complex N-heterocycle synthesis via iron-catalyzed, direct C–H bond amination. Science 340, 591–595 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Hennessy ET, Liu RY, Iovan DA, Duncan RA & Betley TA Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci 5, 1526–1532 (2014). [Google Scholar]
- 23.Liu Y et al. Nonheme iron-mediated amination of C(sp3) –H bonds. Quinquepyridine-supported iron-imide/nitrene intermediates by experimental studies and DFT calculations. J. Am. Chem. Soc 135, 7194–7204 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Lu H, Subbarayan V, Tao J & Zhang XP Cobalt(II)-catalyzed intermolecular benzylic C–H amination with 2,2,2-trichloroethoxycarbonyl azide (TrocN3). Organometallics 29, 389–393 (2010). [Google Scholar]
- 25.Lu H, Hu Y, Jiang H, Wojtas L & Zhang XP Stereoselective radical amination of electron deficient C(sp3) –H bonds by Co(II)-based metalloradical catalysis: direct synthesis of α-amino acid derivatives via α-C–H amination. Org. Lett 14, 5158–5161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gephart RT III & Warren TH Copper-catalyzed sp3 C–H amination. Organometallics 31, 7728–7752 (2012). [Google Scholar]
- 27.Fructos MR, Trofimenko S, Díaz-Requejo MM & Pérez PJ Facile amine formation by intermolecular catalytic amidation of carbon–hydrogen bonds. J. Am. Chem. Soc 128, 11784–11791 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Bergsten TM, & Groves JT Manganese-catalyzed late-stage aliphatic C–H azidation. J. Am. Chem. Soc 137, 5300–5303 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Karimov RR, Sharma A & Hartwig JF Late stage azidation of complex molecules. ACS Cent. Sci 2, 715–724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruppel JV; Kamble RM & Zhang XP Cobalt-catalyzed intramolecular C–H amination with arylsulfonyl azides. Org. Lett 9, 4889–4892 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Safari N et al. Rapid and efficient synthesis of metallophthalocyanines in ionic liquid. J. Porphyrins Phthalocyanines 9, 256–261 (2005). [Google Scholar]
- 32.Du Bois J 2,2,2-Trichloroethoxysulfonamide. e-EROS Encyclopedia of Reagents for Organic Synthesis (2009).
- 33.Asensio G, González-Núñez ME, Bernardini CB, Mello R & Adam W Regioselective oxyfunctionalization of unactivated tertiary and secondary C–H bonds of alkylamines by methyl(trifluoromethyl)dioxirane in acid medium. J. Am. Chem. Soc 115, 7250–7253 (1993). [Google Scholar]
- 34.Howell JM, Feng K, Clark JR, Trzepkowski LJ & White MC Remote oxidation of aliphatic C—H bonds in nitrogen-containing molecules. J. Am. Chem. Soc 137, 14590–14593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee M & Sanford MS Platinum-catalyzed, terminal selective C(sp3) –H oxidation of aliphatic amines. J. Am. Chem. Soc 137, 12796–12799 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor RD, MacCoss M & Lawson ADG Rings in drugs. J. Med. Chem 57, 5845–5859 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Stepan AF, Mascitti V, Beaumont K & Kalgutkar AS Metabolism-guided drug design. Med. Chem. Commun 4, 631–652 (2013). [Google Scholar]
- 38.Cernak T, Dykstra KD, Tyagarajan S, Vachal P & Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Lund BW et al. Discovery of a potent, orally available, and isoform-selective retinoic acid β2 receptor agonist. J. Med. Chem 48, 7517–7519 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Alibert S et al. Effects of a series of dihydroanthracene derivatives on drug efflux in multidrug resistant cancer cells. Eur. J. Med. Chem 38, 253–263 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Chambers MS et al. Spiropiperidines as high-affinity, selective σ ligands. J. Med. Chem 35, 2033–2039 (1992). [DOI] [PubMed] [Google Scholar]
- 42.Wikström H et al. N-substituted 1,2,3,4,4a,5,6,10b-octahydrobenzo[f]quinolines and 3-phenylpiperidines: effects on central dopamine and σ receptors. J. Med. Chem 30, 2169–2174 (1987). [DOI] [PubMed] [Google Scholar]
- 43.Morgan BP et al. Discovery of potent, nonsteroidal, and highly selective glucocorticoid receptor antagonists. J. Med. Chem 45, 2417–2424 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Krishnegowda G et al. Synthesis and biological evaluation of a novel class of isatin analogs as dual inhibitors of tubulin polymerization and Akt pathway. Bioorg. Med. Chem 19, 6006–6014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hille UE, Zimmer C, Vock CA & Hartmann RW First selective CYP11B1 inhibitors for the treatment of cortisol-dependent diseases. ACS Med. Chem. Lett 2, 2–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang X, Song Z, Xu C, Yao Q & Zhang A (D,L)-10-Camphorsulfonic-acid-catalysed synthesis of diaryl-fused 2,8-dioxabicyclo[3.3.1]nonanes from 2-hydroxychalcones and naphthol derivatives. Eur. J. Org. Chem 418–425 (2014). [Google Scholar]
- 47.Lu H, Jiang H, Hu Y, Wojtas L & Zhang XP Chemoselective intramolecular allylic C–H amination versus C=C aziridination through Co(II)-based metalloradical catalysis. Chem. Sci 2, 2361–2366 (2011). [Google Scholar]
- 48.Luo Y-R Comprehensive Handbook of Chemical Bond Energies Ch. 3 (CRC Press; 2007). [Google Scholar]
- 49.Greenwood NN & Earnshaw A Chemistry of the Elements (Second Edition). Ch. 24 (Butterworth-Heinemann, Oxford, 1997). [Google Scholar]
- 50.Chen MS & White MC A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science 318, 783–787 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.