

Abstract
A series of compounds containing pyrrolidine and pyrrolizidine cores with appended hydrophobic substituents were prepared as constrained analogs of FTY720 and phytosphingosine. The effect of these compounds on the viability of cancer cells, on downregulation of the nutrient transport systems, and on their ability to cause vacuolation was studied. An attempt to inhibit HDACs with some phosphate esters of our analogs was thwarted by our failure to reproduce the reported inhibitory action of FTY720-phosphate.
Keywords: Cytotoxicity, Vacuolation, Nutrient transporter, Protein phosphatase 2A, ring-constrained sphingosines
Graphical Abstract

1. Introduction
Cancer chemotherapy remains an enigmatic and challenging endeavor. In spite of heroic efforts and impressive advances on many fronts, major obstacles such as resistance and toxicity plague the search for effective drugs [1–4]. Compounds that exploit the metabolic differences between cancer and normal cells provide an alternative to toxic systemic chemotherapies or therapies targeting oncogenic signal transduction cascades [5–7]. FTY720 (Gilenya), an approved drug for the treatment of multiple sclerosis [8] is also known to have a cytotoxic effect against cancer cells [9]. However, when phosphorylated it exerts profound cardiovascular effects at higher doses [10]. We have previously shown that pyrrolidines related to ring-constrained analogs of synthetic sphingolipids such as FTY720 (Gilenya, fingolimod) 1 (Figure 1) and natural phytosphingosine 2 kill cancer cells by interfering with one or more nutrient transport systems required for sustenance [11–13]. This strategy of “starving cancer cells to death” has been effectively demonstrated in vitro and in vivo with the analog 3 [14], which unlike FTY720, is not phosphorylated in vivo and avoids the cardiovascular effects induced by FTY720-phosphate through its interaction with sphingosine-1-phosphate receptors [15–20].
Figure 1.

Reported anticancer compounds and a new proposed congener A
We have shown that four of the nutrient acquisition mechanisms used by mammalian cells, namely transporter-mediated import of amino acids and glucose, receptor-mediated LDL uptake and processing, autophagy, and macropinocytosis, are inhibited by analog 3 [14]. Remarkably, cytotoxicity is limited to cancer cells, most likely because non-transformed cells can adapt to the stress caused by nutrient deprivation by altering their metabolic program. The ability of analog 3 to kill cancer cells at ca.2 μM doses is attributed in part to PP2A activation which restricts access to nutrients by down-regulating amino acid and glucose transporters from the cell surface and blocks lysosomal fusion (visualized as extensive vacuolation, Supplementary Figure 1) [14]. Variations of chain length, stereochemistry, and functional group manipulations were also performed to establish thresholds of activity for each of three phenotypes: viability, transporter loss, and vacuolation [13]. The favorable activity profile and promising pharmacological properties of analog 3, including water solubility, encouraged us to study related analogs with a view to better understanding the role of the structural and functional features that contribute to cancer cell killing. As a first objective, we were particularly interested to find new structural variants of the original analog 3 that were differentiated with regard to down-regulation and/or vacuolation, the latter property being an indication of a compound’s ability to block lysosomal fusion. We initially prepared analogs in which the extremity of the C-3-aryloctyl chain was modified. As a second objective, we considered transposing the C-3-aryloctyl chain in analog 3 to the C-2 position of the pyrrolidine ring and including polar substituents within geometric proximity to the nitrogen atom. A third objective was to determine if these new analogs now carrying the aryloctyl chain containing polar groups but branched at the C-2-position of the pyrrolidine ring, could inhibit histone deacetylases (HDACs) [21–23]. Considering the position of the nitrogen atom in the pyrrolidine ring, we speculated that placing a proximal keto group could possibly result in bidentate metal coordination in a cellular environment to target zinc-dependent metalloenzymes, thereby leading to a dual mode of antiproliferative activity on cancer cells in addition to activation of PP2A. Following this notion, we were encouraged that in 2014 Spiegel et al. had reported that FTY720-phosphate was an inhibitor of HDACs by virtue of an increase in acetylation of lysine residues [24–25]. However, Gardner et al. were unable to confirm this in a different cell type [26]. Nevertheless, we considered a generic compound represented by structure A as a constrained C-2-substituted pyrrolidine variant of FTY720 that would combine the attributes of the lead compound 3 while also having the potential to function as an inhibitor of HDACs (Figure 1).
2. Results and discussion
2.1. New C-3 arylchain-modfied variants of analog 3
As previously reported, constrained analog 3 was similarly active to FTY720 (Table 1) while lacking the dose-limiting cardiotoxicity associated with FTY720 [11–14]. In an effort to further probe the structural characteristics of analog 3 vis-a-vis cytotoxicity, nutrient transporter down-regulation, and vacuolation, we turned our attention to the hydrophobic aryloctyl appendage.
Table 1.
Cytotoxicity, CD98 down-regulation and vacuolation profiles of the new C-3-arylalkyl chain variants of analog 3

| Comp. | IC50 (μM) [95% CI] | %CD98 down-regulation | Vacuolation score | |||||
|---|---|---|---|---|---|---|---|---|
| 2.5 μM | 10 μM | 40 μM | 2.5 μM | 10 μM | 40 μM | |||
| 1 | 2.1 | [1.9, 2.4] | 30 | 63 | n.d. | +++ | +++ | n.d. |
| 3 | 2.1 | [2.0, 2.2] | 41 | 66 | n.d. | ++ | +++ | n.d. |
| 4 | 10.9 | [8.7, 13.7] | 11 | 14 | 51 | n.d. | 0 | 0 |
| 5 | 10.2 | [8.7, 12.0] | 5 | 18 | 61 | n.d. | 0 | 0 |
| 6 | 8.4 | [7.0, 10.1] | 5 | 33 | 61 | n.d. | 0 | 0 |
| 7 | 15.1 | [11.2, 20.3] | −15 | −8 | 23 | n.d. | 0 | 0 |
Inspired by the structure of the immunosuppressant KRP-203 (Figure 2) [27] we wanted to investigate the effect of inserting a benzyl ether moiety at the extremity of the aryloctyl chain as shown in compound 4 (Table 1) (see Experimental Section).
Figure 2.
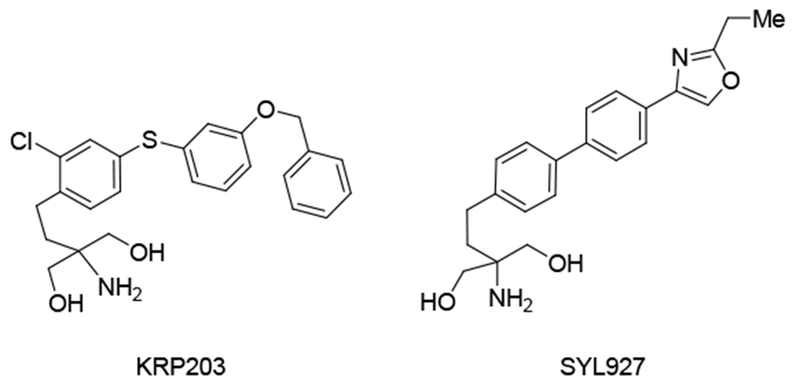
Immunosuppressant KRP-203 and S1P1 agonist SYL927
Next, we introduced a 2-methyl oxazoline moiety at the extremity of the octyl chain, as found in the selective S1P1 agonist SYL927 [28] to obtain analogs 5 and 6. Cytotoxicity assays measured as IC50 showed a 4-7 fold reduction in potency for compounds 4, 5, 6 and 7 compared to analog 3 (Table 1). The loss of activity of the 2-octylketone 7 indicated that polar groups at the extremity of the octyl chain are detrimental to activity. These analogs down-regulated the nutrient transporter-associated protein CD98 at high concentrations, but failed to vacuolate cells even at 40 μM, a dose where CD98 down-regulation and cell killing were robust, suggesting that they lacked the ability to block lysosomal fusion. In view of these results, we decided not to pursue further modifications within the 3-aryloctyl appendage of the lead compound 3.
2.2. Extended C-2 modified variants of analog 3
Being cognizant that the pyrrolidine core had to maintain its basic character for cytotoxicity [11–14], we next sought to probe the positioning of the aryloctyl side chain within the pyrrolidine ring. To this end, we considered branching at C-2 to generate a series of 2-substituted pyrrolidines with extended chains. Anticipating the need for a polar group within the chain for possible metal chelation and HDAC inhibition, we initially considered placing a ketone at the beta and gamma positions next to the pyrrolidine ring (Table 2). We were pleased that the modified variants in this new series exhibited cytotoxicity similar to analog 3. They also down-regulated nutrient transporters and vacuolated at concentrations near their IC50, suggesting that they most likely share the same mechanism of action as analog 3 (Table 2).
Table 2.
Cytotoxicity, CD98 down-regulation and vacuolation profiles of the C-2 modified variants of analog 3 (as HCl salts)

| Comp. | IC50 (μM) [95% CI] | %CD98 down-regulation | Vacuolation score | |||||
|---|---|---|---|---|---|---|---|---|
| 2.5 μM | 10 μM | 40 μM | 2.5 μM | 10 μM | 40 μM | |||
| 3 | 2.1 | [2.0, 2.2] | 42 | 66 | n.d. | ++ | +++ | n.d. |
| 8 | 2.5 | [2.3, 2.8] | 28 | 57 | n.d. | + | +++ | n.d. |
| 9 | 2.8 | [2.6, 3.0] | 41 | 63 | n.d. | ++ | +++ | n.d. |
| 10 | 3.0 | [2.7, 3.3] | 16 | 58 | n.d. | 0 | +++ | n.d. |
| 11 | 2.1 | [1.5, 2.8] | 22 | 46 | n.d. | +++ | +++ | n.d. |
| 12 | 2.0 | [1.6, 2.6] | 44 | 60 | n.d. | ++ | +++ | n.d. |
| 13 | 4.0 | [3.5, 4.6] | 8 | 46 | n.d. | 0 | +++ | n.d. |
| 14 | 12.2 | [11.4, 13.0] | 0 | 32 | 68 | 0 | 0 | + |
| 15 | 2.1 | [1.9, 2.3] | 15 | 52 | n.d. | ++ | +++ | n.d. |
| 16 | 2.2 | [2.1,2.3] | 40 | 63 | n.d. | ++ | +++ | n.d. |
Compounds 8, 9, and 10 were prepared from the Weinreb amide derivative of L-homoproline 8a previously reported by Georg et al. [29] (Scheme 1). Treatment of 8a with 3 equivalents of octylphenylmagnesium bromide in Et2O at 0 °C led to benzylic ketone 8b as a versatile common intermediate. The presence of by-products from the reagent necessitated careful chromatography of the crude reaction product affording 62% yield of pure 8b. Removal of the N-Boc group with 4 N HCl in dioxane afforded 8 as a mixture of enantiomers due to rapid racemization in methanol or water (vide infra). Ketone 8b was reduced with NaBH4 to give the corresponding benzylic alcohol 8c as a 4:1 diastereomeric mixture that could be easily separated by column chromatography.
Scheme 1.

Synthesis of 2-substituted pyrrolidines
Although the stereochemistry of each diastereomer was not determined, the major diastereomer was converted to the methyl ether, then deprotected to afford product 10. The crude diastereomeric mixture of 8c was also catalytically hydrogenated at atmospheric pressure in ethanol, affording product 9 after final removal of the N-Boc protective group.
Products 11 and 12 were obtained starting from the 4-substituted homoprolines 11a and 12a respectively (Scheme 2) (see Supporting Information for the synthesis of 11a and 12a). The corresponding Weinreb amides 11b and 12b were treated with octylphenylmagnesium bromide to give ketones 11c and 12c with acceptable yields. Careful chromatographic purification to separate by-products resulting from the Grignard reagent followed by treatment with TBAF and acid led to products 11 and 12. Products 13 and 15 were prepared starting from 13a (Scheme 3) (see Supporting Information for the synthesis of 13a) and commercially available 1-(tert-butyl) 2-methyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)pyrrolidine-1,2-dicarboxylate. Reduction of the ester group to the corresponding aldehyde with DIBAL-H, followed by a Wittig reaction with methyl(triphenylphosphoranylidene)acetate, and catalytic hydrogenation afforded the ester intermediates 13b and 15a. The corresponding Weinreb amides were subsequently reacted with octylphenylmagnesium bromide to give ketones 13c and 15b, in 37% and 54% yield respectively. Removal of the OTBDPS and N-Boc groups afforded products 13 and 15.
Scheme 2.

Synthesis of 2- and 4-substituted pyrrolidines
Scheme 3.

Synthesis of C-2 substituted extended pyrrolidines
We observed that 11 and 12 epimerized at the C-2 position when dissolved in protic solvents. In the presence of D2O at room temperature, deuterium was incorporated at C-2 confirming a fast enolization followed by β-elimination and ring closure. Keto pyrrolidine 12 was stable only in very acidic conditions (pH ≤ 3 in H2O), while epimerization was extremely fast in basic conditions. The incorporation of deuterium is complete in 30 min at pH = 10, and in 3 hours at pH = 7. A similar behavior had already been observed for the chlorpromazine-like central nervous system depressant Su17595A, [30], the muscle relaxant tolperisone [31], and the antiarrhythmic drug moricizine [32], albeit at a slower rate.
When the keto group was further removed from the pyrrolidine ring by extending the chain length as in 13, the desired ketone was found to be in equilibrium with an azabicyclic salt resulting from intramolecular iminium ion formation (2:1 mixture in H2O) (13d), which upon reduction with NaBH4 led to 14 (Scheme 4, B). The same behavior was observed in the case of compound 15.
Scheme 4.

Epimerization and cyclization processes observed
Surprisingly the new bicyclic derivative 14 maintained a reasonable cytotoxic activity (IC50 12.2 μM). We therefore decided to further investigate this new structural analog and we prepared the bicyclic enantiopure pyrrolizidine 17 bearing the aryloctyl appendage on C-3, in analogy with our lead compound 3, as well as the monocyclic variant 18, to check the effect of the substituent position and the nature of the azacycles (Scheme 5).
Scheme 5.

Pyrrolizidine and pyrrolidine syntheses
We started the synthesis from the readily available (S)-prolinal which was arylated and reoxidized to the corresponding ketone since the addition of the corresponding aryl Grignard on the Weinreb amide resulted in decomposition of the reagent without conversion of the substrate (Scheme 5). Subsequent addition of the lithium enolate of ethyl acetate delivered 17c as a mixture of diastereomers that could not be separated at this stage. The ester was further reduced to the alcohol and tosylated, which when warmed up to 105 °C in toluene, underwent a spontaneous deprotection/cyclisation, leading to the formation of the bicyclic structure 17e and its epimer (epi-17e) as tosylate salts in a ratio of 2:1.
The diastereomers were separated at this stage, delivering the major product 17e in 38% yield. Suzuki coupling of 17e followed by hydrogenation of the resulting alkene delivered 17. The moderate yield over the two last steps was attributed to the highly sensitive benzylic alcohol decomposing easily under acidic conditions.
The racemic compound 18 was easily accessed from N-Boc 3-pyrrolidinone which was arylated with the octylphenylmagnesium bromide and deprotected under acidic conditions.
Interestingly, compound 17 proved to be more active than 14 with an IC50 in the same range as the monocyclic C-2 substituted variants while 18 was less active than 17 (Figure 3), indicating that the increased steric hindrance generated by the pyrrolizidine was tolerated.
Figure 3.

Pyrrolizidine and pyrrolidine analogs
2.3. Repositioning the keto group in extended C-2 modified variants of analog 3
In considering an alternative position of the keto group in the chain, we placed it on the α-position of C-2 branched aryloctyl pyrrolidine analogs (Table 3). This would avoid the issues of partial epimerization issues encountered due to beta elimination and ring closure as described above, although the basicity of the pyrrolidine nitrogen could be diminished due to the inductive effect of the carbonyl group [33]. In general, the cytotoxic activity of the C-2 ketoaryl compounds was significantly reduced compared to 3 and to the corresponding extended keto variants (compare Tables 2 and 3). This indicated that a carbonyl group adjacent to the pyrrolidine nitrogen atom on a chain at C-2 was not well tolerated, whereas alcohols 25 and 26 retained activity similar to the lead compound 3.
Table 3.
Cytotoxicity, CD98 down-regulation and vacuolation profiles of the C-2 substituted α-keto and α-hydroxy pyrrolidines

| Comp. | IC50 (μM) [95% CI] | down-regulation | Vacuolation score | |||||
|---|---|---|---|---|---|---|---|---|
| 2.5 μM | 10 μM | 40 μM | 2.5 μM | 10 μM | 40 μM | |||
| 3 | 2.1 | [2.0, 2.2] | 42 | 66 | n.d. | ++ | +++ | n.d. |
| 19 | <40 | −21 | −4 | 33 | 0 | 0 | +++ | |
| 20 | 28.1 | [26.1, 30.2] | 2 | 9 | 48 | 0 | 0/+ | +++ |
| 21 | 15.3 | [14.6, 15.9] | 12 | 51 | 41 | 0 | +++ | +++ |
| 22 | 19.6 | [18.7, 20.5] | 0 | 19 | 61 | 0 | 0 | +++ |
| 23 | 18.6 | [15.5, 22.3] | 13 | 23 | 59 | 0 | 0 | +++ |
| 24 | 17.1 | [14.5, 20.2] | 11 | 16 | 54 | 0 | 0 | +++ |
| 25 | 3.0 | [2.6, 3.5] | 19 | 62 | n.d. | + | +++ | n.d. |
| 26 | 1.8 | [1.7, 2.0] | 9 | 62 | n.d. | 0 | ++ | n.d. |
Nevertheless, all of the C-2 keto compounds were able to down-regulate nutrient transporters at elevated concentrations. The same trend held for vacuolation, as 21 reached maximum vacuolation at 10 μM (Table 3). The negative influence of the α-keto group in the chain was further seen by the 5-10 fold decrease in cytotoxicity when comparing compounds 20 and 21 to 16. It is also notable that, at the concentration where compound 26 kills 50% of cells, no down-regulation of nutrient transporter proteins or vacuolation was observed, suggesting an alternative mode of action.
Compounds 20, 22 and 23, as well as the corresponding reduction products 16 and 25 were synthetized as single enantiomers from the intermediates 20b and 22b (Scheme 6). The Weinreb amides 20b and 22b were reacted with octylphenylmagnesium bromide to give the corresponding ketones, which were deprotected to give 20c and 22c. Treatment with acid afforded the pyrrolidines 20 and 22 respectively.
Scheme 6.

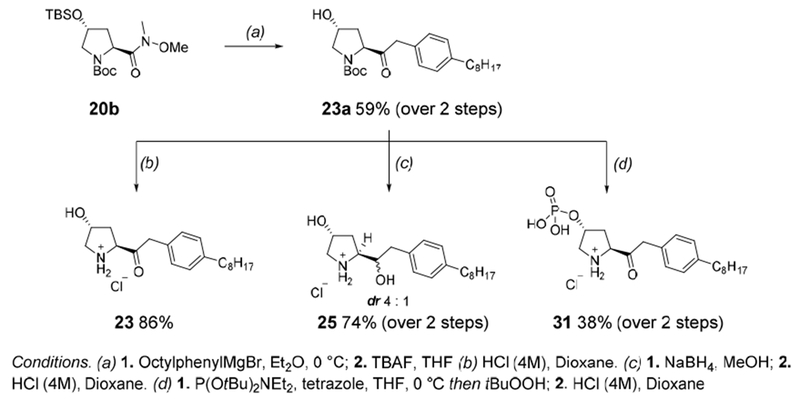
Syntheses of the α-keto and α-hydroxy C2-substituted pyrrolidines and their phosphate esters
Pd/C catalyzed hydrogenation of the keto group in 22c followed by N-Boc deprotection led to 16 in modest yield. Following a similar protocol, we prepared the extended ketone intermediate 23a that was transformed to 23 and 25, the latter consisting of a 4:1 mixture of diastereomers (Scheme 6).
As previously stated, we proposed to prepare constrained C-2 substituted α-hydroxy pyrrolidine analogs of FTY720 bearing a phosphate group to test them as HDAC inhibitors. To this end we prepared phosphate esters 29 and 31 as well as their enantiomers 30 and 32 (not shown) by standard methods (Scheme 6).
2.4. Anticipated HDAC activity presents a conundrum
The nuclear zinc-dependent metalloprotease enzymes known as histone deacetylases have been the subject of extensive studies over the past decades because they play a major role in regulating gene expression [34]. Chromatin remodeling affects the accessibility of DNA to transcription factors and thus plays a central role in controlling gene expression and determining cellular phenotypes. The acetylation and deacetylation of N-acetyl lysine residues catalyzed by histone acetylases (HATs) and histone deacetylases (HDACs) respectively, is a tightly controlled process that, if perturbed, can lead to cancer. [34] Consequently, the search for HDAC inhibitors as drugs to combat cancer has been of considerable interest. [21–23] Inspired by the structure of the natural product trichostatin A (TSA) (27) [35], Breslow and coworkers [36] developed the simple suberoylanilide hydroxamic acid (SAHA) (28), which is marketed under the trade name Zolinza (Vorinostat) (Figure 4).
Figure 4.
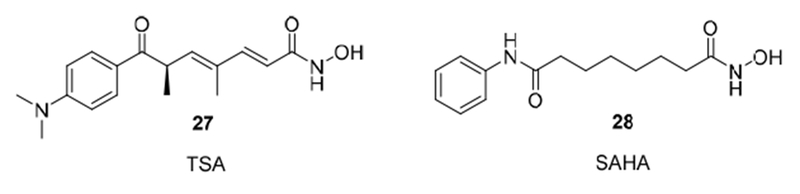
Classical HDAC inhibitors
Previous studies from our laboratory have focused on exploring the importance of the chain length and incorporation of an a-alkoxy chain as well as stereochemical features on a SAHA motif on HDAC inhibitory activity [37–38]. Relying on molecular modeling and the crystal structure of HDAC 8 [39] we reported on a series of macrocyclic inhibitors with a nanomolar activity profile against a number of class I and class II HDACs [40].
As previously mentioned, we were intrigued by the reports by Spiegel and coworkers [24–25] that FTY720-phosphate and sphingosine-1-phosphate [41] are inhibitory toward HDACs. In these studies, nuclear SphK2 was required to observe FTY720- and sphingosine-1-phosphate-dependent increases in several histone acetylation marks. The (S)-phosphate formed in vivo from FTY720 would mimic the natural sphingosine phosphate intracellularly and act as a ‘synthetic’ inhibitor. Their results showed that FTY720-P inhibited recombinant HDACs 1, 2, 3 and 8, approaching the activity of SAHA. Using the reported crystal structure of HDAC 2, the Spiegel group conducted molecular docking studies of FTY720-P on the active site and concluded that the binding mode was very similar to SAHA and sphingosine-1-phosphate. They hypothesized that the juxtaposition of the primary amino group and the hydroxymethyl group bearing the (S)-configured phosphate in FTY720 might act similarly to the hydroxamic acid group in SAHA and the phosphate in S1P. They further invoked a favorable π-π interaction of the phenyl group in FTY720-P with Phe206 and Phe151 (Figure 5).
Figure 5.

Proposed binding mode of FTY720-phosphate (Spiegel) and a hypothetical pose for a synthetic C-2-keto arylalkyl pyrrolidine 3-phosphate (32)
Encouraged by these highly intriguing results from the Spiegel group, we were poised to test phosphate analogs 29-32 sharing similar interactions as suggested by a docking study using FITTED against HDACs. [42–43]. However, prior to undertaking this initiative, we wanted to see if these phosphates exhibited any cytotoxic activity. While it was not surprising that the phosphate esters 29, 30, 31, and 32 were totally inactive in down-regulation and vacuolation tests compared to their hydroxy pyrrolidine ketone progenitors 20, 21, 23 and 24, as the charged phosphate should not be able to enter the cell, we were surprised to find that these analogs were cytotoxic at IC50 12.9 μM, 12.8 μM, 25.0 μM and 25.3 μM respectively (Table 4). The cytotoxicity of these phosphate esters over the unphosphorylated progenitors despite the absence of transporter loss or vacuolation suggests that they could act through a distinct mechanism, possibly targeting a receptor on the cell surface.
Table 4.
Cytotoxicity, CD98 down-regulation and vacuolation profiles of phosphate ester analogs

| Comp. | IC50 (μM) [95% CI] | %CD98 down-regulation | Vacuolation score | |||||
|---|---|---|---|---|---|---|---|---|
| 2.5 μM | 10 μM | 40 μM | 2.5 μM | 10 μM | 40 μM | |||
| 3 | 2.1 | [2.0, 2.2] | 42 | 66 | n.d. | ++ | +++ | n.d. |
| 29 | 12.9 | [11.9, 14.0] | n.d. | 3 | -8 | 0 | 0 | 0 |
| 30 | 12.8 | [11.7, 14.0] | n.d. | -9 | 0 | 0 | 0 | 0 |
| 31 | 25.0 | [18.1, 34.4] | n.d. | 3 | 0 | n.d. | 0 | 0 |
| 32 | 25.3 | [19.5, 32.9] | n.d. | 5 | 8 | n.d. | 0 | 0 |
We then focused on establishing the parameters to test HDAC activity. Pleasingly, the reported activity of SAHA was confirmed in both our in vitro and cell-based test systems (85% inhibition at 2 μM concentration). However, FTY720-P, reported to exhibit 60-75% inhibition at 5 μM by Spiegel, [24–25] was inactive up to 20 μM in our hands despite the fact that its integrity and phosphorylation were confirmed by UPLC-ESI-MS/MS (Supplemental Figures 2A, B and 3 and data not shown). Assays performed using 12, 25, 26, 31, and 32 that employed SAHA as a positive control failed to detect HDAC inhibition (Supplemental Figure 2B, C). Examination of histone acetylation in intact cells also failed to confirm a role for FTY720-P as an HDAC inhibitor (Supplemental Figure 3). Since other groups have reported difficulty detecting increased acetylation of specific lysine residues [26], we utilized antibodies against multiple acetylation sites on both H3 and H4; however, no changes in histone acetylation were detected in cells treated with FTY720-P despite consistent and robust increases in acetylation in cells treated with SAHA. Clearly, this disappointing outcome presented an unexplained conundrum that led us to refrain from conducting further HDAC inhibition tests with additional compounds in this series. Given that the parent compound FTY720-P did not inhibit HDAC activity, our intended objective of achieving dual-action inhibitors was not realized.
3. Conclusions
In conclusion, we have reported that placing the aryloctyl group found in the anticancer analog 3 in the C-2 position of the pyrrolidine maintains cytotoxic activity. However, introduction of a carbonyl group in between the aryloctyl appendage and the pyrrolidine ring resulted in variable levels of activity depending on its position along the chain. Placing the carbonyl group two or three carbon atoms removed from the pyrrolidine ring was generally better tolerated with an activity profile only slightly diminished compared to the lead analog 3, whereas attempts to bring it closer were detrimental to the activity, likely due to a decrease of the nitrogen basicity. Our attempts to mimic the HDAC inhibiting activity of FTY720-phosphate as reported by Spiegel and coworkers with a series of constrained C-2 keto pyrrolidine analogs and their phosphate esters were thwarted by the fact that in our hands FTY720-phosphate did not act as an inhibitor. Thus, our search for a dual-action anticancer agent derived from a synthetic pyrrolidine-based sphingolipid was only partially validated with a new series of C-2 alkyl and C-2 ketoalkyl aryloctyl pyrrolidines. Gratifyingly, these maintained cytotoxicity, nutrient transporter down-regulation, and vacuolation activity profiles equal to that of the lead analog 3 with the notable exception of 4, 5, 6, and 7, which do not vacuolated even at high concentrations, and 26, which triggers changes in endolysosomal trafficking at elevated doses but appears to kill cells by binding to an additional target.Our efforts also uncovered pyrrolizidines with appended aryloctyl chains as new cytotoxic agents. Studies are in progress to better understand the anti-neoplastic actions of synthetic sphingolipids and to identify their direct protein targets in cells.
4. Experimental section
4.1. Chemistry: General information
All reactions involving moisture sensitive compounds were performed in flame-dried glassware under a positive pressure of dry, oxygen free, argon and in dry solvents. Anhydrous solvents were distilled under a positive pressure of argon before use and dried by standard methods. THF, ether, CH2Cl2 and toluene were dried by the SDS (Solvent Delivery System). Commercial grade reagents were used without further purification. Silica column chromatography was performed on 230-400 mesh silica gel. Thin layer chromatography (TLC) was carried out on glass-backed silica gel plates. Visualisation was effected by UV light (254 nm) or by staining with potassium permanganate solution, cerium ammonium molybdate or p-anisaldehyde followed by heating. 1H and 13C NMR spectra were recorded on Bruker AV-400 and AV-500 MHz spectrometers at room temperature (298 K). Chemical shifts are reported in parts per million (ppm) referenced from CDCl3 (δH: 7.26 ppm and δC: 77.0 ppm). Coupling constants (J) are reported in Hertz (Hz). Multiplicities are given as multiplet (m), singlet (s), doublet (d), triplet (t), quartet (q), quintet (quin.) and broad (br.). Infrared spectra were recorded on a FT-IR spectrometer and are reported in reciprocal centimetres (cm−1). Optical rotations were determined on an Anton Paar MCP 300 polarimeter at 589 nm. Specific rotations are given in units of 10−1 deg cm2 g−1. High resolution mass spectra (HRMS) were performed by the “Centre régional de spectroscopie de masse de l’Université de Montréal” with electrospray ionisation (ESI) coupled to a quantitative time-of-flight (TOF) detector.
General procedure A for N-Boc deprotection
HCl (500 μL, 4M in dioxane, excess) was added to substrate in dry dioxane. The reaction was stirred at rt until disappearance of the starting material by TLC analysis. The solution was then concentrated in vacuo in several cycles co-distilling with dry dioxane.
General procedure B for removal of silyl ethers
TBAF (1.1 eq., 1.0 M in THF) was added to a solution of substrate in dry THF (C = 0.06 M). The reaction was then stirred at rt until disappearance of the starting material by TLC analysis. The solution was diluted with saturated aq. NaHCO3 solution and EtOAc. The aqueous layer was extracted ×2 with EtOAc. The organic layers were combined, washed x1 with brine, dried over Na2SO4, filtered, concentrated.
General procedure C for addition of Grignard reagents
A 3-neck round-bottomed flask equipped with a thermometer and a condenser was flame-dried and flushed with Ar. The flask was then charged with Mg0 (1.1 eq.), a single I2 crystal and another vacuum/Ar cycle was performed. Et2O (C = 0.65 M) was added resulting in a bright orange suspension of Mg0 pellets. Phenyl octyl bromide (1.0 eq.) was then added in one portion and the suspension was heated via a heatgun until the internal temperature reached 32 °C and stabilized for 5-10 seconds, indicating that the Grignard formation had started. The reaction was stirred at rt until disappearance of the starting material by 1H NMR analysis (e.g. ≈ 1h). The Grignard solution (3.0 eq., C = 0.65 M) was then syringed to another flask containing substrate (1.0. eq.) in dry Et2O (C = 0.05 M). The solution was stirred at rt until disappearance of the starting material by TLC analysis. Saturated aqueous NH4Cl solution was added and the aqueous layer was extracted x2 with EtOAc. The organic layers were collected, washed x1 brine, dried over Na2SO4, filtered, concentrated in vacuo.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(3-hydroxyprop-1-yn-1-yl)phenyl)-5-oxopyrrolidine-1-carboxylate (4b) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-3-(4-bromophenyl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate 4a (182 mg, 0.30 mmol, 1.0 equiv.) was dissolved in dry DMF (1mL) and Et3N (3 mL), then propargyl alcohol (0.14 mL, 2.40 mmol, 8.0 equiv.) was added. Inside the solution was bubbling a flow of Ar for 20 min before to add sequentially CuI (17 mg, 0.09 mmol, 0.3 equiv.) and Pd(PPh3)4 (52 mg, 0.05 mmol, 1.5 equiv.). The bubbling of Ar was continued for further 15 min, then the mixture was warmed to 70 °C and stirred for 18 h in a sealed flask. The mixture was extracted with an aqueous NH4OH 1 M solution (100 mL) mixed with brine (100 mL) and EtOAc (100 mL × 4 times). The combined organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude was purified by silica gel column chromatography with hexane/EtOAc (gradient from 8:2 to 6:2) to afford the title compound 4b as a brown oil (68 mg, 39%). Rf: 0.54 (Hexane/EtOAc 6:4). [α]20D +12.9° (c 0.5, CHCl3). IR (neat), νmax: 3435, 3165, 3071, 2927, 2856, 2167, 2079, 1778, 1748, 1713, 1366, 1149, 1107, 1029, 882, 700. 1H NMR (CDCl3, 500 MHz), δ: 7.70-7.60 (m, 4H), 7.48-7.36 (m, 8H), 7.09 (d, J = 8.2 MHz, 2H), 4.53-4.47 (m, 2H), 4.08-4.07 (m, 1H), 3.97 (dd, J = 10.6 MHz, J = 4.4 MHz, 1H), 3.80 (dd, J = 12.9 MHz, J = 2.6 MHz, 1H), 3.48 (broad d, J = 9.4 MHz, 1H), 3.19 (dd, J = 17.9 MHz, J = 9.4 MHz, 1H), 2.55 (dd, J = 17.9 MHz, J = 2.6 MHz, 1H), 1.85-1.75 (m, 1H), 1.42 (s, 9H), 1.08 (s, 9H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 173.9, 149.7, 144.5, 135.7, 135.6, 133.0, 132.6, 130.1, 128.1, 128.0, 126.6, 121.7, 87.8, 85.2, 83.4, 66.6, 64.3, 51.8, 39.8, 38.7, 28.1, 27.0, 19.4, 1.2 ppm. HRMS (ESI) calcd. for C35H42NO5Si+ (M+H)+: 584.28268, found: 584.28328.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(3-hydroxypropyl)phenyl)pyrrolidine-1-carboxylate (4c) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(3-hydroxyprop-1-yn-1-yl)phenyl)-5-oxopyrrolidine-1-carboxylate 4b (52 mg, 0.09 mmol) was dissolved in EtOH (0.80 mL) and Pd/C (10%, 48 mg, 0.05 mmol, 0.5 equi.) was added. The air was pumped out of the flask and replaced by H2. Upon completion (24 h), the reaction mixture was filtered through celite. The solvent was removed under reduced pressure to afford a colorless oil. The crude was dissolved in dry THF (2 mL) and the solution was cooled to 0 °C. Borane dimethyl sulfide complex (0.05 mL, 0.53 mmol, 6.0 equiv.) was then added and the reaction was warmed up to room temperature and stirred for 18 h. The mixture was quenched with H2O (1 mL) and the solvent was removed under reduced pressure. The residue was extracted with an aqueous NaHCO3 saturated solution (50 mL) and CH2Cl2 (50 mL × 4 times). The combined organic layers were dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford 4c as a pale yellow oil (51 mg, 98% over two steps). Rf: 0.58 (Hexane/EtOAc 6:4). [α]20D −6.0° (c 0.5, CHCl3). IR (neat), νmax: 3461, 3070, 3047, 2958, 2929, 2858, 2166, 2115, 2010, 1692, 1589, 1514, 1472, 1454, 1426, 1391, 1364, 1254, 1166, 1106, 1070, 1033, 1006, 987, 923, 856, 821, 772, 737, 700, 643, 608. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.70-762 (m, 4H), 7.46-7.34 (m, 6H), 7.15-7.06 (m, 4H), 4.20-3.35 (m, 7H), 2.74-2.64 (m, 1H), 2.59-2.52 (m, 1H), 2.31-2.17 (m, 1H), 1.95-1.83 (m, 2H), 1.68-1.55 (m, 1H), 1.50 (s, 3H), 1.33 (s, 6H), 1.06 (s, 9H), 0.98-0.92 (m, 1H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 154.4, 154.3, 141.1, 140.2, 135.8, 133.7, 133.5, 129.8, 129.7, 128.8, 127.9, 127.8, 127.7, 127.6, 127.4, 127.2, 79.5, 65.7, 65.5, 63.7, 62.5, 47.3, 46.6, 46.4, 45.5, 37.8, 34.3, 33.1, 32.0, 31.8, 28.7, 28.6, 27.0, 24.7, 19.5, 19.4, 14.0 ppm. HRMS (ESI) calcd. for C35H48NO4Si+ (M+H)+: 574.33471, found: 574.33298.
(2S,3R)-3-(4-(3-(Benzyloxy)propyl)phenyl)-2-(hydroxymethyl)pyrrolidin-1-ium chloride (4) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(3-hydroxypropyl)phenyl)pyrrolidine-1-carboxylate 4c (34 mg, 0.06 mmol, 1.0 equiv.) was dissolved in dry THF (0.50 mL) and the solution was cooled to 0 °C. To this solution NaH (60% in mineral oil, 12 mg, 0.30 mmol, 5.0 equiv.) was slowly added in one portion, then benzyl bromide (0.03 mL, 0.24 mmol, 4.0 equiv.) was added dropwise. The reaction mixture was stirred for 2 h, then quenched with the addition of an aqueous NH4Cl saturated solution (1 mL) and extracted with EtOAc (4× 30 mL). The combined organic layers were washed x1 with brine (30 mL), dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford a pale yellow oil. This oil was dissolved in dry THF (1 mL) and the solution was cooled to 0 °C. Tetrabutylammonium fluoride solution (1M in THF, 0.30 mL, 0.30 mmol, 5.0 equiv.) was added and the reaction mixture was warmed to room temperature and stirred for 18 h. The reaction mixture was quenched with an aqueous NaHCO3 saturated solution (20 mL) and extracted three times with CH2Cl2 (30 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford a pale yellow oil. To this crude a HCl 1M solution in dioxane (0.50 mL, 0.50 mmol, 8.0 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h, then the solvent was removed under reduced pressure. The crude mixture was purified by silica gel column chromatography with CH2Cl2/MeOH (gradient from 9:1 to 6:4) to give a pale yellow oil. This oil was dissolved in water, filtered through a plastic syringe filter (pore size: 0.45 μm) and lyophilized to afford the title compound 4 as a white solid (20 mg, 93% over three steps). Rf: 0.72 (CH2Cl2/MeOH 8:2). [α]20D −19.2° (c 0.1, MeOH). IR (neat), νmax: 3254, 2922, 2848, 2496, 1719, 1595, 1516, 1494, 1476, 1452, 1427, 1391, 1353, 1307, 1265, 1206, 1174, 1117, 1067, 1046, 998, 920, 858, 841, 812, 791, 745, 699. 1H NMR (MeOD, 500 MHz), δ: 7.37-7.18 (m, 9H), 4.50 (s, 2H), 3.75 (t, J = 5.6 MHz, 2H), 3.71-3.53 (m, 2H), 3.49 (J = 6.0 MHz, 2H), 3.45-3.35 (m, 1H), 3.35-3.25 (m, 1H), 2.70 (t, J = 7.2 MHz, 2H), 2.53-2.40 (m, 1H), 2.30-2.18 (m, 1H), 1.94-1.85 (m, 2H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 141.5, 138.4, 135.7, 128.9, 128.0, 127.6, 127.3, 127.2, 72.6, 72.2, 71.1, 69.0, 67.1, 60.8, 58.3, 44.7, 44.6, 42.4, 32.5, 31.5, 31.2 ppm. HRMS (ESI) calcd. for C21H28NO2+ (M+H)+: 326.21146, found: 326.21200.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(7-hydroxyhept-1-yn-1-yl)phenyl)-5-oxopyrrolidine-1-carboxylate (5b) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-3-(4-bromophenyl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate 5a (436 mg, 0.72 mmol, 1.0 equiv.) was dissolved in dry DMF (3 mL) and Et3N (1 mL), then 1-heptynol (0.18 mL, 1.44 mmol, 2.0 equiv.) was added. Inside the solution was bubbling a flow of Ar for 20 min before adding CuI (41 mg, 0.22 mmol, 0.3 equiv.) and Pd(PPh3)4 (125 mg, 0.11 mmol, 0.15 equiv.). The bubbling of Ar was continued for further 15 min, then the mixture was warmed to 70 °C and stirred for 18 h in a sealed flask. An aqueous NH4OH 1 M solution (100 mL) mixed with brine (100 mL) was added and the mixture was extracted with EtOAc (4× 100 mL). The combined organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude was purified by silica gel column chromatography with hexane/EtOAc (gradient from 8:2 to 3:7) to afford the title compound 5b as a brown oil. (345 mg, 75%). Rf: 0.51 (Hexane/EtOAc 5:5). [α]20D +12.3° (c 0.5, CHCl3). IR (neat), νmax: 3489, 3166, 2932, 2858, 2154, 1781, 1748, 1713, 1366, 1303, 1255, 1149, 1072, 822, 700. 1H NMR (CDCl3, 500 MHz), δ: 7.70-7.64 (m, 4H), 7.48-7.40 (m, 8H), 7.37 (d, J = 7.4 MHz, 2H), 7.10 (d, J = 7.4 MHz, 2H), 4.09 (broad s, 1H), 4.00 (dd, J = 10.6 MHz, J = 4.0 MHz, 1H), 3.83 (d, J = 10.6 MHz, 1H), 3.67 (t, J = 6.4 MHz, 2H), 3.51 (d, J = 8.9 MHz, 1H), 3.22 (dd, J = 17.8 MHz, J = 9.6 MHz, 1H), 2.58 (d, J = 17.8 MHz, 1H), 2.43 (t, J = 6.4 MHz, 2H), 1.86 (broad s, 1H), 1.68-1.50 (m, 4H), 1.44 (s, 9H), 1.11 (s, 9H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 174.0, 149.6, 143.3, 135.6, 135.5, 132.9, 132.6, 132.2, 130.0, 128.0, 127.9, 126.3, 123.0, 90.6, 83.2, 80.2, 66.6, 64.2, 62.7, 39.7, 38.6, 32.3, 28.5, 28.0, 26.9, 25.1, 19.4, 19.2 ppm. HRMS (ESI) calcd. for C39H50NO5Si+ (M+H)+: 640.34528, found: 640.34597.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(6 oxohexyl)phenyl)pyrrolidine-1-carboxylate (5c) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(7-hydroxyhept-1-yn-1-yl)phenyl)-5-oxopyrrolidine-1-carboxylate 5b (312 mg, 0.49 mmol, 1.0 equiv.) was dissolved in EtOH (2.5 mL) and Pd/C (10%, 164 mg, 0.15 mmol, 0.3 equiv.) was added. The air was pumped out of the flask and replaced by H2. Upon completion (18 h), the reaction mixture was filtered through Celite. The solvent was removed under reduced pressure to afford a colorless oil. The crude was dissolved in dry THF (5 mL) and the solution was cooled to 0 °C, then borane dimethyl sulfide complex (0.18 mL, 1.94 mmol, 4.0 equiv.) was added and the reaction was allowed to warm to room temperature and stirred for 24 h. The mixture was quenched with H2O (1 mL) and the solvent was removed under reduced pressure. An aqueous NaHCO3 saturated solution (80 mL) was added and the solution was extracted with CH2Cl2 (4× 80 mL), The combined organic layers were dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford a slight yellow oil. This crude was used directly for the next step without further purification. A solution of dimethyl sulfoxide (0.08 mL, 1.18 mmol, 2.4 equiv.) in CH2Cl2 (2 mL) was added dropwise, via cannula, over 10 min to a stirred solution of oxalyl chloride (0.05 mL, 0.59 mmol, 1.2 equiv.) in CH2Cl2 (2 mL) at −78 °C. The mixture was stirred at −78 °C for 15 min and then a solution of the previous alcohol in CH2Cl2 (3 mL) was added dropwise, via cannula, over 10 min. The mixture was stirred at −78 °C for 1 h and then Et3N (0.25 mL, 1.76 mmol, 3.6 equiv.) was added dropwise over 5 min. The mixture was warmed to 0 °C over a period of 3.5 h, then quenched with an aqueous NaHCO3 saturated solution (50 mL) and warmed to room temperature. The separated aqueous layer was extracted with CH2Cl2 (50 mL × 3 times), and the combined organic layers were dried over Na2SO4 and concentrated in vacuum to leave a yellow oil which was purified by silica gel column chromatography with hexane/EtOAc (gradient from 9:1 to 8:2) to afford the title compound 5c as a colourless oil (220 mg, 72% over three steps). Rf: 0.59 (Hexane/EtOAc 8:2). [α]20D −9.7° (c 0.3, CHCl3). IR (neat), νmax: 3072, 3050, 2951, 2831, 2740, 1732, 1692, 1426, 1390, 1164, 1105, 1070, 1034, 822, 701. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 9.76 (s, 1H), 7.71-7.63 (m, 4H), 7.46-733 (m, 6H), 7.16-7.06 (m, 4H), 4.20-3.35 (m, 6H), 2.62-2,55 (m, 2H), 2.42 (t, J = 7.2 MHz, 2H), 2.32-2.18 (m, 1H), 1.99-1.85 (m, 1H), 1.70-1,57 (m, 4H), 1.55-1.42 (m, 4H), 1.41-1.30 (m, 9H), 1.07 (s, 9H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 202.8, 154.3, 154.2, 141.1, 140.8, 135.6, 133.6, 133.4, 129.7, 129.6, 128.6, 127.8, 127.8, 127.7, 127.3, 127.2, 79.4, 79.1, 65.6 65.4, 63.6, 62.1, 60.4, 53.4, 47.2, 46.5, 46.3, 45.4, 43.9, 35.4, 32.9, 31.9, 31.3, 29.1, 29.0, 28.6, 28.4, 26.9, 22.0, 19.4, 19.3 ppm. HRMS (ESI) calcd. for C39H54NO4Si+ (M+H)+: 628.38166, found: 628.38194.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-((E)-7-(2-methyloxazol-4-yl)hept-6-en-1-yl)phenyl)pyrrolidine-1-carboxylate (5d) (See Supp. Info for a Scheme)
A solution of Lithium bis(trimethylsilyl)amide (1 M in THF, 0.49 mL, 0.49 mmol, 2 equiv.) was added over 5 min via syringe to a stirred solution of dimethyl((2-methyloxazol-4-yl)methyl)phosphonate). The oxazole phosphonate derivative was prepared by using standard procedures from the known 4-hydroxymethyl-2-methyloxazole [44] (101 mg, 0.49 mmol, 2.0 equiv.) in dry THF (2 mL) at −78 °C. The resulting orange solution was stirred at −78 °C for 30 min and then a solution of the aldehyde derivative 5c (154 mg, 0.25 mmol, 1.0 equiv.) in dry THF (2 mL) was added via cannula. The mixture was allowed to warm slowly to room temperature over 5 h, stirred overnight, and then quenched with an aqueous NH4Cl saturated solution (20 mL) and diluted with EtOAc (50 mL). The organic extract was washed with brine (50 mL), then dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography with hexane/EtOAc (gradient from 9:1 to 8:2) to afford the title compound 5d as a colourless oil (88 mg, 51%). Rf: 0.36 (Hexane/EtOAc 85:15). [α]20D −15.2° (c 0.5, CHCl3). IR (neat), νmax: 3072, 3045, 2956, 2855, 2032, 2022, 1692, 1654, 1516, 1488, 1447, 1410, 1391, 1270, 1070, 982, 742, 700. 1H NMR (CDCl3, 400 MHz, mixture of rotamers), δ: 7.79-7.66 (m, 4H), 7.46-7.25 (m, 7H), 7.15-7.05 (m, 4H), 6.40 (dt, J = 15.6 MHz, J = 7.0 MHz, 1H), 6.17 (d, J = 15.6 MHz, 1H), 4.25-3.35 (m, 5H), 2.63-2.52 (m, 2H), 2.44 (s, 3H), 2.35-2.15 (m, 3H), 2.00-1.85 (m, 1H), 1.70-1.25 (m, 16H), 1.08 (s, 9H) ppm. 13C NMR (CDCl3, 100 MHz, mixture of rotamers), δ: 161.6, 154.4, 141.2, 139.2, 135.7, 133.6, 132.9, 129.8, 128.7, 127.8, 127.2, 118.2, 79.4, 72.7, 65.7,64.7, 63.6, 48.1, 46.6, 35.6, 32.9, 32.0, 31.5, 29.3, 29.1, 28.7, 28.5, 27.0, 19.4, 14.0 ppm. HRMS (ESI) calcd. for C44H59N2O4Si+ (M+H)+: 707.42386, found: 707.42492.
(2S,3R)-2-(Hydroxymethyl)-3-(4-((E)-7-(2-methyloxazol-4-yl)hept-6-en-1-yl)phenyl)pyrrolidin-1-ium chloride (5) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-((E)-7-(2-methyloxazol-4-yl)hept-6-en-1-yl)phenyl)pyrrolidine-1-carboxylate 5d (27 mg 0.04 mmol, 1.0 equiv.), was dissolved in dry THF (1 mL) and the solution was cooled to 0 °C. Tetrabutylammonium fluoride solution (1M in THF, 0.08 mL, 0.08 mmol, 2.0 equiv.) was added and the reaction mixture was warmed to room temperature and stirred for 24 h. The reaction mixture was quenched with an aqueous NaHCO3 saturated solution (20 mL) and extracted with CH2Cl2 (30 mL × 3 times). The combined organic layers were washed with brine (50 mL), dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford a pale yellow oil. To this crude a HCl 1M solution in dioxane (0.50 mL, 0.50 mmol, 13 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h, then the solvent was removed under reduced pressure. The crude mixture was purified by silica gel column chromatography with CH2Cl2/MeOH (gradient from 9:1 to 6:4) to give a yellow oil. This oil was dissolved in water, filtered through a plastic syringe filter (pore size: 0.45 μm) and lyophilized to afford the title compound 5 as a white solid (15 mg, 96% over two steps). Rf: 0.59 (CH2Cl2/MeOH 8:2). [α]20D −16.5° (c 0.2, MeOH). IR (neat), νmax: 3331, 3254, 2929, 2857, 2029, 2020, 1654, 1515, 1487, 1449, 1407, 1270, 1079, 982, 742, 705, 576. 1H NMR (MeOD, 500 MHz), δ: 7.63 (s, 1H), 7.27 (d, J = 11.0, MHz, 2H), 7.22 (d, J = 11.0 MHz, 2H), 6.38-6.29 (m, 1H), 6.21 (d, J = 15.8 MHz, 1H), 3.88-3.75 (m, 1H), 3.70-3.53 (m, 3H), 2.63 (t, J = 7.7 MHz, 2H) 2.53-2.42 (m, 1H), 2.45 (s, 3H), 2.30-2.15 (m, 3H), 1.70-1.57 (m, 2H), 1.55-1.30 (m, 6H) ppm. 13C NMR (MeOD, 125 MHz), δ: 162.3, 142.3, 138.7, 135.5, 134.3, 132.4, 128.8, 127.1, 117.7, 67.1, 58.2, 44.7, 44.5, 35.0, 32.4, 32.3, 31.2, 28.8, 28.7, 28.6, 12.1 ppm. HRMS (ESI) calcd. for C23H33N2O2+ (M+H)+: 369.25365, found: 369.25509.
(2S,3R)-2-(Hydroxymethyl)-3-(4-(7-(2-methyloxazol-4-yl)heptyl)phenyl)pyrrolidin-1-ium chloride (6) (See Supp. Info for a Scheme)
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-((E)-7-(2-methyloxazol-4-yl)hept-6-en-1-yl)phenyl)pyrrolidine-1-carboxylate 5d (34 mg, 0.05 mmol, 1.0 equiv.) was dissolved in freshly distilled Et3N (1 mL) and Pd/C (10%, 20 mg, 0.02 mmol, 0.4 equiv.) was added. The air was pumped out of the flask and replaced by H2. Upon completion (6 h), the reaction mixture was filtered through celite. The solvent was removed under reduced pressure to afford a colorless oil. This oil was dissolved in dry THF (1 mL) and the solution was cooled to 0 °C. Tetrabutylammonium fluoride solution (1M in THF, 0.10 mL, 0.10 mmol, 2.0 equiv.) was added and the reaction mixture was warmed to room temperature and stirred for 24 h. The reaction mixture was quenched with an aqueous NaHCO3 saturated solution (30 mL) and extracted with CH2Cl2 (30 mL × 4 times). The combined organic layers were washed with brine (60 mL), dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford a pale yellow oil. To this crude a HCl 1M solution in dioxane (0.50 mL, 0.50 mmol, 10 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h, then the solvent was removed under reduced pressure. The crude mixture was purified by silica gel column chromatography with CH2Cl2/MeOH (gradient from 9:1 to 6:4) to give a pale yellow oil. This oil was dissolved in water, filtered through a plastic syringe filter (pore size: 0.45 μm) and lyophilized to afford the title compound 6 as a white solid (17 mg, 88% over three steps). Rf: 0.59 (CH2Cl2/MeOH 8:2). [α]20D −22.9° (c 0.2, MeOH). IR (neat), νmax: 3390, 3192, 2924, 2855, 2344, 2244, 2166, 2038, 1709, 1654, 1607, 1515, 1381, 1227, 1181, 1065, 1032, 1019, 819, 723, 599, 543. 1H NMR (DMSO, 500 MHz), δ: 9.86 (broad s, 1H), 9.02 (broad s, 1H), 7.67 (s, 1H), 7.25 (d, J = 8.1 MHz, 2H), 7.16 (d, J = 8.1 MHz, 2H), 3.62-3.43 (m, 3H), 3.42-3.35 (m, 1H), 3.25-3.12 (m, 3H), 2.56-2.52 (m, 2H) 2.39-2.35 (m, 1H) 2.36 (s, 3H), 2.34-2.25 (m, 1H), 2.07-1.98 (m, 1H), 1.68 (m, 4H), 1.32-1.20 (m, 9H) ppm. 13C NMR (DMSO, 125 MHz), δ: 160.7, 141.3, 139.7, 136.7 134.3, 128.6, 127.5, 66.5, 62.8, 58.5, 48.6, 44.3, 44.2, 34.7, 32.5, 31.0, 28.8, 28.7, 28.5, 27.7, 25.3, 13.5 ppm. HRMS (ESI) calcd. for C23H35N2O2+ (M+H)+: 371.26930, found: 371.26964.
tert-Butyl (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(7-oxooct-1-yn-1-yl)phenyl)pyrrolidine-1-carboxylate (7b) (See Supp. Info for a Scheme)
A solution of commercially available oct-7-yn-2-one A (176 mg, 1.42 mmol, 2 eq.) in dry DMF (500 μL) was added to a solution of 7a (422 mg, 0.71 mmol) in dry DMF (1.2 mL) and Et3N (727 μL). The resulting mixture was degassed by bubbling argon for 20 min, before adding CuI (41 mg, 0.231 mmol, 0.3 eq.) and Pd(PPh3)4 (82 mg, 0.071 mmol, 0.1 eq.). After having been degassed again for additional 15 min, the reaction was stirred for 48 hours at 70 °C. Afterwards, the mixture was diluted with a combination of NH4OH 1 M (25 mL) and brine (50 mL) and the product was extracted with EtOAc (4 × 30 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:6, Rf: 0.09) to give 7b as a yellow oil (236 mg, 52%). [α]25D −0.8 (c 1.55, CHCl3). IR (neat), νmax: 2931, 2858, 1690, 1510, 1472, 1427, 1391, 1364, 1240, 1166, 1107, 1070, 987, 823, 773, 740, 702, 605, 541, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.67-7.62 (m, 4 H), 7.42-7.35 (m, 6 H), 7.31 (t, J = 9.1 Hz, 2H), 7.07 (d, J = 7.5 Hz, 2 H), 4.13 (dd, J = 10.1, 3.4 Hz, 0.4 H), 3.89 (br. s, 0.4 H), 3.79-3.75 (m, 1.8 H), 3.72-3.69 (m, 1 H), 3.64-3.59 (m, 1 H), 3.56-3.52 (m, 0.4 H), 3.44-3.38 (m, 1 H), 2.49 (t, J = 7.3 Hz, 2 H), 2.42 (t, J = 6.9 Hz, 2 H), 2.30-2.22 (m, 1 H), 2.15 (s, 3 H), 1.93-1.86 (m, 1 H), 1.77-1.72 (m, 2 H), 1.63-1.58 (m, 2 H), 1.50 (s, 3.6 H), 1.33 (s, 5.4 H), 1.06 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 208.7, 154.2, 143.3, 142.7, 135.6, 133.7, 133.5, 133.4, 133.3, 131.8, 129.7, 129.6, 127.7, 127.3, 127.1, 122.1, 89.5, 80.7, 79.5, 79.2, 65.4, 65.1, 63.5, 62.1, 47.0, 46.5, 46.4, 45.6, 43.2, 32.7, 31.7, 29.9, 28.6, 28.4, 28.1, 26.8, 23.0, 19.2 ppm. HRMS (ESI) calcd. for C40H52NO4Si (M+H)+ 638.36601, found 638.36681.
tert-Butyl (2S,3R)-2-(hydroxymethyl)-3-(4-(7-oxooctyl)phenyl)pyrrolidine-1-carboxylate (7c) (See Supp. Info for a Scheme)
7b (200 mg, 0.31 mmol) was dissolved in EtOAc (5 mL) and Pd/C (10%, 37 mg) was added to the resulting solution. The air was removed from the flask under vacuum and replaced with hydrogen (balloon). The reaction was vigorously stirred overnight at room temperature. Afterwards, the mixture was filtered through a celite pad, washing with EtOAc. The collected solution was concentrated in vacuo, affording the intermediate (2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(7-oxooctyl)phenyl)pyrrolidine-1-carboxylate a colorless oil (201 mg, 99%). [α]25D +8.5 (c 0.87, CHCl3). IR (neat), νmax: 2929, 2856, 1692, 1514, 1472, 1427, 1392, 1365, 1254, 1166, 1108, 987, 856, 822, 772, 740, 701, 607, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.73-7.63 (m, 4 H), 7.42-7.35 (m, 6 H), 7.15-7.08 (m, 4 H), 4.16-4.13 (m, 0.4 H), 3.90 (br. s, 0.4 H), 3.84-3.76 (m, 1.8 H), 3.72-3.69 (m, 1 H), 3.66-3.60 (m, 1 H), 3.58-3.54 (m, 0.4 H), 3.44-3.39 (m, 1 H), 2.59-2.55 (m, 2 H), 2.41 (t, J = 7.4 Hz, 2 H), 2.30-2.21 (m, 1 H), 2.13 (s, 3 H), 1.95-1.88 (m, 1 H), 1.62-1.55 (m, 4 H), 1.50 (s, 3.6 H), 1.33 (br. s, 9.4 H), 1.06 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 209.2, 154.3, 154.2, 141.0, 140.9, 135.6, 134.8, 133.8, 133.6, 133.5, 133.4, 129.7, 129.6, 128.6, 127.7, 127.3, 127.1, 79.4, 79.0, 65.5, 65.3, 63.5, 62.1, 47.1, 46.4, 46.2, 45.3, 43.7, 35.4, 32.9, 31.9, 31.3, 29.8, 29.1, 29.0, 28.6, 28.5, 28.4, 28.0, 26.9, 26.5, 23.7, 19.4. 19.3 ppm. HRMS (ESI) calcd. for C40H56NO4Si (M+H)+ 642.3973, found 642.40020.
(2S,3R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-(4-(7-oxooctyl)phenyl)pyrrolidine-1-carboxylate (50 mg, 0.078 mmol) was submitted to general procedure B. The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.29) to give 7c as a colorless oil (31 mg, 99%). IR (neat), νmax: 3402, 2928, 2855, 1690, 1666, 1514, 1454, 1394, 1365, 1251, 1165, 1115, 1084, 1054, 968, 854, 772, 535 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.15-7.11 (m, 4 H), 3.91-3.88 (m, 1 H), 3.76-3.73 (m, 2 H), 3.61 (dd, J = 11.5, 6.9 Hz, 1 H), 3.34 (td, J = 10.5, 6.4 Hz, 1 H), 2.90 (br. s, 1 H), 2.58-2.55 (m, 2 H), 2.41 (t, J = 7.4 Hz, 2 H), 2.13 (s, 3 H), 1.99-1.91 (m, 1 H), 1.62-1.54 (m, 4 H), 1.50 (s, 9 H), 1.34-1.30 (m, 4 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 209.3, 156.5, 141.6, 138.0, 128.7, 127.5, 80.5, 67.0, 65.7, 47.5, 47.1, 43.7, 35.4, 32.8, 31.2, 29.9, 29.0, 28.5, 23.7 ppm. HRMS (ESI) calcd. for C24H37NO4Na (M+Na)+ 426.26150, found 426.26007.
8-(4-((2S,3R)-2-(hydroxymethyl)pyrrolidin-3-yl)phenyl)octan-2-one hydrochloride (7) (See Supp. Info for a Scheme)
Prepared according to general procedure A, starting from 7c (20 mg, 0.050 mmol). The crude was recrystallized from EtOAc to give product 7 as a white solid (16 mg, 94%). For biological testing a portion of the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D +16.4 (c 0.55, CHCl3). IR (neat), νmax: 3277, 2922, 2848, 1707, 1589, 1517, 1464, 1401, 1367, 1332, 1216, 1161, 1110, 1064, 1020, 965, 928, 819, 775, 716, 654, 634, 591, 542, 503 cm−1. 1H NMR (CD3OD, 500 MHz), δ: 7.27 (d, J = 8.2 Hz, 2 H), 7.22 (d, J = 8.2 Hz, 2 H), 3.77 (q, J = 6.2 Hz, 1 H), 3.66-3.60 (m, 2 H), 3.58 (ddd, J = 11.8, 8.8, 3.3 Hz, 1 H), 3.41 (ddd, J = 11.8, 9.9, 7.3 Hz, 1 H), 3.31-3.27 (m, 1 H), 2.64-2.61 (m, 2 H), 2.50-2.45 (m, 3 H), 2.28-2.20 (m, 1 H), 2.14 (s, 3 H), 1.66-1.60 (m, 2 H), 1.59-1.53 (m, 2 H), 1.37-1.32 (m, 4 H) ppm. 13C NMR (CD3OD, 125 MHz), δ: 210.7, 142.2, 135.5, 128.8, 127.1, 67.1, 58.2, 44.7, 44.5, 42.8, 35.0, 32.4, 31.1, 28.6, 28.4, 23.4 ppm.
tert-Butyl (S)-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (8b)
Prepared according to general procedure C, starting from 8a [25] (300 mg, 1.10 mmol). The crude was purified by flash column chromatography (EtOAc/hexane 1:6, Rf: 0.17) to give 8b as a colorless oil (274 mg, 62%). [α]25D −22.9 (c 1.3, CHCl3). IR (neat), νmax: 2924, 2854, 1680, 1606, 1455, 1391, 1365, 1277, 1169, 1116, 1012, 989, 772, 545 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.96-7.87 (m, 2 H), 7.25 (d, J = 7.5 Hz, 2 H), 4.34-4.29 (m, 1 H), 3.74 (br. d, J = 14.8 Hz, 0.5 H), 3.47 (br. d, J = 15.2 Hz, 0.5 H), 3.40 (br. s, 1 H), 3.32 (br. s, 1 H), 2.85-2.73 (m, 1 H), 2.64 (br. s, 2 H), 2.03 (br. s, 1 H), 1.90-1.79 (m, 2 H), 1.75 (br. s, 1 H), 1.64-1.57 (m, 2 H), 1.45 (s, 9 H), 1.30-1.22 (m, 10 H), 0.86 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.8, 198.3, 154.4, 154.3, 149.1, 148.7, 134.6, 128.7, 128.5, 128.4, 79.7, 79.2, 54.5, 54.3, 46.7, 46.2, 43.7, 43.0, 36.0, 31.9, 31.3, 31.1, 30.3, 29.4, 29.3, 29.2, 28.6, 23.6, 22.8, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 402.30027, found 402.29965.
(S)-1-(4-Octylphenyl)-2-(pyrrolidin-2-yl)ethan-1-one hydrochloride (8)
Prepared according to general procedure A, starting from 8b (100 mg, 0.25 mmol). The crude was purified by flash column chromatography (EtOH/CH2Cl2 1:4, Rf: 0.45) to give product 8 as a white solid (84 mg, 99%). Note: the product racemized spontaneously when dissolved in MeOH or H2O. For biological testing a portion of this solid was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D −39.1 (c 0.23, CHCl3). IR (neat), νmax: 2921, 2852, 1678, 1605, 1589, 1466, 1377, 1222, 1188, 1032, 976, 914, 822, 770, 569 cm−1. 1H NMR (CDCl3, 400 MHz), δ: 9.52 (br. s, 2 H), 7.86 (d, J = 8.1 Hz, 2 H), 7.18 (d, J = 7.8 Hz, 2 H), 4.17-4.09 (m, 1 H), 3.89 (dd, J = 18.4, 5.9 Hz, 1 H), 3.50 (dd, J = 18.4, 6.8 Hz, 1 H), 3.40 (t, J = 7.2 Hz, 2 H), 2.61-2.57 (m, 2 H), 2.37-2.29 (m, 1 H), 2.10-1.95 (m, 2 H), 1.81-1.70 (m, 1 H), 1.59-1.54 (m, 2 H), 1.30-1.24 (m, 10 H), 0.88 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 196.8, 149.6, 133.6, 128.7, 128.4, 56.0, 45.0, 40.5, 36.0, 31.9, 31.0, 30.6, 29.7, 29.4, 29.3, 29.2, 23.6, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C20H32NO (M)+ 302.24784, found 302.24782.
tert-Butyl (2S)-2-(2-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate (8c)
NaBH4 (4.9 mg, 0.13 mmol, 1.5 eq.) was added to a solution of 8b (35 mg, 0.087 mmol) in MeOH (3 mL) at 0 °C. The resulting mixture was stirred for 2 hours at the same temperature. Afterwards, the reaction was quenched with brine (1 mL), the MeOH was removed in vacuo and the product was extracted with EtOAc (4 × 4 mL). The organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:6, then EtOAc/hexane 1:4 Rf: 0.38 and 0.19) to give 8c diast1 (7 mg, 20%) and 8c diast2 (28 mg, 80%) as colorless oils. 8c diast1: [α]20D −8.0 (c 0.2, MeOH). IR (neat), νmax: 3406, 2923, 2851, 1723, 1671, 1397, 1245, 1168, 1104, cm−1. 1H NMR (CDCl3, 300 MHz), δ: 7.28 (d, J = 7.9 Hz, 2 H), 7.13 (d, J = 8.0 Hz, 2 H), 5.33 (br. s, 1 H), 4.64-4.57 (m, 1 H), 4.33-4.25 (m, 1 H), 3.37 (t, J = 6.6 Hz, 2 H), 2.60-2.54 (m, 2 H), 2.03-1.93 (m, 2 H), 1.91-1.87 (m, 2 H), 1.72-1.67 (m, 2 H), 1.62-1.54 (m, 2 H), 1.49 (s, 9 H), 1.25 (br. s, 10 H), 0.87 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (CDCl3, 75 MHz), δ: 156.7, 141.6, 141.5, 128.2, 125.6, 80.0, 69.8, 54.0, 46.6, 46.3, 35.6, 31.9, 31.5, 31.2, 29.7, 29.5, 29.3, 28.5, 23.6, 22.7, 14.1 ppm. HRMS (ESI) calcd. for C25H41NO3Na (M+Na)+ 426.29787, found 426.29919. 8c diast2 [α]20D −52.5 (c 0.8, MeOH). IR (neat), νmax: 3413, 2924, 2854, 1668, 1393, 1365, 1247, 1168, 1103, 849, 772, 557 cm−1. 1H NMR (CDCl3, 300 MHz), δ: 7.26 (d, J = 7.8 Hz, 2 H), 7.13 (d, J = 7.7 Hz, 2 H), 4.74 (br. s, 1 H), 4.10 (br. s, 1 H), 3.31 (br. s, 2 H), 2.60-2.55 (m, 2 H), 2.14 (br. s, 1 H), 2.05-1.93 (m, 1 H), 1.89-1.79 (m, 2 H), 1.69 (br. s, 2 H), 1.61-1.54 (m, 2 H), 1.46 (s, 9 H), 1.30-1.26 (m, 10 H), 0.87 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (CDCl3, 75 MHz), δ: 155.4, 142.3, 141.6, 128.3, 125.5, 79.7, 72.5, 55.7, 46.4, 46.3, 35.6, 32.4, 31.9, 31.5, 29.7, 29.5, 29.3, 29.2, 28.5, 23.8, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C25H41NO3Na (M+Na)+ 426.29787, found 426.29907.
(R)-2-(4-Octylphenethyl)pyrrolidine hydrochloride (9)
8c (12 mg, 0.0297 mmol) was dissolved in EtOH (3 mL) and Pd/C (10%, 7 mg) was added to the resulting solution. The air was removed from the flask under vacuum and replaced with hydrogen (balloon). The reaction was vigorously stirred overnight at room temperature. Afterwards, the mixture was filtered through a celite pad, washing with EtOH. The collected solution was concentrated in vacuo, affording the intermediate tert-Butyl (R)-2-(4-octylphenethyl)pyrrolidine-1-carboxylate as a colorless oil (9 mg, 78%). [α]20D −36.0 (c 0.45, CHCl3). IR (neat), νmax: 2924, 2853, 1694, 1514, 1455, 1391, 1364, 1254, 1169, 1100, 771 cm−1. 1H NMR (CDCl3, 400 MHz, mixture of rotamers), δ: 7.09 (s, 4 H), 3.85 (br. s, 0.4 H), 3.75 (br. s, 0.6 H), 3.41 (br. s, 0.8 H), 3.32 (br. s, 1.2 H), 2.58-2.54 (m, 4 H), 2.14 (br. s, 0.4 H), 2.04-1.87 (m, 1.6 H), 1.85-1.80 (m, 2 H), 1.72 (br. s, 1 H), 1.63-1.55 (m, 3 H), 1.45 (s, 9 H), 1.32-1.24 (m, 10 H), 0.88 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (CDCl3, 75 MHz, major rotamer), δ: 154.6, 140.3, 139.2, 128.3, 128.1, 79.0, 56.9, 46.1, 36.4, 35.5, 32.4, 31.9, 31.6, 30.6, 29.7, 29.5, 29.4, 29.2, 28.6, 23.2, 22.7, 14.1 ppm. HRMS (ESI) calcd. for C25H41NO2K (M+K)+ 426.27744, found 426.27543.
tert-Butyl (R)-2-(4-octylphenethyl)pyrrolidine-1-carboxylate (9 mg, 0.023 mmol) was submitted to general procedure A. The crude was purified by flash column chromatography (EtOH/CH2Cl2 1:8, Rf: 0.16) to give product 9 as a white solid (7 mg, 93%). For biological testing a portion of this solid was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D −4.0 (c 0.35, CHCl3). IR (neat), νmax: 2921, 2852, 2751, 1591, 1514, 1455, 1418, 1042, 815, 722, 554 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 9.69 (br. s, 1 H), 9.19 (br. s, 1 H), 7.12 (d, J = 8.0 Hz, 2 H), 7.05 (d, J = 8.0 Hz, 2 H), 3.56-3.48 (m, 1 H), 3.44-3.37 (m, 1 H), 3.35-3.30 (m, 1 H), 2.80-2.74 (m, 1 H), 2.71-2.65 (m, 1 H), 2.55-2.52 (m, 2 H), 2.37-2.30 (m, 1 H), 2.15-2.08 (m, 1 H), 2.06-1.97 (m, 2 H), 1.96-1.88 (m, 1 H), 1.72-1.64 (m, 1 H), 1.59-1.53 (m, 2 H), 1.29-1.25 (m, 10 H), 0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 140.9, 137.2, 128.6, 128.3, 60.0, 44.8, 35.5, 34.0, 32.5, 31.9, 31.6, 30.4, 29.7, 29.5, 29.4, 29.3, 23.5, 22.7, 14.1 ppm. HRMS (ESI) calcd. for C20H34N (M)+ 288.26858, found 288.26992.
(2S)-2-(2-Methoxy-2-(4-octylphenyl)ethyl)pyrrolidine hydrochloride (10)
NaH (1.3 mg, 60% dispersion in mineral oil, 0.033 mmol, 1.2 eq.) was added to a solution of 8c diast2 (11 mg, 0.027 mmol) in dry THF (1 mL) at 0 °C. The resulting mixture was stirred at the same temperature for 1 h, before adding methyl iodide (5 μL, 0.081 mmol, 3 eq.). Then, the reaction was stirred at room temperature for 3 h, before being quenched with water (1 mL). The product was extracted with EtOAc (3 × 2 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:6, Rf: 0.16) to give the intermediate tert-Butyl (2S)-2-(2-methoxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate as a colorless oil (7 mg, 64%). [α]25D −77.1 (c 0.35, CHCl3). IR (neat), νmax: 2924, 2854, 1692, 1454, 1391, 1364, 1251, 1170, 1102, 771 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.23-7.13 (m, 4 H), 4.11 (br. s, 1 H), 4.02 (br. s, 0.5 H), 3.93 (br. s, 0.5 H), 3.39 (br. s, 0.5 H), 3.29 (br. s, 1.5 H), 3.16 (s, 3 H), 2.60-2.57 (m, 2 H), 2.27 (br. s, 1 H), 1.82-1.73 (m, 2 H), 1.62-1.56 (m, 3 H), 1.46 (s, 9 H), 1.30-1.26 (m, 10 H), 0.88 (t, J = 6.9 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 154.5, 142.2, 139.5, 128.4, 126.5, 81.6, 79.0, 78.7, 56.3, 54.7, 46.3, 46.1, 42.6, 35.7, 31.9, 31.5, 30.9, 30.3, 29.7, 29.5, 29.3, 29.2, 28.6, 23.7, 23.2, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C26H44NO3 (M+H)+ 418.33210, found 418.33141.
tert-Butyl (2S)-2-(2-methoxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate (6 mg, 0.014 mmol) was submitted to general procedure A. The crude was purified by flash column chromatography (EtOH/CH2Cl2 1:10, Rf: 0.20) to give product 10 as a white solid (3 mg, 60%). For biological testing a portion of this solid was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D −55.0 (c 0.20, CHCl3). IR (neat), νmax: 2921, 2851, 2766, 1459, 1107, 1033, 827, 722, 564 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 10.53 (br. s, 1 H), 8.54 (br. s, 1 H), 7.19 (d, J = 8.1 Hz, 2 H), 7.14 (d, J = 8.1 Hz, 2 H), 4.35 (dd, J = 10.2, 2.9 Hz, 1 H), 3.91 (br. s, 1 H), 3.51-3.46 (m, 1 H), 3.38-3.33 (m, 1 H), 3.20 (s, 3 H), 2.59-2.56 (m, 2 H), 2.28-2.21 (m, 2 H), 2.07-2.03 (m, 2 H), 1.95-1.92 (m, 1 H), 1.73-1.67 (m, 1 H), 1.61-1.55 (m, 2 H), 1.30-1.24 (m, 10 H), 0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 143.2, 137.3, 128.7, 126.4, 82.4, 58.9, 56.5, 44.5, 40.1, 35.7, 31.9, 31.4, 30.7, 29.7, 29.5, 29.3, 29.2, 23.1, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C21H36NO (M)+ 318.27914, found 318.28009.
tert-Butyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(2-methoxy-2-oxoethyl)pyrrolidine-1-carboxylate (11a)
Commercially available 1-(tert-butyl) 2-methyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)pyrrolidine-1,2-dicarboxylate (200 mg, 0.56 mmol, 1.0 eq.) was dissolved in MeOH (1 mL) and an aqueous LiOH (330 μL, 1M, 1.5 eq.) was added. The solution was stirred at 45 °C for 3h whereby TLC analysis indicated that the reaction had gone to completion. A 5% (w/w) aqueous HCl solution was added dropwise until pH = 2, whereby a white precipitate was formed. The mixture was extracted with Et2O (2 × 5 mL). The resulting organic layer was collected, dried over Na2SO4, filtered and concentrated to afford an incolore oil which was brought to the next step without further purification.
The incolore oil (120 mg, 0.35 mmol) was dissolved back in THF (2 mL) then Et3N (97 μL, 0.70 mmol, 2 eq.) and isobutylchloroformate (58 μL, 0.310 mmol, 1.6 eq.) were added. The resulting mixture was stirred at room temperature for 1 h. Afterwards, the reaction was cooled down to 0 °C and a freshly prepared solution of CH2N2 in Et2O was added dropwise until the resulting mixture remained bright yellow. Then, the reaction was stirred for 1h at 0 °C and for 30 min at room temperature, adding further CH2N2 any time the mixture had turned back to colorless. After a persistant yellow color remained, the flask was cooled down again to 0 °C and a 0.5 M solution of acetic acid in water was slowly added until the mixture turned colorless. Then, the layers were separated and the aqueous one was extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:3, Rf: 0.21) to give the diazo intermediate as a pale yellow oil. This intermediate was redissolved in dry MeOH (2 mL) and a solution of silver benzoate (16 mg, 0.07 mmol, 0.2 eq.) in Et3N (97 μL, 0.70 mmol, 2 eq.) was added to this mixture under an argon atmosphere. Then, the flask was wrapped in aluminum foil and the reaction was refluxed for 2 h. Afterwards, the mixture was left to reach room temperature, filtered through a celite pad washing with abundant EtOAc and concentrated.The residue was purified by flash column chromatography (hexane/EtOAc 8:2 Rf: 0.28) to give 11a as a colorless oil (77 mg, 59% over 2 steps). [α]25D −74.1 (c 0.78, CHCl3). IR (neat), νmax: 2929, 1739, 1693, 1472, 1152, 1108, 853, 774 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 4.32-4.28 (p, J = 4.3 Hz, 1 H), 4.24-4.16 (m, 1 H), 3.65 (s, 3 H), 3.43-3.33 (m, 2 H), 2.99-2.86 (dd, J = 11.1, 4.6 Hz, 1 H), 2.37 (m, 1 H), 2.09 (m, 1 H), 1.84 (m, 1 H), 1.44 (s, 9 H), 0.85 (s, 9 H), 0.04 (s, 6 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 172.0, 171.9, 155.0, 154.9, 79.9, 79.5, 70.2, 69.6, 55.1, 54.7, 53.0, 51.6, 41.2, 40.5, 39.6, 38.9, 28.6, 25.8, 18.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 374.23680, found 374.23637.
tert-Butyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate (11b)
Isopropyl magnesium chloride (240 μL, 2 M in THF, 0.48 mmol, 6 eq.) was added dropwise to a solution of 11a (30.0 mg, 0.08 mmol) and N,O-dimethylhydroxylamine (23 mg, 0.24 mmol, 3 eq.) in dry THF (1 mL) at −20 °C. The resulting mixture was allowed to reach 0 °C over 3 h, then, it was stirred at the same temperature overnight. Afterwards, the reaction was quenched by adding a few drops of water. Then, the mixture was filtered on a celite pad washing with abundant EtOAc and concentrated. The residue was purified by flash column chromatography (hexane/EtOAc 7:3 Rf: 0.35) to give 11b as a colorless oil (26 mg, 81%). [α]25D −56.6 (c 0.58, CHCl3). IR (neat), νmax: 2954, 1692, 1390, 1252, 1156, 835 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 4.34-4.26 (m, 2 H), 3.68 (s, 3 H), 3.43-3.32 (m, 2 H), 3.16-3.02 (m, 4 H), 2.46 (bs, 1 H), 2.12 (bs, 1 H), 1.87 (bs, 1 H), 1.45 (s, 9 H), 0.85 (s, 9 H), 0.04 (s, 6 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 172.7, 172.4, 155.0, 79.7, 79.3, 70.3, 69.7, 61.4, 55.1, 54.5, 53.1, 41.4, 40.6, 37.7, 36.7, 32.1, 32.1, 28.6, 28.5, 25.9, 25.9, 18.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 403.26230, found 403.26277.
tert-Butyl (2S,4R)-4-hydroxy-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (11c)
11b was submitted to general procedure C (11 mg, 0.02 mmol). The crude yellow oil was submitted to general procedure B without further purification. The resulting residue was purified by flash column chromatography (hexane/EtOAc 8:2 Rf: 0.33) to give 11c as a yellow oil (5 mg, 63% over 2 steps). [α]25D +50.4 (c 0.25, CHCl3). IR (neat), νmax: 2953, 1856, 1783, 1251, 932, 704 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.89 (bs, 2 H), 7.26-7.24 (m, 2 H), 4.44-4.40 (m, 2 H), 3.90-3.87 (m, 0.57 H), 3.67.3.60 (m, 1 H), 3.46 (m, 0.64 H), 2.89-2.84 (dd, J = 15.5, 9.6 Hz, 1 H), 2.65-2.62 (m, 2 H), 2.22 (m, 2 H), 1.90 (bs, 1 H), 1.62-1.59 (m, 2 H), 1.45 (s, 9 H), 1.32-1.23 (m, 10 H), 0.88-0.85 (t, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.9, 198.2, 171.3, 154.9, 149.3, 149.1, 134.6, 128.8, 128.5, 80.3, 79.7, 69.9, 69.4, 60.5, 54.9, 54.6, 53.3, 43.3, 40.3, 36.1, 32.0, 31.2, 29.5, 29.4, 29.3, 28.6, 22.8, 21.2, 14.3, 14.2 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 418.29519, found 418.29710.
(2S,4R)-4-Hydroxy-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidin-1-ium chloride (11)
11 was synthesized in accordance with the general procedure A, starting from 11c (21 mg, 0.05 mmol). 11 was obtained as a white solid (13 mg, 75%). IR (neat), νmax: 3310, 2921, 1669, 1605, 1277 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of diastereomers), δ: 7.71-7.70 (d, J = 8.2 Hz, 2 H), 7.00-6.99 (d, J = 8.0 Hz, 2 H), 4.51 (s, 1 H), 4.07-4.04 (dd, J = 11.3, 6.4 Hz, 1 H major isomer), 3.99-3.96 (dd, J = 9.0, 6.2 Hz, 1 H minor isomer), 3.41-3.24 (m, 2 H), 2.37-2.05 (m, 3 H), 1.77-1.63 (m, 1 H), 1.36 (m, 2 H), 1.18-1.14 (m, 10 H), 0.78-0.76 (t, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of diastereomers), δ: 198.5, 149.1, 133.5, 128.5, 128.5, 69.1, 69.1, 54.3, 54.0, 52.8, 38.5, 37.7, 35.6, 31.8, 30.8, 29.4, 29.4, 29.3, 22.6, 13.8 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 318.24276, found 318.24284.
1-(tert-butyl) 2-ethyl (2R,4R)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-5-oxopyrrolidine-1,2-dicarboxylate (12a3) and 1-(tert-butyl) 2-ethyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-5-oxopyrrolidine-1,2-dicarboxylate (epi-12a3) (See Supp. Info for a Scheme)
HCl (84 mL, 0.2 N in H2O, 16.8 mmol, 1 eq.) was added dropwise to a solution of 12a1 (5.25 g, 16.8 mmol) in MeOH (125 mL) and the resulting solution was stirred at room temperature for 1 h. Afterwards, a small amount of bromocresol green was added to monitor the pH of the solution and NaBH3CN (2.11 g, 33.6 mmol, 2 eq.) was added portion wise (4 portions over 2 h). Meanwhile, the pH of the solution had been corrected by adding a few drops of HCl (0.2 N in H2O), anytime the pH indicator had turned blue. HCl has always been added in the minimal amount necessary to make the indicator turn back to yellow. The reaction was stirred at room temperature for 48 hours, continuing monitoring and correcting the pH when needed. Eventually, a few drops of NaHCO3 satd. solution were added until the indicator turned blue and the mixture was concentrated in vacuo to remove the organic solvent. Brine (130 mL) was added to the mixture and the product was extracted in EtOAc (3 × 130 mL). The organic layers were dried over Na2SO4, filtered and concentrated. The resulting residue was purified by flash column chromatography (MeOH/CH2Cl2 1:20, Rf: 0.24) to give the intermediate alcohol 12a2 (3.38 g, 70%) as a 2:1 mixture of diasteroisomers. This intermediate was redissolved in dry DMF (60 mL) and imidazole (2.41 g, 35.4 mmol, 3 eq.) was added to the resulting solution. Then, this mixture was cooled down to 0 °C and TBDPSCl (4.60 mL, 17.7 mmol, 1.5 eq.) was added dropwise. The resulting solution was stirred at room temperature for 3 h, before adding EtOH (1 mL) and stirring for additional 30 min. Eventually, the mixture was poured into water (400 mL) and the product was extracted into Et2O (3 × 300 mL). The organic layers were dried over Na2SO4, filtered and concentrated. The resulting residue was purified by flash column chromatography (Et2O/hexane 1:2 Rf: 0.13 and 0.06, then Et2O/hexane 1:1) to give 12a3 (2.98 g, 48%) and epi-12a3 (1.12 g, 18%) as colorless oils. The stereochemistry was assigned by performing NOESY experiments (through space coupling observed between 2-H and 4-H in 12a3, epi-12a3) 12a3: [α]25D +28.3 (c 2.7, CHCl3). IR (neat), νmax: 2931, 1792, 1746, 1718, 1472, 1428, 1369, 1313, 1278, 1188, 1151, 1111, 1007, 966, 913, 848, 822, 734, 702, 613, 504 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.66-7.62 (m, 4 H), 7.43-7.37 (m, 6 H), 4.62 (dd, J = 9.7, 3.2 Hz, 1 H), 4.24 (q, J = 7.1 Hz, 2 H), 4.03 (dd, J = 10.2, 4.8 Hz, 1 H), 3.80 (dd, J = 10.2, 3.4 Hz, 1 H), 2.82-2.77 (m, 1 H), 2.46-2.40 (m, 1H), 2.17-2.12 (m, 1 H), 1.50 (s, 9 H), 1.30 (t, J = 7.1 Hz, 3 H), 1.03 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 173.3, 171.6, 149.3, 135.7, 135.5, 133.2, 132.6, 129.8, 127.7, 83.4, 62.9, 61.6, 57.6, 44.5, 27.8, 26.8, 25.2, 19.2, 14.1 ppm. HRMS (ESI) calcd. for C29H39NO6SiNa (M+Na)+ 548.24389, found 548.24492. epi-12a3: [α]25D +7.7 (c 3.6, CHCl3). IR (neat), νmax: 2931, 1791, 1746, 1718, 1473, 1428, 1369, 1318, 1151, 1109, 1032, 970, 909, 822, 781, 734, 702, 613, 504 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.66-7.61 (m, 4 H), 7.44-7.35 (m, 6 H), 4.50 (dd, J = 8.9, 7.5 Hz, 1 H), 4.22-4.13 (m, 2 H), 3.93 (dd, J = 10.3, 6.7 Hz, 1 H), 3.87 (dd, J = 10.3, 4.1 Hz, 1 H), 2.79 (dddd, J = 9.5, 8.7, 6.7, 4.1 Hz, 1 H), 2.52-2.45 (m, 1H), 2.16 (ddd, J = 13.2, 8.7, 7.5 Hz, 1 H), 1.49 (s, 9 H), 1.25 (t, J = 7.1 Hz, 3 H), 1.04 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 172.7, 171.3, 149.2, 135.6, 135.5, 133.2, 132.9, 129.7, 127.7, 83.5, 62.3, 61.5, 57.6, 45.4, 27.8, 26.7, 24.3, 19.2, 14.0 ppm. HRMS (ESI) calcd. for C29H39NO6SiNa (M+Na)+ 548.24389, found 548.24498.
1-(tert-Butyl) 2-ethyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)pyrrolidine-1,2-dicarboxylate (13a) (See Supp. Info for a Scheme)
LiEt3BH (4.68 mL, 1 M in THF, 4.68 mmol, 1.2 eq.) was added dropwise to a solution of 12a3 (2.05 g, 3.90 mmol) in anhydrous THF (70 mL) at −78 °C under an argon atmosphere and the resulting solution was stirred at the same temperature for 30 min. Afterwards, the reaction was quenched with NaHCO3 satd. sol. (20 mL) and allowed to reach 0 °C, then a few drops of H2O2 30% were added and the mixture was stirred at 0 °C for 20 min. Eventually, the organic solvent was removed under vacuo and the remaining aqueous layer was extracted with CH2Cl2 (3 × 120 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated, affording a colorless oil. This intermediate hemiaminal was redissolved in anhydrous CH2Cl2 (70 mL) and Et3SiH (1.25 mL, 7.8 mmol, 2 eq.) was added to the solution under an argon atmosphere. The resulting mixture was cooled down to −78 °C and BF3OEt2 (481 μL, 3.90 mmol, 1 eq.) was added dropwise. The reaction was stirred at −78 °C for 30 min, before adding NaHCO3 satd. sol. (20 mL) and allowing the mixture to reach room temperature. The product was extracted with CH2Cl2 (3 × 120 mL) and the organic extracts were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:6, Rf: 0.24) to give 13a as a colorless oil (1.52 g, 76% over two steps). [α]25D +24.7 (c 1.5, CHCl3). IR (neat), νmax: 2931, 2858, 1744, 1699, 1473, 1427, 1389, 1365, 1257, 1188, 1110, 1030, 939, 870, 823, 741, 702, 611, 505 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.65-7.63 (m, 4 H), 7.44-7.37 (m, 6 H), 4.34 (dd, J = 7.9, 4.1 Hz, 0.4 H), 4.24-4.14 (m, 2.6 H), 3.73 (dd, J = 10.6, 7.7 Hz, 0.6 H), 3.64-3.59 (m, 2.4 H), 3.29 (dd, J = 10.6, 7.4 Hz, 0.6 H), 3.23, (dd, J = 10.5, 7.4 Hz, 0.4 H), 2.60-2.51 (m, 1 H), 2.13-2.04 (m, 1H), 2.03-1.98 (m, 1 H), 1.47 (s, 3.6 H), 1.42 (s, 5.4 H), 1.30-1.25 (m, 3 H), 1.06 (s, 3.6 H), 1.05 (s, 5.4 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 173.2, 172.9, 154.4, 153.7, 135.5, 133.4, 133.3, 129.7, 127.7, 79.8, 79.7, 64.8, 60.9, 60.8, 59.0, 58.7, 48.9, 48.8, 39.8, 38.9, 33.0, 32.2, 28.4, 28.3, 26.8, 19.2, 14.3, 14.1 ppm. HRMS (ESI) calcd. for C29H42NO5Si (M+H)+ 512.28268, found 512.28059.
(2R,4S)-1-(tert-Butoxycarbonyl)-4-(((tert-butyldiphenylsilyl)oxy)methyl)pyrrolidine-2-carboxylic acid (12a4) (See Supp. Info for a Scheme)
NaOH (290 μL, 1 N in H2O, 0.29 mmol, 1.5 eq.) was added to a solution of 13a (99 mg, 0.19 mmol) in MeOH (1.2 mL) and the resulting mixture was vigorously stirred for 24 h. Afterwards, the organic solvent was concentrated in vacuo and the residue was suspended in brine (20 mL). Afterwards, while gradually acidifying to pH=2 by adding HCl 0.2 N, the product was extracted with CH2Cl2 (6 × 20 mL). The organic layers were dried over Na2SO4, filtered and concentrated, affording 12a4 (93 mg, 99%) as a colorless solid. [α]25D +19.3 (c 0.9, MeOH). IR (neat), νmax: 2929, 1699, 1390, 1366, 1162, 1108, 998, 906, 823, 739, 700, 608, 503 cm−1. 1H NMR (CD3OD, 500 MHz, mixture of rotamers), δ: 7.74-7.66 (m, 4 H), 7.47-7.37 (m, 6 H), 4.30-4.21 (m, 1 H), 3.66-3.57 (m, 3 H), 3.37-3.33 (m, 1 H), 2.57-2.54 (m, 1 H), 2.17-2.11 (m, 1H), 2.06-2.03 (m, 1 H), 1.48 (s, 3.6 H), 1.44 (s, 5.4 H), 1.06 (s, 9 H) ppm. 13C NMR (CD3OD, 125 MHz, mixture of rotamers), δ: 175.9, 154.9, 154.6, 135.9, 135.3, 134.6, 133.1, 129.6, 129.5, 129.0, 127.5, 127.2, 80.0, 79.8, 64.8, 64.6, 59.5, 48.9, 48.6, 39.7, 38.9, 32.8, 32.1, 27.4, 27.2, 26.0, 25.8, 18.7, 18.5 ppm. HRMS (ESI) calcd. for C27H38NO5SiNa (M+Na)+ 506.23332, found 506.23394.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(2-methoxy-2-oxoethyl)pyrrolidine-1-carboxylate (12a) (See Supp. Info for a Scheme)
Et3N (54 μL, 0.388 mmol, 2 eq.) and isobutylchloroformate (40 μL, 0.310 mmol, 1.6 eq.) were added to a solution of 12a4 (94 mg, 0.194 mmol) in anhydrous THF (2 mL) at 0 °C and the resulting mixture was stirred at room temperature for 1 h. Afterwards, the reaction was cooled down to 0 °C and a freshly prepared solution of CH2N2 in Et2O was added dropwise until the resulting mixture remained bright yellow. Then, the reaction was stirred for 1h at 0 °C and for 30 min at room temperature, adding further CH2N2 any time the mixture had turned back to colorless. After a persistant yellow color remained, the flask was cooled down again to 0 °C and a 0.5 M solution of acetic acid in water was slowly added until the mixture turned colorless. Then, the layers were separated and the aqueous one was extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with water (5 mL) and brine (5 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:3, Rf: 0.21) to give the diazo intermediate as a pale yellow oil. This intermediate was redissolved in dry MeOH (2 mL) and a solution of silver benzoate (9 mg, 0.0388 mmol, 0.2 eq.) in Et3N (54 μL, 0.388 mmol, 2 eq.) was added to this mixture under an argon atmosphere. Then, the flask was wrapped in aluminum foil and the reaction was refluxed for 2 h. Afterwards, the mixture was left to reach room temperature, filtered through a celite pad washing with abundant EtOAc and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:4, Rf: 0.27) to give 12a as a colorless oil (54 mg, 55% over two steps). [α]25D +21.2 (c 1.0, CHCl3). IR (neat), νmax: 2931, 2858, 1737, 1692, 1428, 1388, 1365, 1253, 1161, 1109, 823, 740, 702, 611, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.64-7.62 (m, 4 H), 7.44-7.37 (m, 6 H), 4.21 (br. s, 0.5 H), 4.13 (br. s, 0.5 H), 3.67 (s, 3 H), 3.63-3.60 (m, 1 H), 3.58-3.51 (m, 1.5 H), 3.43 (br. s, 0.5 H), 3.25-3.16 (m, 1 H), 2.91, (d, J = 14.2 Hz, 0.5 H), 2.80 (d, J = 14.6 Hz, 0.5 H), 2.50-2.44 (m, 1 H), 2.33 (dd, J = 14.6, 9.7 Hz, 1H), 1.86 (br. s, 1 H), 1.80-1.77 (m, 1 H), 1.46 (s, 9 H), 1.05 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 171.9, 154.3, 154.1, 135.5, 133.5, 129.7, 127.9, 127.7, 79.6, 79.3, 65.3, 54.0, 51.6, 49.1, 39.3, 39.2, 38.7, 38.4, 33.7, 33.1, 28.5, 26.8, 19.2 ppm. HRMS (ESI) calcd. for C29H42NO5Si (M+H)+ 512.28268, found 512.28235.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate (12b)
12a (53 mg, 0.104 mmol) was submitted to the procedure from 11b. The residue was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.30) to give 12b as a colorless oil (47 mg, 84%). [α]25D +23.3 (c 0.6, CHCl3). IR (neat), νmax: 2931, 2857, 1690, 1385, 1364, 1256, 1162, 1109, 1000, 906, 870, 823, 739, 702, 611, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.65-7.62 (m, 4 H), 7.44-7.36 (m, 6 H), 4.24 (br. s, 1 H), 3.68 (s, 3 H), 3.65-3.62 (m, 1 H), 3.56 (br. s, 1.5 H), 3.45 (br. s, 0.5 H), 3.24 (br. s, 1 H), 3.17 (s, 3 H), 3.00, (d, J = 14.7 Hz, 0.5 H), 2.88 (d, J = 13.6 Hz, 0.5 H), 2.52-2.45 (m, 2 H), 1.89-1.81 (m, 2 H), 1.46 (s, 9 H), 1.04 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 172.3, 154.2, 135.6, 135.5, 133.6, 129.7, 127.7, 79.5, 79.1, 65.4, 61.2, 54.0, 49.1, 39.2, 38.4, 36.8, 36.4, 33.8, 32.0, 28.5, 26.8, 19.2 ppm. HRMS (ESI) calcd. for C30H45N2O5Si (M+H)+ 541.30923, found 541.30687.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (12c)
Prepared according to general procedure C, starting from 12b (30 mg, 0.055 mmol). The crude was purified by flash column chromatography (EtOAc/hexane 1:10, Rf: 0.17) to give 12c as a colorless oil (25 mg, 68%). [α]25D +5.3 (c 0.3, CHCl3). IR (neat), νmax: 2924, 2853, 1671, 1606, 1515, 1458, 1390, 1366, 1175, 1111, 823, 739, 701, 611, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.95 (d, J = 7.3 Hz, 1 H), 7.90 (d, J = 6.8 Hz, 1 H), 7.64-7.61 (m, 4 H), 7.43-7.35 (m, 6 H), 7.27-7.26 (m, 2 H), 4.38-4.34 (m, 1 H), 3.72 (d, J = 14.8 Hz, 0.5 H), 3.61 (br. s, 1 H), 3.58-3.53 (m, 1.5 H), 3.50-3.43 (m, 1 H), 3.29-3.25 (m, 0.5 H), 3.22-3.19 (m, 0.5 H), 2.88-2.78 (m, 1 H), 2.65 (br. s, 2 H), 2.54-2.48 (m, 1 H), 1.87-1.75 (m, 2 H), 1.65-1.59 (m, 2 H), 1.47 (s, 4.5 H), 1.45 (s, 4.5 H), 1.31-1.26 (m, 10 H), 1.03 (s, 9 H), 0.88 (t, J = 6.9 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.7, 198.3, 154.4, 154.2, 134.6, 133.5, 129.7, 128.7, 128.5, 128.4, 127.7, 79.7, 79.3, 65.4, 65.2, 54.4, 49.2, 43.9, 43.2, 39.2, 38.4, 36.0, 33.7, 32.7, 31.8, 31.1, 29.7, 29.4, 29.3, 29.2, 28.5, 26.8, 22.6, 19.2, 14.1 ppm. HRMS (ESI) calcd. for C42H60NO4Si (M+H)+ 670.42861, found 670.42981.
2-((2R,4S)-4-(Hydroxymethyl)pyrrolidin-2-yl)-1-(4-octylphenyl)ethan-1-one hydrochloride (12)
Prepared according to general procedure B, starting from 12c (14 mg, 0.021 mmol). The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.28) to give the intermediate tert-Butyl (2R,4S)-4-(hydroxymethyl)-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate as a colorless oil (9 mg, 99%). [α]25D +7.5 (c 0.4, CHCl3). IR (neat), νmax: 3439, 2924, 2854, 1673, 1606, 1394, 1366, 1255, 1173, 1123, 772, 558 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.94 (d, J = 7.7 Hz, 1 H), 7.89 (d, J = 7.9 Hz, 1 H), 7.27-7.24 (m, 2 H), 4.42-4.37 (m, 1 H), 3.72 (d, J = 15.5 Hz, 0.5 H), 3.63 (br. s, 1.5 H), 3.58-3.49 (m, 1.5 H), 3.47 (br. s, 0.5 H), 3.26-3.23 (m, 0.5 H), 3.16-3.13 (m, 0.5 H), 2.91-2.80 (m, 1 H), 2.65 (br. s, 2 H), 2.55-2.46 (m, 1 H), 1.89-1.80 (m, 2 H), 1.64-1.59 (m, 2 H), 1.46-1.40 (m, 9 H), 1.31-1.25 (m, 10 H), 0.88 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.5, 198.0, 154.2, 148.8, 149.0, 134.5, 128.5, 128.4, 127.7, 79.7, 79.5, 65.2, 54.4, 49.2, 49.0, 43.9, 43.2, 39.2, 38.4, 36.0, 33.7, 32.7, 31.8, 31.1, 29.7, 29.4, 29.3, 29.2, 28.5, 26.8, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C26H42NO4 (M+H)+ 432.31084, found 432.30903.
tert-Butyl (2R, 4S)-4-(hydroxymethyl)-2-(2-(4-octylphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (8 mg, 0.019 mmol) was submitted to general procedure A. The crude was purified by flash column chromatography (EtOH/CH2Cl2 1:4, Rf: 0.15) to give product 12 as a white solid (6 mg, 88%). Note: the product epimerized spontaneously on C-2 when dissolved in MeOH or H2O, giving a 1:1 mixture of diasteroisomers. For biological testing a portion of the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. IR (neat), νmax: 3376, 2922, 2852, 1675, 1605, 1570, 1465, 1378, 1282, 1184, 1039, 906, 815, 722, 549 cm−1. 1H NMR (CD3OD, 500 MHz, 1:1 mixture of diasteroisomers), δ: 7.98 (d, J = 8.3 Hz, 4 H), 7.38 (d, J = 8.3 Hz, 4 H), 4.18-4.12 (m, 1 H), 4.09-4.04 (m, 1 H), 3.76-3.65 (m, 4 H, partially deuterated), 3.63-3.58 (m, 2 H), 3.47-3.39 (m, 4 H, partially deuterated), 3.20-3.13 (m, 2 H), 2.74-2.71 (m, 4 H), 2.67-2.58 (m, 2 H), 2.44-2.38 (m, 1 H), 2.22-2.17 (m, 1 H), 1.99 (dt, J = 13.5, 8.4 Hz, 1 H), 1.70-1.62 (m, 5 H), 1.36-1.30 (m, 20 H), 0.91 (t, J = 7.0 Hz, 6 H) ppm. 13C NMR (CD3OD, 125 MHz, 1:1 mixture of diasteroisomers), δ: 197.1, 149.7, 133.6, 128.6, 128.0, 62.4, 61.9, 56.2, 55.3, 39.6, 39.0, 35.5, 32.9, 32.6, 31.6, 30.9, 29.1, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C21H34NO2 (M)+ 332.25841, found 332.25898.
tert-Butyl (2S,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(3-methoxy-3-oxopropyl)pyrrolidine-1-carboxylate (13b)
DIBAL-H (760 μL, 1 M in CH2Cl2, 0.76 mmol, 2 eq.) was added dropwise to a solution of 13a (194 mg, 0.38 mmol) in dry CH2Cl2 (3.5 mL) at −78 °C. The resulting mixture was stirred at the same temperature for 2 hours, before quenching the reaction with MeOH (100 μL). Then, the solution was allowed to reach room temperature and a 2 M solution of potassium sodium tartrate in water (3.5 mL) was added. The resulting mixture was vigorously stirred at room temperature for 30 min, before separating the layers. The aqueous one was extracted with CH2Cl2 (3 × 7 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated, affording a colorless oil. This intermediate aldehyde was redissolved in dry CH2Cl2 (3.5 mL) and methyl(triphenylphosphoranylidene)acetate (191 mg, 0.57 mmol, 1.5 eq.) was added at 0 °C. The resulting solution was stirred for 1 h at 0 °C and for 1 h at room temperature, before being cooled down again to 0 °C and quenched with NH4Cl satd. sol. (3.5 mL). The layers were separated and the aqueous one was extracted with CH2Cl2 (3 × 7 mL). The combined organic layers were washed with water (7 mL) and brine (7 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:4, Rf: 0.30) to give the intermediate tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-((E)-3-methoxy-3-oxoprop-1-en-1-yl)pyrrolidine-1-carboxylate a colorless oil (143 mg, 72%). [α]25D +36.4 (c 1.1, CHCl3). IR (neat), νmax: 2931, 2858, 1725, 1693, 1428, 1387, 1364, 1265, 1162, 1107, 978, 861, 823, 740, 701, 611, 503 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.65-7.62 (m, 4 H), 7.43-7.37 (m, 6 H), 6.87-6.97 (m, 1 H), 5.83 (t, J = 14.5 Hz, 1 H), 4.53 (br. s, 0.4 H), 4.37 (br. s, 0.6 H), 3.75 (s, 1.8 H), 3.73 (s, 1.2 H), 3.61 (d, J = 6.3 Hz, 2 H), 3.60-3.57 (m, 0.6 H), 3.50-3.46 (m, 0.4 H), 3.27 (t, J = 9.4 Hz, 0.6 H), 3.21-3.18 (m, 0.4 H), 2.50-2.41 (m, 1 H), 1.97-1.86 (m, 1 H), 1.80 (dd, J = 6.4, 2.5 Hz, 0.6 H), 1.78 (dd, J = 6.4, 2.5 Hz, 0.4 H), 1.46 (s, 5.4 H), 1.41 (s, 3.6 H), 1.05 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 166.8, 154.2, 154.1, 148.7, 148.4, 135.5, 133.3, 129.7, 127.9, 127.7, 120.0, 79.7, 65.0, 64.9, 57.7, 57.4, 51.6, 49.0, 48.9, 39.3, 38.4, 34.1, 33.4, 28.4, 28.2, 26.8, 19.2 ppm. HRMS (ESI) calcd. for C30H42NO5Si (M+H)+ 524.28270, found 524.28136.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-((E)-3-methoxy-3-oxoprop-1-en-1-yl)pyrrolidine-1-carboxylate (122 mg, 0.23 mmol) was dissolved in MeOH (9 mL) and Pd/C (10%, 29 mg) was added to the resulting solution. The air was removed from the flask under vacuum and replaced with hydrogen (balloon). The reaction was vigorously stirred overnight at room temperature. Afterwards, the mixture was filtered through a celite pad, washing with MeOH. The collected solution was concentrated in vacuo, affording 13b as a colorless oil (122 mg, 99%). [α]20D +20.9 (c 0.9, CHCl3). IR (neat), νmax: 2931, 2857, 1737, 1690, 1427, 1388, 1364, 1254, 1169, 1109, 823, 739, 701, 610, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.70-7.63 (m, 4 H), 7.44-7.37 (m, 6 H), 3.88 (br. s, 0.5 H), 3.80 (br. s, 0.5 H), 3.67 (s, 3 H), 3.58 (d, J = 6.2 Hz, 2 H), 3.49-3.43 (m, 0.5 H), 3.40-3.35 (m, 0.5 H), 3.29-3.24 (m, 0.5 H), 3.22-3.17 (m, 0.5 H), 2.54-2.48 (m, 1 H), 2.32 (br. s, 2 H), 2.04-1.92 (m, 1 H), 1.75-1.66 (m, 3 H), 1.46 (s, 9 H), 1.05 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 173.9, 173.7, 154.7, 154.5, 135.5, 133.5, 129.7, 127.7, 79.4, 79.1, 65.6, 65.4, 56.5, 51.5, 48.9, 39.5, 38.7, 33.4, 32.8, 31.1, 30.2, 30.0, 28.5, 26.8, 19.2, ppm. HRMS (ESI) calcd. for C30H44NO5Si (M+H)+ 526.2983, found 526.2993.
tert-Butyl (2S,4S)-4-(hydroxymethyl)-2-(3-(4-octylphenyl)-3-oxopropyl)pyrrolidine-1-carboxylate (13c)
13b (122 mg, 0.23 mmol) was submitted to the procedure reported for 12b. The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.28) to give the intermediate tert-Butyl (2S,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(3-(methoxy(methyl)amino)-3-oxopropyl)pyrrolidine-1-carboxylate as a colorless oil (111 mg, 87%). [α]25D +20.2 (c 1.1, CHCl3). IR (neat), νmax: 2930, 2857, 1688, 1386, 1364, 1254, 1175, 1109, 997, 823, 740, 702, 611, 504 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.65-7.63(m, 4 H), 7.44-7.36 (m, 6 H), 3.92 (br. s, 0.5 H), 3.83 (br. s, 0.5 H), 3.68 (s, 3 H), 3.59 (d, J = 6.1 Hz, 2 H), 3.51-3.45 (m, 0.5 H), 3.43-3.37 (m, 0.5 H), 3.30-3.25 (m, 0.5 H), 3.17 (br. s, 3.5 H), 2.57-2.51 (m, 1 H), 2.43 (br. s, 2 H), 1.94 (br. s, 1 H), 1.71 (br. s, 3 H), 1.45 (s, 9 H), 1.04 (s, 9 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 174.4, 174.2, 154.7, 135.5, 133.5, 129.6, 127.7, 79.2, 78.9, 65.7, 65.5, 61.1, 56.8, 48.8, 39.5, 38.7, 33.6, 33.0, 32.2, 30.0, 29.8, 29.7, 29.2 28.5, 26.8, 19.2 ppm. HRMS (ESI) calcd. for C31H47N2O5Si (M+H)+ 555.3249, found 555.3268.
tert-Butyl (2S,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(3-(methoxy(methyl)amino)-3-oxopropyl)pyrrolidine-1-carboxylate (34 mg, 0.061 mmol) was submitted in sequence to general procedures C and B. The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.20) to give 13c as a colorless oil (12 mg, 44% over two steps). [α]25D +6.3 (c 0.6, CHCl3). IR (neat), νmax: 3437, 2923, 2854, 1678, 1605, 1391, 1364, 1253, 1174, 1122, 770, 567 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.87 (d, J = 8.3 Hz, 2 H), 7.26-7.24 (m, 2 H), 4.01 (br. s, 0.5 H), 3.95 (br. s, 0.5 H), 3.64-3.60 (m, 2 H), 3.47-3.44 (m, 1 H), 3.31-3.26 (m, 0.5 H), 3.18-3.12 (m, 0.5 H), 3.09-3.02 (m, 0.5 H), 2.96 (br. s, 1.5 H), 2.64 (t, J = 7.6 Hz, 2 H), 2.54 (br. s, 1 H), 2.04 (br. s, 1 H), 1.88-1.74 (m, 3 H), 1.64-1.54 (m, 2 H), 1.41 (s, 9 H), 1.30-1.25 (m, 10 H), 0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 199.8, 199.2, 154.8, 148.8, 148.6, 134.6, 128.6, 128.2, 79.5, 79.2, 64.8, 56.8, 49.0, 48.4, 39.6, 38.8, 36.0, 35.7, 35.3, 33.8, 33.2, 31.8, 31.1, 29.7, 29.6, 29.5, 29.4, 29.2, 29.1, 28.5, 27.5, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C27H44NO4 (M+H)+ 446.32650, found 446.32667.
3-((2S,4S)-4-(Hydroxymethyl)pyrrolidin-2-yl)-1-(4-octylphenyl)propan-1-one hydrochloride (13) and (2S)-2-(hydroxymethyl)-5-(4-octylphenyl)-1,2,3,6,7,7a-hexahydropyrrolizin-4-ium chloride (13d)
Prepared according to general procedure A, starting from 13c (6 mg, 0.013 mmol). The crude was triturated and washed with Et2O, affording 13 as a white solid (4 mg, 80%). Note: in CD3OD the product was slowly but completely converted into the bicyclic salt 13d. On the other hand, in D2O the two species resulted in equilibrium, giving a mixture with a 2:1 constant ratio in favor of the open compound 13. For biological testing a portion of the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. 13: 1H NMR (CD3OD, 500 MHz), δ: 7.94 (d, J = 8.3 Hz, 2 H), 7.33 (d, J = 8.3 Hz, 2 H), 3.75-3.67 (m, 1 H), 3.63-3.53 (m, 2 H), 3.50 (dd, J = 11.8, 8.2 Hz, 1 H), 3.23 (dt, J = 13.9, 6.9 Hz, 2 H, partially deuterated), 3.12 (dd, J = 11.8, 6.8 Hz, 1 H), 2.71-2.67 (m, 2 H), 2.66-2.60 (m, 1 H), 2.21-2.1 (m, 3 H), 1.91 (dt, J = 13.5, 9.1 Hz, 1 H), 1.68-1.61 (m, 2 H), 1.33-1.29 (m, 10 H), 0.89 (t, J = 6.9 Hz, 3 H) ppm. 13d: IR (neat), νmax: 3410, 2923, 2853, 1645, 1605, 1456, 1417, 1373, 1296, 1190, 1045, 811, 566 cm−1. 1H NMR (CD3OD, 500 MHz), δ: 7.93 (d, J = 8.4 Hz, 2 H), 7.56 (d, J = 8.4 Hz, 2 H), 5.09-5.00 (m, 1 H), 4.23-4.17 (m, 1 H), 4.14-4.06 (m, 2 H, partially deuterated), 3.71 (dd, J = 6.0, 1.3 Hz, 2 H), 3.68-3.61 (m, 1 H, partially deuterated), 3.02-2.95 (m, 1 H), 2.80-2.76 (m, 2 H), 2.60 (dt, J = 12.7, 7.6 Hz, 1 H), 2.28 (ddd, J = 12.8, 7.4, 2.0 Hz, 1 H), 2.13-2.02 (m, 1 H), 1.98-1.90 (m, 1 H), 1.72-1.65 (m, 2 H), 1.35-1.29 (m, 10 H), 0.89 (t, J = 6.9 Hz, 3 H) ppm. 13C NMR (CD3OD, 125 MHz), δ: 178.8, 152.6, 131.2, 129.5, 123.6, 75.7, 63.2, 51.4, 43.4, 41.1, 35.6, 31.6, 31.1, 30.7, 29.1, 29.0, 28.9, 26.6, 22.3, 13.0 ppm.
((2S,5S)-5-(4-Octylphenyl)hexahydro-1H-pyrrolizin-2-yl)methanol hydrochloride (14)
NaBH4 (0.5 mg, 0.012 mmol, 1.5 eq.) was added to a solution of 13d (3 mg, 0.008 mmol) in MeOH (300 μL) at 0 °C. The resulting mixture was stirred for 1 hour at the same temperature. Afterwards, the reaction was quenched with HCl 1 N (20 μL) and concentrated. The crude was purified by flash column chromatography (MeOH/CH2Cl2 1:4, Rf: 0.58) to give product 14 as a white solid (3 mg, 99%, dr. 10:1). The stereochemistry was assigned by performing NOESY experiments (through space coupling observed between 2-H and 5-H in the major diasteroisomer). For biological testing the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D −58.7 (c 0.15, MeOH). IR (neat), νmax: 3417, 2923, 2853, 1518, 1456, 1089, 1037, 833, 535 cm−1. 1H NMR (CD3OD, 500 MHz), δ: 7.49 (d, J = 8.1 Hz, 2 H), 7.32 (d, J = 8.1 Hz, 2 H), 4.52-4.46 (m, 1 H), 4.42 (dd, J = 10.9, 6.7 Hz, 1 H), 3.69 (dd, J = 11.1, 5.2 Hz, 1 H), 3.62 (dd, J = 11.1, 5.9 Hz, 1 H), 3.45 (dd, J = 11.8, 6.6 Hz, 1 H), 3.09 (dd, J = 11.8, 10.5 Hz, 1 H), 2.92-2.83 (m, 1 H), 2.67-2.64 (m, 2 H), 2.54-2.51 (m, 1 H), 2.41-2.35 (m, 2 H), 2.09-2.05 (m, 2 H), 2.00-1.92 (m, 1 H), 1.65-1.59 (m, 2 H), 1.32-1.28 (m, 10 H), 0.89 (t, J = 6.9 Hz, 3 H) ppm. 13C NMR (CD3OD, 125 MHz), δ: 145.1, 130.2, 129.2, 127.9, 72.7, 68.6, 60.7, 54.1, 39.5, 35.2, 32.8, 32.7, 31.6, 31.2, 31.1, 29.1, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C22H36NO (M)+ 330.2797, found 330.2793.
tert-Butyl (2R,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(3-methoxy-3-oxopropyl)pyrrolidine-1-carboxylate (15a)
15a was sunmitted to the Horner-Wadsworth-Emmons extension procedure from 13b, starting from commercially available 1-(tert-butyl) 2-methyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)pyrrolidine-1,2-dicarboxylate (200 mg, 0.56 mmol). The residue was purified by flash column chromatography (hexane/EtOAc 8:2 Rf: 0.38) to give the intermediate tert-Butyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-2-((E)-3-methoxy-3-oxoprop-1-en-1-yl)pyrrolidine-1-carboxylate as a colorless oil (150 mg, 69% over 2 steps). [α]25D −3.0 (c 3.15, CHCl3). IR (neat), νmax: 2977, 2926, 2855, 1701, 1396, 1260, 987, 753 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 6.85-6.84 (m, 1 H), 5.90-5.75 (d, J = 15.0 Hz, 1 H), 4.57-4.47 (m, 1 H), 4.35-4.33 (t, J = 7.8 Hz, 1 H), 3.75 (s, 3 H), 3.47-3.36 (m, 2 H), 2.10-2.06 (m, 1 H), 1.85-1.80 (m, 1 H), 1.45 (s, 9 H), 0.89 (s, 9 H), 0.07 (s, 6 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 200.3, 200.0, 166.9, 154.7, 149.3, 148.9, 128.5, 127.6, 127.0, 120.0, 79.9, 70.0, 69.7, 69.5, 56.9, 55.3, 54.8, 51.6, 41.5, 40.6, 36.9, 28.4, 28.2, 25.7, 17.9 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 618.35301, found 618.35381.
tert-Butyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-2-((E)-3-methoxy-3-oxoprop-1-en-1-yl)pyrrolidine-1-carboxylate (150 mg, 0.39 mmol) was dissolved in MeOH (15 mL) and Pd/C (10%, 47 mg) was added to the resulting solution. The air was removed from the flask under vacuum and replaced with hydrogen (balloon). The reaction was vigorously stirred overnight at room temperature. Afterwards, the mixture was filtered through a celite pad, washing with MeOH. The collected solution was concentrated in vacuo, affording 15a as a colorless oil (149 mg, 99%). [α]20D −29.0 (c 0.44, CHCl3). IR (neat), νmax: 2929; 1739; 1693; 1390; 1154; 833; 773 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 4.33-4.32 (q, 1 H), 4.10-3.95 (m, 1 H), 3.66 (s, 3 H), 3.48-3.32 (m, 2 H), 2.30 (bs, 2 H), 2.07-1.96 (m, 2 H), 1.78-1.71 (m, 2 H), 1.45 (s, 9 H), 0.86 (s, 9 H), 0.05 (s, 6 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 174.1, 155.3, 79.6, 79.4, 70.5, 70.0, 55.6, 55.0, 54.7, 51.7, 40.8, 40.1, 31.0, 30.7, 30.5, 30.2, 28.6, 25.9, 18.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 388.25140, found 388.25200.
tert-Butyl (2R,4R)-4-hydroxy-2-(3-(4-octylphenyl)-3-oxopropyl)pyrrolidine-1-carboxylate (15b)
15a (334.0 mg, 0.86 mmol) was submitted to the procedure from 12b. The residue was purified by flash column chromatography (hexane/EtOAc 6:4 Rf: 0.25) to give the intermediate tert-Butyl (2R,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(3-(methoxy(methyl)amino)-3-oxopropyl)pyrrolidine-1-carboxylate as a colorless oil (328 mg, 92%).
tert-Butyl (2R,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(3-(methoxy(methyl)amino)-3-oxopropyl)pyrrolidine-1-carboxylate (100 mg, 0.24 mmol) was submitted in sequence to general procedures C and B. The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6 Rf: 0.33) to give 15b as a yellow oil (61 mg, 59% over 2 steps). [α]25D −19.0 (c 0.40, CHCl3). IR (neat), νmax: 2922, 1672, 1411 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.90-7.88 (d, J = 8.2 Hz, 2 H), 7.29-7.27 (d, J = 9.3 Hz, 2 H), 4.48-4.47 (m, 1 H), 4.12-4.09 (m, 1 H), 3.60-3.58 (d, J = 11.7 Hz, 1 H), 3.47-3.43 (dd, J = 12.0, 4.6 Hz, 1 H), 2.99-2.95 (m, 2 H), 2.69-2.66 (t, J = 6.4 Hz, 2 H), 2.21-2.11 (m, 2 H), 1.91-1.87 (m, 3 H), 1.66-1.63 (m, 2 H), 1.46 (s, 9 H), 1.33-1.28 (m, 10 H), 0.92 (t, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 199.4, 155.2, 148.8, 134.5, 129.5, 128.6, 128.2, 79.6, 70.0, 55.6, 54.7, 40.3, 36.0, 31.9, 31.1, 29.7, 29.6, 29.4, 29.3, 29.2, 28.5, 22.7, 14.1, 14.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 454.29280, found 454.29428.
(2R,4R)-4-Hydroxy-2-(3-(4-octylphenyl)-3-oxopropyl)pyrrolidin-1-ium chloride (15)
15 was synthesized in accordance with the general procedure A, starting from 15b (43 mg, 0.10 mmol). 15 was obtained as a white solid (26 mg, 72%). As for 13, 15 revealed prone to cyclize spontaneously to 15d in protic solvent such as MeOH. IR (neat), νmax: 2977, 2926, 2855, 1701, 1396, 1260, 987, 753 cm−1. 1H NMR (CDCl3, 500 MHz, opened form 15), δ: 7.96-7.94 (d, J = 8.3 Hz, 2 H), 7.35-7.33 (d, J = 8.3 Hz, 2 H), 4.54 (m, 1 H), 3.95-3.90 (m, 1 H), 3.48-3.43 (dd, J = 12.5, 4.1 Hz, 1 H), 3.28-3.19 (m, 3 H), 2.71-2.67 (m, 2 H), 2.26-2.10 (m, 3 H), 1.87-1.80 (m, 1 H), 1.65 (m, 2 H), 1.34-1.29 (m, 10 H), 0.91-0.88 (t, J = 6.85 Hz, 3 H) ppm. 1H NMR (CDCl3, 500 MHz, cyclized form 15d), δ: 8.00-7.98 (d, J = 8.5 Hz, 1 H), 7.59-7.57 (d, J = 8.4 Hz, 2 H), 5.24 (m, 1 H), 4.93-4.90 (m, 1 H), 4.33-4.29 (m, 1 H), 4.17-4.13 (m, 2 H), 3.74 (m, 1 H), 2.83-2.79 (m, 2 H), 2.64, (m, 1 H), 2.36-2.32 (dd, J = 13.1, 6.1 Hz, 1 H), 2.12-2.09 (m, 1 H), 1.97-1.89 (m, 1 H), 1.71 (m, 2 H), 1.36-1.31 (m, 10 H), 0.93-0.90 (t, J = 6.9 Hz, 3 H) ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 618.35301, found 618.35381.
(S)-tert-Butyl 2-(4-bromobenzoyl)pyrrolidine-1-carboxylate (17b)
nBuLi (2.5 M in hexane, 11.4 mL, 24 mmol, 2.0 eq.) was added dropwise via syringe to a stirred and cooled to −78 °C solution of 1,4-dibromobenzene (5.88 g, 24.9 mmol, 2.06 eq.) in dry THF (42 mL) under Ar. Stirring was continued for 30 min before N-Boc-D-prolinal 17a (2.2038 g, 1.0 mmol) in THF (18 mL) was added via syringe over ca 5 min. Stirring was continued for a further 15 min and the mixture was quenched by addition of sat. NH4Cl (20 mL) and H2O (10 mL). The bulk of the THF was then removed in vacuo and the mixture was extracted into Et2O (30 mL), washed once with brine (15 mL) and dried (MgSO4). Evaporation of the solvent and chromatography over SiO2 (2.5 × 40 cm) using 13% EtOAc-hexanes afforded (2S)-tert-butyl 2-((4-bromophenyl) (hydroxy)methyl) pyrrolidine-1-carboxylate as a mixture of diastereomers (2.3 g, 59%).
Dess-Martin periodinane (3.5 g, 8.2 mmol, 1.3 eq.) was added as a solid over ca 2 min to a stirred and cooled (0 °C) solution of (2S)-tert-butyl 2-((4-bromophenyl) (hydroxy)methyl)pyrrolidine-1-carboxylate (mixture of diastereomers, 2.3 g, 6.5 mmol, 1.0 eq.) in CH2Cl2 (24 mL). The flask was capped with a glass stopper and stirring was continued overnight. The mixture was then quenched by the addition of sat. NaHCO3 (10 mL) and H2O (5 mL) and stirring was continued for 30 min. The mixture was then filtered through Celite (2 × 3cm), washing the filter cake with CH2Cl2. The aqueous layer was extracted once with CH2Cl2 (10 mL) and the combined organic was dried (MgSO4), evaporated, and chromatographed over SiO2 (2.5 × 30 cm) using 13% EtOAc-hexanes to afford (S)-tert-butyl 2-(4-bromobenzoyl)pyrrolidine-1-carboxylate 17b (1.6011 g, 70%). HRMS (ESI) calcd. for C16H20BrNO3 (M+Na)+ 376.05188, found 376.05202.
tert-Butyl (S)-2-((R)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate (17c) and tert-butyl (S)-2-((S)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate (epi-17c)
BuLi (2.5 M in hexane, 5.5 mL, 13.7 mmol, 3.0 eq.) was added via syringe to a stirred and cooled to −78 °C solution of i-Pr2NH (1.9 mL, 13.6 mmol, 3.0 eq.) in THF (12 mL). The cooling bath was removed for 10 min and then replaced and stirring was continued for a further 10 min before EtOAc (1.5 mL, 15.4 mmol, 3.4 eq.) was added dropwise via syringe. Stirring was then continued for 45 min before (S)-tert-butyl-2-(4-bromobenzoyl)pyrrolidine-1-carboxylate 17b (1.6011 g, 4.55 mmol, 1.0 eq.) in THF (5 mL + 1 mL rinse) was added at a slow dropwise rate via syringe (ca 15 min). Stirring was then continued for 20 min and then the mixture was quenched by the addition of sat. NH4Cl (5 mL) and H20 (5 mL). The mixture was diluted with Et2O (30 mL) and washed once with H2O (20 mL), once with brine (20 mL) and dried (Na2SO4). Evaporation of the solvent provided (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate 17c and (S)-tert-butyl 2-((R)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate epi-17c as a mixture (1.54 g, 76%), which was used directly in the next step without further purification. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 464.10431, found 464.10432.
tert-Butyl (S)-2-((R)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate (17d) and (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate (epi-17d)
LiBH4 solution (2 M in THF, 1.1 mL, 2.2 mmol, 0.6 eq.) was added via syringe to a stirred and cooled to 0 °C solution of esters (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate 17c and (S)-tert-butyl 2-((R)-1-(4-bromophenyl)-3-ethoxy-1-hydroxy-3-oxopropyl)pyrrolidine-1-carboxylate epi-17c (mixture from previous step, 1.54 g, 3.48 mmol, 1.0 eq.) in THF (10 mL) under Ar. The ice-bath was left in place but not recharged and stirring was continued for 7 h. The mixture was then quenched by the careful addition of H2O (3 mL) and then NaHCO3aq(sat., 5 mL). EtOAc (10 mL) was then added and the biphasic mixture was stirred for 1 h. The aqueous phase was extracted once with EtOAc (10 mL) and the combined organic was washed once with brine (10 mL) and dried (Na2SO4). Evaporation of the solvent and filtration of the residue through a plug of SiO2 (2 × 4 cm) using 40% EtOAc-hexanes (ca 100 mL, TLC control) afforded the alcohols (S)-tert-butyl 2-((R)-1-(4-bromophenyl)-1,3-dihydroxypropyl)pyrrolidine-1-carboxylate and (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-1,3-dihydroxypropyl)pyrrolidine-1-carboxylate as a mixture (1.38 g, 72%).
Et3N (0.93 mL, 6.7 mmol, 1.2 eq.) followed by TsCl (1.16 g, 6.1 mmol, 1.1 eq.) were added to a stirred solution of alcohols (S)-tert-butyl-2-((R)-1-(4-bromophenyl)-1,3-dihydroxypropyl) pyrrolidine −1-carboxylate and (S)-tert-butyl-2-((S)-1-(4-bromophenyl)-1,3-dihydroxypropyl) pyrrolidine-1-carboxylate mixture from previous step (2.22 g, 5.55 mmol, 1.0 eq.) in CH2Cl2 (10 mL). The flask was capped with a glass stopper and stirred for 8 h. The mixture was diluted with CH2Cl2 (10 mL), washed once with H2O (25 mL), and dried (MgSO4). Removal of the solvent in vacuo and chromatography over SiO2 (2.5 × 35 cm) using 10% EtOAc-hexanes afforded the tosylates (S)-tert-butyl 2-((R)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate 17d and (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate epi-17d as a mixture (2.23 g, 72%).
(1R)-1-(4-Bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate (17e) and (1S)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate (epi-17e)
A solution of tosylates (S)-tert-butyl 2-((R)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate 17d and (S)-tert-butyl 2-((S)-1-(4-bromophenyl)-1-hydroxy-3-(tosyloxy)propyl)pyrrolidine-1-carboxylate epi-17d (2.2 g, 4.0 mmol, 1.0 eq.) was stirred for 10h at 110 °C in PhMe (18 mL) under Ar. The solvent was then removed in vacuo and the residue was chromatographed over SiO2 (2.5 × 35 cm) using 10-20% MeOH-CHCl3 to give a faster eluting fraction ((1R,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate) 17e and a slower eluting fraction ((1S’,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate) epi-17e. Mixed fractions were discarded. After removal of the solvent, the faster eluting diastereomer was crystallized from CH2Cl2-t-BuOMe (note 1) to provide ((1R,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate 17e (0.7 g, 38%). The slower eluting fraction was also evaporated to dryness and the solid residue was suspended in CH2Cl2 (3 mL) and filtered through a syringe filter (25 mm, PTFE 0.45 μm) to remove silica washing with three portions of CH2Cl2 (3 ml each) (note 2). The solvent was then removed in vacuo and the solid residue was crystallized from CH2Cl2-t-BuOMe (note 3) to provide (1S,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate epi-17e (0.3 g, 17%). 17e: mp 143 °C. [α]25D +21.0 (c 0.65, MeOH). IR (neat), νmax: 3371, 3673, 1657, 1394, 561 cm−1. 1H NMR (400 MHz, CDCl3), δ: 1.74-1.83 (m, 1 H), 2.05-2.13 (m, 1 H), 2.24-2.33 (m, 2 H), 2.39 (s, 3 H), 2.45 (dd, J = 5.4, 13.4 Hz, 1 H), 2.81 (ddd, J = 7.2, 13.0, 13.0 Hz, 1 H), 3.03-3.09 (m, 1 H), 3.32 (ddd, J = 5.9, 12.8, 12.8 Hz, 1 H), 3.55 (ddd, J = 6.0, 6.0, 11.7 Hz, 1 H), 3.80-3.87 (br s, 1 H), 3.94-3.99 (m, 1 H), 4.38 (app dd, J = 4.5, 8.0 Hz, 1 H), 7.18 (d, J = 8.1 Hz, 2 H), 7.36 (d, J = 8.7 Hz, 2 H), 7.45 (d, J = 8.7 Hz, 2 H), 7.71 (d, J = 8.1 Hz, 2 H), 11.26 (br s, 1 H) ppm. 13C NMR (100 MHz, CDCl3), δ: 21.8, 22.8, 27.5, 42.6, 53.7, 55.7, 79.4, 122.3, 126.1, 127.7, 129.4, 132.0, 140.3, 141.0, 142.0 ppm. LRMS found m/z 282.0. epi-17e: mp = 177.5-178.5 °C. IR (neat), νmax: 1234, 1185, 1009, 815, 696, 568, 478 cm−1. 1H NMR (400 MHz, CDCl3), δ: 1.18-1.26 (m, 1 H), 1.77-1.84 (m, 1 H), 1.93-2.01 (m, 2 H), 2.39 (s, 3 H), 2.53-2.66 (overlapping m, 2 H), 2.97 (ddd, J = 11.4, 9.6, 6.8 Hz, 1 H), 3.26 (dd, J = 11.6, 6.4 Hz, 1 H), 3.98 (ddd, J = 11.3, 6.1, 6.1 Hz, 1 H), 4.31 (ddd, J = 11.9, 11.9, 7.1 Hz, 1 H), 4.61 (dd, J = 10.0, 8.2 Hz, 1 H), 5.80 (br s, 1 H), 7.18 (d, J = 7.9 Hz, 2 H), 7.33 (d, J = 8.6 Hz, 2 H), 7.49 (d, J = 8.6 Hz, 2 H), 7.73 (d, J = 7.9 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3), δ: 4.3, 21.8, 26.0, 29.8, 33.5, 54.2, 57.2, 81.8, 123.1, 126.3, 128.6, 129.3, 132.2, 139.4, 140.9, 141.8 ppm. LRMS found m/z 282.0. Note 1: Diastereomer (1R,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate was dissolved in ca. 10 mL of CH2Cl2 and was then brought to boiling with a heat gun. t-BuOMe (ca 5 mL) was then added and the solution was allowed to crystallize overnight, the flask being left completely open. The next day the flask was capped with a glass stopper and cooled at ca −15 °C (freezer section of fridge) for a further 10 h. Filtration afforded flocculent white needles of ((1R,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate. Note 2: The syringe filter was attached to a syringe and the plunger was removed. The suspension was then transferred to this syringe via Pasteur pipette and forced through the filter by re-inserting the plunger. Note 3: Diastereomer (1S,7aS)-1-(4-bromophenyl)-1-hydroxyoctahydropyrrolizin-4-ium 4-methylbenzenesulfonate was dissolved in ca 10 mL of CH2Cl2 with stirring in an oil bath set at 45 °C. tBu-OMe was added (ca 3mL) and stirring was discontinued. The oil bath was shut off but left in place and the mixture was allowed to crystallize overnight, the flask being left completely open. The next day the flask was capped with a glass stopper and cooled at ca −15 °C (freezer section of fridge) for a further 10 h. Filtration afforded small needles.
(1R)-1-(4-Octylphenyl)hexahydro-1H-pyrrolizin-1-ol hydrochloride (17)
To a solution of catecholborane (135 μL, 1.0 M in THF, 0.135 mmol, 1.5 eq.) was added octyne dropwise (19.9 μL, 0.135 mmol, 1.5 eq.). Gas formation was observed during the addition. The colorless solution was refluxed for 2h then cooled back to rt. In another flask, 17e (41 mg, 0.09 mmol, 1.0 eq.) was dissolved in a biphasic mixture of DME (1.1 mL) and aqueous NaHCO3 (1 M) solution, then the octyne/catecholborane solution was syringed into the flask. Pd(PPh3)4 (3.1 mg, 0.0027 mmol, 0.03 eq.) was added and the overall white suspension was refluxed overnight. Et2O and brine were added and the aqueous layer was extracted x2 with Et2O. The organic layers were collected, dried over Na2SO4, filtered through Celite, concentrated. The crude oil was chromatographed over SiO2 (9:1 DCM/MeOH) to deliver an orange oil (18 mg). The product was engaged in the next step without further purification. The oil (12 mg, 0.038 mmol, 1.0 eq.) was dissolved in MeOH (1.2 mL) and Pd/C was added in one portion (0.4 mg, 0.0038 mmol, 0.1 eq.). The flask was purged ×3 with H2 and the black suspension was stirred for 1h30. Then HCl (9.5 μL, 4 M, 0.038 mmol, 1.0 eq.) was added and the solution was stirred for 2h, then filtered through a pad of Celite and concentrated. The resulting crude was chromatographed on reversed C18 column (0 to 20% MeCN in H2O) to deliver 17 as a colorless oil (9 mg, 29% over two steps). [α]25D −16.0 (c 0.275, MeOH) IR (neat), νmax: 3357, 2923, 1593, 1349 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.14-7.08 (m, 4 H), 4.78 (bs, 1 H), 4.60-4.57 (t, J =, 0.47 H), 4.54-4.50 (t, J = 14.8, 0.53 H), 3.91-3.67 (m, 3 H), 3.54-3.47 (m, 1 H), 2.58-2.54 (m, 2 H), 2.30-2.25 (m, 0.53 H), 2.13-2.09 (m, 0.47 H), 1.84-1.81 (m, 1 H), 1.58-1.56 (m, 2 H), 1.58-1.40 (m, 25 H), 1.29-1.25 (m, 13 H), 0.89-0.86 (t, J = , 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 207.1, 207.1, 154.6, 153.8, 142.2, 141.9, 130.6, 130.3, 129.7, 129.6, 129.0, 128.9, 83.1, 83.1, 83.1, 83.0, 82.9, 80.9, 80.5, 75.7, 75.6, 74.8, 74.8, 63.4, 62.5, 53.8, 53.7, 53.5, 53.5, 47.6, 46.3, 32.0, 30.0, 30.0, 29.6, 29.4, 28.5, 28.4, 22.8, 14.2 ppm. HRMS (ESI) calcd. for C23H37NO3Na (M+H)+ 316.2635, found 316.2644.
tert-Butyl 3-hydroxy-3-(4-octylphenyl)pyrrolidine-1-carboxylate ((±)18b)
(±)18b was synthesized in accordance with the general procedure C, starting from 18a (100 mg, 0.54 mmol). The resulting residue was purified by flash column chromatography (hexane/EtOAc 8:2, Rf: 0.28) to give (±)18b as a pale yellow oil (123 mg, 61%). IR (neat), νmax: 3392, 2923, 1670, 1412, 1134 cm−1. 1H NMR (500 MHz, CDCl3, mixture of rotamers) δ 7.37 (t, J = 6.6 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 3.80 – 3.49 (m, 4H), 2.65 – 2.50 (m, 2H), 2.36 – 2.09 (m, 2H), 1.67 – 1.54 (m, 2H), 1.47 (d, J = 11.1 Hz, 9H), 1.38 – 1.11 (m, 10H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3, mixture of rotamers) δ 154.9, 154.8, 142.8, 140.1, 128.7, 125.2, 125.2, 80.7, 79.9, 79.6, 59.7, 58.8, 45.2, 44.7, 39.6, 38.9, 35.7, 32.0, 31.6, 29.9, 29.6, 29.5, 29.4, 28.7, 22.8, 14.3. HRMS (ESI) calcd. for C23H37NO3Na (M+Na)+ 398.2666, found 398.2681.
3-Hydroxy-3-(4-octylphenyl)pyrrolidin-1-ium chloride ((±)18)
18 was synthesized in accordance with the general procedure A, starting from 18b (25 mg, 0.066 mmol). The resulting residue was triturated with a mixture of CH2Cl2/Et2O (9:1) to give 18 as a pale yellow oil (9.6 mg, 45 %). Rf: 0.18 CH2Cl2/MeOH 9:1, 1% Et3N). IR (neat), νmax: 3385, 2922, 1617, 1379, 1179, 1086 cm−1. 1H NMR (500 MHz, MeOD) δ 7.44 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 3.61 (d, J = 9.8 Hz, 2H), 3.43 (dd, J = 30.3, 11.5 Hz, 2H), 2.68 – 2.55 (m, 2H), 2.43 (dd, J = 23.4, 10.3 Hz, 1H), 2.33 (d, J = 11.0 Hz, 1H), 1.61(d, J = 7.3 Hz, 2H), 1.40 – 1.22 (m, 10H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 142.7, 137.9, 128.3, 125.1, 79.6, 56.6, 48.1, 48.0, 47.8, 47.6, 47.4, 47.3, 47.1, 44.4, 38.1, 35.0, 31.6, 31.3, 29.2, 29.0, 28.9, 22.3, 13.0. HRMS (ESI) calcd. for C18H29NO (M+H)+ 276.2322, found 276.2326.
tert-Butyl (S)-2-(methoxy(methyl)carbamoyl)pyrrolidine-1-carboxylate (19b)
To a −20 °C cooled solution containing 19a (321 mg, 1.4 mmol, 1.0 eq.) and N,O-dimethylhydroxylamine-HCl (205 mg, 2.10 mmol, 1.5 eq.) in dry THF (1 mL) was added iPrMgCl (1.4 mL, 2.8 mmol, 2.0 M in THF, 2.0 eq.) dropwise. The light brown solution was stirred at −10 °C for 20 min whereby additional N,O-dimethylhydroxylamine-HCl (205 mg, 2.10 mmol, 1.5 eq.) was added in one portion followed by iPrMgCl (1.4 mL, 2.8 mmol, 2.0 M in THF, 2.0 eq.) dropwise. The reaction was stirred for 20 additional minuts at −10 °C whereby TLC analysis indicated that the reaction had gone to completion. Saturated aqueous NH4Cl solution (5 mL) was added and the resulting aqueous layer was extracted with EtOAc (2 × 5 mL). The organic layers were collected, dried over Na2SO4, filtered, concentrated to give 19b as a pure incolore oil (321 mg, 89%) which was brought to the next step without further purification (Rf: 0.34 hexanes/EtOAc 5:5). [α]20D – 13.9 (c 1.25, CHCl3). The datas matched those reported in the literature. [45]
tert-Butyl (S)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (19c)
19c was synthesized in accordance with the general procedure C, starting from 19b (100 mg, 0.39 mmol). The resulting residue was purified by flash column chromatography (hexane/EtOAc 8:2 Rf: 0.32) to give 19c as a yellow oil (98 mg, 65%). [α]20D −78.0 (c 0.21, CHCl3). IR (neat), νmax: 2925, 2584, 1687, 1391, 1160 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.88 (dd, J = 19.0, 8.2 Hz, 2H), 7.25 (dd, J = 15.6, 7.8 Hz, 2H), 5.37 – 5.16 (m, 1H), 3.76 – 3.41 (m, 2H), 2.64 (dt, J = 11.2, 7.8 Hz, 2H), 2.37 – 2.22 (m, 1H), 1.99 – 1.85 (m, 3H), 1.67 – 1.55 (m, 2H), 1.46 (s, 4H), 1.36 – 1.20 (m, 16H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.7, 198.2, 154.6, 154.0, 149.1, 149.1, 133.0, 132.9, 128.8, 128.8, 128.8, 128.4, 79.8, 79.7, 61.4, 61.1, 46.9, 46.8, 36.1, 32.0, 31.2, 29.5, 29.4, 29.3, 28.6, 28.3, 23.7, 22.8, 14.2 ppm. HRMS (ESI) calcd. for C24H37NO3 (M+Na)+ 410.26660, found 410.26710.
(S)-2-(4-Octylbenzoyl)pyrrolidin-1-ium chloride (19)
19 was synthesized in accordance with the general procedure A, starting from 19c (27 mg, 0.07 mmol). The resulting residue was triturated with EtOAc to give 19 as a white powder (14 mg, 64%). [α]20D −38.7 (c 0.16, CHCl3). IR (neat), νmax: 2923, 2853, 1686, 1397, 1250, 997 cm−1. 1H NMR (500 MHz, MeOD) δ 8.00 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 8.3 Hz, 2H), 5.34 (dd, J = 9.3, 7.1 Hz, 1H), 3.45 (dt, J = 15.3, 6.1 Hz, 2H), 2.79 – 2.61 (m, 1H), 2.24 – 1.90 (m, 2H), 1.73 – 1.58 (m, 2H), 1.42 – 1.23 (m, 10H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 194.9, 152.9, 132.1, 130.8, 130.7, 64.8, 47.8, 37.3, 33.3, 32.6, 31.4, 30.9, 30.7, 30.6, 25.5, 24.0, 14.7 ppm. HRMS (ESI) calcd. for C19H30NO (M+H)+ 288.23219, found 288.23192.
tert-Butyl (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-2-(methoxy(methyl)carbamoyl)pyrrolidine-1-carboxylate (20b)
20b was synthesized in accordance with the procedure from 19b, starting from 20a (50.0 mg, 0.14 mmol). The crude incolore oil of 20b (54 mg, 95%) was brought to the next step without further purification (Rf: 0.25 hexanes/EtOAc 7:3). [α]25D −14.0 (c 0.50, CHCl3). The spectral datas matched those reported in the litterature.7
tert-Butyl (2R,4S)-4-((tert-butyldimethylsilyl)oxy)-2-(methoxy(methyl)carbamoyl)pyrrolidine-1-carboxylate (21b)
21b was synthesized in accordance with the procedure of its enantiomer 20b, starting from 21a (300 mg, 0.84 mmol). 21b was obtained as a colorless oil (298 mg, 91 %) which was brought to the next step without further purification (Rf: 0.25 hexanes/EtOAc 7:3). [α]25D +13.0 (c 1.00, CHCl3). The spectral datas matched those reported for its enantiomer.
tert-Butyl (2S,4R)-4-hydroxy-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (20c)
20b (200 mg, 0.52 mmol) was submitted in sequence to general procedures C and B. The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6 Rf: 0.35) to give 20c as a yellow oil (135 mg, 65% over 2 steps). [α]25D −6.6 (c 0.27, CHCl3). IR (neat), νmax: 2924, 1686, 1605, 1399, 1158 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.95-7.90 (m, 2 H), 7.31-7.27 (m, 2 H), 5.51-5.48 (t, J = 7.6 Hz, 0.45 H), 5.42-5.39 (t, J = 8.1 Hz, 0.55 H), 4.55 (s, 1 H), 3.80-3.56 (m, 2 H), 2.71-2.68 (m, 2 H), 2.41-2.37 (m, 1 H), 2.09-2.04 (m, 1 H), 1.74-1.62 (m, 3 H), 1.49 (s, 3 H), 1.32-1.26 (m, 15 H), 0.92-0.89 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 199.0, 198.5, 154.9, 154.4, 149.7, 149.6, 133.4, 133.3, 129.2, 129.1, 129.1, 128.7, 80.7, 80.4, 71.0, 70.3, 60.0, 59.7, 55.6, 40.1, 39.3, 36.4, 32.2, 31.5, 31.4, 29.8, 29.6, 29.6, 28.8, 28.5, 23.0, 14.5 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 426.26150, found 426.26245.
tert-Butyl (2R,4S)-4-hydroxy-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (21c)
21b (298 mg, 0.77 mmol) was submitted in sequence to general procedures C and B. The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6 Rf: 0.35) to give 21c as a yellow oil (205 mg, 62% over 2 steps). [α]25D +38.1 (c 1.65, CHCl3). The spectral datas matched those reported for its enantiomer.
(2S,4R)-4-Hydroxy-2-(4-octylbenzoyl)pyrrolidin-1-ium chloride (20)
20 was synthesized in accordance with the general procedure A, starting from 20c (18 mg, 0.07 mmol). 20 was obtained as a white solid (18 mg, 86%). [α]25D −30.6 (c 0.80, CHCl3). IR (neat), νmax: 2923, 2470, 2070, 1596, 1463, 1119, 973 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.99-7.98 (d, J = 8.2 Hz, 1 H), 7.43-7.42 (d, J = 7.9 Hz, 2 H), 5.51-5.47 (dd, J = 10.3, 8.2 Hz, 1 H), 4.61-4.60 (m, 1 H), 3.39 (s, 2 H), 2.73-2.71 (m, 2 H), 2.66-2.62 (dd, J = 12.9, 8.2 Hz, 1 H), 2.07-2.02 (ddd, J = 13.8, 10.4, 4.2 Hz, 1 H), 1.68-1.65 (m, 2 H), 1.34-1.28 (m, 10 H), 0.91-0.88 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 194.6, 162.8, 152.6, 131.8, 130.4, 130.4, 71.3, 63.3, 55.0, 40.5, 37.0, 33.0, 32.2, 30.5, 30.4, 30.3, 23.7, 14.4 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 304.22770, found 304.22860.
(2R,4S)-4-Hydroxy-2-(4-octylbenzoyl)pyrrolidin-1-ium chloride (21)
21 was synthesized in accordance with the general procedure A, starting from 21c (30 mg, 0.07 mmol). 21 was obtained as a white solid (16 mg, 64%). [α]25D +25.6 (c 0.34, CHCl3). The spectral datas matched those reported for its enantiomer.
(3R,5R)-5-(4-Octylbenzyl)pyrrolidin-3-ol (16)
20c (20 mg, 0.050 mmol) was dissolved in EtOH (5 mL) and Pd/C (10%, 24 mg) was added to the resulting solution. The air was removed from the flask under vacuum and replaced with hydrogen (balloon). The reaction was vigorously stirred for 24 hours at room temperature. Afterwards, the mixture was filtered through a celite pad, washing with abundant EtOH, and the collected solution was concentrated in vacuo. The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.35) to give the intermediate tert-Butyl (2R,4R)-4-hydroxy-2-(4-octylbenzyl)pyrrolidine-1-carboxylate as a colorless oil (6 mg, 32%). [α]25D −32.7 (c 0.30, CHCl3). IR (neat), émax 3408, 2923, 2853, 1694, 1668, 1513, 1455, 1393, 1365, 1253, 1153, 1116, 981, 858, 770, 553 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.11-7.04 (m, 4 H), 4.22-4.14 (m, 2 H), 3.50 (br. s, 0.6 H), 3.35 (br. s, 0.4 H), 3.30 (br. s, 1 H), 3.09 (br. s, 1 H), 2.68 (br. s, 0.4 H), 2.63 (br. s, 0.6 H), 2.57-2.54 (m, 2 H), 1.87 (br. s, 2 H), 1.60-1.55 (m, 2 H), 1.52 (s, 9 H), 1.31-1.26 (m, 10 H), 0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 154.9, 141.0, 135.3, 129.3, 128.4, 79.7, 69.7, 69.4, 57.3, 54.5, 40.3, 39.4, 35.6, 31.9, 31.6, 29.5, 29.3, 29.2, 28.6, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C24H39NO3Na (M+Na)+ 412.28222, found 412.28110.
tert-Butyl (2R,4R)-4-hydroxy-2-(4-octylbenzyl)pyrrolidine-1-carboxylate (6 mg, 0.015 mmol) was submitted to general procedure A. The crude was triturated in Et2O to give product 16 as a white solid (5 mg, 100%). For biological testing a portion of the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D +4.0 (c 0.25, MeOH). IR (neat), νmax: 3318, 2920, 2851, 1515, 1437, 1394, 1314, 1266, 1159, 1080, 1063, 1031, 961, 772, 720, 615, 531, 450 cm−1. 1H NMR (CD3OD, 500 MHz), δ: 7.25 (d, J = 8.1 Hz, 2 H), 7.21 (d, J = 8.1 Hz, 2 H), 4.56 (t, J = 4.2 Hz, 1 H), 4.12-4.05 (m, 1 H), 3.50 (dd, J = 12.4, 4.2 Hz, 1 H), 3.19 (d, J = 12.4 Hz, 1 H), 3.05 (d, J = 7.6 Hz, 2 H), 2.63-2.60 (m, 2 H), 2.13 (dd, J = 13.7, 5.8 Hz, 1 H), 1.90 (ddd, J = 13.7, 11.6, 4.2 Hz, 1 H), 1.65-1.59 (m, 2 H), 1.34-1.31 (m, 10 H), 0.92 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CD3OD, 125 MHz), δ: 141.9, 133.6, 128.7, 128.4, 69.0, 60.3, 53.0, 39.5, 37.1, 35.1, 31.6, 31.3, 29.2, 29.0, 28.9, 22.3, 13.0 ppm.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(methoxy(methyl)carbamoyl)pyrrolidine-1-carboxylate (22b)
Isopropyl magnesium chloride (585 μL, 2 M in THF, 1.17 mmol, 6 eq.) was added dropwise to a solution of 13a (100 mg, 0.195 mmol) and N,O-dimethylhydroxylamine (87 mg, 0.89 mmol, 4.5 eq.) in dry THF (1 mL) at −20 °C. The resulting mixture was kept at the same temperature and stirred for 1 h. Afterwards, the reaction was quenched at −20 °C by canulating the solution to an aqueousNH4Cl satd. sol. (5 mL) and the product was extracted with EtOAc (3 × 5 mL). The organic layers were washed with brine (5 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 1:2, Rf: 0.09) to give 22b as a colorless oil (102 mg, 99%). [α]25D +13.6 (c 1.7, CHCl3). IR (neat), νmax: 2931, 2858, 1696, 1472, 1427, 1388, 1365, 1319, 1256, 1164, 1109, 999, 940, 909, 879, 823, 741, 702, 613, 504, 489 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.65-7.63 (m, 4 H), 7.43-7.37 (m, 6 H), 4.75 (d, J = 6.5, 0.5 H), 4.65 (d, J = 6.2, 0.5 H), 3.80-3.78 (m, 0.5 H), 3.77 (s, 1.5 H), 3.70 (s, 1.5 H), 3.69-3.65 (m, 0.5 H), 3.59 (dd, J = 6.1, 1.7 Hz, 2 H), 3.35 (dd, J = 10.5, 7.0 Hz, 0.5 H), 3.29 (dd, J = 10.5, 7.1 Hz, 0.5 H), 3.20 (s, 3 H), 2.69-2.55 (m, 1 H), 2.11 (dt, J = 12.9, 8.8 Hz, 0.5 H), 2.03 (dt, J = 12.9, 9.5 Hz, 0.5 H), 1.95-.1.90 (m, 1 H), 1.46 (s, 4.5 H), 1.42 (s, 4.5 H), 1.05 (s, 4.5 H), 1.04 (s, 4.5 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ; 173.1, 154.5, 153.8, 135.5, 133.4, 133.3, 129.6, 127.7, 79.5, 79.4, 65.0, 61.3, 61.2, 56.7, 56.4, 49.3, 49.1, 39.5, 38.6, 32.7, 32.0, 28.5, 28.4, 26.8, 26.7, 19.2 ppm. HRMS (ESI) calcd. for C29H43N2O5Si (M+H)+ 527.29358, found 527.29360.
tert-Butyl (2R,4S)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (22c)
Prepared by applying in sequence general procedures C and B, starting from 22b (85 mg, 0.16 mmol). The crude was purified by flash column chromatography (EtOAc/hexane 1:1, Rf: 0.16) to give 22c as a colorless oil (32 mg, 48% over two steps). [α]25D +16.6 (c 0.65, CHCl3). IR (neat), νmax: 3434, 2924, 2854, 1687, 1605, 1394, 1365, 1223, 1161, 1129, 998, 882, 769 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ; 7.89 (d, J = 8.1 Hz, 0.8 H), 7.85 (d, J = 8.1 Hz, 1.2 H), 7.27 (d, J = 8.1 Hz, 1.2 H), 7.24 (d, J = 8.1 Hz, 0.8 H), 5.37 (dd, J = 9.4, 2.4 Hz, 0.4 H), 5.24 (dd, J = 9.3, 3.6 Hz, 0.6 H), 3.82-3.75 (m, 1 H), 3.66-3.57 (m, 2 H), 3.33 (dd, J = 10.7, 6.9 Hz, 0.6 H), 3.27 (dd, J = 10.6, 7.8 Hz, 0.4 H), 2.67-2.62 (m, 2 H), 2.57-2.46 (m, 1 H), 2.20 (dt, J = 12.9, 9.0 Hz, 0.6 H), 2.12 (dt, J = 12.2, 9.6 Hz, 0.4 H), 2.03-1.99 (m, 1 H), 1.93 (br. s, 0.4 H), 1.76 (br. s, 0.6 H), 1.66-1.58 (m, 2 H), 1.45 (s, 3.6 H), 1.30-1.27 (m, 10 H), 1.26 (s, 5.4 H), 0.87 (t, J = 6.9 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ; 198.3, 197.8, 154.5, 153.9, 149.1, 132.6, 132.4, 128.7, 128.6, 128.3, 79.9, 79.8, 64.1, 64.0, 61.0, 60.9, 49.3, 48.9, 39.7, 38.8, 36.0, 33.1, 32.2, 31.8, 31.1, 31.0, 29.4, 29.2, 28.5, 28,2, 22.6, 14.1 ppm. HRMS (ESI) calcd. for C25H39NO4Na (M+Na)+ 440.27713, found 440.27771.
((2R,4S)-4-(Hydroxymethyl)pyrrolidin-2-yl)(4-octylphenyl)methanone hydrochloride (22)
Prepared according to general procedure A, starting from 22c (6 mg, 0.014 mmol). The crude was triturated in Et2O to give product 22 as a white solid (5 mg, 100%). For biological testing a portion of the product was dissolved in the minimum amount of HPLC grade water, filtered (pore size = 0.45 μm) and lyophilized. [α]25D +46.4 (c 0.25, CHCl3). IR (neat), νmax: 3370, 2922, 2852, 1683, 1605, 1570, 1464, 1416, 1400, 1373, 1350, 1310, 1263, 1182, 1164, 1092, 1060, 1013, 989, 967, 901, 722, 528 cm−1. 1H NMR (CD3OD, 500 MHz), δ; 8.01 (d, J = 8.3 Hz, 2 H), 7.45 (d, J = 8.3 Hz, 2 H), 5.43 (t, J = 7.9 Hz, 1 H), 3.70 (dd, J = 10.9, 4.7 Hz, 1 H), 3.65, (dd, J = 10.9, 5.0 Hz, 1 H), 3.62 (dd, J = 11.2, 6.8 Hz, 1 H), 3.34-3.30 (m, 1 H), 2.77-2.73 (m, 2 H), 2.60-2.52 (m, 2 H), 2.19-2.13 (m, 1 H), 1.71-1.65 (m, 2 H), 1.37-1.31 (m, 10 H), 0.91 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CD3OD, 125 MHz), δ: 193.1, 151.1, 130.2, 129.0, 63.2, 61.7, 48.0, 39.5, 35.6, 32.3, 31.6, 30.8, 29.1, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C20H32NO2 (M)+ 318.24276, found 318.24265.
tert-Butyl (2S,4R)-4-hydroxy-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate (23a)
Prepared by applying in sequence general procedures C using octyl benzyl bromide [46] then B starting from 20b (500 mg, 1.24 mmol). The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6, Rf: 0.40) to give 23a as a yellow oil (303 mg, 59% over 2 steps). [α]25D −66.5 (c 1.55, CHCl3). IR (neat), νmax: 3433, 2923, 1676, 1394, 1159 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.14-7.09 (m, 4 H), 4.61-4.54 (m, 1 H), 4.35 (bs, 1 H), 3.81 (s, 0.75 H), 3.74-3.67 (m, 1.25 H), 3.63-3.61 (m, 0.63 H), 3.53-3.44 (m, 1.37 H), 2.58-2.55 (t, J = 7.7 Hz, 2 H), 2.08-1.79 (m, 2 H), 1.59-1.56 (m, 2 H), 1.46 (s, 3.51 H), 1.39 (s, 5.19 H), 1.30-1.26 (m, 10 H), 0.89-0.86 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 207.9, 207.2, 154.8, 154.2, 142.0, 141.7, 130.6, 130.3, 129.6, 129.5, 128.8, 128.7, 80.7, 80.3, 70.4, 69.5, 63.4, 62.7, 55.2, 55.2, 47.1, 46.1, 35.6, 31.9, 31.5, 29.5, 29.3, 29.3, 28.3, 22.7, 14.1 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 318.24276, found 318.24270.
tert-Butyl (2R,4S)-4-hydroxy-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate (24a)
Prepared by applying in sequence general procedures C using octyl benzyl bromide [46] then B starting from 21b (484 mg, 1.20 mmol). The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6, Rf: 0.40) to give 24a as a yellow oil (340 mg, 68% over 2 steps). [α]25D +65.3 (c 0.19, CHCl3). The spectral datas matched those reported for its enantiomer.
(2S,4R)-4-Hydroxy-2-(2-(4-octylphenyl)acetyl)pyrrolidin-1-ium chloride (23)
23 was synthesized in accordance with the general procedure A, starting from 23a (25 mg, 0.06 mmol). 23 was obtained as a white powder (18 mg, 86%). [α]25D +91.9 (c 0.09, CHCl3). IR (neat), νmax: 3364, 3191, 2953, 2703, 1716, 1332, 1071, 763, 682 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.19-7.16 (s, 4 H), 4.79-4.75 (dd, J = 7.5 Hz, 1 H), 4.56 (m, 1 H), 3.92 (s, 2 H), 3.32-3.26 (m, 2H), 2.61 (dd, J = 10.8, 7.8 Hz, 2 H), 2.49-2.45 (dd, J = 13.4, 7.7 Hz, 1 H), 2.06-2.00 (ddd, J = 13.5, 11.1, 4.0 Hz, 1 H), 1.61-1.59 (m, 2 H), 1.32-1.28 (m, 10 H), 0.93-0.88 (t, J = 6.98 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 202.3, 201.9, 145.0, 142.0, 129.6, 129.4, 128.5, 128.4, 128.2, 69.6, 68.7, 64.6, 64.2, 53.4, 63.1, 35.1, 31.6, 31.3, 29.2, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 318.24276, found 318.24750.
(2R,4S)-4-Hydroxy-2-(2-(4-octylphenyl)acetyl)pyrrolidin-1-ium chloride (24)
24 was synthesized in accordance with the general procedure A, starting from 24a (17 mg, 0.04 mmol). 24 was obtained as a white powder (15 mg, 98%). [α]25D −80.0 (c 0.04, CHCl3). The spectral datas matched those reported for its enantiomer.
(2S,4R)-4-Hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidin-1-ium chloride (25)
NaBH4 (2.2 mg, 0.058 mmol, 1.2 eq.) was added in one portion to a solution of 23a (20.0 mg, 0.050 mmol, 1.0 eq.) in dry MeOH (0.8 mL). The solution was stirred at rt for 2h. Saturated aqueous NH4Cl solution was added and the resulting aqueous layer was extracted with EtOAc (1 × 2 mL). The organic layers were combined, washed with brine (1 × 2 mL), dried over Na2SO4, filtered, concentrated. The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6, Rf: 0.38) to give the intermediate tert-Butyl (2S,4R)-4-hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate as a colorless oil (15 mg, 75%). [α]20D −11.7 (c 1.02, CHCl3). IR (neat), νmax: 3409, 2923, 1664, 1403, 1160, 990, 771 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.17-7.15 (d, J = 7.8 Hz, 2 H), 7.11-7.09 (d, J = 7.9 Hz, 2 H), 4.40 (bs, 1 H), 4.11-4.09 (m, 1 H), 3.83 (bs, 1 H), 3.65 (bs, 1 H), 3.39-3.36 (dd, J = 12.1, 4.2 Hz, 1 H), 2.82-2.78 (m , 1 H), 2.57-2.54 (m, 3 H), 2.09-2.06 (m, 1 H), 1.89-1.77 (m, 2 H), 1.65 (bs, 1 H), 1.60-1.55 (m, 2 H), 1.46 (s, 9 H), 1.31-1.25 (m, 10 H), 0.89-0.86 (t, J = 7.0 Hz, 3 H), ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 158.1, 157.9, 141.1, 135.6, 129.5, 128.6, 80.9, 80.3, 73.0, 70.0, 55.6, 35.7, 32.0, 31.7, 29.6, 29.5, 29.4, 28.6, 28.5, 22.8, 14.2 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 442.29278, found 442.29415.
tert-Butyl (2S,4R)-4-hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate (6 mg, 0.014 mmol) was submitted to the general procedure A. 25 was obtained as a white powder (5 mg, 98%). IR (neat), νmax: 3363, 2955, 1315, 968, 557 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of diastereomers), δ: 7.20-7.19 (d, J = 8.1 Hz, 2 H), 7.15-7.13 (d, J = 8.1 Hz, 2 H), 4.55-4.53 (m, 1 H), 3.92-3.88 (m, 1 H), 3.79-3.74 (m, 1 H), 3.30-3.29 (m, 2 H), 3.22-3.19 (m, 1 H), 2.83-2.80 (m, 2 H), 2.60-2.57 (t, J = , 2 H), 2.08-2.04 (m, 1 H), 1.97-1.91 (m, 1 H), 1.61-1.58 (m, 2 H), 1.33-1.29 (m, 10 H), 0.91-0.89 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of diastereomers), δ: 142.7, 135.8, 130.7, 129.8, 72.8, 71.2, 63.9, 54.4, 42.2, 38.1, 36.7, 33.2, 33.0, 30.8, 30.6, 30.5, 23.9, 14.6 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 320.25835, found 320.25841.
(2R,4S)-4-Hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidin-1-ium chloride (26)
24a (14 mg, 0.03 mmol) was submitted to the procedure of its enantiomer 23a. The resulting residue was purified by flash column chromatography (hexane/EtOAc 4:6, Rf: 0.38) to give the intermediate tert-Butyl (2R,4S)-4-hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate as a colorless oil (10 mg, 71 %). The spectral datas matched those reported for its enantiomer.
tert-Butyl (2R,4S)-4-hydroxy-2-(1-hydroxy-2-(4-octylphenyl)ethyl)pyrrolidine-1-carboxylate (10 mg, 0.024 mmol) was submitted to the general procedure A. 26 was obtained as a white powder (8 mg, 95 %). The spectral datas matched those reported for its enantiomer.
(2S,4R)-2-(4-Octylbenzoyl)-4-(phosphonooxy)pyrrolidin-1-ium chloride (29)
Di-tert-butyl diethylphosphoramidite (93%, 44 μL, 0.15 mmol, 2.0 eq.) and tetrazole (0.45 M in ACN, 0.22 mmol, 3.0 eq.) were added dropwise to a solution of 20c (30.0 mg, 0.074 mmol, 1.0 eq.) in dry THF (1 mL) at 0°. The resulting mixture was allowed to warm up to rt and stirred for 1.5 hours,. The reaction was cooled back to −30 °C whereby tBuOOH (5.0 M, 0.30 mmol, 4.0 eq.) was added dropwise. The resulting mixture was stirred at −30 °C for 15 minutes and at rt for 15 additional minutes. Afterwards, the reaction was cooled back to 0 °C whereby an aqueous NaHSO3 solution (10% w/w, 2 mL) was added dropwise. The aqueous layer was extracted with EtOAc (3 × 2 mL). The resulting organic layer was washed with brine (2 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (EtOAc/hexane 5:5 + 0.5% Pyridine, Rf: 0.32) to give the intermediate tert-Butyl (2S,4R)-4-((di-tert-butoxyphosphoryl)oxy)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate as a colorless oil (26 mg, 62%). [α]25D −11.7 (c 1.02, CHCl3). IR (neat), νmax: 2977, 2926, 2855, 1701, 1396, 1260, 987, 753 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.91-7.90 (d, J = 8.2 Hz, 0.85 H), 7.87-7.86 (d, J = 8.2 Hz, 1.15 H), 7.28-7.26 (d, J = 8.7 Hz, 1.15 H), 7.25-7.24 (d, J = 8.7 Hz, 0.85 H), 5.47-5.43 (t, J = 8.0 Hz, 0.4 H), 5.37-5.33 (t, J = 8.2 Hz, 0.6 H), 4.91 (m, 1 H), 3.93-3.90 (dd, J = 12.2 Hz, 0.6 H), 3.87-3.85 (m, 0.4 H), 3.75-3.69 (m, 1 H), 2.67-2.62 (m, 2 H), 2.62-2.54 (m, 1 H), 2.09-2.01 (m, 1 H), 1.62 (m, 2 H), 1.50-1.48 (m, 18 H), 1.44 (s, 4 H), 1.30-1.22 (m, 15 H), 0.87 (t, J = 7.4 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 198.5, 198.0, 154.3, 153.7, 149.5, 149.4, 133.1, 133.0, 128.9, 128.9, 128.8, 128.5, 83.1, 83.0, 83.0 83.0, 80.4, 80.2, 75.8, 75.8, 75.2, 75.2, 59.5, 59.2, 53.7, 53.7, 53.4, 53.3, 36.2, 32.0, 31.2, 30.1, 30.0, 29.5, 59.3, 28.5, 28.2, 22.8, 14.2 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 618.35301, found 618.35381.
tert-Butyl (2S,4R)-4-((di-tert-butoxyphosphoryl)oxy)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (25 mg, 0.04 mmol) was submitted to general procedure A. 29 was obtained as a white solid (12 mg, 66%). [α]25D −.29.3 (c 0.37, CHCl3). IR (neat), νmax: 2923, 1685, 1165, 1032, 922, 513 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 8.01-7.99 (d, J = 7.4 Hz, 2 H), 7.42-7.40 (d, J = 7.8 Hz, 2 H), 5.53 (bs, 1 H), 4.98 (bs, 1 H), 3.70 (bs, 1 H), 3.44 (bs, 1 H), 2.99 (bs, 1 H), 2.73-2.70 (t, J = 7.6, 2 H), 2.08 (m, 1 H), 1.67-1.64 (m, 2 H), 1.34-1.25 (m, 10 H), 0.90-0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 193.0, 151.2, 130.3, 129.1, 129.0, 74.2, 61.9, 52.7, 38.0, 35.6, 31.6, 30.8, 29.1, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 384.19344, found 384.19257.
(2S,4R)-2-(4-Octylbenzoyl)-4-(phosphonooxy)pyrrolidin-1-ium chloride (30)
21c (50 mg, 0.12 mmol) was submitted to the procedure of its enantiomer. The residue was purified by flash column chromatography (EtOAc/hexane 5:5 + 0.5% Pyridine, Rf: 0.32) to give the intermediate tert-Butyl (2R,4S)-4-((di-tert-butoxyphosphoryl)oxy)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate as a colorless oil (71 mg, 63%). [α]25D +11.0 (c 0.53, CHCl3). The spectral datas matched those reported for its enantiomer.
tert-Butyl (2R,4S)-4-((di-tert-butoxyphosphoryl)oxy)-2-(4-octylbenzoyl)pyrrolidine-1-carboxylate (27 mg, 0.05 mmol) was submitted to general procedure A. 30 was obtained as a white solid (14 mg, 78%). [α]25D +27.2 (c 0.55, CHCl3). The spectral datas matched those reported for its enantiomer.
(2S,4R)-2-(2-(4-Octylphenyl)acetyl)-4-(phosphonooxy)pyrrolidin-1-ium chloride (31)
23a (68 mg, 0.16 mmol) was submitted to the procedure from 31. The resulting residue was purified by flash column chromatography (hexane/EtOAc 6:4, Rf: 0.37) to give the intermediate tert-Butyl (2S,4R)-4-((di-tert-butoxyphosphoryl)oxy)-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate as a colorless oil (65 mg, 67 %). [α]25D −45.2 (c 0.65, CHCl3). IR (neat), νmax: 2925, 1696, 1393, 1262, 1160, 989 cm−1. 1H NMR (CDCl3, 500 MHz, mixture of rotamers), δ: 7.14-7.08 (m, 4 H), 4.78 (bs, 1 H), 4.60-4.57 (t, J = 8.9, 7.4 Hz, 0.47 H), 4.54-4.50 (t, J = 9.1, 7.7 Hz, 0.53 H), 3.91-3.67 (m, 3 H), 3.54-3.47 (ddd, J = 16.6, 13.4, 3.3 Hz, 1 H), 2.58-2.54 (m, 2 H), 2.30-2.25 (m, 0.53 H), 2.13-2.09 (m, 0.47 H), 1.84-1.81 (m, 1 H), 1.58-1.56 (m, 2 H), 1.58-1.40 (m, 25 H), 1.29-1.25 (m, 13 H), 0.89-0.86 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz, mixture of rotamers), δ: 207.1, 207.1, 154.6, 153.8, 142.2, 141.9, 130.6, 130.3, 129.7, 129.6, 129.0, 128.9, 83.1, 83.1, 83.1, 83.0, 82.9, 80.9, 80.5, 75.7, 75.6, 74.8, 74.8, 63.4, 62.5, 53.8, 53.7, 53.5, 53.5, 47.6, 46.3, 32.0, 30.0, 30.0, 29.6, 29.4, 28.5, 28.4, 22.8, 14.2 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 610.38672, found 610.38470.
tert-Butyl (2S,4R)-4-((di-tert-butoxyphosphoryl)oxy)-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate (30 mg, 0.05 mmol) was submitted to general procedure A. 31 was obtained as a purple paste (12 mg, 57%). [α]25D +4.7 (c 1.35, CHCl3). IR (neat), νmax: 2922, 1724, 1514, 1173, 1009 cm−1. 1H NMR (CDCl3, 500 MHz), δ: 7.17 (m, 4 H), 5.00 (bs, 1 H), 4.82-4.77 (m, 1 H), 3.98-3.92 (s, 2 H), 3.60-3.57 (d, J = 12.7 Hz, 1 H), 3.42-3.39 (d, J = 9.4 Hz, 1 H), 2.83-2.78 (m, 1 H), 2.62-2.58 (m, 2 H), 2.18-2.13 (m, 1 H), 1.60 (m, 2 H), 1.33-1.29 (m, 10 H), 0.92-0.88 (t, J = 10.7 Hz, 3 H) ppm. 13C NMR (CDCl3, 125 MHz), δ: 201.7, 142.0, 141.6, 129.5, 129.5, 129.4, 129.4, 128.5, 128.4, 128.2, 75.0, 74.4, 74.4, 64.5, 64.1, 63.8, 63.3, 52.4, 52.3, 45.1, 35.1, 31.6, 31.3, 29.2, 29.0, 28.9, 22.3, 13.0 ppm. HRMS (ESI) calcd. for C25H40NO3 (M+H)+ 398.20909, found 398.20750.
(2R,4S)-2-(2-(4-Octylphenyl)acetyl)-4-(phosphonooxy)pyrrolidin-1-ium chloride (32)
24a (68 mg, 0.16 mmol) was submitted to the procedure of its enantiomer. The resulting residue was purified by flash column chromatography (hexane/EtOAc 6:4, Rf: 0.37) to give the intermediate tert-Butyl (2R,4S)-4-((di-tert-butoxyphosphoryl)oxy)-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate as a colorless oil (66 mg, 68 %). [α]25D +53.1 (c 0.85, CHCl3). The spectral datas matched those reported for its enantiomer.
tert-Butyl (2R, 4S)-4-((di-tert-butoxyphosphoryl)oxy)-2-(2-(4-octylphenyl)acetyl)pyrrolidine-1-carboxylate (35 mg, 0.06 mmol) was submitted to general procedure A. 32 was obtained as a purple paste (15 mg, 60%). [α]25D −5.0 (c 1.20, CHCl3). The spectral datas matched those reported for its enantiomer.
4.2. Biology methods
4.2.1. Compounds.
Compound stocks (2.5-50 mM) were prepared in water with the exception of 5, 6, 15, 20, 21, 23, 24, 27, 28, 29, and 30, which were prepared in DMSO. FTY720 (S)-Phosphate was purchased from Cayman Chemical Company (cat# 10006408) and prepared in DMSO immediately before use. Vorinostat (suberoylanilide hydroxamic acid, SAHA) was purchased from LC Labs (cat# V-8477) and prepared in DMSO.
4.2.2. Cell Culture Studies.
FL5.12 cells (murine hematopoietic stem cells originally obtained from Craig Thompson, Memorial Sloan Kettering Cancer Center) were maintained at 5-750,000 cells/mL, with a density of 400-600,000 cells/mL the day of treatment. FL5.12 cells were cultured in RPMI 1640 (Mediatech) supplemented with 500 pg/mL recombinant murine IL-3 (cat# 575502, Biolegend), 10% Fetal Bovine Serum (Sigma-Aldrich), 1% HEPES buffer (Mediatech), 2 mM L-Glutamine (Mediatech), 55 μM β-mercaptoethanol (Sigma-Aldrich), and antibiotics. MEFs (murine embryonic fibroblasts generated in-house or obtained from Ashley Snider, Stony Brook School of Medicine) were maintained in DMEM with 4.5 g/L glucose and L-glutamine (Mediatech) supplemented with 10% Fetal Bovine Serum (Sigma-Aldrich) and antibiotics. Cells were screened for Mycoplasma every 3 months using the Look-Out Mycoplasma PCR Detection kit (cat# MP0035, Sigma-Aldrich).
4.2.3. Viability Assays.
FL5.12 cells were treated at 25,000/mL. Viability was determined at 48 h by vital dye exclusion (4’6 diamidino-2-phenylindole (DAPI) or propidium iodide) using flow cytometry. IC50 values were calculated using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA)
4.2.4. Nutrient Transporter Expression.
Surface CD98 (4F2hc, SLC3A2) expression in FL5.12 cells plated at 225-250,000/mL was measured after a 3 h treatment using phycoerythrin (PE)-conjugated anti-mouse CD98 antibody (Biolegend). PE-conjugated rat IgG2a κ antibody (Biolegend) was used as an isotype control. 150-200,000 cells were stained in triplicate in 100 μL FACS block (PBS with 10% FBS and 0.05% NaN3) containing 0.25 μL antibody on ice for 30 min, washed twice with 1 mL FACS wash (PBS containing 2% FCS and 0.05% NaN3), and analyzed on a BD LSR II or FACSCalibur flow cytometer. Data was processed using FlowJo (Treestar), where analysis was limited to viable cells as determined by vital dye exclusion (DAPI) or forward/side scatter.
4.2.5. Vacuolation Assay.
FL5.12 cells were treated at a density of 200,000/mL for 3 hours before being imaged at 100× using brightfield microscopy on a Nikon TE2000-S fluorescence microscope equipped with DIC filters. Vacuoles in at least 5 images were qualitatively evaluated for at least 2 biological replicates in each experimental condition. Scores were assigned as follows: 0 = no vacuoles, + = some cells vacuolated, ++ = most cells have at least 1 vacuole, +++ = all cells heavily vacuolated. Two researchers scored each replicate independently to mitigate any potential biases.
4.2.6. HDAC activity assays.
HDAC activity was measured for recombinant HDAC1 (Cayman, cat# 10011617) or HeLa extract (Enzo, cat# BML-KI137) using the Enzo COLOR DE LYS HDAC colorimetric activity assay kit (cat# BML-AK501-0001) or the Cayman HDAC Fluorometric Activity Assay Kit (cat# 10011563). Reaction mixtures containing acetylated lysine substrate were incubated at 37°C for 15-30 min. After developer was added, plates were incubated for 10-15 min at 37°C and absorbance at 405 nm measured with an Epoch microplate spectrophotometer (BioTek) or 15 min at RT and fluorescence measured with a SpectraMax Gemini XS plate reader (Molecular Devices) with excitation at 355 nm and emission at 460 nm. For fluorescence measurements, each reaction was conducted ± trichostatin A to generate corrected fluorescence values per the Cayman protocol.
4.2.7. Western blot.
Cell lysates were prepared in RIPA lysis buffer (10 mM Tris-Cl (pH 8.0), 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) containing 1× complete protease inhibitor (Pierce cat# 88265). Where indicated, histones were acid extracted in 0.2 N HCl overnight at 4°C. Protein concentration was quantified using Pierce BCA protein assay (Thermo Scientific cat# 23223) and samples prepared in 1× NuPage sample buffer (Invitrogen cat#NP0007) with 5 mM DTT. Samples were run on 4-12% NuPAGE gels (cat# NP0336BOX), transferred to BioTrace NT nitrocellulose membranes (Pall cat# 66485), incubated 1 h in blocking solution (5% bovine serum albumin, 7.7 μM NaN3 in 1× TBST), incubated with primary antibodies (H3K9, cat#9649; H3K18, cat#13998; H3, cat#4499; H4K5, cat#8647; H4K8, cat#2594; H4, cat#2935, all ) overnight at 4°C in blocking solution, and with secondary antibodies (IRDye 800 or 680 conjugates, cat# 926-3211 or 926-6802, LI-COR) for 1 h at RT. Membranes were analyzed on a LI-COR Odyssey SA Imager and analyzed using Image Studio software (LI-COR).
4.2.8. Statistics.
IC50 values are shown with the 95% confidence interval as calculated in GraphPad Prism. CD98 loss is given as the mean of at least 2 biological replicates.
Supplementary Material
Highlights of the submitted manuscript:
Pyrrolizidines inhibit transporters of cell surface expression in cancer cells
Substituents on pyrrolidines affect vacuolation/nutrient transporter inhibition
Probing functional group tolerance of the hydrophobic substituents
Rationale and synthesis of constrained pyrrolidine phosphate esters as dual agents
Acknowledgements
ANM was supported by NIH (R01 GM089919), a Proof of Product grant from UCI Applied Innovation, and a UC CRCC grant (CRR-17-426826) to ALE. ALE was supported by grants from the NIH (R01 GM089919) and CDMRP (W81XWH-11-1-0535). The Montreal group thanks NSERC for financial assistance and the Departmental X-ray service for structure determination. We thank Professor Nicolas Moitessier from McGill University for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Biology figures; Spectroscopic data, X-ray data (See Supporting Informations)
References
- 1.Kroschinsky F, Stolzel F, von Bonin S, Beutel G, Kochanek M, Kiehl M, Schellongowski P, New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management, Crit Care 21 (2017) 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang M, Shen A, Ding J, Geng M, Molecularly targeted cancer therapy: some lessons from the past decade, Trends Pharmacol. Sci. 35 (2014) 41–50 [DOI] [PubMed] [Google Scholar]
- 3.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG, Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer 13 (2013) 714–726 [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–674 [DOI] [PubMed] [Google Scholar]
- 5.Selwan EM, Finicle BT, Kim SM, Edinger AL Attacking the supply wagons to starve cancer cells to death, FEBS Lett 590 (2016)885–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Outschoorn UE., Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP, Cancer metabolism: a therapeutic perspective, Nat. Rev. Clin. Oncol. 14 (2017) 11–31 [DOI] [PubMed] [Google Scholar]
- 7.Luengo A, Gui DY, Vander Heiden MG, Targeting Metabolism for Cancer Therapy, Cell. Chem. Biol, 24 (2017) 1161–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P, Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis, Nat. Rev. Drug. Discov. 9 (2010) 883–897 [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Wang HD, Cong ZX, Zhu JH, Zhou Y, FTY720 for cancer therapy (Review), Oncology Reports 30 (2013) 2571–2578 [DOI] [PubMed] [Google Scholar]
- 10.Camm J, Hla T, Bakshi R, Brinkmann V, Cardiac and Vascular Effects of Fingolimod: Mechanistic Basis and Clinical Implications Am. Heart J. 168 (2014) 623–644 [DOI] [PubMed] [Google Scholar]
- 11.Fransson R, McCracken AN, Chen B, McMonigle RJ, Edinger AL, Hanessian S, Design, Synthesis, and Anti-leukemic Activity of Stereochemically Defined Constrained Analogs of FTY720 (Gilenya), ACS Med. Chem. Lett. 4, (2013), 969–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen B, Roy SG, McMonigle RJ, Keebaugh A, McCracken AN, Selwan E, Fransson R, Fallegger D, Huwiler A, Kleinman MT, Edinger AL, Hanessian S, Azacyclic FTY720 Analogues That Limit Nutrient Transporter Expression but Lack S1P Receptor Activity and Negative Chronotropic Effects Offer a Novel and Effective Strategy to Kill Cancer Cells in Vivo, ACS Chem. Biol. 11 (2016) 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perryman MS, Tessier J, Wiher T, O’Donoghue H, McCracken AN, Kim SM, Nguyen DG, Simitian GS, Viana M, Rafelski S, Edinger AL, Hanessian S, Effects of stereochemistry, saturation, and hydrocarbon chain length on the ability of synthetic constrained azacyclic sphingolipids to trigger nutrient transporter down-regulation, vacuolation, and cell death Bioorg. Med. Chem. 24 (2016) 4390–4397 [DOI] [PubMed] [Google Scholar]
- 14.Kim SM, Roy SG, Chen B, Nguyen TM, McMonigle RJ, McCracken AN, Zhang Y, Kofuji S, Hou J, Selwan E, Finicle BT, Nguyen TT, Ravi A, Ramirez MU, Wiher T, Guenther GG, Kono M, Sasaki AT, Weisman LS, Potma EO, Tromberg BJ, Edwards RA, Hanessian S, Edinger AL, Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways, J. Clin. Invest. 126 (2016) 4088–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung N, Mao C, Heitman J, Hannun YA, Obeid LM, Phytosphingosine as a specific inhibitor of growth and nutrient import in Saccharomyces cerevisiae, J. Biol. Chem. 276 (2001) 35614–21 [DOI] [PubMed] [Google Scholar]
- 16.Skrzypek MS, Nagiec MM, Lester RL, Dickson RC, Inhibition of amino acid transport by sphingoid long chain bases in Saccharomyces cerevisiae J. Biol. Chem. 273 (1998) 2829–34 [DOI] [PubMed] [Google Scholar]
- 17.Welsch CA, Roth LW, Goetschy JF, Movva NR, Genetic, biochemical, and transcriptional responses of Saccharomyces cerevisiae to the novel immunomodulator FTY720 largely mimic those of the natural sphingolipid phytosphingosine, J. Biol. Chem. 279 (2004) 36720–31 [DOI] [PubMed] [Google Scholar]
- 18.Guenther GG, Peralta ER, Rosales KR, Wong KR, Y. S, Siskind LJ & Edinger AL Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A 105, (2008) 17402–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romero Rosales K, Singh G, Wu K, Chen J, Janes MR, Lilly MB, Peralta ER, Siskind LJ, Bennett MJ, Fruman DA, Edinger AL, Sphingolipid-based drugs selectively kill cancer cells by down-regulating nutrient transporter proteins, Biochem. J. 439 (2011) 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barthelemy C, Barry AO, Twyffels L, André B, FTY720-induced endocytosis of yeast and human amino acid transporters is preceded by reduction of their inherent activity and TORC1 inhibition, Scientific Reports 7 (2017) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Körner M, Tibes U, 5 Histone Deacetylase Inhibitors: A Novel Class of Anti-Cancer Agents on its Way to the Market, Pro. Med. Chem. 46 (2008) 205. [DOI] [PubMed] [Google Scholar]
- 22.Atadja PW, HDAC Inhibitors and Cancer Therapy, In: Gasser S, Li E (eds) Epigenetics and Disease. Progress in Drug Research, Springer, Basel, 67 (2011) [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Fang H, Xu W, Strategies in Developing Promising Histone Deacetylase Inhibitors, Med.Chem.Rev. 30 (2009) 585–202 [DOI] [PubMed] [Google Scholar]
- 24.Hait NC, Wise LE, Allegood JC, O’Brien M, Avni D, Reeves TM, Knapp PE, Lu J, Luo C, Miles MF, Milstien S, Lichtman AH, Spiegel S, Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory, Nat. Neuro. 17 (2014) 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hait NC, Avni D, Yamada A, Nagahashi M, Aoyagi T, Aoki H, Dumur CI, Zelenko Z, Gallagher EJ, Leroith D, Milstien S, Takabe K, Spiegel S The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERalpha expression and enhances hormonal therapy for breast cancer, Oncogenesis 4 (2015) e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardner NM, Riley RT, Showker JL, Voss KA, Sachs AJ, Maddox JR, Gelineau-van Waes JB, Elevated Nuclear and Cytoplasmic FTY720-Phosphate in Mouse Embyronic Fibroblasts Suggests the Potential for Multiple Mechanisms in FTY720-Induced Neural Tube Defects, Toxicological Sciences, 150 (2016) 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu H, Takahashi M, Kaneko T, Murakami T, Hakamata Y, Kudou S, Kishi T, Fukushi K, Iwanami S, Kuriyama K, Yasue T, Enosawa S, Matsumoto K, Takeyoshi I, Morishita Y, Kobayashi E, KRP-203, a novel synthetic immunosuppressant, prolongs graft survival and attenuates chronic rejection in rat skin and heart allografts, Circulation 2 (2004) 222–229H. [DOI] [PubMed] [Google Scholar]
- 28.Xiao Q, Jin J, Wang X, Hu J, Xi M, Tian Y, Yin D, Synthesis, identification, and biological activity of metabolites of two novel selective S1P1 agonists, Bioorg. Med. Chem. 24 (2016) 2273–2279 [DOI] [PubMed] [Google Scholar]
- 29.Kimball FS, Turunen BJ, Ellis KC, Himes RH, Georg GI, Enantiospecific synthesis and cytotoxicity of 7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one enantiomers, Bioorg. Med. Chem. 16 (2008) 4367–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollica JA, Smith JB, Nunes IM, Govan HK, Kinetics of the decomposition of a Mannich base, J. Pharm. Sci. 59 (1970) 1770–1774 [DOI] [PubMed] [Google Scholar]
- 31.Orgován G, Tihanyi K, Noszál B, NMR analysis, protonation equilibria and decomposition kinetics of tolperisone, J. Pharm. Biomed. Anal. 50 (2009) 718–723 [DOI] [PubMed] [Google Scholar]
- 32.King S-YP, Sigvardson KW, Dudzinski J, Torosian G, Degradation kinetics and mechanisms of moricizine hydrochloride in acidic medium, J. Pharm. Sci. 81 (1992) 586–591 [DOI] [PubMed] [Google Scholar]
- 33.Morgenthaler M, Schweizer E, Hoffmann-Roder A, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerli D, Schneider J, Diederich F, Kansy M, Muller K, Predicting and tuning physicochemical properties in lead optimization: Amine basicities, Chem. Med. Chem. 2 (2007) 1100–1115 [DOI] [PubMed] [Google Scholar]
- 34.Chen HP, Zhao YT, Zhao TC, Histone deacetylases and Mechanisms of Regulation of Gene Expression (Histone Deacetylase in cancer), Crit. Rev. Oncog. 20 (2015) 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida M, Kijima M, Akita M, Beppu T, Potent and Specific Inhibitors of Mammalian Histone Deacetylase Both in Vivo and in Vitro by trichostatin A, J. Biol. Chem. 265 (1990) 17174–17179 [PubMed] [Google Scholar]
- 36.Marks PA, Breslow R, Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug, Nat. Biotechnol. 1 (2007) 84–90 [DOI] [PubMed] [Google Scholar]
- 37.Hanessian, Auzzas L, Giannini G, Marzi M, Cabri W, Barbarino M, Vesci L, Pisano C, ω-Alkoxy Analogues of SAHA (Vorinostat) as Inhibitors of HDAC. A Study of Chain length, Bioorg. Med. Chem. Lett. 17 (2007) 6261–6265 [DOI] [PubMed] [Google Scholar]
- 38.Hanessian S, Auzzas L, Larsson A, Zhang J, Giannini G, Gallo G,, Ciacci A, Cabri W, Vorinostat-like Molecules as Structural, Stereochemical, and Pharmacological Tools, ACS.Med.Chem.Lett. 1 (2010) 70–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vannini A, Volpari C, Gallinari P, Jones P. Mattu M, Carfi A, DeFrancesco R, Steinkühler C, DiMarco S, Substrate Binding to Histone Deacetylase as Shown by Crystal Structure of HDAC8-Substrate Complex, EMBO, Rep. 8 (2007) 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanessian S, Auzzas L, Larsson A, Matera R, Baraldi A, Giannini G, Cabri W, Battistuzzi G, Gallo G, Ciacci A, Vesci L, Pisano C, Non-natural Macrocyclic Inhibitors of Histone Deacetylase, J. Med.Chem. 53 (2010) 8387–8399 [DOI] [PubMed] [Google Scholar]
- 41.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S, Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate, Science, 325 (2009) 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moitessier N, Pottel J, Therrien E, Englebienne P, Liu Z, Tomberg A, Corbeil CR, Medicinal Chemistry Projects Requiring Imaginative Structure-Based Drug Design Methods, Acc. Chem. Res. 49 (2016) 1646–1657 [DOI] [PubMed] [Google Scholar]
- 43.Pottel J, Therrien E, Gleason JL, Moitessier N, Docking Ligands into Flexible and Solvated Macromolecules. 6. Development and Application to the Docking of HDACs and other Zinc Metalloenzymes Inhibitors, J. Chem. Inf. Model. 54 (2014) 254–265 [DOI] [PubMed] [Google Scholar]
- 44.Entwistle DA, Jordan SI, Montgomery J, Pattenden G, Total Synthesis of Oxazole-Based Virginiamycin Antibiotics: 14,15-Anhydropristinamycin IIB, Synthesis (1998) 603–612 [Google Scholar]
- 45.Huy P, Schmalz H-G, Practical One-Pot Double Functionalizations of Proline, Synthesis 6 (2011) 954–960 [Google Scholar]
- 46.Radl S, Doubsky J, A Method for the Preparation of Fingolimod, WO2013185740 (2013) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.