

SUMMARY:
Molecular epidemiology uses the distribution and organization of a pathogen’s DNA to understand the distribution and determinants of disease. Since the biology of DNA for eukaryotic pathogens differs substantially from that of bacteria, the analytic approach to their molecular epidemiology can also differ. While many of the genotyping techniques presented earlier in this series, Advances in Molecular Epidemiology of Infectious Diseases, can be applied to eukaryotes, the output must be interpreted in the light of how DNA is distributed from one generation to the next. In some cases, parasite populations can be evaluated in ways reminiscent of bacteria. They differ, however, when analyzed as sexually reproducing organisms, where all individuals are unique, but the genetic composition of the population does not change unless a limited set of events occurs. It is these events (migration, mutation, non-random mating, selection, genetic drift) that are of interest. At a given time, not all of them are likely to be equally important, so the list can easily be narrowed down to understand the driving forces behind the population as it is now and even what it will look like in the future. The main population characteristics measured to assess these events are differentiation and diversity, interpreted in the light of what is known about the population from observation. The population genetics of eukaryotes is important for planning and evaluation of control measures, surveillance, outbreak investigation, and monitoring the development and spread of drug resistance.
INTRODUCTION
The study of molecular epidemiology of parasitic infections and their vectors is meant to answer the same types of questions as for bacterial or viral infections. As with bacteria and viruses, the molecular epidemiology of eukaryotic infections follows the distribution and dynamics of microbial DNA. The key difference, however, is precisely this biology, which at times demands a distinct approach to molecular epidemiologic investigation of infections caused by eukaryotic organisms. In bacterial reproduction, each individual passes down an essentially identical copy of all the DNA to the next generation. Some eukaryotic pathogens also reproduce in this way, while many others reproduce sexually for at least part of their life cycle, and in a few it is not entirely clear (100). An asexually reproducing individual generates a clone of itself, but can also exhibit promiscuous horizontal gene transfer, which is a major source of variation and adaptation (99). This is not sex, however. Sex is the biologically-necessary programmed recombination (crossing over) and random shuffling (reassortment) of chromosomal DNA in the process of reproduction. This results in an enormous reservoir of variation. For example, for schistosomes with 8 pairs of chromosomes, and considering there are some differences on each chromosome of the pair, reassortment alone offers >65,000 possible combinations of alleles with each mating. This of course pales besides the human with 46 chromosomes and 7 × 1013 potential combinations. This is in addition to variation from exchange of genetic segments with crossing-over. In nature, bacteria are heterogenous conglomerates or communities (62, 99), but when they cause disease, especially in epidemics, it is generally a clone1 that is responsible and that will be tracked. Sexual reproduction in some protozoa, many parasitic worms, and most vectors never results in a clone (with the exception of identical twins). There is genetic conservation, however, within a group of organisms that tends to breed together. In genetics, this is the working definition of a population. For sexually reproducing organisms, the population is the epidemiologic unit to track. Within the group, allele frequencies and thus traits are conserved under well-defined conditions. The unique power of the genetics of populations is that it reflects not only present individuals, but also the population’s past and the future potential for subsequent generations (35). Many parasites exhibit both sexual and asexual modes of reproduction, but these life stages are often distributed in different hosts. Treatment of their molecular epidemiology is doubly complex, but can be simplified for many questions by considering their biology just in the human host. As with most genetics, the output from population genetics has to be interpreted within the context of the question being asked and the biology and epidemiology of organisms being studied. The whole field of population genetics is perhaps the most complex area of genetics, but it rests on simple precepts. This review outlines the basic models used in population genetics that are directly applicable to problems of public health epidemiology. A list of abbreviations and terms in bold type is provided at the end of this chapter.
DEFINING GENOTYPE IN EUKARYOTIC ORGANISM
Some expressions may not be familiar to some readers, so it is important to define these early and in simple terms. A glossary is provided with this review. One of the dividing lines between the DNA of bacteria and sexually reproducing parasites and vectors of human disease is their physical structure and organization. Sexually reproducing organisms will pass some portion of their life cycle where their chromosomes exist as nearly identical pairs (diploid). Some organisms, malaria for example, also have only one copy of each chromosome (haploid) during their asexual stage, and this is the stage that infects humans. A similar location on each of the chromosomes of a diploid individual or those of different haploid individuals is a locus, and differences between loci (nucleotides, insertions, deletions, copies) are alleles (Figure 1).
Figure 1.
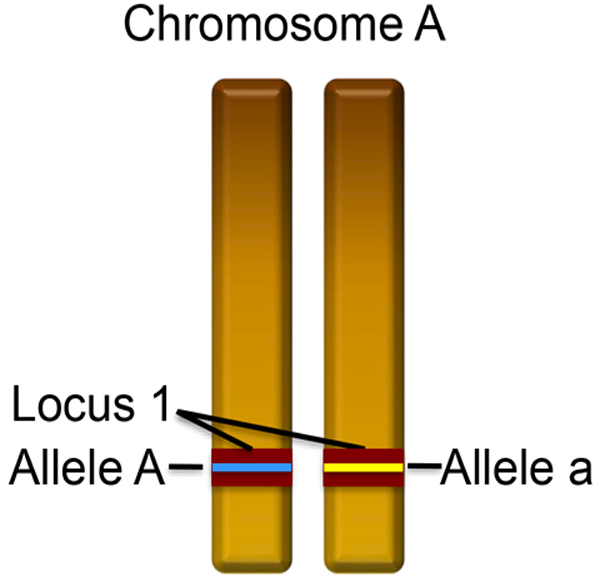
There are 2 nearly identical copies of all chromosomes for diploid organisms, except those responsible for determining sex. The corresponding location on each chromosome is a locus. Specific differences or similarities within a locus are alleles. The identity of both alleles at a locus is the genotype. In this case the genotype is Aa, i.e. heterozygous. Were it AA, the genotype would be homozygous.
The geometry of DNA also strongly differentiates eukaryotes from bacteria (Figure 2). There are a number of exceptions, but only a framework for understanding essential differences is presented here. Most prokaryotes have a single circular chromosome2, whereas even the simplest eukaryotes (yeast) have at least 16 linear chromosomes. A specific marker on a bacterial chromosome will be transmitted at reproduction together with any other marker or trait in the genome. The same also occurs with an asexually reproducing eukaryote despite having multiple linear chromosomes. A marker on the genome of a sexually reproducing eukaryote, by contrast, will have a 50% chance of being transmitted away from any marker it is not very close to. Because of crossing-over, markers near each other can be linked, i.e. occur together more frequently than expected by random chance in subsequent generations. This is physical linkage of the loci. At the population level, some loci that are not near each other can also be associated non-randomly due to demography or population history. The non-random association between loci is termed linkage disequilibrium (LD). This is a statistical result and not necessarily recognition of a physical relationship. The labeling of each allele present at the same locus on each chromosome constitutes the genotype. A locus with the same polymorphism at the same site on each of the chromosome is homozygous, and with a different polymorphism is heterozygous.
Figure 2.
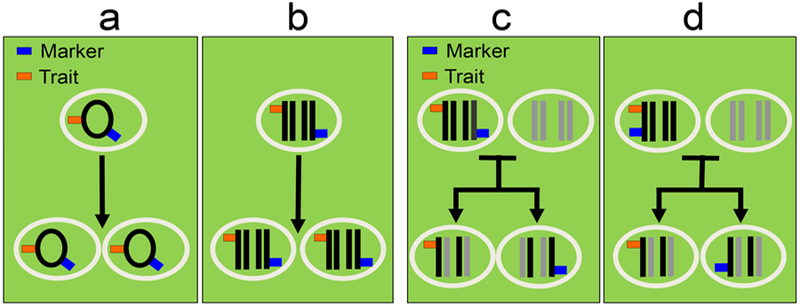
(left panel) Asexual organisms or organelles: a) Circular genome with bacterial reproduction; b) diploid linear genome and asexual eukaryotic reproduction. Every time the marker is present, the trait genotype is present.
(right panel) Sexual organisms: Sexual reproduction between organisms with diploid chromosomes. c) reassortment; d) recombination and reassortment. Every time the marker is present, the trait genotype may or may not be present.
OPTIONS FOR MOLECULAR EPIDEMIOLOGY OF EUKARYOTES
Study asexual parasites and clonal groups
Use markers in organelles that behave like bacteria (e.g. mitochondria)
Use a marker close to the trait of interest (if known)
Use many markers throughout the genome or perform sequencing
Study the whole group of organisms in which the trait is present (population genetics)
HARDY-WEINBERG EQUILIBRIUM: THE POPULATION NULL HYPOTHESIS
A population has a mathematical definition based on allele frequencies, which ultimately contributes to the development of tools for key measures of differentiation and diversity. Allele frequencies can differentiate populations, and genotypic frequencies can do so with even greater resolution. The Hardy-Weinberg Equilibrium describes a relationship between allelic frequency and genotypic frequency. It is a simple mathematical relationship that is the definition of a population. If we use the letters “A” and “a” to represent different alleles at a single diallelic locus and “p” and “q” to represent their respective frequencies, a population with p=0.8 and q=0.2 is clearly different from a population where p=0.2, q=0.8, especially where this kind of result is found at multiple loci. Allele frequencies are not always the most sensitive measure of differentiation, however. The same allele frequency may still be found in what are clearly distinct populations if assessed for genotypic frequencies. Alleles combine to form genotypes, so allelic frequencies are closely related to genotypic frequencies. For a diallelic locus where we know the frequency of each allele, the sum of these frequencies is 1 or (p + q = 1). For sexually reproducing organisms, the next generation arises from the combination of alleles from a pool of males with alleles from a pool of females. If we imagine that individuals from these pools will pair at random, the subsequent distribution of alleles in genotypes is equivalent to flipping 2 coins. For independent, random events the probability of 2 events occurring simultaneously is the product of their frequencies [(p + q)female × (p + q)male = 1]. The genotypic frequencies of the offspring for such a population should be p2 + 2pq + q2, if all assumptions are met, where p2 and q2 are the frequencies for the homozygotes and 2pq the heterozygotes. This expression is the Hardy-Weinberg equilibrium (HWE). This simple quadratic equation is the basis for all population genetics even when it is not measured directly. It represents the expected genotypic frequencies for a given set of allelic frequencies. It is one of the most stable mathematical relationships in nature. It is so much the expectation that when not observed in sequencing projects, it can suggest sequencing errors. It is the null hypothesis and mathematical definition of a stable population at equilibrium; nothing is happening.
The relationship HWE describes is true under a set of 5–10 assumptions that represent the most important factors that influence population genetic structure. The 5 most common assumptions are that there is:
-
1)
Random mating (panmixia, assortative mating)
-
2)
No selection
-
3)
No migration
-
4)
An infinite population
-
5)
No mutation
It is rare to have any of these assumptions met in nature, but the proportions are so resilient that the assumptions have to be severely violated to disturb this relationship. Even if they are violated, the proportions will be reestablished within 1–2 generations once the population is stabilized. As with most models, the underlying assumptions are the most important aspects. They are the basis for most conversations in population genetics.
MARKERS
Microsatellites, single nucleotide polymorphisms (SNPs) and sequencing are currently the marker types most employed in population genetics. Microsatellites are short tandem repeats of 2–8 nucleotides (reviewed in (29)). Microsatellites have fallen out of favor in studies of statistical genetics or gene finding, since SNPs and sequencing provide better resolution at the level of individuals. Microsatellites, however, remain important in population genetics since they are mostly neutral for selection and have higher allelic richness and information content than single SNPs, and individuals are not the focus of population genetics. The microsatellite’s rapid mutation rate (10−2 ─ 10−5 per generation) and stepwise mode of mutation can limit their application to questions that extend over short time scales and to certain statistical approaches. SNPs have lower rates of mutation (10−8) in eukaryotes, often are diallelic, are ten times more abundant (48, 114), and have high processivity and scorability. Sequencing essentially provides a very dense panel of SNPs and identifies rare variants as well as structural polymorphisms (copy number, insertions/deletions).
Mitochondrial and ribosomal DNA markers are much less abundant, less polymorphic, and thus less informative than microsatellites or SNPs. Some are under selection and in the case of mitochondrial DNA, the genome is haploid (only 1 copy of chromosomal DNA) and may or may not have sex-specific inheritance depending on the species (50). They are useful for phylogeny studies, may be more economical to use in laboratories with limited capabilities and are sometimes combined with other markers. Some aspects of malaria such as multiplicity of infection or populations on a continental scale are studied as asexually reproducing organisms, which they are in the human host. The ideal markers for this turn out to be from organelle DNA since these structures behave like bacteria. The analysis then is based on markers usually scored simply as present or absent as for DNA barcodes (102).
MEASURES OF DIFFERENTIATION AND DIVERSITY
Evolution and conservation are the areas most often addressed by using population genetics are. These two areas deal with essentially the same phenomenon, but at different time scales, thus the questions, the approaches, and the interpretation will differ depending on the nature of the problem. The relevant public health questions in population genetics focus on identity and dynamics of the group rather than individuals, extend over short time scales and are directed at the control or extinction of a parasite or vector. Who’s sleeping with whom, modes of reproduction, evolution or the last common ancestor are all important in different contexts. These issues may be useful to help explain anomalies and can influence interpretation, but they are rarely answers to issues of control or intervention. Understanding how diverse a population is or the degree of difference between populations, combined with good study design, will contribute directly to determining the impact of control measures, host or parasite demographics, resistance, risk, resilience or fragility of the population.
The field of population genetics depends heavily on mathematical analyses, some simple and some very sophisticated, to answer these questions. Mathematical treatments of all of the indices and statistics of differentiation and diversity can be easily obtained online, from textbooks or publications and will not be included here. Fortunately for the mathematically challenged, many open source, individual computer programs and modules are available in R. The risk that goes with all readymade programs is a failure to understand what is being asked or the assumptions and limitations of the approach being taken. A list of some frequently used programs is provided at the end of this review (Table 1).
Table 1. Selected Population Genetics Programs.
| Program | Functions | Platform | Update | Authors | URL |
|---|---|---|---|---|---|
| Admixture | Clustering | Windows | 2015 | Alexander etal. |
https://www.genetics.ucla.edu/software/admixture/ |
| Arlequin (123) †‡ |
HWE, Fst, AMOVA |
Windows Mac Linux |
2015 | Excoffier, and Lischer |
http://cmpg.unibe.ch/software/arlequin35/ |
| Bottleneck (127) † |
Expected heterozygosity |
Windows | 1999 | Cornuet et al. | http://www1.montpellier.inra.fr/CBGP/software/Bottleneck/bottleneck.html |
| diveRsity | HWE, Fst, Ar | R package | 2017 | Keenan | http://diversityinlife.weebly.com/ |
| DnaSP (124) ‡ |
Fst, Gst | Windows Mac Linux |
2018 | J. Rozas et al. | http://www.ub.edu/dnasp/ |
| estMOl (30) |
Haplotype generation |
Windows | 2014 | Assefa et al. | http://pathogenseq.lshtm.ac.uk |
| Fluctuate‡ | Ne | Windows Mac Linux |
2002 | Kuhner | http://evolution.genetics.washington.edu/lamarc.html |
| FSTAT | HWE, Fst, Rst | Windows | 2005 | J Goudet | http://www2.unil.ch/popgen/softwares/fstat.htm |
| Genealex (121) † |
HWE, Ae, Fst, Rst, Gst, G’st, Jost’s D, Nm and AMOVA |
Windows Mac |
2016 | Peakall and Smouse |
http://biology-assets.anu.edu.au/GenAIEx/ |
| Genepop (122) |
HWE, Nm, Fst* |
Online Windows Mac Linux Unix |
2017 | Raymond and Rousset |
http://genepop.curtin.edu.au/ |
| Genetix | HWE, Fst, Gst | Windows | 2004 | K Belkhir., etal. | http://kimura.univmontp2.fr/genetix/ |
| MLNE (126) † |
Ne, m, Nm | Windows | 2003 | Wang | https://www.zsl.org/science/software/mine |
| mmod (129) |
Dest, G”ST and φ’ST |
R package | 2017 | Winter et al. | https://github.com/dwinter/mmod |
| Pegas (130) |
HWE, Fst, AMOVA |
R package | 2017 | Paradis | https://cran.r-project.org/web/packages/pegas/index.html |
| Popgen (131) |
HWE, Ar | R package | 2016 | Kamvar | http://popgen.nescent.org/ |
| PopGen Report (128) |
HWE, summary statistics, Dest, G”st and φ’st |
R package | 2015 | Adamack and Gruber |
http://www.popgenreport.org/ |
| Poptree2 (113) |
GST, standardized GST, and Jost’s D |
Online, Windows, DOS, UNIX |
2014 | N. Takezaki, M. Nei, and K. Tamura |
http://www.med.kagawa-u.ac.jp/~genomelb/takezaki/poptree2/index.html |
| SpadeR† | Jost’s D | Online R package |
2015 | Chao | http://chao.stat.nthu.edu.tw/wordpress/ |
| SPAGeDi (125) |
Fst, Gst, Rst | Windows Mac Linux |
2015 | Hardy and Vekemans |
http://ebe.ulb.ac.be/ebe/SPAGeDi.html |
| Structure (96) |
Clustering | Windows Mac |
2012 | Pritchard et al. | http://web.stanford.edu/group/pritchardlab/structure.html |
| TFPGA | HWE, Fst, genetic distance |
Windows | 2000 | Miller | http://www.marksgeneticsoftware.net/tfpga.htm |
Most programs perform additional analyses, but the emphasis here is for measures of differentiation and diversity discussed in this part. All programs accept discrete genotypes, except DnaSP. Some (†) accept allele frequency data and others (‡) raw DNA sequences. Programs producing estimates of Fst using Weir and Cockerham (121) are indicated by (*). Additional lists of programs can be found in Excoffier et al. (33) or at websites:
- Population genetics in R (http://grunwaldlab.github.io/Population_Genetics_in_R/)
- University of Washington (https://courses.washington.edu/popgen/Software.htm)
- Softlinks (http://softlinks.amnh.org/popgen.html)
POPULATION DIFFERENTIATION INDICES
In addition to the Hardy-Weinberg equilibrium, populations can be further differentiated by other statistical tests. These are based on comparing population heterozygosity (Fst, Gst, G’st) or the effective number of alleles (Jost’s D).
Fst, Gst, G’st: In the 1950’s, Sewell Wright and Gustave Malécot developed a family of indices (the fixation indices) one of which (the Fst) describes the likelihood of homozygosity (fixation) at a single diallelic locus based on heterozygosity of a subpopulation (s) compared to the total population (t). Theoretically, values should range from 0 (identity—every individual is genotypically the same) to1 (no similarity—every individual is genotypically different). Nei extended the Fst to handle the case of more than two alleles and developed the Gst (84). Although the term “Fst” is often used in the literature, formally most studies today perform the Gst. When highly polymorphic loci, such as microsatellites, are genotyped, the Gst can severely underestimate differentiation and will not range from 0 – 1. Hedrick adjusted the range of values for the Gst by dividing it by its maximum possible value given the markers used and forcing the range back to 0 – 1 (49). This is the G’st. The G’st makes possible the full numerical range of differentiation. Fst and Gst relate to inherent properties of populations, contain evolutionary information, and approximate how close populations are to fixation (59). Fst-like measures have been and continue to be widely used to describe population structure, and their characteristics and behavior are well known. There are additional related indices (e. g. φst (34), AMOVA, Rst (106), θ (121)) that address other aspects of differing genetic models, unequal sample sizes, accounting for haploid genomes, mechanisms of mutation and selection.
Jost’s D. This is sometimes referred to as simply D, but since there are numerous other D’s related to genetics and statistics Jost’s D (after its creator, Lou Jost (58)) will be used here. There can be logical inconsistencies for estimating differentiation based directly on heterozygosity. Ratios of pooled subpopulations to total population diversities tend toward zero when the subpopulation diversity is high (58). Unlike those based directly on heterozygosity, Jost’s D is based on the effective allele number (see below). It has the property of yielding a linear response to changes in allele frequencies and is independent of subpopulation diversity. Unlike Fst, Gst and similar indices, Jost’s D does not carry information relevant to the evolutionary processes responsible for the present composition of a subpopulation. It is described by supporters and detractors as only a measure of differentiation (122). It was never meant to do more.
Whitlock provides one of the best comparisons of these 2 approaches:
“This (Jost’s) D differs from Fst in a fundamental definitional way: Fst measures deviations from panmixia, while D measures deviations from total differentiation. As a result, their denominators differ, and thus, the two indices can behave quite differently. D indicates the proportion of allelic diversity that lies among populations, while Fst is proportional to the variance of allele frequency among populations. D is more related to the genetic distance between populations than to the variance in allele frequencies; it may be preferable to call D a genetic distance measure
(122).”
There has been controversy about the use of these different types of indices. There should not be. They address different but related questions and resolve different analytic problems. It should be recognized that the G’st and Jost’s D yield similar results when the number of populations is small and the markers have a small number of alleles. The G’st and Jost’s D have given similar results in our own studies using microsatellites (16) and in simulation (122) with G’st giving values slightly higher than those for Jost’s D. Some authors recommend calculating both Gst and Jost’s D, in part to satisfy everyone and in part to obtain the useful information about population diversity their departure may provide.
In relation to public health, most questions about parasites and vectors deal with near term events of 2–20 generations, rather than events occurring on an evolutionary scale. The result we often seek is simply to know how different the allelic composition of one group is compared to another. In addition, the population or subpopulation structures may be such that there cannot be strong assumptions of HWE. The use of other markers, such as SNPs and sequencing may require other approaches such as principal component or factor analysis (41). Apart from differentiation, individuals can be clustered into populations by model-based, structured association methods used in programs such as Structure (96) and Admixture (6). These programs use approximation to HWE between populations as part of their clustering rule. They do provide a value for Fst to quantify the differences between the clusters formed.
DIVERSITY
Diversity like differentiation has myriad formulations and interpretations. The simplest expression is mean heterozygosity (H). For microsatellite data, this is usually high due to the intrinsically high mutation rate of these markers, and markers with higher variability are usually selected. Allelic richness (Ar) is simply a count of the number of alleles at each locus. Differences in sample size and marker choice will necessarily result in differences in allele number. This can be adjusted for by statistical methods such as rarefaction (66) to standardize sample sizes between comparison groups. The effective allele number (Ae) is also a measure of diversity and needs no adjustment for sample size. The maximum heterozygosity under HWE occurs when the frequency of each of the alleles at a locus are equal. The Ae represents the number of alleles with equal frequencies in an ideal population that will produce the same heterozygosity as that of the actual population.
The most informative measure of diversity is the effective population size (Ne), a concept also introduced by Sewell Wright. It addresses the essential reason that diversity is important, namely, it reflects the strength of genetic drift. Genetic drift is the effect of random transmission of alleles during reproduction to succeeding generations. When numbers of reproducing individuals are small, the genetic composition of the population of offspring can differ, by chance, from what is expected given the composition of the parents. For examples, if two coins are flipped, it would not be that unusual for both to come up heads. If a thousand coins are flipped, the ratio of heads to tails will always be very near the expected 50:50 ratio. Thus, genetic drift is like what happens when few coins are flipped. Chance variation from the expected distribution is stronger when populations are small or reduced.
Ne can represent the number of actively breeding individuals in the population (inbreeding Ne) or the number of individuals in an ideal population needed to reconstitute the diversity in an actual population (variance Ne). It is almost always less than the census population (Nc). It is a key value in conservation genetics and population genetics in general, since it reflects the history and potential of a population. Increasing drift (decreasing Ne) tends to neutralize the force of directional selection, permits retention of deleterious mutations and hampers the ability of populations to adapt to stresses (47). Like differentiation, there are several approaches to Ne that can produce different values and are designed to measure different aspects of diversity.
Despite its importance, Ne can be difficult to estimate in wild populations due to uncertainties of the demographic, genetic ,and biological context (92, 120). It can be affected by sample size, overlapping generations, sampling interval, sex ratios, gene flow, age-structure, variation in family size, fluctuating population size or selection. Increasing the numbers of markers is less important than large samples for accurate estimates of Ne. Sampling the number of individuals that represent as much as 10% of the value of Ne has been recommended (92). Its interpretation can also be uncertain. Estimated Ne has been used as an aid in predicting extinction using the concept of a minimum viable population size. Some have suggested that a population at an Ne of 50–500 will experience extinction in the short- or long-run, respectively (40). Others have argued that this might occur at Ne = 5000 (64). While it is clear that lack of diversity has an impact on extinction (110), it is also clear that there cannot be a single number or range for the minimum viable population size for all populations (39, 116). In any case, theory suggests that there is a number defined by the amount of genetic drift below which populations are likely to go extinct on their own. Determination of the range for this number will be context-specific and will require multiple species-specific studies under multiple conditions. This kind of analysis might contribute to developing a stopping rule as control measures approach elimination.
APPLICATION OF MOLECULAR TECHNIQUES TO THE EPIDEMIOLOGY OF EUKARYOTIC PATHOGENS
As we emphasize throughout reviews in this series, the molecular method employed to assess the epidemiology of a pathogen or vector depends on the question being asked and the biology of the organism’s DNA. Many protozoa employ asexual reproduction, and the tools applied to their study resemble the pattern recognition approaches used for bacteria: DNA fingerprinting, rep-PCR, ERIC, spoligotyping etc. Although this part has emphasized population genetics, the study of malaria and other protozoa with similar life histories may present a special case relative to helminths. While malaria is a sexually reproducing organism in the mosquito, in humans it reproduces asexually, and its multiplicity of infection (MOI) is rarely more than 3 even when based on genomic sequence (10) and usually less than 2. Despite infection with multiple genotypes, yet these infections remain simple, so that several simple marker systems and analyses suffice for some questions. Helminths, by contrast, tend not to reproduce asexually in humans, and infections are acquired one or a few individuals at a time over years (111). This produces a rich mix of multilocus genotypes, a situation not easily addressed by analysis of banding patterns on a gel. To differentiate species, or individuals of the same species, or to identify resistance when the gene(s) responsible are known, requires few markers and a read-out for presence or absence. Depending on the question, however, larger numbers of polymorphic markers and standard population genetics approaches offer better differentiation, an understanding of the reasons for diversity and population histories, as much for malaria as for helminths.
A major role for molecular techniques in infection control programs is for detection. In many cases, PCR is more sensitive (68) than microscopy and permits differentiation of closely related species, such as the filarial worms (74), hookworm species (127) or Leishmania (14, 55). Beyond this, simple prevalence or incidence of infection alone may not reveal local differences in the stability of the parasite population or its response to control measures. An analysis of population genetics with adequate sampling better reflects the whole population, since the genetics represents sampling from a much wider range of individuals and over several generations. The pattern of transmission of DNA over generations in itself provides information about the relationship of multiple unseen members of the species. For example, reduction in population size may be accompanied by increasing LD where inbreeding and genetic drift result in reduced diversity (105). An increase in differentiation from one time period to the next may indicate a new introduction or an epidemic burst from a resident subpopulation. Similarity between geographic isolates (low Fst) also suggests a high degree of movement between regions, i.e. high gene flow. Interpretation will still depend on the context: presence of control measure, change in morbidity or prevalence, a change in vectors, new hosts etc. Each of these conditions may require a different public health response. Some examples of the application of molecular methods to address problems of parasite epidemiology are given in this section.
Molecular methods become essential for the assessment of population status when transmission is declining in order to improve detection, to focus attention on “hot spots”, to evaluate the impact of interventions and to scan for evidence of resistance. This section is a partial review of studies that apply molecular techniques to questions of public health and parasites. This section also has a heavy emphasis on malaria and some helminths. Schistosoma mansoni and Plasmodium vivax are addressed in greater detail in later reviews, but a few examples are included here. Given the importance of P. falciparum for global health, a large amount of attention has been focused on this pathogen.
Distinguishing recrudescence from re-infection and determining multiplicity of infection
For studies of treatment efficacy, new infections must be distinguished from persistence of the pre-treatment infection. For most P. falciparum studies, the highly polymorphic loci merozoite surface proteins 1 and 2 (msp1, msp2), and the glutamate-rich protein (glurp) have become standard tools for infection control (75, 108). They are specifically recommended by the WHO (36) to accompany trials of drug efficacy, and there is an extensive literature on their application for this purpose (2, 3, 22, 46, 52, 75, 80, 82, 83, 91, 101, 103, 118).
In addition to distinguishing persistence from reinfection, msp 1, msp 2 and glurp are commonly used to determine MOI. Prior to molecular analyses, human infections with malaria were often considered completely clonal. We now know that infection with multiple genotypes is common, albeit with a small number of individuals, so that MOI ranges from 1–3. MOI is used as an indicator of the intensity of transmission and diversity (4). While this set of markers clearly has utility and often performs the purpose for which it was designed, it has some significant technical and analytic limitations. To reduce cost, these markers are often analyzed on agarose gels. The joint amplification of these markers in multiplex assays, and the resolution available on agarose gels likely underestimates diversity compared to their detection by capillary electrophoresis (75). There is also inherent variability in agarose gel electrophoresis that needs to be taken into account when establishing criteria for band sizes (83, 101). Even under ideal conditions, they tend to underestimate MOI compared to sequencing approaches (10). As control programs achieve lower prevalence, the loss of diversity can further decrease the ability to differentiate relapse from new infections locally (91), while genetic differentiation between foci of infection increases (79). The small number of markers also does not permit a higher order analysis of population dynamics such as effective population size or differentiation.
Distinguishing epidemic from endemic disease occurrence
Reduce variability is a key characteristic of founder, bottlenecked and outbreak populations (107). With bacteria and some protozoa (9), an outbreak often means the presence of a clonal population. In an outbreak of P. falciparum in Cape Verde reported by Arez et al., the population was genetically homogeneous based on the msp 1, msp 2, glurp loci. Since the area was previously one of very low transmission, this suggested that the increased incidence was due to a single introduction and single initial transmission, given that the parasite needs to complete a sexual cycle for continued transmission. More commonly, for outbreaks with sexually reproducing organisms, diversity is low but not clonal and LD is high. These characteristics have previously identified a spike in P. vivax infections in the Amazon basin as an epidemic circulating on top of endemic transmission (12).
Diversity can also be used in other ways to interpret population structure. Where there is a low level of endemic circulation, there will be low diversity, fewer multiple infections, and high LD due to higher amounts of inbreeding. This was the expectation in a survey of 130 samples from 7 districts in Bangladesh, where malaria is considered hypoendemic based on the number of symptomatic cases. Instead, a relatively high MOI and high heterozygosity, more typical of hyperendemic regions of Africa, were observed and this is consistent with a much larger population than expected. This indicated to the authors that the current national strategy should now include asymptomatic cases, since they likely contributed to the overall size of the population and persistence (4).
Distinguishing pathovars vs commensal flora or saprophytes
Distinguishing pathogenic varieties (pathovars) of parasites from non-pathogenic ones is primarily a question of distinguishing species This task, therefore, employs marker systems that provide simple “present” or “absent” responses. A classic problem in the diagnosis of amebiasis (a complex of conditions including severe dysentery and liver abscess) is how to distinguish Entamoeba histolytica (the principal cause), from E. moshkovskii (an uncommon cause) and E. dispar (a commensal). They are all morphologically indistinguishable. Serologic tests have been developed, which are less sensitive than PCR for 16 S rDNA (68, 93). The diagnosis of large tapeworms is similarly difficult primarily due to a newly identified species Taeina asiatica. For many decades, dogma held that there were 2 species of tapeworms infecting humans in the genus Taenia. Differentiation was important, since the species using the pig as an intermediate host (T. solium) produced an invasive intermediate form causing serious neurologic disease in humans (cysticercosis), while the species developing in cattle (T. saginata) did not. These species could be distinguished morphologically. In the early 1990’s, however, a parasite with the morphological features of T. saginata and infecting pigs was found to be invasive for humans (30). This new species was designated T. asiatica. Sequences within the valine tRNA and NADH genes were used to design a species-specific PCR capable of distinguishing these 3 important species. The complete sequence of the mitochondrial genome (56, 57) provided a level of resolution that allowed in-depth analysis of the population status and dynamics of this new pathogen. These studies also showed the close phylogenetic similarity of T. asiatica to T. saginata (18) as well as evidence of hybridization, which, unhappily, may indicate a path for rescuing characteristics of the pathogenic species from extinction (5).
Ascarid nematode worms have the ability to infect a variety of vertebrates, but it is uncertain whether worms from different hosts represent different species. When the pig pathogen, Ascaris suum, infects humans, the larvae can be invasive and may migrate aberrantly to viscera, including to the central nervous system. The morphologically identical A. lumbricoides is not invasive. The two can be differentiated by PCR amplification of the ribosomal internal transcribed spacer and by using a commercial kit based on NADH dehydrogenase subunit 5, yet there is still controversy (24, 67, 94). Molecular studies have not been sufficient to resolve this question, since it has not been clear what is meant by “species” in this case and to what degree it matters.
In addition to distinguishing pathogens from commensals, molecular techniques have also been used to differentiate more pathogenic strains from lesser pathogens within the same species. Infection with Onchocerca volvulus is a cause of blindness and a debilitating dermatitis, but blindness and other severe complications are more common with the savanna form than with stains found in the forest. In transition zones and in a landscape being shaped by human activity these geographic descriptions do not always correspond to the parasite strain being transmitted. Several investigators (31, 32, 74, 128) helped develop a system for differentiating species and strains of Onchocerca based on PCR amplification of a highly repeated 150 bp element followed by hybridization with a strain-specific probe (O-150). This marker system has been widely adopted by the WHO’s Onchocerciasis Control Program to estimate the potential for severe disease in multiple populations of the parasite (126). The use of a single marker will not carry implications for status of the population in and of itself, but combined with demographic data can indicate migration events (90) and inform the planning of control/elimination measures (44, 69, 119).
Identifying new hosts and modes of transmission
Many helminthic parasites distribute their development among different hosts, and the epidemiology of the infection is greatly influenced by the biology of these hosts. Further, the host range of a given parasite is not always clear. In several recent cases, the zoonotic contribution to human disease could only be appreciated by molecular techniques. Morphologically similar parasites may infect human and/or animal hosts without belonging to the same species (15). In other cases, there may be new or unappreciated hosts, and these must be verified using molecular techniques. The elimination of Guinea worm suffered a recent set back when it became apparent that dogs were previously unappreciated or had become major hosts of the parasite (20, 28). Whole genome sequence verified the identity of the parasite in both humans and dogs as the same guinea worm species, Dracunculus medinensis. This necessitated a re-analysis of the elimination strategy. Similarly, sequence of Hymenolepis nana from humans and rats demonstrated that the same species was being transmitted and confirmed rodent involvement in the life cycle (71) with attendant implications for control measures.
In Denmark, human infection with ascarid worms is uncommon (1%), but is commonly found in Danish pigs (25%). By PCR-RFLP analysis, parasites from infected Danes clustered with the local pig parasites rather than human infections elsewhere in the world (85). Thus, the few observed human infections in Denmark were native A. suum and not imported cases of A. lumbricoides. A similar association was observed in the United Kingdom (13). The implication for public health is that in these countries, whether or not there are separate species, human infection results from a zoonosis, and control in the human population should target control in pigs.
Finally, S. japonicum, a trematode, unlike the other major human schistosomes is primarily zoonotic. The risk to humans tracks with the biology of the other infected species. Rudge et al. (96) observed local geographic clustering of S. japonicum base on microsatellite genotypes and Fst. They also clustered with parasites infecting the predominant local non-human host species. This suggested that rodents might represent a more important reservoir that initially thought in some areas. The highest gene flow was between humans and domestic ruminants as suggested by low Fst values and Bayesian clustering with the program Structure (96). Identification of the principal local hosts by a molecular approach contributes to planning local control measures.
Tracking geographic and temporal distribution of infectious disease agents
Panels of 23 or 24 SNP markers for P. falciparum (25, 95) have been derived from bacteria-like organelles, such as mitochondria and apicoplasts. These cellular structures have the advantage of not being subject to sexual recombination (112), and markers can be analyzed simply as present or absent analogous to supermarket barcode labels. DNA barcoding was originally developed as a taxonomic tool. The 23 SNP barcode panel was able to classify most isolates by continent of isolation with 92% predictive accuracy. It was able to determine that the origin of a mystery isolate that infected a Dutch resident living near Amsterdam’s international airport originated to the continent of Africa, but was unable to determine if the source was from East or West Africa. A limitation of such markers is their resolution, since the questions are often over a continental scale and resolution deteriorates with increasing diversity. A subsequent panel of 42 nuclear SNPs forms a barcode may have better resolution and is able to distinguish between populations from Brazil/French Guiana, Ethiopia and Sri Lanka (11).
Genomic sequencing, while currently the most costly approach, has the advantage of providing higher resolution. For P. vivax, most studies have used sequences from the mitochondrial genome (23, 43, 51, 54, 98). Paradoxically, while differentiating regional populations, mitochondrial sequences were less useful for differentiating continental origins (98). Mitochondrial genome sequence were critical for identifying the origin for the 1993 re-emergence of vivax malaria in South Korea as coming from a regional source (54) rather than a more distant importation.
In and of itself, geographic origin may not be a key epidemiologic factor, unless associated with a pathogen phenotype, a new invasion, or as a factor in evaluation or planning for a control program. The study of longitudinal microsatellite diversity of P. vivax populations has been able to resolve many questions about the nature of transmission (37). In the Amazon basin, for example, increased incidence during a three-year study was associated with diminished genetic diversity signaling a localized outbreak by a limited number of strains. As the incidence fell, diversity returned to pre-outbreak levels (12). By contrast, during a resurgence of vivax malaria in Greece (109) and Sri Lanka (45) high parasite diversity indicated contribution from multiple strains consistent with rapid expansion of the local population. In the context of declining prevalence, this suggested multiple introductions, rather than clonal expansion. Options for control and prevention differ for these 2 situations.
Understanding the distribution of resistance
Molecular tools for the study of the epidemiology of drug resistance rely primarily on identification of the gene or genes involved. The paradigm of “one drug, one resistance gene”, however, clearly does not always reflect reality for parasites any more than it does for bacteria. Anti-malarials is the category of most concern at present. For chloroquine and the combination sulfadoxine and pyrimethamine (Fansidar) there is a well-defined and limited number of genes responsible. By contrast, artemisinin resistance is the result of a summation of several mutations in multiple genes with key variants tipping the balance toward resistance (72, 77). These patterns correspond to what have been termed, respectively, hard selective sweeps, where resistance arises on one or a few genes, and soft selective sweeps, which are the result of the summation and concentration of multiple pre-existing characteristics to result in a resistant phenotype. Even for what appears to be hard selective sweeps, multiple mutations are required in 2 genes for resistance to chloroquine (pfcrt and pfmdr1) (124) and sulfadoxine/pyrimethamine (pfdhps and pfdhfr) (reviewed in (123)). The epidemiology of antimalarials, then, depends on the genetic mechanism for the development of resistance. Mutations within the kelch motif protein 13 gene serve as markers for artemisinin resistance, at least in Asia (72). An approach for detection of resistance without identification of a responsible gene are discussed in a later review.
Since 1996, the WHO has recommended national surveillance for antimalarial resistance every 2 years. Initially the advocated methodology was purely based on monitoring response to treatment (125), but in 2009 standard PCR was added for a more sensitive measure of infection and to distinguish persistence from reinfection. There is as yet no standard recommendation for surveillance based purely on identification of resistance genes. A surveillance program for malaria and resistance genes revealed that subclinical infections were more frequent than symptomatic cases (OR 3.7) and served as a likely reservoir for chloroquine resistance genes pfcrt and pfmdr1 (19). The existence and importance of this reservoir could only be uncovered by a molecular method. Molecular surveillance of specific polymorphisms was also used to demonstrate high degrees of susceptibility (61, 70, 73, 87), sustained resistance (60) or high risk for resistance (89) in specific countries. In each case, ongoing molecular surveillance is suggested as an early warning mechanism for the arrival or development of resistance. An excellent resource for the distribution of artemisinin, chloroquine and sulfadoxine/pyrimethamine resistance mutants is the Worldwide Antimalarial Resistance Network website (http://www.wwarn.org). Detailed and updated interactive maps and tables are provided. Similar approaches are taken using equivalent P. vivax genes (76).
A limited number of markers is sufficient to track resistance within a country and across the globe when the gene(s) is known. Some of these studies allow assessment for resistance arising within a population, such as the assessment of mutations contributing to artemisinin resistance in migrants in Myanmar (89) or geographic clustering of sulfadoxine/pyrimethamine resistance in Republic of Congo (115). Another dimension to the potential for the spread of resistance is the degree of gene flow between populations. Since resistance usually arises in one or a few areas, the mobility of parasite genotypes in general will predict the rapidity and extent of spread of a resistant phenotype. Based on using multiple microsatellite markers, low differentiation indices (Fst ~0.02) predict the ready dissemination of any new resistance characteristic that arrives in areas of Mali (88) or the Sudan (1). In a similar way, measures of gene flow are useful for planning the release of modified mosquitoes in vector control programs (65).
Anderson et al. used SNPs to provide a new much higher estimate of Ne to study the development of artemisinin resistance in Southeast Asia (7). They were able to estimate that mutations in the most important resistance locus (kelch13) arose hundreds of times, implying that the drugs used in combination with artemisinin were likely to be only partially effective. This lent support to the use of 3 effective drugs for treatment.
Few resistance genes against antihelminthics have been identified, but the number of drugs in common use is small to begin with.. Recently, a gene for resistance to the drug oxamniquine was found (117), but this drug is no longer commonly used. The target of albendazole, a key drug for nearly all nematode infections, is β-tubulin. In veterinary practice, resistance is becoming common, since it relies on point mutations within this gene. Surveillance for some of these mutations in human parasites showed them to be high following treatment (27). Decreased efficacy, however, was observed only in Trichuris trichuria and not in hookworm or Ascaris lumbricoides, where the relevant mutants were few.
Planning control strategies
The very structure of populations is a useful guide for planning control measures. The ability of parasites to migrate into a control area can be gauged by looking at differentiation as an indication of gene flow. In the Comoros archipelago, gene flow for P. falciparum was high between all 4 islands (Fst <0.05), except the most southern of them (>0.10). This was still not considered sufficient degree of isolation to prevent rapid repopulation. The authors felt that the best strategy would be for control measures initiated simultaneously on all islands to avoid rapid repopulation (97). Similar degrees of gene flow with similar recommendations was observed on islands in Lake Victoria (81). A survey across West Africa likewise demonstrated extensive gene flow (most Fst values <0.03) and for that reason the authors were pessimistic about the development of a fragmented landscape that would facilitate elimination without very extensive fall in prevalence (78). The distribution of P. falciparum in Malaysia stands in contrast to West Africa. Here endemicity has been declining for some time, and there are high levels of differentiation between sites and elevated LD suggesting elevated inbreeding (8, 79). While the fragmentation suggests an island-by-island approach may be successful for malaria control, the increased LD and even clonal expansion in some areas may make monitoring difficult, since it will become more difficult to distinguish recrudescence from reinfection where individuals are highly related.
Surveillance and monitoring response to intervention
Molecular techniques contribute to the measurement of the success of control programs by increasing the sensitivity of detection and assessing the impact on target populations. In addition to morbidity reduction, the aim of many programs today is to eliminate transmission, which requires increasingly more sensitive methods of detection. Lamberton et al. (63) assessed the impact of ivermectin treatment on transmission of onchocerciasis in Ghana where treatment and control programs have been ongoing for 3–24 years, depending on locale. Using species-specific ribosomal markers and the O-150 marker on captured black flies, they observed a negative correlation between the prevalence of O. volvulus in the vector and duration of the treatment program, yet there was still vector positivity after 24 years of control efforts. This meant treatment with ivermectin alone was unlikely lead to elimination. This emphasized the need for new approaches with triple combination therapy, which is showing remarkable ability to maintain long-term elimination in humans (38) and is now recommended by the WHO.
Senegal intensified its malaria control efforts starting in 2006 through their National Malaria Control Program (NMCP), with apparent marked reduction in transmission. The long-term effect on the population could be tracked through analysis of 24 microsatellite markers (25, 95) as well as complete sequencing of a sample of parasites. SNPs developed from the sequences were optimized into sets of barcodes, and the extent of similarity and identity were used to model rates of reproduction. The pattern of inbreeding and extensive regions of LD mirrored the significant reduction from 2006–2010. Re-expansion of unique barcodes in 2013–2014 were consistent with the rebound observed in clinical incidence data (26).
In a similar fashion, Ingasia in Kenya surveyed P. falciparum populations from the 2 well-defined ecological zones of lowland and highland transmission as well as Eastern versus Western Kenya (53). Of particular importance, this group wanted to assess the effect on the population of the introduction of artemether–lumefantrine. The study employed 12 microsatellite loci genotyped in >200 isolates from 5 sites. In Eastern Kenya, where the infection has been in decline due to intensive treatment and use of bed nets, parasite diversity was low. Otherwise, there was high diversity elsewhere in the country suggesting high effective population size and little effect. In the highlands there was high within population LD, but overall differentiation between sites was moderate (Fst 0.072). In Kenya, where gene flow is high, the prevalence of antimalarial resistance genes was comparable across geographic and ecological regions as predicted from population structure (17).
Not all efforts at malaria control have been associated with evidence of a genetic impact on the parasite population. Another study on falciparum malaria in Kenya employed 8 microsatellite loci to assess parasite genetic structure over a 10 year period following the introduction of insecticide treated bed nets (42). The expectation was for a decrease in diversity, but no significant changes in multiplicity of infection, heterozygosity or LD. Differentiation between the populations over this period remained low at an average Fst of 0.04. The high diversity and low differentiation suggested that high gene flow helped maintain the population in this area. Haiti was another country where stepped up efforts to control and eliminate malaria occurred especially following the 2010 earthquake. Microsatellite analysis with 12 neutral loci indicated high gene flow, high diversity and little evidence for a genetic bottleneck (21). The approach to control needed to be modified.
Finally, Malawi instituted a multipronged control effort directed at vector control with indoor residual spraying, widespread use of bed nets, rapid diagnosis and treatment. Parasites were sampled 2 years before and twice during a 5-year period following implementation of these measures. Many of the methods mentioned in this review were applied to evaluate the impact of these efforts. No changes were noted for MOI, heterozygosity, genotypic richness, multilocus LD and Ne. There was some modest reduction in heterozygosity for multiple-genotype infections (104), but the general conclusion was that these efforts were insufficient to effect significant change in the overall population and that other approaches should be incorporated. It was also possible that the sample size gave insufficient pIower to detect changes in all of the parameters measured. The report of this study further acknowledged 2 other studies of malaria (45, 86) with similar poor responses when the starting prevalence was very high.
CONCLUDING REMARKS
Some problems in the study of molecular epidemiology of eukaryotic pathogens can be addressed with a few markers and categorical interpretations for their presence or absence (e.g. MOI, savannah vs forest forms, pathovar vs non-pathovar). Markers for antimicrobial resistance sometimes fall into this category, but as with bacteria, we are finding resistance can be complex and multigenic traits in parasites. The application of population genetics to the study of parasite epidemiology is more complex, but promises enormous rewards in bringing to light processes at work within the pathogen community. Every control program whose aim is elimination should apply this kind of analysis to assess success and understand failure. Otherwise, these programs operate blindly with respect to the parasite’s perspective on how the outcome was achieved.
A population genetic analysis is not a perfect crystal ball, but contributes important clues about the continued risk of transmission, the quality of the intervention (coverage), as well as with what resources the parasites as a group responded (drug resistance, immigration, reservoirs, force of infection, persistence) and consequently, how best to approach control or elimination.
KEY POINTS.
Asexual reproduction usually produces a clone; sexual reproduction never does.
A population is a group of organisms that tends to breed together
Allele and genotype frequencies describe populations
Allelic/genotypic frequencies and traits tend to be maintained within groups of interbreeding organisms (derived from the Hardy-Weinberg equilibrium)
Gross changes in allelic/genotypic frequencies are due to a limited set of events
Allele and genotype frequencies can be used to infer population histories
Indices and statistics can be used to compare & assess population history and to project population dynamics
Terms and Abbreviations
- Ae (effective allele number)
A measure of diversity. It is related to heterozygosity by Ae = 1/(1-Hexp)
- Allele
Points of variation at homologous loci
- Ar (Allelic richness)
A measure of diversity. The total number of different alleles in a population
- Diallelic
having 2 alleles (types of nucleotide variation) at the given locus
- Diploid
The status of organisms or cells that have 2 homologous chromosome
- Crossing over
During gametogenesis in sexually reproducing organisms, homologous segments of different chromosomes are exchanged
- Gene flow
The geographic movement of genes due to migration and reproduction of organisms
- Genetic drift
Gamete and zygote formation in sexual reproduction are probabilistic, so expected combinations appear at expected frequencies when the numbers are large. When the numbers are small unusual combinations occur. Populations with small numbers will develop unexpected allelic and genotypic frequencies over time. For this reason infinite population size is one of the criteria for the HWE
- Haploid
The status of organisms or cells with 1 copy of each
- Heterozygosity
The percent of a population that is heterozygous at a given locus or the average percent over all loci. This is often taken to be one measure of diversity, but is dependent on the kind of marker used. Microsatellites often have high heterozygosity (50–80%), while SNP heterozygosity is generally less than 50%
- Homozygote/Heterozygote
For diploid organisms, homozygotes have no variation between DNA sequence or structure at homologous loci, while heterozygotes differ
- HWE (Hardy-Weinberg Equilibrium)
A description of the relationship of allele frequency to genotypic frequency in an ideal population. The null hypothesis in population genetics
- Linkage
The degree to which loci within the genome are inherited randomly due to their proximity. Loci that are close to each other tend to be transmitted together to the next generation
- LD (Linkage disequilibrium)
The observation of the joint occurrence of alleles at 2 or more loci more often than expected given their frequency. The concept refers to families and populations and involves demographic and evolutionary processes more than the physical geography of loci in the genome (reviewed in (105)). The expectation under HWE is that there will be no association between alleles on different loci
- Locus
An identified region of a chromosome
- Ne (effective population size)
A measure of diversity. The sensitivity of a population to genetic drift, the number of breeding pairs in a population or the minimum number of individuals in an ideal population required to reconstitute the diversity in a real population
- MOI
multiplicity of infection
- Panmixia
The state where all individuals have an equal chance of reproducing with all others
- Pathovars
Pathogenic varieties
- Reassortment
The mixing of genetic material in the process of reproduction. In sexually reproducing organisms this includes the random assignment of chromosomes to the haploid sex cells (gametes) as well as the process of exchanging homologous sections of chromosomes (see crossing-over). In viruses, such as influenza, segments from one viral strain or species may be mixed and segregated with those of a different strain
Footnotes
See Advances In this series, Molecular Epidemiology Of Infectious Diseases Part 2, for a discussion of the meaning of “clone.”
Leptospira spp,. Vibrio cholera and some others are exceptions with 2 circular chromosomes. Several species also possess a linear chromosome.
References
- 1.Abdel-Muhsin AA, Mackinnon MJ, Awadalla P, Ali E, Suleiman S, Ahmed S, Walliker D, and Babiker HA. 2003. Local differentiation in Plasmodium falciparum drug resistance genes in Sudan. Parasitology 126:391–400. [DOI] [PubMed] [Google Scholar]
- 2.Adjuik M, Agnamey P, Babiker A, Borrmann S, Brasseur P, Cisse M, Cobelens F, Diallo S, Faucher JF, Garner P, Gikunda S, Kremsner PG, Krishna S, Lell B, Loolpapit M, Matsiegui PB, Missinou MA, Mwanza J, Ntoumi F, Olliaro P, Osimbo P, Rezbach P, Some E, and Taylor WR. 2002. Amodiaquine-artesunate versus amodiaquine for uncomplicated Plasmodium falciparum malaria in African children: a randomised, multicentre trial. Lancet 359:1365–72. [DOI] [PubMed] [Google Scholar]
- 3.Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, and White N. 2004. Artesunate combinations for treatment of malaria: meta-analysis. Lancet 363:9–17. [DOI] [PubMed] [Google Scholar]
- 4.Alam MS, Elahi R, Mohon AN, Al-Amin HM, Kibria MG, Khan WA, Khanum H, and Haque R. 2016. Plasmodium falciparum Genetic Diversity in Bangladesh Does Not Suggest a Hypoendemic Population Structure. Am J Trop Med Hyg 94:1245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ale A, Victor B, Praet N, Gabriel S, Speybroeck N, Dorny P, and Devleesschauwer B. 2014. Epidemiology and genetic diversity of Taenia asiatica: a systematic review. Parasit Vectors 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexander DH, Novembre J, and Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson TJ, Nair S, McDew-White M, Cheeseman IH, Nkhoma S, Bilgic F, McGready R, Ashley E, Pyae Phyo A, White NJ, and Nosten F. 2016. Population Parameters Underlying an Ongoing Soft Sweep in Southeast Asian Malaria Parasites. Mol Biol Evol 34:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anthony TG, Conway DJ, Cox-Singh J, Matusop A, Ratnam S, Shamsul S, and Singh B. 2005. Fragmented population structure of Plasmodium falciparum in a region of declining endemicity. J Infect Dis 191:1558–64. [DOI] [PubMed] [Google Scholar]
- 9.Arez AP, Snounou G, Pinto J, Sousa CA, Modiano D, Ribeiro H, Franco AS, Alves J, and do Rosario VE. 1999. A clonal Plasmodium falciparum population in an isolated outbreak of malaria in the Republic of Cabo Verde. Parasitology 118 ( Pt 4):347–55. [DOI] [PubMed] [Google Scholar]
- 10.Assefa SA, Preston MD, Campino S, Ocholla H, Sutherland CJ, and Clark TG. 2014. estMOI: estimating multiplicity of infection using parasite deep sequencing data. Bioinformatics 30:1292–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baniecki ML, Faust AL, Schaffner SF, Park DJ, Galinsky K, Daniels RF, Hamilton E, Ferreira MU, Karunaweera ND, Serre D, Zimmerman PA, Sa JM, Wellems TE, Musset L, Legrand E, Melnikov A, Neafsey DE, Volkman SK, Wirth DF, and Sabeti PC. 2015. Development of a single nucleotide polymorphism barcode to genotype Plasmodium vivax infections. PLoS Negl Trop Dis 9:e0003539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batista CL, Barbosa S, Da Silva Bastos M, Viana SA, and Ferreira MU. 2015. Genetic diversity of Plasmodium vivax over time and space: a community-based study in rural Amazonia. Parasitology 142:374–84. [DOI] [PubMed] [Google Scholar]
- 13.Bendall RP, Barlow M, Betson M, Stothard JR, and Nejsum P. 2011. Zoonotic ascariasis, United Kingdom. Emerg Infect Dis 17:1964–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bensoussan E, Nasereddin A, Jonas F, Schnur LF, and Jaffe CL. 2006. Comparison of PCR assays for diagnosis of cutaneous leishmaniasis. J Clin Microbiol 44:1435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bimi L, Freeman AR, Eberhard ML, Ruiz-Tiben E, and Pieniazek NJ. 2005. Differentiating Dracunculus medinensis from D. insignis, by the sequence analysis of the 18S rRNA gene. Ann Trop Med Parasitol 99:511–7. [DOI] [PubMed] [Google Scholar]
- 16.Blank WA, Test MR, Liu SF, Lewis FA, and Blanton RE. 2010. Long-term genetic stability and population dynamics of laboratory strains of Schistosoma mansoni. J Parasitol 96:900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonizzoni M, Afrane Y, Baliraine FN, Amenya DA, Githeko AK, and Yan G. 2009. Genetic structure of Plasmodium falciparum populations between lowland and highland sites and antimalarial drug resistance in Western Kenya. Infect Genet Evol 9:806–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowles J, and McManus DP. 1994. Genetic characterization of the Asian Taenia, a newly described taeniid cestode of humans. Am J Trop Med Hyg 50:33–44. [PubMed] [Google Scholar]
- 19.Brown T, Smith LS, Oo EK, Shawng K, Lee TJ, Sullivan D, Beyrer C, and Richards AK. 2012. Molecular surveillance for drug-resistant Plasmodium falciparum in clinical and subclinical populations from three border regions of Burma/Myanmar: cross-sectional data and a systematic review of resistance studies. Malar J 11:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callaway E 2016. Dogs thwart effort to eradicate Guinea worm. Nature 529:10–1. [DOI] [PubMed] [Google Scholar]
- 21.Carter TE, Malloy H, Existe A, Memnon G, St Victor Y, Okech BA, and Mulligan CJ. 2015. Genetic Diversity of Plasmodium falciparum in Haiti: Insights from Microsatellite Markers. PLoS One 10:e0140416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cattamanchi A, Kyabayinze D, Hubbard A, Rosenthal PJ, and Dorsey G. 2003. Distinguishing recrudescence from reinfection in a longitudinal antimalarial drug efficacy study: comparison of results based on genotyping of msp-1, msp-2, and glurp. Am J Trop Med Hyg 68:133–9. [PubMed] [Google Scholar]
- 23.Culleton R, Coban C, Zeyrek FY, Cravo P, Kaneko A, Randrianarivelojosia M, Andrianaranjaka V, Kano S, Farnert A, Arez AP, Sharp PM, Carter R, and Tanabe K. 2012. The origins of African Plasmodium vivax; insights from mitochondrial genome sequencing. PLoS One 6:e29137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.da Silva Alves EB, Conceicao MJ, and Leles D. 2016. Ascaris lumbricoides, Ascaris suum, or “Ascaris lumbrisuum”? J Infect Dis 213:1355. [DOI] [PubMed] [Google Scholar]
- 25.Daniels R, Volkman SK, Milner DA, Mahesh N, Neafsey DE, Park DJ, Rosen D, Angelino E, Sabeti PC, Wirth DF, and Wiegand RC. 2008. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J 7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daniels RF, Schaffner SF, Wenger EA, Proctor JL, Chang HH, Wong W, Baro N, Ndiaye D, Fall FB, Ndiop M, Ba M, Milner DA Jr., Taylor TE, Neafsey DE, Volkman SK, Eckhoff PA, Hartl DL, and Wirth DF. 2015. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci U S A 112:7067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diawara A, Halpenny CM, Churcher TS, Mwandawiro C, Kihara J, Kaplan RM, Streit TG, Idaghdour Y, Scott ME, Basanez MG, and Prichard RK. 2013. Association between response to albendazole treatment and beta-tubulin genotype frequencies in soil-transmitted helminths. PLoS Negl Trop Dis 7:e2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eberhard ML, Ruiz-Tiben E, Hopkins DR, Farrell C, Toe F, Weiss A, Withers PC Jr., Jenks MH, Thiele EA, Cotton JA, Hance Z, Holroyd N, Cama VA, Tahir MA, and Mounda T. 2014. The peculiar epidemiology of dracunculiasis in Chad. Am J Trop Med Hyg 90:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellegren H 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–45. [DOI] [PubMed] [Google Scholar]
- 30.Eom KS, and Rim HJ. 1993. Morphologic descriptions of Taenia asiatica sp. n. Korean J Parasitol 31:1–6. [DOI] [PubMed] [Google Scholar]
- 31.Erttmann KD, Meredith SE, Greene BM, and Unnasch TR. 1990. Isolation and characterization of form specific DNA sequences of O. volvulus. Acta Leiden 59:253–60. [PubMed] [Google Scholar]
- 32.Erttmann KD, Unnasch TR, Greene BM, Albiez EJ, Boateng J, Denke AM, Ferraroni JJ, Karam M, Schulz-Key H, and Williams PN. 1987. A DNA sequence specific for forest form Onchocerca volvulus. Nature 327:415–7. [DOI] [PubMed] [Google Scholar]
- 33.Excoffier L, and Heckel G. 2006. Computer programs for population genetics data analysis: a survival guide. Nat Rev Genet 7:745–58. [DOI] [PubMed] [Google Scholar]
- 34.Excoffier L, Smouse PE, and Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falconer D 1960. Introduction to Quantitative Genetics Ronald Press Company, New York. [Google Scholar]
- 36.Felger I, and Snounou G. 2008. Recommended Genotyping Procedures (RGPs) to identify parasite populations World Health Organization. Medicines for Malaria Venture [Google Scholar]
- 37.Ferreira MU, and Rodrigues PT. 2014. Tracking malaria parasites in the eradication era. Trends Parasitol 30:465–6. [DOI] [PubMed] [Google Scholar]
- 38.Fischer PU, King CL, Jacobson JA, and Weil GJ. 2017. Potential Value of Triple Drug Therapy with Ivermectin, Diethylcarbamazine, and Albendazole (IDA) to Accelerate Elimination of Lymphatic Filariasis and Onchocerciasis in Africa. PLoS Negl Trop Dis 11:e0005163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flather CH, Hayward GD, Beissinger SR, and Stephens PA. 2011. Minimum viable populations: is there a ‘magic number’ for conservation practitioners? Trends Ecol Evol 26:307–16. [DOI] [PubMed] [Google Scholar]
- 40.Franklin I, and Frankham R. 1998. How large must populations be to retain evolutionary potential? Animal Conservation 1:69–73. [Google Scholar]
- 41.Fumagalli M, Vieira FG, Korneliussen TS, Linderoth T, Huerta-Sanchez E, Albrechtsen A, and Nielsen R. 2013. Quantifying population genetic differentiation from next-generation sequencing data. Genetics 195:979–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gatei W, Gimnig JE, Hawley W, Ter Kuile F, Odero C, Iriemenam NC, Shah MP, Howard PP, Omosun YO, Terlouw DJ, Nahlen B, Slutsker L, Hamel MJ, Kariuki S, Walker E, and Shi YP. 2015. Genetic diversity of Plasmodium falciparum parasite by microsatellite markers after scale-up of insecticide-treated bed nets in western Kenya. Malar J 13 Suppl 1:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelabert P, Sandoval-Velasco M, Olalde I, Fregel R, Rieux A, Escosa R, Aranda C, Paaijmans K, Mueller I, Gilbert MT, and Lalueza-Fox C. 2016. Mitochondrial DNA from the eradicated European Plasmodium vivax and P. falciparum from 70-year-old slides from the Ebro Delta in Spain. Proc Natl Acad Sci U S A 113:11495–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez RJ, Cruz-Ortiz N, Rizzo N, Richards J, Zea-Flores G, Dominguez A, Sauerbrey M, Catu E, Oliva O, Richards FO, and Lindblade KA. 2009. Successful interruption of transmission of Onchocerca volvulus in the Escuintla-Guatemala focus, Guatemala. PLoS Negl Trop Dis 3:e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gunawardena S, Ferreira MU, Kapilananda GM, Wirth DF, and Karunaweera ND. 2014. The Sri Lankan paradox: high genetic diversity in Plasmodium vivax populations despite decreasing levels of malaria transmission. Parasitology 141:880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Happi CT, Gbotosho GO, Sowunmi A, Falade CO, Akinboye DO, Gerena L, Kyle DE, Milhous W, Wirth DF, and Oduola AM. 2004. Molecular analysis of Plasmodium falciparum recrudescent malaria infections in children treated with chloroquine in Nigeria. Am J Trop Med Hyg 70:20–6. [PubMed] [Google Scholar]
- 47.Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, Waples RS, Ruegg K, and Palstra F. 2010. Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25:438–49. [DOI] [PubMed] [Google Scholar]
- 48.Hartl DL 2014. Essential Genetics, 6th ed. Jones & Bartlett Learning, Burlington, MA. [Google Scholar]
- 49.Hedrick PW 2005. A standardized genetic differentiation measure. Evolution 59:1633–8. [PubMed] [Google Scholar]
- 50.Hoeh WR, Blakley KH, and Brown WM. 1991. Heteroplasmy suggests limited biparental inheritance of Mytilus mitochondrial DNA. Science 251:1488–90. [DOI] [PubMed] [Google Scholar]
- 51.Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, Vallejo AF, Herrera S, Arevalo-Herrera M, Fan Q, Wang Y, Cui L, Lucas CM, Durand S, Sanchez JF, Baldeviano GC, Lescano AG, Laman M, Barnadas C, Barry A, Mueller I, Kazura JW, Eapen A, Kanagaraj D, Valecha N, Ferreira MU, Roobsoong W, Nguitragool W, Sattabonkot J, Gamboa D, Kosek M, Vinetz JM, Gonzalez-Ceron L, Birren BW, Neafsey DE, and Carlton JM. 2016. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet 48:953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imwong M, Jindakhad T, Kunasol C, Sutawong K, Vejakama P, and Dondorp AM. 2015. An outbreak of artemisinin resistant falciparum malaria in Eastern Thailand. Sci Rep 5:17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ingasia LA, Cheruiyot J, Okoth SA, Andagalu B, and Kamau E. 2016. Genetic variability and population structure of Plasmodium falciparum parasite populations from different malaria ecological regions of Kenya. Infect Genet Evol 39:372–80. [DOI] [PubMed] [Google Scholar]
- 54.Iwagami M, Hwang SY, Fukumoto M, Hayakawa T, Tanabe K, Kim SH, Kho WG, and Kano S. 2010. Geographical origin of Plasmodium vivax in the Republic of Korea: haplotype network analysis based on the parasite’s mitochondrial genome. Malar J 9:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jara M, Adaui V, Valencia BM, Martinez D, Alba M, Castrillon C, Cruz M, Cruz I, Van der Auwera G, Llanos-Cuentas A, Dujardin JC, and Arevalo J. 2013. Real-time PCR assay for detection and quantification of Leishmania (Viannia) organisms in skin and mucosal lesions: exploratory study of parasite load and clinical parameters. J Clin Microbiol 51:1826–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeon HK, Kim KH, and Eom KS. 2007. Complete sequence of the mitochondrial genome of Taenia saginata: comparison with T. solium and T. asiatica. Parasitol Int 56:243–6. [DOI] [PubMed] [Google Scholar]
- 57.Jeon HK, Lee KH, Kim KH, Hwang UW, and Eom KS. 2005. Complete sequence and structure of the mitochondrial genome of the human tapeworm, Taenia asiatica (Platyhelminthes; Cestoda). Parasitology 130:717–26. [DOI] [PubMed] [Google Scholar]
- 58.Jost L 2008. GST and its relatives do not measure differentiation. Molecular Ecology 17:4015–4026. [DOI] [PubMed] [Google Scholar]
- 59.Jost L, Archer F, Flanagan S, Gaggiotti O, Hoban S, and Latch E. 2018. Differentiation measures for conservation genetics. Evol Appl:preprint online [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kateera F, Nsobya SL, Tukwasibwe S, Hakizimana E, Mutesa L, Mens PF, Grobusch MP, van Vugt M, and Kumar N. 2016. Molecular surveillance of Plasmodium falciparum drug resistance markers reveals partial recovery of chloroquine susceptibility but sustained sulfadoxine-pyrimethamine resistance at two sites of different malaria transmission intensities in Rwanda. Acta Trop 164:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koukouikila-Koussounda F, Jeyaraj S, Nguetse CN, Nkonganyi CN, Kokou KC, Etoka-Beka MK, Ntoumi F, and Velavan TP. 2017. Molecular surveillance of Plasmodium falciparum drug resistance in the Republic of Congo: four and nine years after the introduction of artemisinin-based combination therapy. Malar J 16:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krause DJ, and Whitaker RJ. 2015. Inferring speciation processes from patterns of natural variation in microbial genomes. Syst Biol 64:926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamberton PH, Cheke RA, Winskill P, Tirados I, Walker M, Osei-Atweneboana MY, Biritwum NK, Tetteh-Kumah A, Boakye DA, Wilson MD, Post RJ, and Basanez MG. 2015. Onchocerciasis transmission in Ghana: persistence under different control strategies and the role of the simuliid vectors. PLoS Negl Trop Dis 9:e0003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lande R 1995. Mutation and conservation. Conserv. Biol 9:782–791. [Google Scholar]
- 65.Lanzaro G, and Tripet F. 2003. Gene flow among populations of Anopheles gambiae: A critical review, p. 109–132. In Takken W and TW S (ed.), Ecological Aspects for the Application of Genetically Modified Mosquitoes Frontis Press, Wageningen, the Netherlands. [Google Scholar]
- 66.Leberg PL 2002. Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11:2445–9. [DOI] [PubMed] [Google Scholar]
- 67.Leles D, Gardner SL, Reinhard K, Iniguez A, and Araujo A. 2012. Are Ascaris lumbricoides and Ascaris suum a single species? Parasit Vectors 5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Llewellyn S, Inpankaew T, Nery SV, Gray DJ, Verweij JJ, Clements AC, Gomes SJ, Traub R, and McCarthy JS. 2016. Application of a Multiplex Quantitative PCR to Assess Prevalence and Intensity Of Intestinal Parasite Infections in a Controlled Clinical Trial. PLoS Negl Trop Dis 10:e0004380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lloyd MM, Gilbert R, Taha NT, Weil GJ, Meite A, Kouakou IM, and Fischer PU. 2015. Conventional parasitology and DNA-based diagnostic methods for onchocerciasis elimination programmes. Acta Trop 146:114–8. [DOI] [PubMed] [Google Scholar]
- 70.Lucchi N, Komino F, Okoth S, Goldman I, Onyona P, Wiegand R, Juma E, Shi Y, Barnwell J, Udhayakumar V, and Kariuki S. 2015. In vitro and molecular surveillance for antimalarial drug resistance in Plasmodium falciparum parasites in Western Kenya reveals sustained artemisinin sensitivity and increased chloroquine sensitivity. Antimicrob Agents Chemother 59:7540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Macnish MG, Morgan-Ryan UM, Monis PT, Behnke JM, and Thompson RC. 2002. A molecular phylogeny of nuclear and mitochondrial sequences in Hymenolepis nana (Cestoda) supports the existence of a cryptic species. Parasitology 125:567–75. [DOI] [PubMed] [Google Scholar]
- 72.MalariaGEN Plasmodium falciparum Community Project. 2016. Genomic epidemiology of artemisinin resistant malaria. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menegon M, Nurahmed AM, Talha AA, Nour BY, and Severini C. 2016. Molecular surveillance of antimalarial drug resistance related genes in Plasmodium falciparum isolates from Eritrea. Acta Trop 157:158–61. [DOI] [PubMed] [Google Scholar]
- 74.Meredith SE, Lando G, Gbakima AA, Zimmerman PA, and Unnasch TR. 1991. Onchocerca volvulus: application of the polymerase chain reaction to identification and strain differentiation of the parasite. Exp Parasitol 73:335–44. [DOI] [PubMed] [Google Scholar]
- 75.Messerli C, Hofmann NE, Beck H, and Felger I. 2016. Critical evaluation of molecular monitoring in malaria drug efficacy trials and pitfalls of length-polymorphic markers. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mint Lekweiry K, Ould A Boukhary Mohamed Salem, Gaillard T, Wurtz N, Bogreau H, Hafid JE, Trape JF, Bouchiba H, Ould Ahmedou Salem MS, Pradines B, Rogier C, Basco LK, and Briolant S. 2012. Molecular surveillance of drug-resistant Plasmodium vivax using pvdhfr, pvdhps and pvmdr1 markers in Nouakchott, Mauritania. J Antimicrob Chemother 67:367–74. [DOI] [PubMed] [Google Scholar]
- 77.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, and Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mobegi VA, Loua KM, Ahouidi AD, Satoguina J, Nwakanma DC, Amambua-Ngwa A, and Conway DJ. 2012. Population genetic structure of Plasmodium falciparum across a region of diverse endemicity in West Africa. Malar J 11:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mohd Abd Razak MR, Sastu UR, Norahmad NA, Abdul-Karim A, Muhammad A, Muniandy PK, Jelip J, Rundi C, Imwong M, Mudin RN, and Abdullah NR. 2016. Genetic Diversity of Plasmodium falciparum Populations in Malaria Declining Areas of Sabah, East Malaysia. PLoS One 11:e0152415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mugittu K, Adjuik M, Snounou G, Ntoumi F, Taylor W, Mshinda H, Olliaro P, and Beck HP. 2006. Molecular genotyping to distinguish between recrudescents and new infections in treatment trials of Plasmodium falciparum malaria conducted in Sub-Saharan Africa: adjustment of parasitological outcomes and assessment of genotyping effectiveness. Trop Med Int Health 11:1350–9. [DOI] [PubMed] [Google Scholar]
- 81.Mulenge FM, Hunja CW, Magiri E, Culleton R, Kaneko A, and Aman RA. 2016. Genetic diversity and population structure of Plasmodium falciparum in Lake Victoria Islands, a region of intense transmission. Am J Trop Med Hyg 95:1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mutabingwa TK, Anthony D, Heller A, Hallett R, Ahmed J, Drakeley C, Greenwood BM, and Whitty CJ. 2005. Amodiaquine alone, amodiaquine+sulfadoxinepyrimethamine, amodiaquine+artesunate, and artemether-lumefantrine for outpatient treatment of malaria in Tanzanian children: a four-arm randomised effectiveness trial. Lancet 365:1474–80. [DOI] [PubMed] [Google Scholar]
- 83.Mwingira F, Nkwengulila G, Schoepflin S, Sumari D, Beck HP, Snounou G, Felger I, Olliaro P, and Mugittu K. 2011. Plasmodium falciparum msp1, msp2 and glurp allele frequency and diversity in sub-Saharan Africa. Malar J 10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nei M 1973. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nejsum P, Parker ED Jr., Frydenberg J, Roepstorff A, Boes J, Haque R, Astrup I, Prag J, and Skov Sorensen UB. 2005. Ascariasis is a zoonosis in Denmark. J Clin Microbiol 43:1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nkhoma SC, Nair S, Cheeseman IH, Rohr-Allegrini C, Singlam S, Nosten F, and Anderson TJ. 2012. Close kinship within multiple-genotype malaria parasite infections. Proc Biol Sci 279:2589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Norahmad NA, Mohd Abd Razak MR, Abdullah NR, Sastu UR, Imwong M, Muniandy PK, Saat MN, Muhammad A, Jelip J, Tikuson M, Yusof N, Rundi C, Mudin RN, and Syed Mohamed AF. 2016. Prevalence of Plasmodium falciparum molecular markers of antimalarial drug resistance in a residual malaria focus area in Sabah, Malaysia. PLoS One 11:e0165515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nsimba B, Jafari-Guemouri S, Malonga DA, Mouata AM, Kiori J, Louya F, Yocka D, Malanda M, Durand R, and Le Bras J. 2005. Epidemiology of drug-resistant malaria in Republic of Congo: using molecular evidence for monitoring antimalarial drug resistance combined with assessment of antimalarial drug use. Trop Med Int Health 10:1030–7. [DOI] [PubMed] [Google Scholar]
- 89.Nyunt MH, Wang B, Aye KM, Aye KH, Han JH, Lee SK, Han KT, Htut Y, and Han ET. 2017. Molecular surveillance of artemisinin resistance falciparum malaria among migrant goldmine workers in Myanmar. Malar J 16:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ogunrinade A, Boakye D, Merriweather A, and Unnasch TR. 1999. Distribution of the blinding and nonblinding strains of Onchocerca volvulus in Nigeria. J Infect Dis 179:1577–9. [DOI] [PubMed] [Google Scholar]
- 91.Oyebola MK, Idowu ET, Olukosi YA, Iwalokun BA, Agomo CO, Ajibaye OO, Tola M, and Otubanjo AO. 2014. Genetic diversity and complexity of Plasmodium falciparum infections in Lagos, Nigeria. Asian Pac J Trop Biomed 4:S87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Palstra FP, and Ruzzante DE. 2008. Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17:3428–47. [DOI] [PubMed] [Google Scholar]
- 93.Parija SC, Mandal J, and Ponnambath DK. 2014. Laboratory methods of identification of Entamoeba histolytica and its differentiation from look-alike Entamoeba spp. Trop Parasitol 4:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peng W, and Criscione CD. 2012. Ascariasis in people and pigs: new inferences from DNA analysis of worm populations. Infect Genet Evol 12:227–35. [DOI] [PubMed] [Google Scholar]
- 95.Preston MD, Campino S, Assefa SA, Echeverry DF, Ocholla H, Amambua-Ngwa A, Stewart LB, Conway DJ, Borrmann S, Michon P, Zongo I, Ouedraogo JB, Djimde AA, Doumbo OK, Nosten F, Pain A, Bousema T, Drakeley CJ, Fairhurst RM, Sutherland CJ, Roper C, and Clark TG. 2014. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun 5:4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pritchard JK, Stephens M, and Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rebaudet S, Bogreau H, Silai R, Lepere JF, Bertaux L, Pradines B, Delmont J, Gautret P, Parola P, and Rogier C. 2010. Genetic structure of Plasmodium falciparum and elimination of malaria, Comoros archipelago. Emerg Infect Dis 16:1686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rodrigues PT, Alves JM, Santamaria AM, Calzada JE, Xayavong M, Parise M, da Silva AJ, and Ferreira MU. 2014. Using mitochondrial genome sequences to track the origin of imported Plasmodium vivax infections diagnosed in the United States. Am J Trop Med Hyg 90:1102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosen MJ, Davison M, Bhaya D, and Fisher DS. 2015. Microbial diversity. Fine-scale diversity and extensive recombination in a quasisexual bacterial population occupying a broad niche. Science 348:1019–23. [DOI] [PubMed] [Google Scholar]
- 100.Rougeron V, De Meeus T, Kako Ouraga S, Hide M, and Banuls AL. 2010. “Everything you always wanted to know about sex (but were afraid to ask)” in Leishmania after two decades of laboratory and field analyses. PLoS Pathog 6:e1001004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rouse P, Mkulama MA, Thuma PE, and Mharakurwa S. 2008. Distinction of Plasmodium falciparum recrudescence and re-infection by MSP2 genotyping: a caution about unstandardized classification criteria. Malar J 7:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schindel DE, and Miller SE. 2005. DNA barcoding a useful tool for taxonomists. Nature 435:17. [DOI] [PubMed] [Google Scholar]
- 103.Sirima SB, Tiono AB, Gansane A, Diarra A, Ouedraogo A, Konate AT, Kiechel JR, Morgan CC, Olliaro PL, and Taylor WR. 2009. The efficacy and safety of a new fixed-dose combination of amodiaquine and artesunate in young African children with acute uncomplicated Plasmodium falciparum. Malar J 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sisya TJ, Kamn’gona RM, Vareta JA, Fulakeza JM, Mukaka MF, Seydel KB, Laufer MK, Taylor TE, and Nkhoma SC. 2015. Subtle changes in Plasmodium falciparum infection complexity following enhanced intervention in Malawi. Acta Trop 142:108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Slatkin M 2008. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Slatkin M 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smith JM, Smith NH, O’Rourke M, and Spratt BG. 1993. How clonal are bacteria? Proc Natl Acad Sci U S A 90:4384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Snounou G, and Beck HP. 1998. The use of PCR genotyping in the assessment of recrudescence or reinfection after antimalarial drug treatment. Parasitol Today 14:462–7. [DOI] [PubMed] [Google Scholar]
- 109.Spanakos G, Alifrangis M, Schousboe ML, Patsoula E, Tegos N, Hansson HH, Bygbjerg IC, Vakalis NC, Tseroni M, Kremastinou J, and Hadjichristodoulou C. 2013. Genotyping Plasmodium vivax isolates from the 2011 outbreak in Greece. Malar J 12:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spielman D, Brook BW, and Frankham R. 2004. Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci U S A 101:15261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stothard JR, Sousa-Figueiredo JC, Betson M, Adriko M, Arinaitwe M, Rowell C, Besiyge F, and Kabatereine NB. 2011. Schistosoma mansoni Infections in young children: when are schistosome antigens in urine, eggs in stool and antibodies to eggs first detectable? PLoS Negl Trop Dis 5:e938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Su XZ 2014. Tracing the geographic origins of Plasmodium falciparum malaria parasites. Pathog Glob Health 108:261–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takezaki N, Nei M, and Tamura K. 2014. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tautz D 1989. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res 17:6463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taylor SM, Antonia AL, Parobek CM, Juliano JJ, Janko M, Emch M, Alam MT, Udhayakumar V, Tshefu AK, and Meshnick SR. 2013. Plasmodium falciparum sulfadoxine resistance is geographically and genetically clustered within the DR Congo. Sci Rep 3:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Traill LW, Bradshaw CJ, and Brooka BW. 2007. Minimum viable population size: A meta-analysis of 30 years of published estimates. Biol Conserv 139:159–166. [Google Scholar]
- 117.Valentim CL, Cioli D, Chevalier FD, Cao X, Taylor AB, Holloway SP, Pica-Mattoccia L, Guidi A, Basso A, Tsai IJ, Berriman M, Carvalho-Queiroz C, Almeida M, Aguilar H, Frantz DE, Hart PJ, LoVerde PT, and Anderson TJ. 2013. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 342:1385–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van Ha N, Dyk Dao L, and Rabinovich SA. 2002. Use of nested PCR for differential diagnosis of falciparum malaria reinfection and relapse in drug-resistant patients. Bull Exp Biol Med 134:379–81. [DOI] [PubMed] [Google Scholar]
- 119.Vlaminck J, Fischer PU, and Weil GJ. 2015. Diagnostic tools for onchocerciasis elimination programs. Trends Parasitol 31:571–82. [DOI] [PubMed] [Google Scholar]
- 120.Wang J, and Caballero A. 1999. Developments in predicting the effective size of subdivided populations. Heredity 82:212–226. [Google Scholar]
- 121.Weir BS, and Cockerham CC. 1984. Estimating F-statistics for analysis of population structure. Evolution 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- 122.Whitlock MC 2011. G’ST and D do not replace FST. Mol Ecol 20:1083–91. [DOI] [PubMed] [Google Scholar]
- 123.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, and Meshnick SR. 2002. Epidemiology of drug-resistant malaria. Lancet Infect Dis 2:209–18. [DOI] [PubMed] [Google Scholar]
- 124.Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, and Su XZ. 2002. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418:320–3. [DOI] [PubMed] [Google Scholar]
- 125.World Health Organization. 2009. Methods for surveillance of antimalarial drug efficacy. WHO [Google Scholar]
- 126.World Health Organization. 1995. Onchocerciasis and its control 852. WHO [Google Scholar]
- 127.Zhan B, Li T, Xiao S, Zheng F, and Hawdon JM. 2001. Species-specific identification of human hookworms by PCR of the mitochondrial cytochrome oxidase I gene. J Parasitol 87:1227–9. [DOI] [PubMed] [Google Scholar]
- 128.Zimmerman PA, Dadzie KY, De Sole G, Remme J, Alley ES, and Unnasch TR. 1992. Onchocerca volvulus DNA probe classification correlates with epidemiologic patterns of blindness. J Infect Dis 165:964–8. [DOI] [PubMed] [Google Scholar]