

Abstract
Iron-catalyzed cross-coupling reactions have attracted significant research interest, as they offer numerous favorable features compared with cross-coupling reactions with precious metal catalysis. While this research has contributed to an empirical understanding of iron-catalyzed cross-coupling, the underlying fundamental mechanisms of reaction and structures of catalytically active species have remained poorly defined. The lack of such detail can be attributed to the difficulties associated with studying such iron-catalyzed reactions, where unstable paramagnetic intermediates abound. Recently, the combined application of physical-inorganic spectroscopic methods, concomitant organic product analysis, and air- and temperature-sensitive inorganic synthesis has yielded the most detailed insight currently available on reactivity and mechanism in iron-catalyzed cross-coupling. This Perspective highlights this approach and the limitations of the contributing techniques as well as some of the key features of the catalytic reactions studied and lessons learned.
1. INTRODUCTION
The formation of C–C bonds is of fundamental importance for the production of pharmaceuticals, organic natural products, and biologically active molecules for health-related research. After initial reports by Kharasch in 1941,1 the pioneering work of Jay Kochi in the 1970s showcased iron as an effective catalyst for C–C cross-coupling reactions.2–4 Kochi demonstrated that simple ferric salts catalyze the stereoselective generation of C–C bonds between alkenyl halides and Grignard reagents (Scheme 1). Although iron showed significant promise for further development, it was kept out of the cross-coupling spotlight for decades by the concurrent discovery of group 10 metal-catalyzed cross-coupling methods, which provided greater ease of handling and development (i.e., bench-stable catalytic protocols). More recently, renewed interest in iron-catalyzed cross-coupling has emerged as a result of not only its low toxicity and high natural abundance but also its complementary reactivity compared with late d-block metals (i.e., group 10 metals). In the late 1990s, Cahiez sparked renewed interest in iron-catalyzed cross-coupling by addressing limitations of Kochi’s original protocol, such as the requirement of excess electrophile, and significantly broadened the substrate scope.5 These feats were accomplished using N-methylpyrrolidone (NMP) as an additive. Subsequent reaction method development using bisamine (e.g., N,N,N′,N′-tetramethyl-1,2-ethylenediamine (TMEDA)),6–8 N-heterocyclic carbene (NHC),9–13 and bisphosphine (e.g., spin-control-intended ortho-phenylenedi-phosphine (SciOPP),14–16 1,2-bis(diphenylphosphino)-benzene (dpbz),17,18 and 1,2-bis(diphenylphosphino)ethane (dppe)19) ligands have continued to broaden the substrate scope and improve the selectivity, as have additional ligand-free iron salt cross-coupling methods.20 More recent examples, including Nakamura’s enantioselective cross-coupling of α-chloroesters and aryl Grignard reagents21 and Bedford’s substrate-directed Suzuki biaryl cross-coupling,22 demonstrate the areas of ongoing interest and development in iron-catalyzed cross-coupling. Overall, these reports reflect the broad potential of iron to catalyze cross-coupling.
Scheme 1.

Selected Examples of Iron-Catalyzed Cross-Coupling Methods
The resurgence of the development of iron-catalyzed cross-coupling has been fraught with numerous challenges, and several areas require significant improvement.23 Examples include the current requirements for large amounts of toxic NMP cosolvent,5,24 the lack of broadly applicable methods for stereoselective cross-couplings, and the need to broaden the scope of nucleophiles and electrophiles.13,25 C(sp3)–C(sp3) cross-couplings remain a challenging yet desirable transformation for metal-catalyzed cross-coupling as a whole.26 The development of new methodologies would broaden the utility to include more complex substrates such as those used for generating natural products. Furthermore, expansion beyond the typically required organometallic reagents (i.e., organomagnesium and organozinc reagents) to include milder, less nucleophilic reagents such as unactivated boronic acids would likely expand the electrophile scope and promote more widespread adoption of iron-catalyzed cross-coupling. Toward addressing these limitations, a detailed understanding of active catalyst structure, mechanism, and ligand and additive effects can provide the basis to both improve current catalytic systems and inspire the development of new catalysts and methodologies that will greatly expand the scope and utility of iron in C–C cross-coupling. However, because of the challenge of dynamically monitoring (i.e., as a function of time) and deconvoluting the possible iron species formed in situ as well as understanding the nuances present in each catalytic system, a drastically different approach from that traditionally utilized to evaluate speciation and mechanism for palladium and other group 10 metals is required. This Perspective focuses on the physical-inorganic approach that has led to the most detailed insight into iron speciation and mechanism in cross-coupling currently available, with emphasis on the key insights obtained as well as critical observations and potential pitfalls that have been identified.
2. MECHANISTIC STUDIES: CHALLENGES AND APPROACH
The lack of mechanistic insight that often exists for iron-catalyzed reactions (including cross-coupling) reflects the formidable obstacles associated with characterizing iron species, including the multifaceted nature of their electronic structures.23 Physical-organic methods, including investigation of kinetics, radical clock experiments, and deuterium isotope labeling studies, have provided some insight into the underlying mechanisms of many reactions. However, the lack of direct probing of the iron species generated in situ greatly limits the utility of these results. As a result of these limitations, no consensus has been reached regarding the mechanism or active catalyst structure across various iron-catalyzed cross-coupling reactions. Even aspects as basic as the oxidation state along a given reaction manifold have proven contentious. This lack of consensus and fundamental understanding underscores (1) the broad range of plausible reaction mechanisms for paramagnetic iron complexes (including both one- and two-electron pathways) and (2) the experimental challenges associated with characterizing paramagnetic iron complexes. These challenges have historically presented an insurmountable barrier to understanding the basic chemistry that governs iron-catalyzed cross-coupling, precluding the rational, mechanistic-driven catalyst development that has proven widely successful in palladium chemistry.27,28
In contrast to palladium-catalyzed cross-couplings, a much broader array of iron redox couples (Fe(I)/Fe(III), Fe(0)/Fe(II), Fe(−II)/Fe(0), and Fe(II)/Fe(III)) and spin states have been proposed to be active for iron-catalyzed cross-couplings.27 This myriad of possible oxidation and spin states of iron has led to hesitation in proposing a generalized mechanism. An accompanying challenge that restricts a more detailed study is the inherent paramagnetism of many of the iron species.23 This paramagnetism makes tracking of iron intermediates by NMR spectroscopy challenging, and quantification of the iron species is typically inaccurate, especially in systems lacking supporting ligands with distal protons from the iron nuclei. Another complication is the air and thermal sensitivity of most reactive iron intermediates. Notably, analogous challenges have historically existed in mechanistic studies of iron-catalyzed reactions in bioinorganic chemistry. To address these challenges, a combination of physical-inorganic spectroscopic methods, including 57Fe Mössbauer, electron paramagnetic resonance (EPR), and magnetic circular dichroism (MCD) spectroscopy (and other more specialized methods such as X-ray absorption spectroscopy (XAS)) have been successfully employed. Combined with additional techniques such as X-ray diffraction (XRD), active-site mutations, and theoretical studies, these spectroscopic methods have enabled the elucidation of the coordination environment of the iron active sites of many different enzymatic proteins as well as the development of detailed structure–function correlations and mechanistic insights.29
Inspired by bioinorganic studies of systems involving iron, an experimental approach involving the application of these physical-inorganic spectroscopies with an emphasis on the direct correlation of changes in organic product distributions with iron speciation has been utilized and proven to be a powerful technique for studying iron-catalyzed cross-coupling reactions (Figure 1).30 Central to this approach is the use of 57Fe Mössbauer spectroscopy as a primary screening tool for determination of the number of iron species present in situ and their relative amounts. 57Fe Mössbauer spectroscopy is ideally suited for tackling these critical questions because the total number of iron species is readily observable. This is in stark contrast to other spectroscopic methods based on spin state (EPR) or methods that can yield significant ambiguity in resolving mixtures of iron species (XAS) in the absence of complementary measurements. In addition, the key spectroscopic parameters for 57Fe Mössbauer spectroscopy (e.g., isomer shift and quadrupole splitting) provide insight into the electronic structure. For example, the isomer shift (δ) reflects the oxidation state and bonding properties of the iron complex, such as covalency between iron nuclei and surrounding ligands as well as the electronegativity of the ligands.29 The quadrupole splitting (ΔEQ) is similarly reflective of the oxidation state and bonding properties as well as the spin state and molecular symmetry. This quadrupole splitting gives rise to the characteristic doublet pattern observed in many 57Fe Mössbauer spectra. Although these parameters are highly characteristic of a given iron complex, comparing them between different reaction manifolds should be highly discouraged when significant differences such as different coordinating ligands (e.g., N vs O donor ligands) are present. Changing the coordination environment and ligand donor strength can drastically affect the overall parametric trend. Additional care must be taken in comparing solid-state parameters to those of iron species generated in situ, as significant distortions can occur when the solid iron complex is dissolved in a coordinating solvent (e.g., THF or Et2O).
Figure 1.

Iron mechanistic toolbox combining physical-inorganic spectroscopies, low-temperature synthesis, and concurrent iron speciation/reactivity studies.
While 57Fe Mössbauer spectroscopy provides a critical starting point for studying speciation, this method is most powerful when combined with additional spectroscopies to obtain more detailed insight into the electronic and geometric properties of iron species generated in situ. One such method, EPR spectroscopy, provides valuable information on the spin states of paramagnetic metal species, allowing for the oxidation state of a given metal center to be inferred. However, selection rules restrict EPR spectroscopy to odd-electron systems (i.e., Kramer’s doublets) in the commonly employed perpendicular-mode geometry.29 Furthermore, even for parallel-mode EPR studies, integer-spin iron sites require specific zero-field splitting parameters for observable EPR activity. These selection rules limit the observation of a vast number of open-shell iron species that may be present during catalysis (integer-spin systems). Recently, many high-spin (S = 2) iron(II) precatalysts have been implicated in C–C cross-coupling, for which traditional X-band, perpendicular-mode EPR spectroscopy cannot be utilized to probe the ground-state electronic structure and alternative spectroscopic techniques (e.g., MCD spectroscopy) must be employed. However, when an appropriate odd-electron iron complex is observed by EPR spectroscopy, significant information about the nature of the unpaired electrons (iron- vs ligand-centered) can be determined from the observed g values, including the spin state (and, by inference, often the oxidation state). Because of spin relaxation effects, liquid helium EPR measurements are required to ensure that all of the EPR-active iron sites are observable. As an example, whereas an S = ½ iron species would be EPR-active at room temperature, an S = 5/2 site would not be observable except by very low temperature measurements and could inadvertently be ruled out as a potential in situ-formed iron species. Additionally, the extreme sensitivity of EPR spectroscopy allows for the detection of iron species at very low concentrations (i.e., below 1% of the total iron present in solution for typical cross-coupling reactions). Therefore, spin integration using a standard is essential in determining the relative concentrations of in situ-generated iron species that are EPR-active. Spin integration of the EPR signal also allows for the determination of the relative lifetimes and consumption with respect to substrate conversion into product using time-resolved freeze-quench EPR spectroscopy combined with concurrent GC analysis. This can provide supporting evidence for the reactivity and possible on-cycle nature of EPR-active iron species during catalysis or, alternatively, determine whether they are instead off-cycle or unreactive.
The limitation of traditional EPR methods to studies of only Kramer’s doublet paramagnetic iron species can be overcome through the utilization of MCD spectroscopy. Notably, any S > 0 iron species is readily observable by MCD. In this method, the differential absorption of left- and right-circularly polarized light in an external magnetic field induces Zeeman splitting and mixing of ground- and excited-state sublevels, ultimately leading to spectroscopic features that enable a complete description of the ligand field and charge-transfer transitions of a paramagnetic iron species via a C-term mechanism.29 Importantly, C-term signals are particularly sensitive for probing the electronic transitions of paramagnetic iron centers while suppressing electronic transitions from diamagnetic compounds such as the organic substrates and products and intraligand transitions that might otherwise dominate traditional electronic absorption measurements. Lastly, because of the saturation behavior of individual spin systems, variable-temperature, variable-field (VTVH) MCD spectroscopy is often utilized to determine the ground-state spin Hamiltonian parameters of the iron center (i.e., the spin state). This is a very powerful experiment for the unambiguous determination of the spin states of integer-spin systems, including freeze-trapped intermediates, which are often not sufficiently stable for more traditional methods used for the determination of effective magnetic moments (e.g., the Evans method, NMR spectroscopy, and SQUID magnetometry).
While the spectroscopic methods outlined above are highly amenable to the study of paramagnetic iron species formed in situ, complementary information on the absolute configuration of the iron complex from single-crystal XRD is highly desirable and often essential, as ambiguous structural information often results from these spectroscopic methods. However, the instability of iron organometallic intermediates in cross-coupling presents a significant challenge to XRD measurements, requiring care in the generation and handling of crystals, often through strict control of the reaction temperature or specialized cryogenic handling procedures and equipment. Fortunately, such studies can be successfully performed with sufficient steadfastness and care, leading to the identification of completely unanticipated iron coordination environments. For example, an iron(I) monomeric structure was proposed and widely accepted as a likely catalytically active species from EPR spectroscopic studies.4 However, by means of low-temperature synthesis and cryogenic handling, the iron cluster [Fe8Me12]− characterized by this S = ½ EPR signal was isolated (see Figure 3), where the iron centers are antiferromagnetically coupled to produce the broad S = ½ signal.31 Without XRD, such an unusual structural motif would not have been proposed or believed.
Figure 3.

X-ray structure and EPR spectrum of Kochi’s observed S = ½ iron complex. Adapted from ref 31. Copyright 2016 American Chemical Society.
While experimental spectroscopic and structural data are key to determining iron speciation and understanding electronic structure, such studies alone are not sufficient to define the roles of identified iron species in catalysis. Instead, directly relating iron speciation and organic product distributions dynamically through time-resolved freeze-trapping and chemical-quench GC analysis is required to definitively identify reactive iron species. This approach has been found to be a very powerful method for experimental elucidation of key mechanistic insights, such as how the electronic structure of the iron species affects the reactivity. By contrast, numerous mechanistic studies have utilized a more indirect and static approach (with respect to monitoring iron speciation) by synthesizing stable precatalytic species and testing their reactivities by monitoring the production of organic products. This reflects an inherent disconnect between potential iron speciation, which may be more complex or dynamic upon redissolution (vide infra), and organic product distributions. The combination of freeze-trapped spectroscopic monitoring and chemical-quenched GC analysis of organic product distributions allows for the determination of the relative rates of reactivity and selectivity of individual iron complexes. This provides for a more definitive determination of the active catalytic species as well as on- and off-cycle iron species. Furthermore, tracking the lifetimes of these unstable intermediates using freeze-trapped methods helps optimize isolation and crystallization conditions.
Beyond the core methods and approaches described above for evaluating iron speciation, reactivity, and mechanism, it should be noted that other spectroscopic methods, such as NMR and XAS, have also been employed (sometimes in combination with XRD studies) in mechanistic studies of iron-catalyzed cross-coupling reactions.32–35 Excellent examples of the use of NMR spectroscopy and XRD were recently reported in the mechanistic investigations of TMEDA in iron-catalyzed cross-coupling, in which the isolation and characterization of numerous iron species were reported and the reactivity toward electrophilic substrates could be further evaluated.32,33 Stoichiometric reactions involving iron complexes and electro-philes to produce cross-coupled products demonstrated the relative reactivity and selectivity of TMEDA-ligated complexes and homoleptic iron ferrate complexes containing TMEDA coordinated to the magnesium cation. While NMR studies are useful (especially when the spectra of isolated, structurally characterized species can be determined), several challenges limit its utility as a primary screening tool for in situ-formed iron species. For example, one difficulty in testing the relative reactivity of these complexes using NMR spectroscopy is the challenge in quantifying the amount of iron remaining after reaction with the substrate. This limitation can result in significant ambiguity in tracking the relative amounts and rates of reaction of individual iron complexes. Additionally, the effect of line broadening and overall spectral crowding adds to the complexity of interpreting the remaining iron. The combination of NMR studies with a broader array of inorganic spectroscopic methods, such as 57Fe Mössbauer spectroscopy, would enable a more complete mechanistic understanding and identification of the iron species present during this cross-coupling reaction. As mentioned previously, XAS has also been utilized in mechanistic investigations of iron-catalyzed cross-coupling reactions, although less frequently. Specifically, X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) experiments can be utilized to determine oxidation states and local coordination environments of metal-centered coordination complexes, including for in situ analysis in the absence of freeze-trapping.34,35 While XAS studies are a powerful method for evaluating electronic structures of pure species, the use of this method as a primary screening tool to monitor potentially complex mixtures of iron complexes formed in situ should be avoided because the obtained spectra reflect a statistically weighted average of all of the species present in solution, the complexity of which is extremely difficult to determine or dissect in the absence of additional information on the number of iron species and relative concentrations. Therefore, such methods are most insightful when used in combination with other spectroscopic methods such as Mössbauer spectroscopy that enable determination of the number and relative amounts of iron species present in solution. Thus, as complementary spectroscopic tools, XANES and EXAFS can provide important additional information on the geometric and electronic structure of iron species present during catalysis.
Lastly, while spectroscopic and XRD studies are essential for studying iron-catalyzed cross-coupling reactions in order to obtain direct experimental evidence in support of specific in situ-formed and reactive iron species, quantum-chemical approaches using density functional theory (DFT) are also employed to generate molecular orbital diagrams and analyze experimentally obtained spectra. Spectral analysis through time-dependent DFT (TD-DFT) is utilized extensively for the assignment of features observed in EPR and MCD spectra to metal–ligand/ligand–metal charge-transfer (MLCT/LMCT) or d–d transitions. Additionally, reaction coordinate studies are often used to evaluate potential reaction pathways, including the key C–C bond formation step in catalysis. As an example, recent reports from both Gutierrez36 and Nakamura37 have utilized DFT to implicate energetically plausible reactive monomeric iron(I) species in an iron- catalyzed asymmetric C(sp2)–C(sp3) cross-coupling reaction. While these theoretical models predict interesting possible precatalytic and intermediate iron species, such studies have not provided sufficient supporting experimental evidence of the presence of the proposed iron species. Ultimately, direct synthetic and spectroscopic studies of this reaction will be required to determine the viability of the proposed reactive iron species and mechanistic pathways. Despite these limitations, such reaction coordinate calculations provide alternative mechanistic hypotheses for these systems, greatly enriching the literature and inspiring future experimentally driven mechanistic studies.
3. INSIGHTS INTO SPECIATION, REACTIVITY, AND MECHANISM IN IRON-CATALYZED CROSS-COUPLING
The use of physical-inorganic spectroscopic methods and XRD studies, including the combination of time-resolved freeze-trapped spectroscopic studies and chemical-quenched GC analysis, has revealed the existence of many insights and caveats present in iron-catalyzed cross-coupling reactions that inform future mechanistic studies as well as the development of new iron-catalyzed reaction protocols. Several key results from these studies over the past few years are presented in the following sections.
3.1. Low-Valent Iron Is Not Required for Effective Cross-Coupling Catalysis.
Electron-rich, low-valent iron species (i.e., oxidation states below 2+) have long been hypothesized as likely active species in iron-catalyzed cross-coupling reactions. These species were proposed to form in situ as a result of the strongly reducing conditions utilized in catalysis. However, recent reports utilizing well-defined iron(II)–ligand complexes as precatalysts have enabled further investigations of whether low-valent iron species are accessible with these stabilizing ligand scaffolds. Notably, bishalide iron(II)–SciOPP complexes have been shown to be highly effective in the cross-coupling of mesityl, phenyl, and alkynyl Grignard reagents with alkyl halides.15,16 Mechanistic investigations of these reactions identified iron(II)–SciOPP complexes as on-cycle reactive intermediates producing cross-coupled products with rates and selectivity representative of the catalytic reaction itself (Figure 2). In fact, low-valent iron complexes accessible in situ were found to be catalytically incompetent. The iron(0)–SciOPP complex Fe(η6-biphenyl)-(SciOPP) was demonstrated not to generate cross-coupled product but instead to re-enter the catalytic cycle by regeneration of the iron–SciOPP bishalide species through reaction with alkyl halide while generating a nondesired organic side product. Similar studies of alkyl–alkyl cross-coupling involving iron–NHC complexes led to an analogous conclusion of reactive iron(II) species for cross-coupling and a potential Fe(II)/Fe(III) redox couple.38 Other spectroscopically observed low-valent iron(I) species were determined to be off-cycle through reaction studies with substrate. While iron-catalyzed cross-coupling methods may exist that utilize low-valent iron for on-cycle catalysis, low-valent iron is not a prerequisite for achieving effective cross-couplings.
Figure 2.

Time-resolved, freeze-trapped 80 K 57Fe Mössbauer spectra of the reaction of FeMes2(SciOPP) and 20 equiv of 1-bromodecane to generate FeBrMes(SciOPP) and the cross-coupled product. Adapted from ref 42. Copyright 2014 American Chemical Society.
3.2. Multinuclear Iron Species as a New Reaction Paradigm.
Unlike systems that involve well-defined ligands, iron speciation in simple iron-salt-catalyzed cross-couplings has proven to be even more challenging because of the large variety of iron species that can be generated under catalytically relevant conditions. One example is the reduction pathway of iron(III) salts in the presence of MeMgBr.31,39 Upon the addition of MeMgBr to iron(III) salts, several iron species are generated that have been identified structurally. These include the first report of tetramethylferrate(III), [FeMe4]−, which was isolated and fully characterized. This species was found to be unreactive with the electrophile but capable of generating an S = ½ species upon warming from −80 °C to room temperature. Through the investigation of this S = ½ species, first reported by Kochi and originally believed to be a monomeric iron(I) complex,4 an unprecedented multimeric iron cluster identified as [Mg(THF)5Cl][Fe8Me12] (Figure 3) was found to correspond to this S = ½ signal. Critically, this iron–methyl cluster was found to be highly reactive toward electrophile and, with subsequent addition of MeMgBr, could selectively generate the cross-coupled product while simultaneously regenerating the cluster. The utilization of multi-nuclear iron species for cross-coupling represents a new paradigm for achieving effective catalysis that clearly merits further investigations utilizing well-defined multinuclear iron precatalysts.
3.3. The Devil Is in the Details of the Reaction Protocol.
Subtle differences in reaction protocols have been found to strongly influence the distribution of iron species formed in situ. For example, slow addition of the nucleophile is performed to improve the selectivity for organic products in many iron-catalyzed cross-coupling methods. In fact, using these catalytically relevant rates of nucleophile addition keeps the relative ratio of nucleophile to iron quite low during catalysis. In the iron–SciOPP study, fast addition of MesMgBr was found to promote the displacement of SciOPP and generate a homoleptic iron ferrate complex, [FeMes3]− (Figure 4). However, upon slow addition (consistent with catalysis) in the presence of electrophile, the low relative ratio of MesMgBr and iron–SciOPP deterred the formation of this homoleptic species. Beyond this system, many iron-catalyzed reactions are highly sensitive to the protocols employed, including the need for inert atmospheric conditions, the use of slow addition of strongly nucleophilic coupling partners, the effects of additives, and the drastic changes in reactivity seen when the catalytic solvent is changed. Thus, investigations of speciation and mechanism must carefully reproduce the exact reaction protocols employed in the catalysis, as model reactions circumventing protocol requirements may radically change the iron speciation and hinder identification of the role of the protocol in obtaining high cross-coupling yields.
Figure 4.

Dependence of iron speciation on the addition of MesMgBr in iron-catalyzed cross-coupling of MesMgBr and alkyl halides.
3.4. Homocoupled Nucleophile as a Probe of Oxidation State.
Identification of the oxidation state of active iron species during catalysis has long been one of the most significant challenges in mechanistic studies of iron-catalyzed cross-couplings. Because of the extremely high reactivity and thermal instability of many of the organoiron species that may form in situ, indirect methods have been employed to try and circumvent these issues. One example of a popular workaround to estimate the average oxidation state of iron is monitoring the homocoupling of the nucleophilic coupling partner, most commonly a Grignard reagent.
Recently it was demonstrated that care must be taken when using this approach to estimate the iron oxidation state, as the results can be misleading in systems where chemical quenching of transmetalated iron species affords homocoupled nucleophile.40 For example, while the ability of the bisphenylated iron–SciOPP complex to undergo reductive elimination and form Fe(η6-biphenyl)(SciOPP) was demonstrated, use of the previously reported chemical quench on the in situ-generated, and confirmed, bisphenyl complex resulted in ~50% biphenyl formation with respect to iron despite the fact that all of the iron was iron(II) prior to the chemical quench.40 Similarly, the iron(II)–NHC bisalkyl complex (Figure 5), formed in situ for alkyl–alkyl cross-coupling, also generated homocoupled nucleophile in ~70% yield with respect to iron when the reported chemical quench was used.40 Together, these studies demonstrated how homocoupled nucleophile may not be reflective of the iron oxidation state when reported chemical quenches are utilized, and hence, experiments that directly measure iron species should be utilized to determine the electronic structure (and oxidation state) of the in situ-formed iron species.
Figure 5.

Precatalyst generation step for iron-NHC catalyzed alkyl–alkyl cross coupling including freeze-trapped EPR spectrum which is unable to observe the majority of in situ iron and the crystal structure of bis-alkyl iron-NHC species found to be the major precatalyst species generated in solution. Adapted with permission from ref 38, published by the Royal Society of Chemistry.
3.5. Ambiguity from Single Spectroscopies in Isolation.
The use of a single spectroscopic technique without the aid of complementary experimental data to evaluate iron speciation and investigate iron-catalyzed cross-coupling methods can be misleading. One spectroscopic method commonly utilized in isolation is EPR spectroscopy, which is often a first port of call when investigating iron-catalyzed systems. There are many good reasons for this, including the accessibility of EPR instrumentation in academic research, the extremely high sensitivity of the method, and the potential to preliminarily rule a variety of possible iron species in or out depending on the observed (or indeed lack of) EPR signal and the observed g values. With regard to the last point, the observation of S = ½ species by various groups across multiple iron-catalyzed cross-coupling reactions has resulted in the proposal of an iron(I)–iron(III) catalytic cycle in many systems.4,13,41 However, such an S = ½ signal could result from many different types of iron species, including mononuclear iron(I), low-spin mononculear iron(III), an antiferrocoupled dimer, or an iron species with even higher nuclearity, to highlight a few possibilities. Indeed, an S = ½ EPR signal was observed by Kochi in a seminal report on the cross-coupling of alkenyl bromides with MeMgBr using simple ferric salts.4 This resulted in his original proposal of an iron(I)–iron(III) catalytic cycle. However, the S = ½ signal observed for this system has subsequently been demonstrated to be consistent with the formation of an [Fe8Me12]− cluster. This species would have been impossible to predict had it not been isolated and structurally characterized.31 Also worthy of note is the fact that spin integration indicated that the resulting EPR signal constituted, within error, all the iron in solution. However, because of the significant broadening of the EPR signal, definitive confirmation of the [Fe8Me12]− cluster as the main species formed in situ was not sufficient. As a result, MCD spectroscopy was used to provide confirmation.
Similarly, an S = ½ EPR signal was observed for an alkyl–alkyl cross-coupling system developed by Cárdenas and coworkers, also resulting in the proposal of an iron(I) active species during catalysis.13 Upon further investigation, specifically, spin integration of the EPR spectra, the S = ½ species was found to constitute <0.5% of all iron in solution (Figure 5).38 The concentration of this species was not observed to change upon reaction with the electrophile during either stoichiometric reactivity tests or catalysis, suggesting that it may well be an off-cycle species. This scenario highlights the critical importance of performing spin integration of EPR spectra when evaluating iron speciation using EPR spectroscopic data (i.e., to determine whether one is looking at most or almost none of the total iron present in solution).
In contrast to EPR spectroscopy, 57Fe Mössbauer spectroscopy is not limited to specific electronic configurations of iron to be observable. However, it is not devoid of its own pitfalls in interpretation of ambiguous data in terms of oxidation state and spin state. Although the spectroscopic parameters obtained are related directly to the oxidation state of the iron species, they should be interpreted with caution because electronic perturbations from differing coordination environments (i.e., ligands) can cause significant deviations from linear tracking of the oxidation and spin states with the isomer shift and quadrupole splitting parameters. In investigations of the stoichiometric reactivity of the bisphosphine precatalyst FeCl2(SciOPP), the addition of 1 or 2 equiv of MesMgBr resulted in the observation of two new iron species.42 While the bischloride precatalyst has parameters δ = 0.94 mm/s and ΔEQ= 2.69 mm/s, the new iron species had parameters of δ = 0.52 mm/s and ΔEQ= 2.12 mm/s and δ = 0.28 mm/s and ΔEQ= 3.67 mm/s, respectively (Figure 6). These marked changes, in particular the very low isomer shift of the species observed after addition of 2 equiv of MesMgBr, may have led to the presumption that reduction of the iron center had taken place. However, these species were subsequently isolated and identified as the mono- and bis-transmetalated iron–SciOPP complexes (FeBrMes(SciOPP) and FeMes2(SciOPP), respectively), with no change in oxidation state having occurred.
Figure 6.

80 K 57Fe Mössbauer spectrum containing the major species FeMes2(SciOPP) and FeBrMes(SciOPP) (Ar = 3,5-tert-butylphenyl). Adapted from ref 42. Copyright 2014 American Chemical Society.
3.6. Iron Speciation May Be Neither Simple…nor Static.
It is natural to hypothesize a reaction mechanism or rationalize the reactivity of a given system on the basis of a single reactive species or well-defined and discrete speciation, especially when there is a lack of spectroscopic insight. While this has been demonstrated to be the case in some systems, others have displayed complex and dynamic speciation. For example, the reaction of FeBr2(SciOPP) with 1 equiv of Grignard reagent would presumably generate a single major species. This is indeed the case upon addition of 1 equiv of MesMgBr, which generated FeBrMes(SciOPP) as the only detectable species.42 By contrast, the addition of 1 equiv of the alkynyl Grignard reagent (TIPS)ethynylmagnesium bromide resulted in a complex four-component mixture after only 1 min (Figure 7).43 This distribution of iron species was also dynamic, evolving over the course of 30 min, illustrating the value and requirement of time-resolved spectroscopy at various time points over the course of a reaction.
Figure 7.

Time-resolved, freeze-quenched 80 K 57Fe Mössbauer spectra of the reaction of FeBr2(SciOPP) (Ar = 3,5-tert-butylphenyl) with 1 equiv of (TIPS)ethynylmagnesium bromide at room temperature. Adapted from ref 43. Copyright 2017 American Chemical Society.
The iron speciation upon reaction with the alkynyl Grignard reagent was subsequently shown to be highly solvent-dependent with regard to the species generated and their subsequent redistribution. These drastic differences upon changing the identity of the nucleophile highlight how the presumption that relatively similar catalytic systems will behave analogously can be deceptive.
3.7. Multiple Iron Species Can Be Present That Are Reactive toward Electrophiles.
As highlighted above, iron speciation can be more complex than is often anticipated. There may be many species present, several of which may display reactivity with the relevant electrophilic coupling partner. Because of this possibility, the examination of the reactivity of an isolated organometallic iron species to assess its potential role as a reactive intermediate, especially without the aid of in situ spectroscopic data, can be problematic in the absence of analogous reactivity studies on all of the iron species present in situ. Many novel and reactive iron organometallic species have been isolated, and a common first experiment is aimed at establishing their efficacy as precatalysts for the relevant catalytic transformation. This can certainly establish whether the given species is off-cycle but little else. Stoichiometric reactivity with the corresponding coupling partner is more revealing, although both the distribution of products and the relative rate of reaction should be compared with that observed for the catalytic reaction itself. For instance, when the cross-coupling of phenyl-based nucleophiles with alkyl halides using FeCl2(SciOPP) was investigated, three species were found to form in situ.40 These were identified as the mono- and bisphenyl iron(II)–SciOPP species and the iron(0) complex Fe(η6-biphenyl)(SciOPP). Upon treatment of each species with excess halocycloheptane, all proved to be reactive. However, upon examination of their selectivities and lower-limit rates of reaction, clear differences were evident (Table 1). It should be noted that the product yields reflect two turnovers of FePh2(SciOPP).
Table 1.
Reactivity Studies of Phenylated Iron(II)–SciOPP Species with Bromocycloheptane
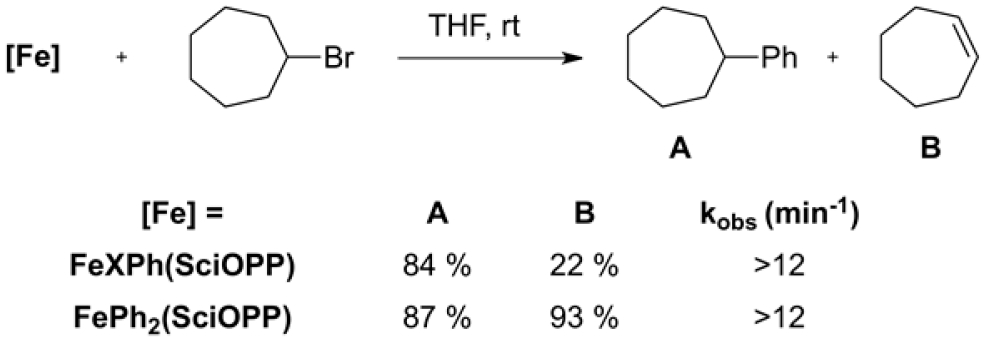
|
Fe(η6-biphenyl)(SciOPP) displayed sluggish reactivity, generating the corresponding alkene and reforming FeCl2(SciOPP). This reactivity suggests that it is an off-cycle species but can re-enter the catalytic cycle through this side reaction. Indeed, this iron(0) complex could be used as a precatalyst for the reaction, although longer reaction times were required to obtain yields comparable to that for the analogous reaction using FeCl2(SciOPP) as the precatalyst. By comparison, both mono- and bisphenyl iron(II)–SciOPP displayed high reactivity toward bromocycloheptane, generating the cross-coupled product at rates consistent with the catalytic reaction. It was noted that despite similar lower limits for the rate of reaction with the electrophile, the reaction of FeXPh(SciOPP) displayed greater selectivity toward the cross-coupled product, consistent with catalysis. These experimental observations, along with additional in situ speciation using freeze-trapped spectroscopic studies, are consistent with the catalytic protocols: slow addition of Grignard reagent for the Kumada cross-coupling and the relatively low rate of transmetalation from the borate nucleophile in the Suzuki–Miyaura variant. When these protocols are utilized, formation of the monophenyl species FeXPh(SciOPP) is favored, yielding optimal selectivity for the cross-coupled product.
3.8. Solvents and Additives Can Drastically Change Iron Speciation.
The solvent, cations, and additives used for a given reaction play a key role and have been found to exert substantial effects on the stabilization of individual iron species. This is most evident when examining a series of species that were generated from the reaction of simple iron salts with methyl nucleophiles. In their influential report, Fürstner et al.44 were able to isolate tetramethylferrate(II) from the reaction of FeCl3 with MeLi in Et2O at low temperature (Figure 8). This distorted tetrahedral complex contains three methyl groups that have additional contacts to three lithium ions as well as elongated iron–carbon bonds compared with the remaining methyl group. By contrast, the analogous reaction of ferric chloride with MeMgBr in THF at low temperatures produced tetramethylferrate(III), [FeMe4]−.39 The crystal structure of this distorted square-planar ferrate complex contains a noncoordinating [MgCl(THF)5]+ countercation. This ferrate was subsequently shown to react further upon warming to form the aforementioned [Fe8Me12]− cluster with the same noncoordinating countercation.31 As previously mentioned, this cluster was also demonstrated to be the reactive and catalytically relevant species formed in situ.
Figure 8.

Methylferrate species generated from the reaction of FeCl3 with MeLi or MeMgBr under various reaction conditions.
One of the most prominent additives used in iron-catalyzed cross-coupling reactions is NMP, which has been demonstrated to improve the reactivity when simple iron salts are used.5,24 The proposed role of NMP has ranged from a simple cosolvent to a ligand for iron. When a reaction similar to those above is carried out using Fe(acac)3 and MeMgBr in the presence of excess NMP, a trimethylferrate(II) species, [FeMe3]−, is formed instead of [Fe8Me12]–45. This ferrate species features a magnesium dication coordinated to six molecules of NMP through the carbonyl oxygen atoms. Identification of this ferrate as the major species formed in situ as well as its selective and rapid reactivity with the electrophile to form the cross-coupled product was also demonstrated.
This collection of methylferrate species illustrates how subtle changes in the reaction conditions (identity of the cation and coordinating ability of the solvent or additive) can dramatically alter the accessible iron species. Although model complexes can provide valuable frames of reference as to the potential iron species formed in situ during catalytic reactions, they should nonetheless be approached with caution because of the significance such changes can exhibit.
3.9. Nucleophiles Are Ligands Too.
The nucleophilic coupling partner in cross-coupling reactions tends to be overlooked when considering possible effects on iron speciation. When the catalytic system is largely the same (e.g., iron salt and ligand used), it is easy to assume that the mechanism, species formed, and reactive species will be analogous. However, this negates large changes in both sterics and electronics, which consequently change the accessibility of the iron species. This is evident from an examination of the reactions of FeCl2(SciOPP) with various Grignard reagents. First, upon comparison of the aryl Grignard reagents MesMgBr and PhMgBr, a variety of differences are observed.40,42 In both cases the mono- and bis-transmetalated analogues can be generated. For the bulkier mesityl group, two distinct geometries are observed.42 The FeMes2(SciOPP) complex exhibits a distorted square-planar geometry, whereas the FeXMes(SciOPP) complex (X = Cl, Br) is a distorted tetrahedron. The differences in geometry are reflected in their reactivities toward the electrophile. The bismesityl complex reacts readily with the electrophile to selectively produce the cross-coupled product, unlike the monomesityl species. With the less bulky phenyl groups, a distorted tetrahedral geometry is adopted for both the FeXPh(SciOPP) and FePh2(SciOPP) complexes.40 Additionally, the bisphenyl complex is also generated in ~30% yield as a distorted square-pyramidal solvent adduct when the reaction is carried out in THF. In contrast to the stark difference in reactivity between the mesityl complexes, both the mono- and bisphenyl analogues are reactive toward the electrophile, with the monophenyl complex reacting more selectively as discussed in previous sections.
The addition of a large excess of MesMgBr (20 equiv) to FeCl2(SciOPP) highlights an additional difference in reactivity between the mesityl and phenyl substituents. In the case of MesMgBr, this results in the formation of [FeMes3]− in ~60% yield, with free ligand being observed and the remaining iron present as FeMes2(SciOPP).42 On the other hand, an excess of PhMgBr results in the formation of the FePh2(SciOPP) complex.40 While this complex will eventually react further to form the iron(0) complex, no ferrate species or free ligand is observed.
Within the alkyl–alkyl cross-coupling system developed by Cardenas and co-workers, the identity of the nucleophile was determined to be essential for forming the reactive iron species.13 The Grignard reagent contained a 1,3-dioxanyl motif (Figure 9) that was demonstrated to be essential for optimal reactivity, compared with unproductive side reactions with the cyclohexyl analogue.38 The effectiveness of this Grignard reagent stems from its ability to form a chelate to the iron center. The isolated complex containing the most catalytically effective NHC ligand (IMes) displayed this chelation mode with both bound equivalents of the Grignard reagent. Changing the NHC ligand to SIPr had a dramatic effect on both the resulting precatalyst structure and the ensuing catalysis. Structurally, the resulting bisalkyl iron–NHC complex differs from the IMes analogue in that the chelation of the alkyl groups is impeded, likely by the increased steric bulk of the SIPr ligand. One of the alkyl groups remains chelated to the iron center, whereas the second is in an open configuration. These structural differences are reflected in the catalytic performance of this complex, with the majority of the electrophile having undergone β-hydride elimination and protodehalogenation rather than the intended cross-coupling. The implication of this is that chelation of the alkyl moiety to the iron center prevents β-hydride elimination and resulting side reactions.
Figure 9.

Bisalkyl iron–NHC species generated in the precatalyst formation step and their performance for catalysis. Adapted with permission from ref 38, published by the Royal Society of Chemistry.
3.10. Not Just a Single Mechanism, Even for Elementary Steps.
From the systems studied so far, multiple mechanisms have already been identified. In all cases these involve the formation of a transmetalated iron species, which subsequently reacts with the electrophile. In the case of ligand-supported systems that have been studied in detail to date, including the bisphospine SciOPP and NHCs, initial reaction of the precatalyst salt and ligand with the Grignard reagent produces the transmetalated iron species. This is illustrated with a simplified catalytic cycle for the cross-coupling of MesMgBr and n-octyl bromide using (SciOPP)Fe(II) bishalide as the precatalyst (Scheme 2A). Frequently the degree of transmetalation is attenuated by mechanical control of the rate of addition of the Grignard reagent. The transmetalated iron(II)–SciOPP bismesityl complex then reacts directly with n-octyl bromide to produce the cross-coupled product and the mono-transmetalated iron(II)–SciOPP complex. This carbon–carbon bond-forming step is proposed to proceed by single electron transfer, generating an iron(III) intermediate and an organic radical that recombine through a radical rebound. However, there is more to be done to establish whether this is indeed the case. The under-transmetalated iron species can subsequently undergo transmetalation once again with an additional equivalent of MesMgBr to regenerate the catalytically active bismesityl complex. An analogous mechanism has been demonstrated to be in operation for the iron(II)–SciOPP-catalyzed cross-coupling of phenyl and alkynyl nucleophiles, iron(II)–NHC-catalyzed alkyl–alkyl cross-coupling, and the simple ferric-salt-catalyzed cross-coupling of MeMgBr in the presence of NMP as an additive.
Scheme 2.

Examples of Mechanistic Pathways in Iron-Catalyzed Cross-Coupling
Alternatively, without the use of supporting ligands, two distinct mechanisms appear to be in operation with MeMgBr as the nucleophile. In the presence of NMP, the trimethylferrate species can be formed. However, without NMP the formation of the [Fe8Me12] cluster is observed. These ferrates are both reactive toward the electrophile, although they react by different means. In the case of the trimethylferrate, as with the ligand-supported systems, direct reaction with electrophile to produce the cross-coupled product and under-transmetalated iron species is operable. The trimethylferrate species can be regenerated upon reaction with additional MeMgBr. By contrast, the [Fe8Me12]− cluster does react with the electrophile to presumably form an adduct, the identity of which remains to be determined, although consumption of the cluster has been observed without the generation of the cross-coupled product (Scheme 2B). However, in the presence of an additional equivalent of MeMgBr, the cluster is reformed along with generation of the cross-coupled product.
The fact that distinct reactivity and mechanisms have already been observed within these systems highlights that one broadly encompassing mechanism is not in operation for iron-catalyzed cross-coupling reactions. Although this may currently seem to be a disadvantage because of the lack of uniformity for rational design, this could be an advantage as different reaction manifolds may deliver complementary reactivity such as reactivity toward different classes of electrophilic coupling partners. Additionally, these reactions effectively operate in the reverse order to the mechanism generally held for palladium-catalyzed equivalents. Specifically, palladium-catalyzed systems generally proceed by initial oxidative addition of the metal center to the electrophile followed by transmetalation of the nucleophilic coupling partner to the metal center, after which reductive elimination can occur. With these iron-catalyzed systems, transmetalation is the initial step to form the reactive organoiron species. These can then react directly with the electrophile to produce the cross-coupled product. While lowvalent species, including iron(0) and small quantities of potential iron(I) complexes, are observed on the basis of the presence of S = ½ signals in the EPR spectra, these have so far proved to be either off-cycle or unreactive.
4. CONCLUSIONS AND FUTURE OUTLOOK
While significant advances in our understanding of iron-catalyzed cross-coupling have occurred over the past 5 years, many challenges and unknowns remain. Most notable are the numerous other reaction protocols, including ligands and additives, that have yet to be investigated in such a detailed manner. This work is especially necessary because of the aforementioned lack of a single underlying mechanism and, hence, the possibility that other completely distinct reaction mechanisms may be effective and operative for catalysis. These possibilities include that of low-valent reactive species, such as iron(0) or iron(I), and less sterically demanding ligands giving rise to 2:1 adducts with the iron center. The nature of the carbon–carbon bond-forming step is another area where further work is required. An example is the identity of the presumed electrophile adduct formed upon reaction of the [Fe8Me12]− cluster. This bond-forming step is an area where computational studies could be invaluable, with the reactive species identified along with the products, and reaction coordinate studies may deliver insight not otherwise obtainable by experimental means to bolster overall understanding of the reaction mechanism as a whole.
Ligand design for iron-catalyzed cross-coupling may require reconsideration, with targeted design based on recent insight. While the ligands used to date, such as bisphosphines and NHCs, were potentially used on the basis of their prior success with palladium-catalyzed systems, they have proven effective but are likely not ideal because of the fundamentally different underlying mechanisms. The rate of addition of the Grignard reagent generally must be controlled to avoid over-transmetalation and displacement of the ligand, yielding a less selective reactive species and therefore a less selective overall reaction. Although a significant challenge, the development of ligands with greater binding affinity for the iron center would be desirable in order to simplify the reaction protocol because the ligand would not be so easily displaced, thereby potentially circumventing the need for the slow addition of the Grignard reagent. Another avenue to achieve this goal would be identifying or developing nucleophilic coupling partners that transmetalate the iron center more slowly. Although the Suzuki–Miyaura variants with iron require an organometallic activator, the system that has been studied does demonstrate a much lower rate of transmetalation compared with the equivalent Grignard reagent. This shows the promise for further development of iron-catalyzed Suzuki–Miyaura cross-coupling reactions, potentially without the need for organo-metallic activators, and is an area that warrants future work to potentially deliver iron-catalyzed cross-coupling reactions that may be more widely adopted.
The isolation and reactivity of the [Fe8Me12]− cluster demonstrate how well-defined multimetallic species offer a new class of compounds to be targeted and investigated. Already having demonstrated a different mode of reaction compared with the monometallic [FeMe3]− ferrate, this type of species could deliver access to catalytic manifolds and reactions that are otherwise inaccessible through mononuclear species.
ACKNOWLEDGMENTS
Financial support from the National Institutes of Health (R01GM111480 to M.L.N.) is gratefully acknowledged.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Kharasch MS; Fields EK J. Am. Chem. Soc 1941, 63, 2316–2320. [Google Scholar]
- (2).Tamura M; Kochi JK J. Am. Chem. Soc 1971, 93, 1487–1489. [Google Scholar]
- (3).Neumann SM; Kochi JK J. Org. Chem 1975, 40, 599–606. [Google Scholar]
- (4).Smith RS; Kochi JK J. Org. Chem 1976, 41, 502–509. [Google Scholar]
- (5).Cahiez G; Avedissian H Synthesis 1998, 1998, 1199–1205. [Google Scholar]
- (6).Nakamura M; Matsuo K; Ito S; Nakamura EJ Am. Chem. Soc 2004, 126, 3686–3687. [DOI] [PubMed] [Google Scholar]
- (7).Cahiez G; Duplais C; Moyeux A Org. Lett 2007, 9, 3253–3254. [DOI] [PubMed] [Google Scholar]
- (8).Guérinot A; Reymond S; Cossy J Angew. Chem., Int. Ed 2007, 46, 6521–6524. [DOI] [PubMed] [Google Scholar]
- (9).Bedford RB; Betham M; Bruce DW; Danopoulos AA; Frost RM; Hird MJ Org. Chem 2006, 71, 1104–1110. [DOI] [PubMed] [Google Scholar]
- (10).Hatakeyama T; Nakamura MJ Am. Chem. Soc 2007, 129, 9844–9845. [DOI] [PubMed] [Google Scholar]
- (11).Hatakeyama T; Hashimoto S; Ishizuka K; Nakamura MJ Am. Chem. Soc 2009, 131, 11949–11963. [DOI] [PubMed] [Google Scholar]
- (12).Ghorai SK; Jin M; Hatakeyama T; Nakamura M Org. Lett 2012, 14, 1066–1069. [DOI] [PubMed] [Google Scholar]
- (13).Guisan-Ceinos M; Tato F; Bunuel E; Calle P; Cárdenas DJ Chem. Sci 2013, 4, 1098–1104. [Google Scholar]
- (14).Hatakeyama T; Hashimoto T; Kondo Y; Fujiwara Y; Seike H; Takaya H; Tamada Y; Ono T; Nakamura MJ Am. Chem. Soc 2010, 132, 10674–10676. [DOI] [PubMed] [Google Scholar]
- (15).Hatakeyama T; Okada Y; Yoshimoto Y; Nakamura M Angew. Chem., Int. Ed 2011, 50, 10973–10976. [DOI] [PubMed] [Google Scholar]
- (16).Hatakeyama T; Fujiwara Y.-i.; Okada Y; Itoh T; Hashimoto T; Kawamura S; Ogata K; Takaya H; Nakamura M Chem. Lett 2011, 40, 1030–1032. [Google Scholar]
- (17).Hatakeyama T; Kondo Y; Fujiwara Y-I; Takaya H; Ito S; Nakamura E; Nakamura M Chem. Commun 2009, 1216–1218. [DOI] [PubMed] [Google Scholar]
- (18) (a).Bedford RB; Huwe M; Wilkinson MC Chem. Commun 2009, 600–602. [DOI] [PubMed] [Google Scholar]; (b) Adams CJ; Bedford RB; Carter E; Gower NJ; Haddow MF; Harvey JN; Huwe M; Cartes MÁ ; Mansell SM; Mendoza C; Murphy DM; Neeve EC; Nunn JJ Am. Chem. Soc 2012, 134, 10333–10336. [DOI] [PubMed] [Google Scholar]
- (19).Bedford RB; Carter E; Cogswell PM; Gower NJ; Haddow MF; Harvey JN; Murphy DM; Neeve EC; Nunn J Angew. Chem., Int. Ed 2013, 52, 1285–1288. [DOI] [PubMed] [Google Scholar]
- (20).Nagano T; Hayashi T Org. Lett 2004, 6, 1297–1299. [DOI] [PubMed] [Google Scholar]
- (21).Jin M; Adak L; Nakamura MJ Am. Chem. Soc 2015, 137, 7128–7134. [DOI] [PubMed] [Google Scholar]
- (22).O’Brien HM; Manzotti M; Abrams RD; Elorriaga D; Sparkes HA; Davis SA; Bedford RB Nat. Catal 2018, 1, 429–437. [Google Scholar]
- (23).Fürstner A ACS Cent. Sci 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fürstner A; Leitner A Angew. Chem., Int. Ed 2002, 41, 609–612. [Google Scholar]
- (25).Dongol KG; Koh H; Sau M; Chai CL L. Adv. Synth. Catal 2007, 349, 1015–1018. [Google Scholar]
- (26).Tasker SZ; Standley EA; Jamison TF Nature 2014, 509, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sherry BD; Fürstner A Acc. Chem. Res 2008, 41, 1500–1511. [DOI] [PubMed] [Google Scholar]
- (28).Fürstner A; Martin R Chem. Lett 2005, 34, 624–629. [Google Scholar]
- (29).Solomon EI; Lever ABP Inorganic Electronic Structure and Spectroscopy; Wiley-Interscience: New York, 2006. [Google Scholar]
- (30).Carpenter SH; Neidig ML Isr. J. Chem 2017, 57, 1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Muñoz SB III; Daifuku SL; Brennessel WW; Neidig ML J. Am. Chem. Soc 2016, 138, 7492–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Noda D; Sunada Y; Hatakeyama T; Nakamura M; Nagashima HJ Am. Chem. Soc 2009, 131, 6078–6079. [DOI] [PubMed] [Google Scholar]
- (33).Bedford RB; Brenner PB; Carter E; Cogswell PM; Haddow MF; Harvey JN; Murphy DM; Nunn J; Woodall CH Angew. Chem., Int. Ed 2014, 53, 1804–1808. [DOI] [PubMed] [Google Scholar]
- (34).Huang Z; Zhang D; Lee J-F; Lei A Chem. Commun 2018, 54, 1481–1484. [DOI] [PubMed] [Google Scholar]
- (35).Schoch R; Desens W; Werner T; Bauer M Chem. - Eur. J 2013, 19, 15816–15821. [DOI] [PubMed] [Google Scholar]
- (36).Lee W; Zhou J; Gutierrez OJ Am. Chem. Soc 2017, 139, 16126–16133. [DOI] [PubMed] [Google Scholar]
- (37).Sharma AK; Sameera WMC; Jin M; Adak L; Okuzono C; Iwamoto T; Kato M; Nakamura M; Morokuma KJ Am. Chem. Soc 2017, 139, 16117–16125. [DOI] [PubMed] [Google Scholar]
- (38).Fleischauer VE; Muñoz SB III; Neate PGN; Brennessel WW; Neidig ML Chem. Sci 2018, 9, 1878–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Al-Afyouni MH; Fillman KL; Brennessel WW; Neidig ML J. Am. Chem. Soc 2014, 136, 15457–15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Daifuku SL; Kneebone JL; Snyder BER; Neidig ML J. Am. Chem. Soc 2015, 137, 11432–11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bedford RB; Brenner PB; Carter E; Carvell TW; Cogswell PM; Gallagher T; Harvey JN; Murphy DM; Neeve EC; Nunn J; Pye DR Chem. - Eur. J 2014, 20, 7935–7938. [DOI] [PubMed] [Google Scholar]
- (42).Daifuku SL; Al-Afyouni MH; Snyder BER; Kneebone JL; Neidig ML J. Am. Chem. Soc 2014, 136, 9132–9143. [DOI] [PubMed] [Google Scholar]
- (43).Kneebone JL; Brennessel WW; Neidig ML J. Am. Chem. Soc 2017, 139, 6988–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fürstner A; Martin R; Krause H; Seidel G; Goddard R; Lehmann CW J. Am. Chem. Soc 2008, 130, 8773–8787. [DOI] [PubMed] [Google Scholar]
- (45).Muñoz SB; Daifuku SL; Sears JD; Baker TM; Carpenter SH; Brennessel WW; Neidig ML Angew. Chem., Int. Ed 2018, 57, 6496–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]