

Abstract
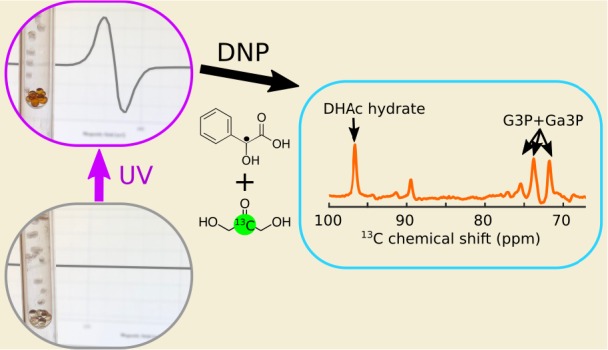
Whether for 13C magnetic resonance studies in chemistry, biochemistry, or biomedicine, hyperpolarization methods based on dynamic nuclear polarization (DNP) have become ubiquitous. DNP requires a source of unpaired electrons, which are commonly added to the sample to be hyperpolarized in the form of stable free radicals. Once polarized, the presence of these radicals is unwanted. These radicals can be replaced by nonpersistent radicals created by the photoirradiation of pyruvic acid (PA), which are annihilated upon dissolution or thermalization in the solid state. However, since PA is readily metabolized by most cells, its presence may be undesirable for some metabolic studies. In addition, some 13C substrates are photosensitive and therefore may degrade during the photogeneration of a PA radical, which requires ultraviolet (UV) light. We show here that the photoirradiation of phenylglyoxylic acid (PhGA) using visible light produces a nonpersistent radical that, in principle, can be used to hyperpolarize any molecule. We compare radical yields in samples containing PA and PhGA upon photoirradiation with broadband and narrowband UV–visible light sources. To demonstrate the suitability of PhGA as a radical precursor for DNP, we polarized the gluconeogenic probe 13C-dihydroxyacetone, which is UV-sensitive, using a commercial 3.35 T DNP polarizer and then injected this into a mouse and followed its metabolism in vivo.
Introduction
Hyperpolarization by dissolution dynamic nuclear polarization (DNP) can enhance the magnetic resonance (MR) signals of molecules in solution by up to 5 orders of magnitude.1 As the list of molecules that have been hyperpolarized increases every year, so do applications across organic and polymer chemistry2 as well as biomedicine.3 DNP is based on the transfer of spin polarization from unpaired electrons of stable free radicals to nuclei at cryogenic temperatures. Stable radicals such as trityls, nitroxides, and 1,3-bisdiphenylene-2-phenylallyl (BDPA) are admixed with the sample containing the molecule to be hyperpolarized.4 A consequence of having to introduce stable radicals is that they accelerate nuclear spin relaxation, which may cause significant signal loss during the dissolution process. A concern for applications in the biomedical field, where dissolution DNP has seen a rapid translation from the laboratory to the clinic,5−7 is the potential radical toxicity. Medical regulatory bodies currently demand that radicals are filtered out prior to injection into a human subject,8,9 which adds to the complexity of the process and becomes a potential failure point before the release of the solution for injection.
Nonpersistent photoinduced free radicals generated by ultraviolet (UV) light irradiation of pyruvic acid (PA) have been proposed as an alternative to the persistent radicals used in dissolution DNP.10 Recently it has been demonstrated that these nonpersistent radicals can be annihilated inside a frozen sample by warming it to ∼200 K. After this point, the nuclear spin hyperpolarization can persist for several hours,11 making it possible to transport the sample for use at a distant location. Several 13C-labeled metabolic substrates have been hyperpolarized using PA as a precursor molecule for the photogeneration of radicals.12,13 However, when PA itself is not one of the substrates of interest,10,13 metabolic interference caused by its presence may be undesirable. Furthermore, PA cannot be considered to be a universal polarizing agent because some metabolic substrates are photosensitive and can degrade during exposure to UV light when generating the radical. One example of this problem is [2-13C]dihydroxyacetone (DHAc), which has been previously hyperpolarized with OX063 trityl radical and used to study gluconeogenesis, glycolysis, and fatty acid synthesis in the liver.14−16 DHAc absorbs light below 300 nm, leading to sample degradation.17 Finally, dimethyl sulfoxide (DMSO), which is frequently used as a glassing agent for the preparation of DNP samples, is also photosensitive.
The purpose of this work is to demonstrate that phenylglyoxylic acid (PhGA) can also be used as an efficient radical precursor for DNP. PhGA has an excellent safety profile and no reported effect on metabolism on the time scale of the hyperpolarized 13C MR experiments.18 Because of its extended absorption into the visible spectrum, a radical can be photogenerated in PhGA at longer wavelengths and can therefore be used in conjunction with molecules that are sensitive to UV light. We show that [2-13C]DHAc can be hyperpolarized using the photogenerated radical of PhGA and a commercial 3.35 T HyperSense polarizer and that the resulting solution can be used for 13C MR metabolic studies in vivo.
Experimental Section
All chemicals were purchased from Sigma-Aldrich (Haverhill, U.K.), and data were processed in MATLAB (Mathworks, Natick, MA, USA), unless stated otherwise.
UV–Vis Absorption Measurements
The ultraviolet–visible (UV–vis) absorption spectra of PhGA, PA, DHAc, and DMSO in water (146 mM, 600 μL) were recorded using an Ocean Optics USB2000+ spectrometer and a DH-2000-BAL UV–vis–NIR light source (Halma PLC, Amersham, U.K.).
To estimate the efficiency of broadband and narrowband UV–vis light sources in generating photoinduced radicals in PA and PhGA, the power profile of each source (provided by the manufacturer) was multiplied by the absorption spectrum of PA and PhGA between 300 and 420 nm (Figure 1). The integral of the multiplied spectra was used to compare the effective light intensity provided by each source.
Figure 1.
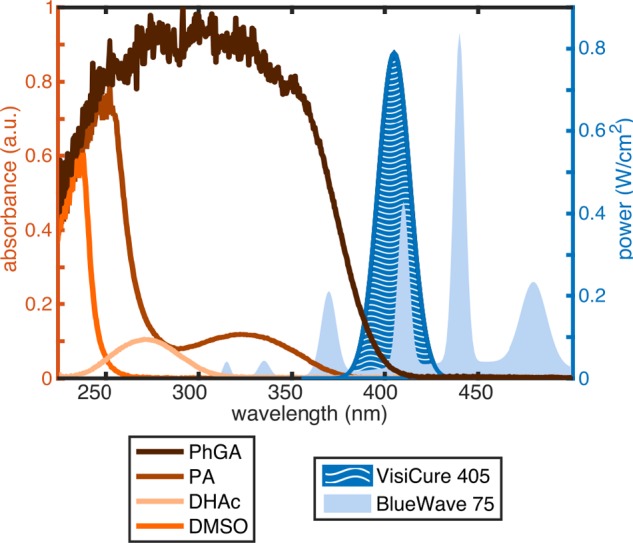
UV–vis absorption spectra (left axis) and light source power distributions (right axis). Power profiles were provided by the manufacturer (Dymax Europe GmbH, Wiesbaden, Germany).
Broadband and Narrowband Photoirradiation
To investigate the effects of photoirradiation on the selected radical precursors, two solutions were prepared: neat PA diluted to 7.0 M in a mixture of 1:1 glycerol/water (v/v) and sonicated for 5 min at 40 °C and a sample containing 7.1 M PhGA in a mixture of 1:1 glycerol/water (v/v) sonicated for 5 min at 40 °C. To study the relationship among the PhGA concentration, light source, and radical yield, the 7.1 M PhGA sample was diluted to 50, 25, and 7% of the original PhGA concentration in a mixture of 1:1 glycerol/water (v/v). Additionally, the effect of the light sources on two photosensitive solutions was tested: neat dimethyl sulfoxide (DMSO) and a sample containing 8.0 M DHAc dissolved in 2H2O were sonicated for 10 min at 40 °C.
Frozen beads of each solution were formed by dispensing droplets from a syringe into an ESR quartz dewar flask (Wilmad-Lab Glass WG-850-B-Q, Goss Scientific, Crewe, U.K.) filled with liquid nitrogen (LN2). The beads were photoirradiated using either a broadband source (Dymax BlueWave 75, Dymax Europe GmbH, Wiesbaden, Germany) or a narrowband source (Dymax BlueWave LED VisiCure 405 nm, Dymax Europe GmbH, Wiesbaden, Germany) operating at maximum power. From this point onward these two sources are referred to as BlueWave 75 and VisiCure 405. The standard used for UV irradiation is described in ref (19). Note that transmission through the quartz dewar was the same across the entire range of wavelengths used in this study.20
X-Band ESR Measurements and Radical Concentration Estimation
ESR spectra of single frozen beads inside the quartz dewar filled with LN2 were acquired using a benchtop X-band ESR spectrometer (MiniScope MS5000, Magnettech GmbH, Berlin, Germany). ESR parameters were optimized to resolve the hyperfine structure of the spectra and then kept constant throughout all of the experiments (number of accumulations = 1, B = [323–353] mT, sweep time = 20 s, modulation amplitude = 0.1 mT, modulation frequency = 100 kHz, and microwave power = 0.2 mW). Sequential ESR spectra were acquired for each bead after different irradiation times to obtain a radical build-up curve (Supporting Information Figure 2). The effect of deuterating PhGA on the ESR line width was investigated by substituting protonated PhGA in water for perdeuterated PhGA (d5-PhGA) in 2H2O. The synthesis of d5-PhGA is detailed in the Supporting Information.
Following ESR signal acquisition, the frozen bead was extracted from the quartz dewar, inserted into an Eppendorf tube, and weighed to estimate the volume of the bead. The radical concentration was determined by comparing the double integration of the first derivative ESR spectrum, corrected for bead volume (∼4 μL), with a calibration curve obtained from beads of known concentrations of 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPOL, Supporting Information Figure 1). Each measurement was repeated at least twice.
Dynamic Nuclear Polarization
Four samples were prepared for dissolution DNP: (sample 1) 8 M [2-13C]DHAc and 1 M PhGA in water; (sample 2) 8 M [2-13C]DHAc, 1 M PhGA, and 1.2 mM gadoteric acid (Gd3+, Dotarem, Guerbet, Roissy, France) in water; (sample 3) 8 M [2-13C]DHAc, 1 M d5-PhGA, and 1.2 mM Gd3+ in 2H2O; and (sample 4) 8 M [2-13C]DHAc, 21 mM OX063 trityl radical (Albeda Research Aps, Copenhagen, Denmark), and 1.2 mM Gd3+ in 1:3 DMSO/water (v/v). All solutions were sonicated for 5 min at 50 °C before the addition of Gd3+. The choice of PhGA concentration in samples 1–3 was based on the fact that hyperpolarized DHAc had previously been polarized with 21 mM OX063,14−16 a radical with a similar ESR line width to that obtained after the photoirradiation of PhGA, and that a similar concentration of the photogenerated radical (18–20 mM) can be obtained if the samples are doped with 1 M PhGA.
Between 7 and 11 beads of samples 1–3 were simultaneously irradiated for 200 s with the VisiCure 405 source. The beads were then placed into a standard HyperSense sample holder in contact with LN2 to preserve the radical and rapidly inserted into a HyperSense polarizer operating at 3.35 T and 1.25 K (Oxford Instruments, Abingdon, U.K). A microwave (μ-wave) sweep between 94.07 and 94.22 GHz (10 MHz steps, 10 min μ-wave irradiation/step) was performed for samples 2–4 and for a sample containing 7 M [1-13C]PA in 1:1 glycerol/water (v/v) irradiated for 400 s with the BlueWave 75 source. The background signal measured at each μ-wave frequency was subtracted from all μ-wave sweeps. For dissolution DNP, samples were polarized with the μ-wave source set to 94.110 GHz (except the sample used for the in vivo experiment, which was polarized at 94.205 GHz) and 100 mW for a period of 1.5–2 h. The samples were dissolved with 6 mL of a phosphate saline buffer (PBS).
Hyperpolarized 13C MR in a Phantom at 7 T
Liquid-state 13C MR acquisitions were carried out in a small-animal horizontal bore 7 T MR scanner (Agilent, Palo Alto, CA) using a 42-mm-diameter bird-cage 1H/13C transmit volume coil and a 20-mm-outer-diameter 13C receive surface coil (Rapid Biomedical GmbH, Rimpar, Germany). The temporal evolution of the hyperpolarized 13C signal was recorded in a 3 mL phantom starting 16 ± 1 s after dissolution using the following parameters: repetition time = 1 s, nominal flip angle = 9°, pulse width = 400 μs (sinc pulse truncated to five lobes), spectral width = 32 kHz. T1 relaxation time constants were determined by fitting an exponential decay function to the data and were corrected for the flip angle. (See the Supporting Information for details on the mathematical formula.)
The thermal equilibrium 13C MR signal was acquired with the same parameters but a longer repetition time (∼5 × T1), and the 13C hyperpolarization at the time of the first acquisition was calculated as the ratio between the hyperpolarized and thermal signals multiplied by the theoretical equilibrium polarization at 7 T and 298 K.
In Vivo Hyperpolarized 13C MR at 7 T
Procedures were performed in compliance with project and personal licenses issued under the United Kingdom Animals (Scientific Procedures) Act, 1986 and were approved by the Cancer Research UK, Cambridge Institute Animal Welfare and Ethical Review Body. A female C57B6 mouse (body weight = 32.2 g) was anesthetized with 2% isoflurane. Its body temperature was maintained at 37 °C, and it was placed inside the 7 T MR system. The coil setup was identical to the one used for the phantom experiments (a 42-mm-diameter bird-cage 1H/13C transmit volume coil and a 20-mm-outer-diameter 13C receive surface coil; Rapid Biomedical GMBH, Rimpar, Germany), with the receive surface coil placed over the liver of the mouse. Positioning of the surface coil was confirmed with sagittal, coronal, and axial T2-weighted 1H images.
Photoirradiated beads (∼30 μL) from sample 2 were polarized as described above with μ-wave irradiation at 94.205 GHz and dissolved in 6 mL of PBS. This solution (400 μL) was injected into the mouse via a tail vein over a period of 3 s. 13C MR acquisition started 12 s after the injection. The parameters used for 13C MR acquisition were the following: repetition time = 0.2 s, nominal flip angle = 15°, pulse width = 2 ms (sinc pulse truncated to five lobes), spectral width = 6 kHz, and total acquisition time = 80 s. The transmitter was centered at 72.3 ppm, and every 10th acquisition it was switched to 214 ppm for a single acquisition before returning to 72.3 ppm. Therefore, the spectral region around 72.3 ppm was sampled 360 times (every 0.2 s), and the region around 214 ppm was sampled 40 times (every 2 s). This high temporal resolution acquisition strategy was chosen to maximize the signal-to-noise ratio of the summed spectra assuming an in vivo T1 of 10 s for the metabolites. (See the analysis presented in ref (21).) A similar scheme has previously been used for the detection of glucose metabolism in vivo.22,23 The volume coil excited the whole body, and reception was coil-selective.
Liquid-State 13C MR at 14.1 T
Following the hyperpolarized 13C phantom experiments, the samples obtained after dissolution were mixed with 10% 2H2O, and their thermally polarized 13C MR spectrum was measured in a 600 MHz vertical-bore Bruker spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany). To further investigate the impact of photoirradiation on the solutions containing PhGA, the 13C spectra of two additional samples prepared by dissolving either a nonirradiated bead or a 200-s-irradiated bead (using VisiCure 405) in 600 μL PBS (10% 2H2O) were recorded for comparison. All four samples were measured with the following parameters: repetition time = 6 s, nominal flip angle = 30°, pulse width = 2.6 μs (hard pulse), and spectral width = 38 kHz. T1 values were measured using an inversion recovery sequence.
Results
UV–Vis Absorption and Light Source Power Profiles
The UV–vis spectra of the two precursors studied here show that PhGA has a broad absorption between 200 and 400 nm while PA has a weaker and narrower absorption at around λmax = 320 nm corresponding to the n−π* electronic transition characteristic of α-ketoacids.24 Metabolic substrate DHAc has an absorption peak at around λmax = 270 nm, and the DMSO solvent has one at around λmax = 240 nm (Figure 1).
The power dependence of the two light sources on wavelength was obtained from the manufacturer’s manual and is overlaid on Figure 1. While the total output power density was nearly the same for both light sources (19 W/cm2 for BlueWave 75 and 16.8 W/cm2 for VisiCure 405), the power distribution across the spectrum was radically different. On the basis of the multiplied spectra calculated from the UV–vis absorption of PA and PhGA and the wavelength-dependent power distribution of each light source, the BlueWave 75 light source was expected to be more effective for radical generation in PA while the VisiCure 405 source should be better suited for radical generation in PhGA since the ratio areabroadband/area405 nm is much larger for PA than for PhGA (15 vs 2.8).
Broadband and Narrowband Photoirradiation
PhGA and PA were irradiated with either the BlueWave 75 broadband light source or the VisiCure 405 narrowband light source. The associated ESR spectra are displayed in Figure 2a,b. The scheme of the photochemical pathway for radical production in PhGA and PA at 77 K is shown in the Supporting Information. Frozen PA and PhGA beads turned a yellow-orange color after irradiation, which became darker as the irradiation time increased (Figure 3a).
Figure 2.

X-band ESR spectra of frozen droplets in LN2, photoirradiated with a broadband (BlueWave 75) or a narrowband (VisiCure 405) light source. (a) PhGA (7.1 M) in 1:1 water/glycerol (v/v). (b) PA (7.0 M) in 1:1 water/glycerol (v/v). (c) DMSO (neat). (d) DHAc (8.0 M) in 2H2O. The spectral structures in panels b and c are caused by the hyperfine coupling to the proton spins of the methyl groups. The units on the y axis are the same in all four plots.
Figure 3.
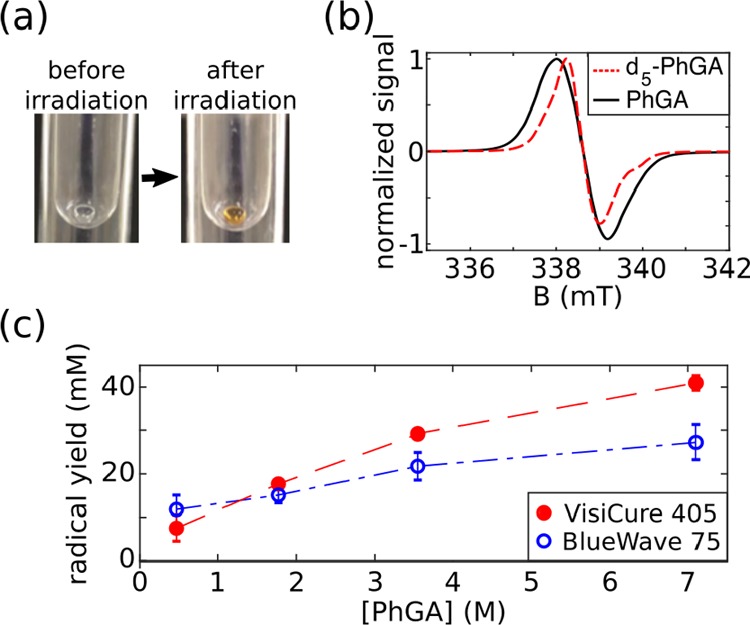
Photoirradiation of PhGA dissolved in 1:1 glycerol/water (v/v). (a) A 4 μL bead of PhGA in LN2 before and after photoirradiation. (b) ESR spectrum of unlabeled PhGA and d5-PhGA. (c) Maximum radical yield vs PhGA concentration using either the broadband (blue open circle) or the narrowband light source (red dots). The radical yield is reported as the mean ± standard deviation across three measurements.
Table 1 summarizes the radical yield for each precursor and light source. BlueWave 75 yielded 3.3 times more radical and was 3 times faster than VisiCure 405 when PA was the precursor. Conversely, the irradiation of PhGA with VisiCure 405 resulted in a 1.6-fold increase in radical yield compared to broadband irradiation, without a reduction in the build-up time. The radical yield increased with PhGA concentration (Figure 3c). It is noticeable that irradiation with either BlueWave 75 or the narrowband light source produced similar radical yields in solutions with PhGA concentrations below 1.77 M. However, irradiation with BlueWave 75 generated lower radical yields at higher PhGA concentrations.
Table 1. Maximum Radical Yield after Photoirradiation.
| precursor | radical yield (mM) BlueWave 75 | radical yield (mM) VisiCure 405 |
|---|---|---|
| PhGA (7.1M) | 25.7 ± 3.7 | 40.4 ± 5.8 |
| PA (7.0M) | 77.7 ± 11.2 | 23.8 ± 3.4 |
The ESR line width of PhGA was about 3 times narrower than that of PA. Perdeuteration of PhGA (d5-PhGA) resulted in an ∼10% narrower ESR line width than protonated PhGA (fwhm = 1.30 ± 0.05 mT vs fwhm = 1.45 ± 0.05 mT, Figure 3b) but did not change the radical yield.
Photosensitive metabolite DHAc and glassing agent DMSO were also irradiated with both light sources. Irradiation of DMSO and DHAc with BlueWave 75 produced radicals that were clearly observed in the ESR spectra, while irradiation with VisiCure 405 did not generate any observable radical (Figure 2c,d).
DNP of [2-13C]DHAc with a PhGA-Derived Radical
The samples containing 8 M [2-13C]DHAc and 1 M PhGA (or d5-PhGA) in water were strongly acidic (pH = 0.5 ± 0.5). A 200 s irradiation with VisiCure 405 was sufficient to reach a plateau in radical production with a yield of 18.1 ± 2.5 mM in the samples with and without Gd3+. (The variability of five measurements was ±0.5 mM.) No ESR signal originating from the photoirradiation of a 1 mM solution of Gd3+ in water could be detected (data not shown).
The width of the μ-wave spectrum of samples containing radical generated by the photolysis of PhGA was noticeably broader than the spectrum of the PA sample containing OX063 trityl radical but narrower than that for photoirradiated PA (Figure 4). However, unlike OX063, the μ-wave spectrum of the PhGA-derived radical was strongly asymmetric, with approximately 1.7 times higher 13C polarization when the μ-wave frequency is set to the value corresponding to the optimal negative polarization, namely, 94.205 ± 0.005 GHz. The build-up rate was, however, identical for both the negative and positive optimal frequencies, and there was no noticeable difference when unlabeled PhGA was replaced with d5-PhGA (Supporting Information Figure 3).
Figure 4.
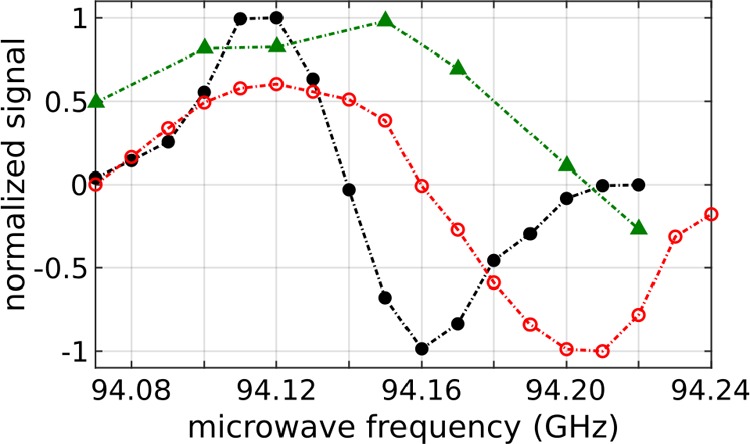
Solid-state microwave spectra at 3.35 T and 1.25 K: (open red circles) photoirradiated [2-13C]DHAc solution (8 M) in H2O doped with 1 M PhGA (18 mM radical); (black dots) [2-13C]DHAc solution (8 M) in H2O/DMSO (v/v) doped with 21 mM OX063; (green triangles) photoirradiated [1-13C]PA solution (7 M) in H2O/glycerol (65 mM radical).
Hyperpolarized [2-13C]DHAc
The build-up time constants as well as the liquid-state 13C polarization were measured for all of the [2-13C]DHAc samples (Table 2). The build-up time constant in samples containing PhGA was not affected by the addition of Gd3+ or the perdeuteration of PhGA. However, the liquid-state polarization increased by 1.7-fold upon addition of 1.2 mM Gd3+. The polarization values shown in Table 2 were measured approximately 16 s after the start of the dissolution process, which means that the estimated 13C polarization at the time of dissolution was ∼8% for the samples doped with Gd3+ and polarized with the μ-wave frequency set to 94.110 GHz. On the basis of the solid-state data, the 13C polarization at the time of dissolution should reach ∼14% if this sample was negatively polarized at 94.205 GHz.
Table 2. DNP Results Obtained with 8 M [2-13C]DHAc Samples Polarized Using a HyperSense Polarizer with Liquid-State Polarizations Measured 16 s after Dissolution at 7 T.
| [2-13C] DHAc doped with | build-up time constant (s) | liquid-state 13C polarization (%) |
|---|---|---|
| (1) PhGA | 1033 ± 82 | 3.1 ± 0.8 |
| (2) PhGA and Gd3+ | 1023 ± 97 | 5.1 ± 1.2 |
| (3) d5-PhGA and Gd3+ | 862 ± 81 | 4.7 ± 1.0 |
| (4) Trityl and Gd3+ | 1611 ± 353 | 16.0 ± 4.0 |
After dissolution, the pH was neutral in all cases (pH = 7.0 ± 0.5). The 13C longitudinal relaxation time constant (T1) of [2-13C]DHAc with PhGA was 35.2 ± 1.8 s at 7 T and 19.9 ± 1.0 s at 14.1 T. Radical quenching was confirmed by making seven frozen beads from the dissolved sample and averaging 100 ESR measurements to compensate for the dilution following dissolution. As confirmation, no ESR signal was observed.
[2-13C]DHAc (213.9 ppm) and [2-13C]DHAc hydrate (96.6 ppm) dominated the hyperpolarized 13C spectrum recorded in the 7 T MR scanner (Figure 5a). The two peaks at 199.4 and 175.3 ppm correspond to the resonances of [1-13C]PhGA and [2-13C]PhGA, respectively. No recombination products from the quenched radical were detected in the hyperpolarized 13C spectrum. A few impurities from the [2-13C]DHAc sample were observed at chemical shifts of 91.6, 94.9, 108.1, 105.9, and 112.7 ppm. The same small quantities of impurities were also detected in the same solution before it had been UV-irradiated or polarized by DNP (Figure 5b-iii). No additional peaks were detected in the thermally polarized spectra of the samples that had undergone photoirradiation or photoirradiation and subsequent DNP (Figure 5b).
Figure 5.
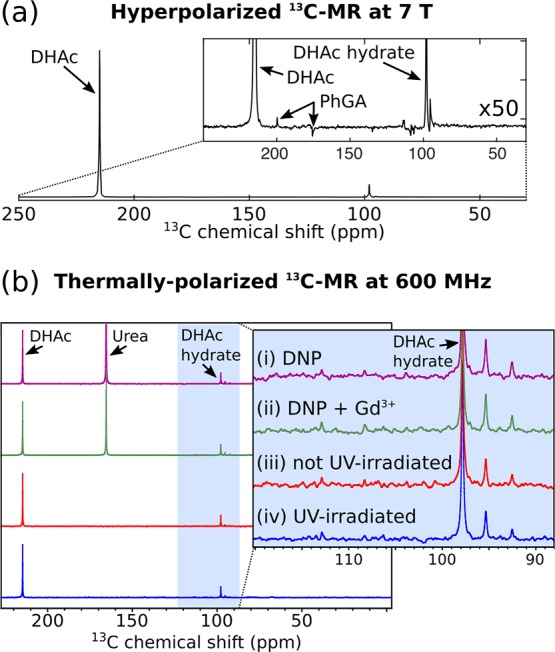
(a) Representative 13C MR spectrum (sum of 180 spectra) of a hyperpolarized solution containing 40 mM [2-13C]DHAc, 5 mM PhGA, and 6 μM Gd3+ in PBS. The MR acquisition started 16 s after the beginning of the dissolution process with a repetition time of 1 s and a nominal flip angle of 9°. (b) 13C MR spectra of thermally polarized solutions at 300 K and 14.1 T. All samples contained [2-13C]DHAc (40 mM-70 mM) and PhGA (4.5–8.0 mM) in PBS with 10% 2H2O. [13C]urea was added to samples i and ii after dissolution as a reference. These samples were made from frozen beads of 8 M [2-13C]DHAc and 1 M PhGA in water. (i) Frozen beads had been irradiated using a narrowband light source (VisiCure 405 nm) for 200 s and dissolution DNP performed on them. (ii) 1.2 mM Gd3+ had been added to the sample prior to photoirradiation and dissolution DNP. (iii) The frozen beads were dissolved in PBS without photoirradiation or DNP. (iv) The frozen beads were irradiated for 200 s and melted in PBS, but they did not undergo dissolution DNP.
In comparison, a sample prepared with [2-13C]DHAc doped with 21 mM OX063 and 1.2 mM Gd3+ polarized nearly 1.6 times slower than the samples with the PhGA-derived radical but reached three times higher liquid-state polarization levels (Table 2). Interestingly, the T1 at 7 T after dissolution of the [2-13C]DHAc sample with OX063 was 29.5 ± 0.5 s, ∼5 s shorter than that of the sample with a nonpersistent PhGA-derived radical. It was also slightly shorter at 14.1 T, where T1 was 17.3 ± 1.4 s for [2-13C]DHAc with OX063. Note that the presence of 6 μM of Gd3+ in the solution did not affect the T1 of [2-13C]DHAc.
In Vivo 13C MR of Hyperpolarized [2-13C]DHAc Metabolism
The 13C MR spectrum after the intravenous injection of 400 μL of a hyperpolarized solution containing 40 mM [2-13C]DHAc, 5 mM PhGA, and 6 μM Gd3+ into a 32.2 g mouse is shown in Figure 6. The injection of this solution did not cause any observable changes in the heart rate or respiratory pattern of the mouse. Since this sample was negatively polarized at 94.205 GHz, the 13C polarization was estimated to be 8.7% at the time of injection, which is 1.7 times higher than the 13C polarization recorded for the same sample positively polarized at 94.110 GHz (Table 2). Besides the injected DHAc and its hydrate, metabolic products were detected between 69 and 77 ppm. Note that the resonance appearing at ∼89 ppm has previously been reported in earlier in vivo studies,15 but it has not been assigned. The peaks at 73.8 and 71.8 ppm could be seen for the first 20 s (first 90 acquisitions), and DHAc could be monitored for approximately 60 s (29 acquisitions). The T1 of [2-13C]DHAc was determined to be 15.8 ± 0.3 s in vivo.
Figure 6.

(a) Axial and sagittal T2-weighted images through the mouse with approximate position of the surface coil. (Red dots represent the cross sections of the coil.) (b) 13C MR spectrum acquired in vivo at 7 T following the intravenous injection of 400 μL of a hyperpolarized solution into a mouse. The solution was composed of 40 mM [2-13C]DHAc, 5 mM PhGA, and 6 μM Gd3+ in PBS. The previously reported resonance detected at ∼89 ppm has not been assigned. DHAc region: sum of 40 spectra. Region with the metabolic products of DHAc and DHAc hydrate: sum of the first 70 spectra.
Discussion
PhGA as a Radical Precursor for DNP
Nonpersistent radicals can be created through UV–vis light irradiation of a precursor molecule. Although this is a simple procedure, the intense light used for the photogeneration of radicals may affect and degrade other molecules that are present in the sample. This is not the case for acetate or butyrate, which can be used in conjunction with PA,12,13 but some solvents (e.g., DMSO) and some metabolic substrates that are used with hyperpolarized 13C MR are photosensitive in the UV range. For example, most ketoacids, such asPA, absorb light at around 325 nm. DHAc, a metabolite that enters gluconeogenesis at the level of the trioses, has an absorption spectrum centered at 270 nm. We have shown that the irradiation of DHAc and DMSO with a broadband UV–vis source leads to the creation of radicals that are stable at 77 K. These radicals not only may affect the DNP process but also may represent the unwanted degradation of these molecules. We have demonstrated that this degradation can be avoided by using a narrowband light source in the visible spectrum in conjunction with PhGA as the radical precursor, which has an absorption spectrum that extends above 400 nm. The photochemistry of the radical generation upon photoirradiation of PhGA and PA at 77 K is shown in Supporting Information Figure 1.
A comparison between the efficiency of the two light sources in photogenerating radicals in PA and PhGA was made on the basis of the wavelength-dependent power profile of each light source and the absorption spectrum of the two precursors. This simple analysis assumes that radical generation is proportional to the density of photons absorbed by the precursors at each wavelength. Such a comparison is only qualitative and most likely depends on many other factors such as light penetration and precursor concentration. Nevertheless, it reproduces the features observed experimentally, namely, that while the broadband light source is more effective in PA radical photogeneration, the difference between the narrowband and the broadband light sources for PhGA radical photogeneration is relatively small, especially for samples containing ∼1 M precursor, which was the concentration used to polarize [2-13C]DHAc.
The X-band ESR line width of photoinduced PhGA radicals is 1.4 mT, which is comparable to the line width of the OX063 trityl radical and much narrower than the photoirradiated PA and TEMPOL line widths (Supporting Information Figure 4), with the latter nitroxyl radical having also previously been used to hyperpolarize 13C substrates by dissolution DNP.21 The four peaks observed in the ESR spectrum of PA (Figure 2b) are due to coupling of the unpaired electron in C2 with the methyl group. The PhGA-derived radical exhibits a narrower line width than the PA-derived radical because the methyl group is substituted by a phenyl group, thereby increasing the distance between the unpaired electron localized around the ketone carbon and the neighboring protons, consequently reducing the hyperfine coupling. However, the non-negligible g anisotropy of the photoinduced PhGA radical leads to a larger spread of the ESR absorption line compared to OX063, especially at the high fields where DNP is performed. Because narrow ESR line width radicals are generally more efficient for DNP,25 especially at 3.35 T, PhGA was expected to be a competitive candidate for hyperpolarized 13C MR experiments using the HyperSense polarizer. Although the maximum 13C polarization obtained with photoinduction was lower than the one obtained with OX063, it was larger than what can typically be obtained with TEMPOL using the Hypersense.26,27
The μ-wave spectrum acquired from samples polarized with photoirradiated PhGA was nearly as narrow as that of the sample doped with OX063 but was clearly asymmetric because of the g anisotropy of the PhGA-derived radical. Interestingly, no improvement in 13C polarization or no significant change in the μ-wave spectrum (Supporting Information Figure 3) could be detected using d5-PhGA, despite its 10% narrower ESR line width at the X-band as compared to that of unlabeled PhGA. Again, this is most likely due to the non-negligible g anisotropy of the PhGA-derived radical, which is exacerbated at 3.35 T as compared to X-band ESR measurements typically performed at 0.3 T.
At 3.35 T, the optimal concentration for the OX063 trityl radical is 15–21 mM,15,28 and that of TEMPOL is 30–50 mM.29 Since the ESR line width of the PhGA-derived radical is only slightly broader than that of OX063, it was assumed that a 15–25 mM radical should be photogenerated in the PhGA samples in order to polarize 13C-enriched substrates using a HyperSense polarizer. This radical concentration can be achieved by irradiating 1 M PhGA for 150–200 s with a light source. Increasing the PhGA concentration in solution increased the photogenerated radical concentration up to 40 mM.
Previous studies showed that by adding ∼1 mM Gd3+ to a trityl-doped sample 13C polarization could be greatly improved, possibly because Gd3+ causes a shortening in the T1 of the DNP-active electron spin in the low-temperature regime.26,27 We observed a similar trend in [2-13C]DHAc samples prepared with the PhGA-derived radical and doped with Gd3+, with a nearly 2-fold increase in 13C liquid-state polarization.
The radical yield with photoirradiated PhGA depended on pH. The highest radical yield was obtained when the solution containing the precursors was acidic, below the pKa of PhGA (pKa = 2.3), and it decreased as the pH was increased. A similar effect has been observed for UV-irradiated aqueous solutions of PA.30 Therefore, PhGA is a radical precursor that is better adapted for acidic formulations of DNP samples. Conversely, TEMPOL cannot be used to polarize acidic samples because it spontaneously degrades.
PhGA has itself been shown to be an interesting probe for detecting hydrogen peroxide using hyperpolarized 13C MR.18 The proposed method herein could obviously be used to hyperpolarize 13C-PhGA samples following photoirradiation with visible light. The presence of a 13C spin on the ketone carbon will broaden the ESR spectrum through an additional hyperfine coupling and hence will most likely reduce its efficiency for DNP at 3.35 T, but as already shown with 13C-PA, this can be mitigated by increasing the magnetic field of the polarizer and using μ-wave frequency modulation.27
Hyperpolarization of [2-13C]DHAc with a PhGA-Derived Radical
The highest liquid-state 13C polarization, measured in a phantom 16 s after dissolution and obtained after having positively polarized a [2-13C]DHAc sample containing a photogenerated PhGA radical, was 5%. The estimated liquid-state 13C polarization in the negatively polarized sample that was injected into the mouse for in vivo application was 8.7% 16 s after dissolution. This is approximately 2 times lower than the maximum 13C polarization obtained with the OX063 radical. In the framework of a potential clinical study, part of this difference could be recovered thanks to the fact that no filtration or quality control for residual radicals would be required, possibly allowing for a more rapid release of the dose for injection. In addition, because they can be annihilated in the solid state,11 nonpersistent photogenerated radicals can enable storage and transport, which would certainly benefit future clinical studies. It is also likely that the 13C polarization could be improved by performing DNP at higher magnetic fields and/or lower temperatures as well as by modulating the μ-wave frequency, as was previously demonstrated with the photoinduced radical in PA.12,19,31
An additional advantage of photogenerated radicals is the longer lifetime of 13C hyperpolarization in the liquid state. The 13C T1 of [2-13C]DHAc measured at 7 and 14.1 T immediately after dissolution was 15–20% longer in the solution prepared with PhGA than in the solution containing 0.1 mM OX063. This is a significant but not large change because the dominant relaxation mechanism at high magnetic fields is due to chemical shift anisotropy (CSA). However, at lower magnetic fields, such as in the stray magnetic field experienced by the solution during transfer between the polarizer and MR system, paramagnetic relaxation dominates,32 and the difference in T1 between the two types of solutions is expected to be much greater. Therefore, in a clinical setting where the transfer time is much longer and the solution might be located in a magnetic field as low as earth’s magnetic field, quenching the radical will help to maintain the 13C polarization until the solution is injected.33 Removing paramagnetic relaxation of the hyperpolarized nuclei will also benefit other research fields that use dissolution DNP as a signal enhancement method, such as low-field NMR and the study of long-lived singlet states.34,35
The impurities observed in the hyperpolarized 13C MR spectra of [2-13C]DHAc were also observed in a thermally polarized sample that had been neither photoirradiated nor hyperpolarized. This shows that the molecule of interest stays intact during both the photoirradiation and the dissolution DNP processes.
By analogy to what has been observed with photoirradiated beads of frozen PA, the only expected recombination products of the PhGA-derived radical are CO2 and benzoic acid.10 Electrospray ionization mass spectrometry analysis showed benzoic acid as the sole product of PhGA photoirradiation (Supporting Information Figure 5). This is further supported by the results of Lippert et al., who showed that, when reacted with hydrogen peroxide, the only product of hyperpolarized [2-13C]PhGA is [1-13C]benzoic acid.18
13C MR Measurements of Hyperpolarized [2-13C]DHAc Metabolism in Vivo
PhGA is a moderate acid (pKa = 2.3) with LD50 = 180 mg/kg in mice36 and is excreted via urinary elimination.37 In fact, a pharmacokinetic study where PhGA was injected intravenously into rats confirmed that PhGA did not metabolize and that it was excreted via urinary elimination within ∼50 min (apparent first-order rate of elimination of PhGA is ∼0.1025 min–1).38 The injection of 400 μL of the hyperpolarized [2-13C]DHAc solution (50 mM) containing 5 mM PhGA (∼LD50/20) into a 32 g mouse did not affect the monitored physiological parameters. Nevertheless, daily exposure to large doses of PhGA (above 200 mg/kg) should be avoided because of potential neurotoxicity due to its amination product, α-phenlyglycine, which leads to the depletion of striatal dopamine.39 Less than 100 μM CO2 and benzoic acid was expected to be present in the solution following recombination of the PhGA-derived radical. The CO2 most likely escaped during the dissolution process, and such a low concentration of benzoic acid is below the doses used therapeutically in humans.40
Many liver pathologies, such as nonalcoholic fatty-liver disease, are characterized by an aberrant glucose metabolism. Hyperpolarized [2-13C]DHAc, which follows gluconeogenesis, glycolysis, and fatty acid synthesis, has previously been used to study hepatic metabolism. For example, the metabolic products of hyperpolarized [2-13C]DHAc in perfused mouse livers reported on differences between fed and fasted metabolic states.14 Hepatic metabolic changes after a fructose injection were also detected in vivo using hyperpolarized [2-13C]DHAc.16
Here, the main metabolic product of hyperpolarized [2-13C]DHAc in the liver, glycerol 3-phosphate, was clearly seen in the 13C MR spectrum as a doublet (75.7 and 71.8 ppm), with glyceraldehyde 3-phosphate in the middle at 73.8 ppm. Smaller peaks were also detected, which are probably metabolites of the gluconeogenic and glycolytic pathways, including lactate, glucose, and glycerol, as reported in a 1H-decoupled spectrum of a perfused liver.14 Although it has been observed in previous in vivo studies,16 the [2-13C]phosphoenolypyruvate resonance (∼152 ppm) was not observed in this experiment because it was outside the excitation bandwidth of the pulse used for radio frequency excitation.
The 13C T1 of [2-13C]DHAc in vivo was ∼16 s at 7 T. This is significantly shorter than the ∼25 s previously reported in a 3 T MRI scanner,15 supporting the conclusion derived from our phantom experiments that CSA is an important relaxation mechanism at high field. The apparent signal decay time constants of the metabolic products and DHAc hydrate were similar to that reported for rats in vivo.16 The DHAc-to-metabolite ratio was ∼70, which is about 2.5 times higher than that reported in perfused liver.14 This is consistent with the fact that regions outside the liver contribute to the DHAc signal, while its metabolites are mainly produced in the liver. These data show that the method proposed herein can be used for metabolic studies of the liver in vivo.
Conclusions
We have described a method to generate a photoinduced radical for dissolution DNP in samples containing photosensitive molecules. The method was used to hyperpolarize [2-13C]DHAc with radical precursor PhGA using the commercial HyperSense polarizer. The nonpersistent radical generated by the photoirradiation of PhGA was quenched after dissolution, increasing the 13C longitudinal relaxation time. The liquid-state 13C polarization was only half of the maximum polarization obtained with the OX063 trityl radical but was nevertheless sufficient to report on the metabolism of gluconeogenic and glycolytic probe [2-13C]DHAc in vivo. The 13C polarization could be improved by using a higher-field hyperpolarizer and optimizing the photogenerated radical concentration. Photoirradiated PhGA could also become an attractive polarizing agent for the remote production of hyperpolarized 13C molecules since it was demonstrated that nonpersistent photogenerated radicals can be annihilated in the solid state.11
Acknowledgments
The authors thank Dr. De-en Hu, Dominik McIntyre, and Madhu Basetti for technical help and Dr. Michael Tayler (University of Cambridge) and Mark Van Criekinge (University of California, San Francisco) for fruitful discussions. This work is part of a project that has received funding from the European Union’s Horizon 2020 European Research Council (ERC Consolidator Grant) under grant agreement no. 682574 (ASSIMILES). Funding was also received from a Cancer Research UK Programme grant (17242) and from the CRUK-EPSRC Imaging Centre in Cambridge and Manchester (16465). F.K. and S.P. received funding from the European Union’s Horizon 2020 Research and Innovation Program under Marie Sklodowska-Curie grant agreement no. 642773 (EUROPOL). A. Capozzi received funding from the European Union’s Horizon 2020 Research and Innovation Program under Marie Sklodowska-Curie grant agreement no. 713683 (COFUNDfellowsDTU).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b09326.
Supporting figures, a mathematical description of the hyperpolarized signal corrected by the flip angle and T1 relaxation, and details of the perdeuteration of phenylglyoxylic acid (PDF)
The authors declare the following competing financial interest(s): A. Comment is currently employed by General Electric Medical Systems, Inc.
Supplementary Material
References
- Ardenkjaer-Larsen J. H.; Fridlund B.; Gram A.; Hansson G.; Hansson L.; Lerche M. H.; Servin R.; Thaning M.; Golman K. Increase in Signal-to-Noise Ratio of > 10,000 Times in Liquid-State NMR. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (18), 10158–10163. 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Heo G. S.; Zeng H.; Wooley K. L.; Hilty C. Detection of Living Anionic Species in Polymerization Reactions Using Hyperpolarized NMR. J. Am. Chem. Soc. 2013, 135 (12), 4636–4639. 10.1021/ja4001008. [DOI] [PubMed] [Google Scholar]
- Marco-Rius I.; Comment A. In Vivo Hyperpolarized 13C MRS and MRI Applications. In Encyclopedia of Magnetic Resonance; Harris R. K., Wasylishen R. L., Eds.; John Wiley & Sons, Ltd: Chichester, U.K., 2018. [Google Scholar]
- Lumata L.; Ratnakar S. J.; Jindal A.; Merritt M.; Comment A.; Malloy C.; Sherry A. D.; Kovacs Z. BDPA: An Efficient Polarizing Agent for Fast Dissolution Dynamic Nuclear Polarization NMR Spectroscopy. Chem. - Eur. J. 2011, 17 (39), 10825–10827. 10.1002/chem.201102037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. J.; Kurhanewicz J.; Vigneron D. B.; Larson P. E. Z.; Harzstark A. L.; Ferrone M.; van Criekinge M.; Chang J. W.; Bok R.; Park I.; Reed G.; Carvajal L.; Small E. J.; Munster P.; Weinberg V. K.; Ardenkjaer-Larsen J. H.; Chen A. P.; Hurd R. E.; Odegardstuen L.-I.; Robb F. J.; Tropp J.; Murray J. A. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1-13C]Pyruvate. Sci. Transl. Med. 2013, 5 (198), 198ra108. 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle K. M. Imaging Metabolism with Hyperpolarized 13C-Labeled Cell Substrates. J. Am. Chem. Soc. 2015, 137, 6418–6427. 10.1021/jacs.5b03300. [DOI] [PubMed] [Google Scholar]
- Kurhanewicz J.; Vigneron D. B.; Brindle K.; Chekmenev E. Y.; Comment A.; Cunningham C. H.; DeBerardinis R. J.; Green G. G.; Leach M. O.; Rajan S. S.; Rizi R. R.; Ross B. D.; Warren W. S.; Malloy C. R. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13 (2), 81–97. 10.1593/neo.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyperpolarized [13C] Pyruvate Documentation Page | Cancer Imaging Program (CIP). https://imaging.cancer.gov/programs_resources/cancer-tracer-synthesis-resources/hyperpolarized-C13-pyruvate-documentation.htm, accessed Oct 6, 2018.
- Ardenkjaer-Larsen J. H.; Leach A. M.; Clarke N.; Urbahn J.; Anderson D.; Skloss T. W. Dynamic Nuclear Polarization Polarizer for Sterile Use Intent. NMR Biomed. 2011, 24 (8), 927–932. 10.1002/nbm.1682. [DOI] [PubMed] [Google Scholar]
- Eichhorn T. R.; Takado Y.; Salameh N.; Capozzi A.; Cheng T.; Hyacinthe J. N.; Mishkovsky M.; Roussel C.; Comment A. Hyperpolarization without Persistent Radicals for in Vivo Real-Time Metabolic Imaging. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (45), 18064–18069. 10.1073/pnas.1314928110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi A.; Cheng T.; Boero G.; Roussel C.; Comment A. Thermal Annihilation of Photo-Induced Radicals Following Dynamic Nuclear Polarization to Produce Transportable Frozen Hyperpolarized 13C-Substrates. Nat. Commun. 2017, 8, 15757. 10.1038/ncomms15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi A.; Hyacinthe J. N.; Cheng T.; Eichhorn T. R.; Boero G.; Roussel C.; Van Der Klink J. J.; Comment A. Photoinduced Nonpersistent Radicals as Polarizing Agents for X-Nuclei Dissolution Dynamic Nuclear Polarization. J. Phys. Chem. C 2015, 119 (39), 22632–22639. 10.1021/acs.jpcc.5b07315. [DOI] [Google Scholar]
- Bastiaansen J. A. M.; Yoshihara H. A. I.; Capozzi A.; Schwitter J.; Gruetter R.; Merritt M. E.; Comment A. Probing Cardiac Metabolism by Hyperpolarized 13C MR Using an Exclusively Endogenous Substrate Mixture and Photo-Induced Nonpersistent Radicals. Magn. Reson. Med. 2018, 79 (5), 2451–2459. 10.1002/mrm.27122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno K. X.; Satapati S.; DeBerardinis R. J.; Burgess S. C.; Malloy C. R.; Merritt M. E. Real-Time Detection of Hepatic Gluconeogenic and Glycogenolytic States Using Hyperpolarized [2–13C]Dihydroxyacetone. J. Biol. Chem. 2014, 289, 35859–35867. 10.1074/jbc.M114.613265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Rius I.; Cao P.; von Morze C.; Merritt M.; Moreno K. X.; Chang G.-Y.; Ohliger M. a.; Pearce D.; Kurhanewicz J.; Larson P. E. Z.; Vigneron D. B. Multiband Spectral-Spatial RF Excitation for Hyperpolarized [2–13C]Dihydroxyacetone 13C-MR Metabolism Studies. Magn. Reson. Med. 2017, 77 (4), 1419–1428. 10.1002/mrm.26226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Rius I.; von Morze C.; Sriram R.; Cao P.; Chang G.-Y.; Milshteyn E.; Bok R. A.; Ohliger M. A.; Pearce D.; Kurhanewicz J.; Larson P. E. Z.; Vigneron D. B.; Merritt M. Monitoring Acute Metabolic Changes in the Liver and Kidneys Induced by Fructose and Glucose Using Hyperpolarized [2–13C]Dihydroxyacetone. Magn. Reson. Med. 2017, 77 (1), 65–73. 10.1002/mrm.26525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission, D.-G . for H. and C. Opinion on Dihydroxyacetone, 2010. [Google Scholar]
- Lippert A. R.; Keshari K. R.; Kurhanewicz J.; Chang C. J. A Hydrogen Peroxide-Responsive Hyperpolarized 13C MRI Contrast Agent. J. Am. Chem. Soc. 2011, 133 (11), 3776–3779. 10.1021/ja111589a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi A.; Karlsson M.; Petersen J. R.; Lerche M. H.; Ardenkjaer-Larsen J. H. Liquid-State 13C Polarization of 30% through Photo-Induced Non-Persistent Radicals. J. Phys. Chem. C 2018, 122 (13), 7432–7443. 10.1021/acs.jpcc.8b01482. [DOI] [Google Scholar]
- Heraeus. SUPRASIL 1, 2, 3 and SUPRASIL Standard. http://www.unitedlens.com/pdf/suprasil.pdf, accessed July 10, 2018.
- Comment A. Dissolution DNP for in Vivo Preclinical Studies. J. Magn. Reson. 2016, 264, 39–48. 10.1016/j.jmr.2015.12.027. [DOI] [PubMed] [Google Scholar]
- Rodrigues T. B.; Serrao E. M.; Kennedy B. W. C.; Hu D.-E.; Kettunen M. I.; Brindle K. M. Magnetic Resonance Imaging of Tumor Glycolysis Using Hyperpolarized 13C-Labeled Glucose. Nat. Med. 2014, 20 (1), 93–97. 10.1038/nm.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishkovsky M.; Anderson B.; Karlsson M.; Lerche M. H.; Sherry A. D.; Gruetter R.; Kovacs Z.; Comment A. Measuring Glucose Cerebral Metabolism in the Healthy Mouse Using Hyperpolarized 13C Magnetic Resonance. Sci. Rep. 2017, 7 (1), 4–11. 10.1038/s41598-017-12086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapf R. J.; Perkins R. J.; Carpenter B. K.; Vaida V. Mechanistic Description of Photochemical Oligomer Formation from Aqueous Pyruvic Acid. J. Phys. Chem. A 2017, 121 (22), 4272–4282. 10.1021/acs.jpca.7b03310. [DOI] [PubMed] [Google Scholar]
- Comment A.; Merritt M. E. Hyperpolarized Magnetic Resonance as a Sensitive Detector of Metabolic Function. Biochemistry 2014, 53 (47), 7333–7357. 10.1021/bi501225t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardenkjaer-Larsen J. H.; Macholl S.; Johannesson H. Dynamic Nuclear Polarization with Triyls at 1.2 K. Appl. Magn. Reson. 2008, 34, 509–522. 10.1007/s00723-008-0134-4. [DOI] [Google Scholar]
- Lumata L.; Merritt M. E.; Malloy C. R.; Sherry A. D.; Kovacs Z. Impact of Gd3+ on DNP of [1–13C]Pyruvate Doped with Trityl OX063, BDPA, or 4-Oxo-TEMPO. J. Phys. Chem. A 2012, 116 (21), 5129–5138. 10.1021/jp302399f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber J.; Ellner F.; Fridlund B.; Gram A.; Jóhannesson H.; Hansson G.; Hansson L. H.; Lerche M. H.; Månsson S.; Servin R.; Thaning M.; Golman K.; Ardenkjær-Larsen J. H. Generating Highly Polarized Nuclear Spins in Solution Using Dynamic Nuclear Polarization. Nucl. Instrum. Methods Phys. Res., Sect. A 2004, 526, 173–181. 10.1016/j.nima.2004.03.171. [DOI] [Google Scholar]
- Comment A.; van den Brandt B.; Uffmann K.; Kurdzesau F.; Jannin S.; Konter J. A.; Hautle P.; Wenckebach W. T.; Gruetter R.; van der Klink J. J. Design and Performance of a DNP Prepolarizer Coupled to a Rodent MRI Scanner. Concepts Magn. Reson., Part B 2007, 31 (4), 255–269. 10.1002/cmr.b.20099. [DOI] [Google Scholar]
- Rapf R. J.; Dooley M. R.; Kappes K.; Perkins R. J.; Vaida V. PH Dependence of the Aqueous Photochemistry of α-Keto Acids. J. Phys. Chem. A 2017, 121 (44), 8368–8379. 10.1021/acs.jpca.7b08192. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Capozzi A.; Takado Y.; Balzan R.; Comment A. Over 35% Liquid-State 13C Polarization Obtained via Dissolution Dynamic Nuclear Polarization at 7 T and 1 K Using Ubiquitous Nitroxyl Radicals. Phys. Chem. Chem. Phys. 2013, 15 (48), 20819. 10.1039/c3cp53022a. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Mishkovsky M.; Bastiaansen J. A.; Ouari O.; Hautle P.; Tordo P.; van den Brandt B.; Comment A. Automated Transfer and Injection of Hyperpolarized Molecules with Polarization Measurement Prior to in Vivo NMR. NMR Biomed. 2013, 26, 1582–1588. 10.1002/nbm.2993. [DOI] [PubMed] [Google Scholar]
- Comment A. The Benefits of Not Using Exogenous Substances to Prepare Substrates for Hyperpolarized MRI. Imaging Med. 2014, 6 (1), 1–3. 10.2217/iim.13.75. [DOI] [Google Scholar]
- Tayler M. C. D.; Levitt M. H. Paramagnetic Relaxation of Nuclear Singlet States. Phys. Chem. Chem. Phys. 2011, 13 (20), 9128–9130. 10.1039/c1cp20471h. [DOI] [PubMed] [Google Scholar]
- Marco-Rius I.; Tayler M. C. D.; Kettunen M. I.; Larkin T. J.; Timm K. N.; Serrao E. M.; Rodrigues T. B.; Pileio G.; Ardenkjaer-Larsen J. H.; Levitt M. H.; Brindle K. M. Hyperpolarized Singlet Lifetimes of Pyruvate in Human Blood and in the Mouse. NMR Biomed. 2013, 26 (12), 1696–1704. 10.1002/nbm.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigma-Aldrich. Phenylglyoxylic acid. https://www.sigmaaldrich.com/MSDS/MSDS/DisplayMSDSPage.do?country=GB&language=en&productNumber=B13055&brand=ALDRICH&PageToGoToURL=https%3A%2F%2Fwww.sigmaaldrich.com%2Fcatalog%2Fproduct%2Faldrich%2Fb13055%3Flang%3Den, accessed July 9, 2018.
- Guillemin M. P.; Bauer D. Human Exposure to Styrene III. Elimination Kinetics of Urinary Mandelic and Phenylglyoxylic Acids after Single Experimental Exposure. Int. Arch. Occup. Environ. Health 1979, 44, 249–263. 10.1007/BF00381658. [DOI] [PubMed] [Google Scholar]
- Amin Y. M.; Nagwekar J. B. Utilization of Model Compounds to Evaluate Effects of Slight Chemical Modifications on Their Distribution Pharmacokinetic Parameters in Rats and Mechanisms Inferred for Their Transmembrane Transport. J. Pharm. Sci. 1975, 64 (11), 1804–1812. 10.1002/jps.2600641112. [DOI] [PubMed] [Google Scholar]
- Terre’blanche G.; Heyer N.; Bergh J. J.; Mienie L. J.; van der Schyf C. J.; Harvey B. H. The Styrene Metabolite, Phenylglyoxylic Acid, Induces Striatal-Motor Toxicity in the Rat: Influence of Dose Escalation/Reduction over Time. Neurotoxic. Res. 2011, 20, 97–101. 10.1007/s12640-010-9222-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batshaw M. L.; MacArthur R. B.; Tuchman M. Alternative Pathway Therapy for Urea Cycle Disorders: Twenty Years Later. J. Pediatr. 2001, 138, S46–S55. 10.1067/mpd.2001.111836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.