

Abstract
Early events in the formation of the thalamocortical tract remain poorly understood. Recent work has suggested that thalamocortical axons follow a path pioneered by transient thalamic afferents originating from the medial part of the ventral telencephalon. We studied the development of these transient afferents and the thalamocortical tract in mutant mice lacking transcription factors normally expressed in the dorsal thalamus or ventral telencephalon. Pax6 is expressed in the dorsal thalamus, but not in the medial part of the ventral telencephalon, and the thalamocortical tract fails to form inPax6−/− embryos. We found that transient thalamic afferents from the ventral telencephalon do not form in Pax6−/−embryos; this may contribute to the failure of their thalamocortical development. The distribution ofPax6−/− cells inPax6−/−↔Pax6+/+ chimeras supports conclusions drawn from forebrain marker gene expression that Pax6 is not required for the normal development of the medial part of the ventral telencephalon but is required in the dorsal thalamus. Failure of the transient afferent pathway to develop is therefore likely a cell nonautonomous defect reflecting primary defects in the thalamus. We then examined the formation of thalamic afferents and efferents inFoxg1−/− embryos, which lack recognizable ventral telencephalic structures. In these embryos thalamic efferents navigate correctly through the thalamus but fail to turn laterally into the telencephalon, whereas other axons are able to cross the diencephalic/telencephalic boundary. Our results support a role for the ventral telencephalon in guiding the early development of the thalamocortical tract and identify a new role for the transcription factor Pax6 in regulating the ability of the thalamus to attract ventral telencephalic afferents.
Keywords: thalamocortical tract, diencephalon, ventral telencephalon, Pax6, Foxg1, transcription factor, axon guidance, chimera, optic tract, mouse, transient thalamic afferents, tract tracing
The thalamocortical tract supplies the cerebral cortex with innervation from the dorsal thalamus. In the mouse, thalamocortical axons emerge from the dorsal thalamus at around embryonic day 13 (E13) (Braisted et al., 1999; Tuttle et al., 1999;Auladell et al., 2000), approximately coincident with the cessation of neurogenesis in this structure (Angevine, 1970). The axons then navigate ventrally through the thalamus and make a sharp lateral turn, avoiding the hypothalamus and entering the ventral telencephalon to form the internal capsule. The first thalamic axons arrive in the cerebral cortex around E15 and form synaptic connections during subsequent development (Braisted et al., 1999; Tuttle et al., 1999;Auladell et al., 2000). Thalamic growth cones perform several maneuvers on their way to the cortex, and their behavior at each point is defined by the properties of the growth cones and the cells and secreted molecules that they encounter (Braisted et al., 1999,2000; Garel et al., 1999; Tuttle et al., 1999; Pratt et al., 2000;Skaliora et al., 2000).
A model to explain how the thalamocortical tract forms postulates a role for transient axons projecting from cells in the medial part of the ventral telencephalon into the dorsal thalamus. According to this model, these early thalamic afferents guide thalamocortical efferents into the ventral telencephalon (Metin and Godement, 1996; Tuttle et al., 1999). This raises the possibility that although later in development the dorsal thalamus is the source of thalamocortical axons, one of its important early functions is to accept innervation from the ventral telencephalon. In this study we investigated how disturbances inflicted on these tissues by mutating the transcription factorsPax6 or Foxg1 affect the formation of this transient afferent tract and the subsequent trajectory of the thalamocortical tract.
Pax6 is expressed dynamically in the diencephalon, the lateral part of the ventral telencephalon, and the cerebral cortex during the formation of the thalamocortical tract (Stoykova et al., 1996, 2000; Warren and Price, 1997; Hirata et al., 2002). The thalamocortical tract does not form in Pax6−/− mouse (Pratt et al., 2000; Hevner et al., 2002) and rat (Kawano et al., 1999) embryos. We used tract tracing inPax6−/− embryos, immunohistochemistry for Mash1 expression, and analysis of the distribution of Pax6−/−cells in Pax6−/−↔Pax6+/+ chimeras to test the hypothesis that the failure of the thalamocortical tract to form is preceded by a failure of the dorsal thalamus to receive transient afferents from the medial part of the ventral telencephalon.
Foxg1 (formerly BF-1) is expressed in the telencephalon (with highest levels of expression ventrally), in the nasal retina of the eye, and not in the diencephalon. The phenotype ofFoxg1−/− embryos reflectsFoxg1 expression with the telencephalon reduced in size, the eye distorted, and the diencephalon appearing normal. Judging from the expression domains of forebrain marker genes (Foxg1,Pax6, Emx2, and Dlx2), theFoxg1−/− telencephalon lacks recognizable ventral structures but possesses a structural correlate of the cerebral cortex, and the diencephalon is normal (Xuan et al., 1995;Dou et al., 1999; Huh et al., 1999). We examined the trajectories of thalamic and retinal axons inFoxg1−/− embryos to further test the hypothesis that a normal ventral telencephalon is required to guide thalamocortical axons laterally toward the cerebral cortex.
MATERIALS AND METHODS
Pax6 and Foxg1 alleles. BothPax6 alleles (Pax6Sey andPax6Sey-Neu1) used in this study are predicted to cause loss of Pax6 function (Favor et al., 1988; Hill et al., 1991; Quinn et al., 1996).Pax6Sey/Sey,Pax6Sey/Sey-Neu1, andPax6Sey-Neu1/Sey-Neu1 embryos have been reported to have the same phenotypic abnormalities (Quinn et al., 1996) and are denoted Pax6−/−. Replacement of the coding sequences of Foxg1 with LacZ coding sequences generated the Foxg1tm1M allele, which causes loss of Foxg1 function (Xuan et al., 1995; Dou et al., 1999, Huh et al., 1999). TheFoxg1tm1(cre)skm allele used in this study was generated by targeted replacement of Foxg1 coding sequences with Cre recombinase coding sequences [using a targeting vector otherwise identical to that used by Xuan et al. (1995)] (Hebert and McConnell, 2000). The anatomical defects reported for Foxg1tm1M/tm1M embryos were recapitulated in the Foxg1tm1(cre)skm/tm1(cre)skm embryos used in this study, and these are denotedFoxg1−/−.
Animals for tract tracing. Pax6−/− andFoxg1−/− embryos were obtained from Pax6Sey/+ ×Pax6Sey/+ and Foxg1tm1(cre)skm/+ × Foxg1tm1(cre)skm/+ timed matings and were identified by anatomical features described previously (Hill et al., 1991; Xuan et al., 1995; Quinn et al., 1997; Huh et al., 1999). The day of finding a vaginal plug was designated E0.5. Control embryos were obtained from wild-type timed matings or nonhomozygous littermates. The numbers of embryos examined in this study were as follows: injections into dorsal thalamus: E12.5: control, n = 5;Pax6−/−, n = 6; Foxg1−/−,n = 5; E13.5: control, n = 3;Foxg1−/−, n= 3; E14.5: control, n = 3;Pax6−/−, n = 3; Foxg1−/−,n = 3; E15.5: control, n = 3;Pax6−/−, n = 3;Foxg1−/−, n= 2; injections into E15.5 telencephalon: control, n = 9; Foxg−/−,n = 9; injections into E15.5 optic cup: control,n = 3;Foxg1−/−, n= 3. For each embryo both left and right sides of the brain (or both eyes) were injected with dioctadecyltetramethylindocarbocyanine perchlorate (DiI) (Molecular Probes, Eugene, OR).
DiI labeling and imaging. Whole embryos (E12.5, E13.5) or heads (E14.5, E15.5) were fixed overnight at 4°C in 4% paraformaldehyde (PFA) in PBS. For injections into the dorsal thalamus, a coronal cut was made in the head to reveal the caudal end of the thalamus. For injections into the optic cup, the lens of the eye was removed to reveal the surface of the retina. For injections into the telencephalon, cuts were made in the head to reveal the surface of the telencephalon. DiI was applied to exposed telencephalon or thalamus by stabbing several times with a pulled glass pipette coated with microscopic DiI crystals. In the case of the eyes, the optic cup was packed with larger clumps of DiI crystals. Embryos were returned to 4% PFA in PBS in the dark at room temperature for ∼1 month to allow DiI to diffuse along axons. Coronal vibratome sections (200 μm) were counterstained with the nuclear dye TOPRO3 (0.2 μm; Molecular Probes) and cleared by serial transfer through 1:1 glycerol/PBS and 9:1 glycerol/PBS at room temperature until sections sank. Sections were stored at 4°C. Images were acquired using either an epifluorescence microscope equipped with a TRITC filter set and digital camera (Leica Microsystems, Hiedelberg, Germany) or a TCS NT confocal microscope (Leica Microsystems). In epifluorescence images, DiI appears orange and TOPRO3 appears red, and in confocal images DiI appears red and TOPRO3 appears blue.
Mash1 immunohistochemistry. Pax6+/+ andPax6Sey/Sey embryos were produced fromPax6Sey/+ heterozygous timed matings and fixed overnight in 4% PFA/PBS before processing to wax blocks. Wax sections were cut at 10 μm and mounted on poly-l-lysine (Sigma)–coated glass slides. Sections were rehydrated through alcohols to PBS. Antigen unmasking was achieved by microwaving in 10 mm sodium citrate buffer, pH 6.0. Sections were blocked with 10% normal goat serum in 0.1% Triton X-100/PBS. Primary antibody (anti-MASH1, 1:100; BD PharMingen) in blocking solution (20% normal goat serum in 0.1% Triton X-100/PBS) was applied overnight. Secondary antibody (biotin-conjugated goat anti-mouse, 1:200 in blocking solution; Dako) was then applied for 1 hr, before final visualization using streptavidin-conjugated Alexaflour 488 (1:200; Molecular Probes) at room temperature in the dark. Sections were mounted using 10% Vectashield (Vector Laboratories) in 1:1 glycerol/PBS, and images were captured using confocal microscopy.
Production of chimeras. Chimeras were produced as described in Quinn et al. (1996). In brief, eight-cell embryos were obtained from the parental cross Pax6Sey-Neu1/+,Gpi1b/b female ×Pax6+/Sey,Gpi1b/b, Tg/Tg male, where Tg denotes the presence of the reiterated β-globin transgene TgN(Hbb-b1)83Clo (Lo, 1986). Pax6+/+,Pax6Sey-Neu1/+,Pax6+/Sey, andPax6Sey-Neu1/Sey embryos were produced, all of which were Gpi1b/b and contained a single copy of the β-globin transgene (Tg+). Donor embryos for aggregation were obtained from inbred BALB/c mice, which werePax6+/+,Gpi1a/a, and negative for the β-globin transgene (Tg−). Embryos were collected from superovulated females at 2.5 d after coitum and aggregated according to West and Flockhart (1994). Aggregated embryos were cultured overnight, transferred to recipient pseudopregnant F1 homozygous albino andGpi1c/c females (Pax6+/+,Gpi1c/c, Tg−), and allowed to develop to E12.5.
Tail and forelimb samples were taken for colorimetric analysis of GPI1 isotype contribution to give a global proportion of chimerism for each embryo (West and Flockhart, 1994). In all embryos, all cells derived from the eight-cell wild-type embryo wereGpi1a/a, and all cells derived from thePax6Sey-Neu1/+ ×Pax6+/Sey cross wereGpi1b/b. A mean of tail and forelimb GPI1B proportion was determined for each embryo. The genotype of each chimera was determined by PCR and restriction digest analysis of genomic DNA as described in Quinn et al. (1996). The use of two mutant Pax6 alleles,PaxSey (Hill et al., 1991) andPax6Sey-Neu1 (Favor et al., 1988), allowed identification of chimeras homozygous for the Pax6 mutations (Pax6SeyNeu-1/Sey) and those heterozygous (Pax6Sey-Neu1/+ andPax6+/Sey) or wild type (Pax6+/+) at the Pax6 locus.
Identification of cells derived from the progeny of thePax6Sey-Neu1/+ ×Pax6+/Sey crosses was achieved by DNA–DNAin situ hybridization using a digoxygenin (DIG)-labeled probe to the reiterated β-globin transgene (Keighren and West, 1993;Quinn et al., 1996). In brief, the head of each embryo was dissected and fixed in alcohol/acetic acid (3:1, v/v). Samples were processed, embedded in wax blocks, sectioned at 7 μm, and mounted on 3-aminopropyltriethoxysilane (Sigma)-coated slides. Sections were hybridized with DIG-labeled DNA probe, detected with peroxidase-labeled antibody, and visualized by diaminobenzidine staining. Slides were counterstained with hematoxylin and eosin and examined using bright-field and phase-contrast light microscopy.
Analysis of chimeras. Two balanced chimeras (those in which the ratio of cells derived from thePax6Sey-Neu1/+ ×Pax6+/Sey crosses to those from thePax6+/+×Pax6+/+ crosses was ∼1:1 as determined by GPI1B analysis) from thePax6Sey-Neu1/Sey↔Pax6+/+(Pax6−/−↔Pax6+/+) andPax6+/+↔Pax6+/+ genotype groups were selected for detailed analysis. Regions examined were the medial part of the ventral telencephalon and the dorsal thalamus. In each chimera, the number of Tg+ signals and individual cell nuclei in a 250 × 250 μm grid area were counted in two nonconsecutive sections for both the left and right sides of the brain (see Fig. 3 for examples of sections used for analysis). A count of Tg+ signal detected to nuclei counted does not give a true estimate of the proportion of Tg+ cells in a tissue. This is because of tissue-specific differences in the efficiency of detecting Tg+ signals and nuclei, caused by variations in nuclear morphologies and packing densities (Quinn et al., 1996). To correct for this effect we performed identical counts in nonchimeric embryos, in which all cells contained the β-globin transgene, to generate tissue-specific correction factors for those regions examined. These tissue-specific correction factors were as follows: dorsal thalamus, 1.07; medial ventral telencephalon, 1.16. A hybridization index was then calculated for each tissue [hybridization index = (Tg+ signals/nuclei)/tissue specific correction factor]. Because the corrected hybridization index gives a true proportion of Tg+ cells in a particular tissue, this can be compared directly with the global contribution of Tg+ cells as determined by GPI1 genotyping.
Fig. 3.
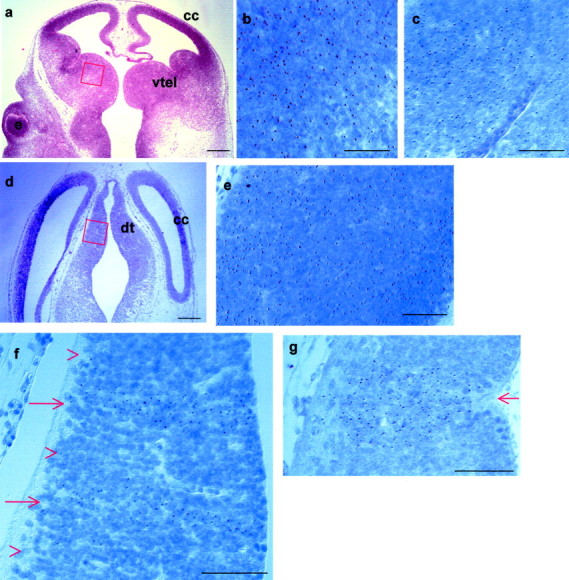
E12.5Pax6−/−↔Pax6+/+ chimeras show an autonomous requirement for Pax6 in the dorsal thalamus but not in the medial part of the ventral telencephalon. a, d, Low magnification of horizontal sections of chimeras like those used to examine the contribution of cells to the medial part of the ventral telencephalon (a) and the dorsal thalamus (d) (red boxes indicate location and orientation of higher magnification fields). b,c, e–g, High magnification of portions of typical fields used for quantification:c, f, g, inPax6−/−,Tg+↔Pax6+/+ chimeras; b,e, inPax6+/+,Tg+↔Pax6+/+ chimeras. In these images medial is to the right, and rostral is at the top. The Tg signals appear asbrown spots in the nuclei (here stainedblue). In the medial part of the ventral telencephalon, bothPax6+/+,Tg+(b) andPax6−/−,Tg+(c) cells intermingle equally well withPax6+/+, Tg− cells. In the dorsal thalamus, althoughPax6+/+,Tg+(e) cells are well mixed with thePax6+/+,Tg−cells,Pax6−/−,Tg+(f, g) cells do not intermingle with their wild-type counterparts and instead form stripes highly enriched for Pax6−/−cells (red arrows in f, g) separated by regions consisting almost exclusively ofPax6+/+ cells (red arrowheads in f). In some cases the larger congregations ofPax6−/− cells seem to have caused the ventricular surface of the dorsal thalamus to buckle inward (red arrow in g).e, Eye; cc, cerebral cortex;dt, dorsal thalamus; vtel, ventral telencephalon. Scale bars: a, d, 500 μm; b, c,e–g, 50 μm.
RESULTS
The ventral telencephalon does not project to the thalamus inPax6−/− embryos
In control embryos at E12.5, DiI injections in the dorsal thalamus labeled a tract coursing through the thalamus in a dorsoventral plane and turning laterally into the ventral telencephalon. A population of retrogradely labeled cell bodies in the ventral telencephalon marked the lateral limit of DiI diffusion at this stage (Fig.1a–d), indicating that thalamic afferents precede thalamocortical efferents. Ventral telencephalic cells projecting to the dorsal thalamus can be identified only by retrograde DiI labeling from the dorsal thalamus, and the number of cells labeled by injections that fill only a part of the dorsal thalamus will always be less than the total number actually projecting. This under-representation is likely to be compounded by the transient nature of the projections, which may mean that at a given time not all of the cells fated to project will be retrogradely labeled from the dorsal thalamus. Examination of our own material and previously published data (Tuttle et al., 1999) shows that retrogradely labeled cells can be scattered over a wide area in the medial part of the ventral telencephalon or densely clustered (Fig.1d), supporting the notion that a large proportion of ventral telencephalic cells do project to the dorsal thalamus. By E14.5 the thalamocortical axons had traversed the ventral telencephalon (Fig. 1e–g). They had turned dorsally into the cerebral cortex by E15.5 (Fig. 1h).
Fig. 1.
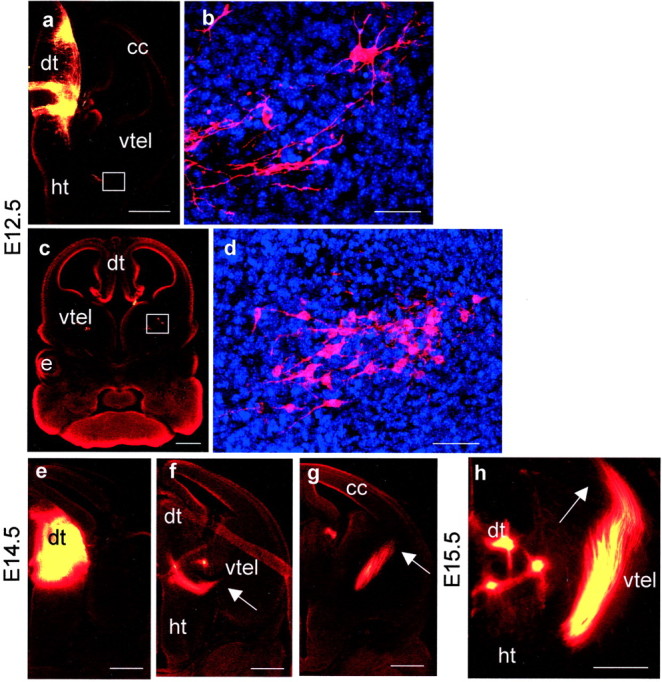
Labeling of thalamic afferents and efferents in control embryos by DiI placement in the dorsal thalamus at E12.5 (a–d), E14.5 (e–g), and E15.5 (h). a, A caudal E12.5 section showing the injection site and axons growing laterally at the diencephalic/telencephalic boundary (white box).b, A higher magnification of the area boxed in a showing retrogradely labeled cell bodies marking the lateral limit of DiI diffusion. c, A more rostral E12.5 section with higher magnification (d) of the area boxed inc showing retrogradely labeled cell bodies in the medial part of the ventral telencephalon. e–g, Caudal to rostral series of sections showing injection site and the trajectory of the thalamocortical tract at E14.5. Arrowsmark the lateral limit of the tract in each section. h, By E15.5 the tract has reached the cerebral cortex (marked witharrow). All sections were cut in the coronal plane.e, Eye; cc, cerebral cortex;dt, dorsal thalamus; ht, hypothalamus;vtel, ventral telencephalon. Scale bars:a, c, e–h, 500 μm; b, d, 50 μm.
No labeled cells or axons were detected in the ventral telencephalon of E12.5–14.5 Pax6−/− embryos after DiI injections into the dorsal thalamus (Fig.2a,f–h) (serial sections from nine embryos were examined). At E14.5, we detected axons descending through the thalamus, but these did not penetrate telencephalic tissues (Fig. 2f–i).
Fig. 2.
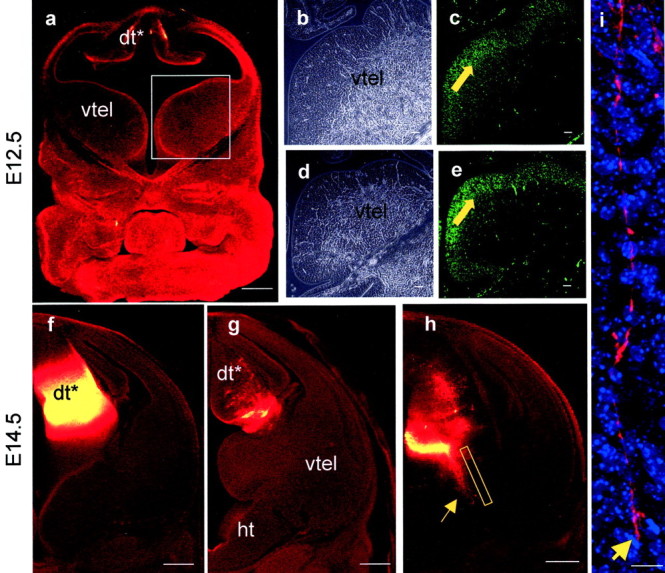
a,f–i, Labeling inPax6−/− embryos after DiI placement in the dorsal thalamus at E12.5 (a) and E14.5 (f–i).a, An E12.5 rostral section, at a level similar to that in Figure 1c, showing that no retrogradely labeled cells were seen in the ventral telencephalon. Boxed areaindicates region of ventral telencephalon shown inb–e. b–e, Mash1 immunohistochemistry in E12.5 Pax6+/+(c) and Pax6−/−(e) ventral telencephalon. In both cases cells expressing Mash1 protein (green) are distributed in the ventricular zone (filled yellow arrows) and not in the central region. b, d, Phase-contrast images corresponding to fluorescent images inc and e. E14.5 caudal (f) and rostral (g) sections showing injection site and the absence of DiI labeling in the ventral telencephalon. h, Thalamic axons descending through the thalamus (indicated by arrow); boxed area in h is shown at higher magnification ini illustrating an axon tipped with a growth cone (arrow in i). dt*,Pax6−/− correlate of thePax6+/+ dorsal thalamus;ht, hypothalamus; vtel, ventral telencephalon. Pax6−/−embryos lack eyes altogether. All sections were cut in the coronal plane. Scale bars: a,f–h, 500 μm;b–e, 50 μm; i, 10 μm.
Mash1-expressing cells occupy their normal positions in the medial part of the Pax6−/− ventral telencephalon
Previous studies have shown that Pax6 is not expressed in the medial part of the ventral telencephalon (Stoykova et al., 1996;Hirata et al., 2002). InPax6−/− embryos, the mRNA expression domains of genes including Nkx2.1 andNetrin1, which normally include the location of cells projecting to the dorsal thalamus (Tuttle et al., 1999), are not altered (Pratt et al., 2000; Stoykova et al., 2000). Expression ofMash1 mRNA, the function of which is required for the formation of these thalamic afferents (Tuttle et al., 1999), also appears unaffected inPax6−/− embryos (Stoykova et al., 2000). Despite these findings, it remained possible that Pax6 is expressed by early progenitors that give rise to ventral telencephalic cells projecting to the thalamus. One consequence of this might be a defect of Mash1 protein expression, and we tested this using Mash1 immunohistochemistry. Cells expressing Mash1 protein occupy their normal positions in the ventricular zone of the ventral telencephalon of E12.5 Pax6−/− embryos (compare expression pattern of Mash1 protein in E12.5Pax6+/+ ventral telencephalon in Fig.2c with that inPax6−/− ventral telencephalon in Fig. 2e). We did not detect any defects in the pattern of Mash1 protein expression inPax6−/− embryos throughout the rostrocaudal axis of the medial ventral telencephalon. This result implies that loss of Pax6 does not impair the ability of the medial part of the ventral telencephalon to develop molecular features associated with and essential for the formation of the transient tract to the dorsal thalamus. The primary defect seen in our DiI tracing experiments is therefore unlikely to be attributable to the disappearance of these cells.
Pax6−/− cells contribute normally to the medial part of the ventral telencephalon but are under-represented and segregate from Pax6+/+ cells in the dorsal thalamus of Pax6−/−↔Pax6+/+ chimeras
It has recently become apparent that the developing telencephalon is a dynamic structure, with newly born cells participating in numerous migratory streams that carry them from their birthplace to their final destination (Wilson and Rubenstein, 2000). Although Pax6 is not expressed in the medial part of the ventral telencephalon, it is expressed more laterally (Stoykova et al., 1996, 2000; Hirata et al., 2002). It is conceivable that cells born in Pax6-expressing territory might migrate to the medial part of the ventral telencephalon before projecting transiently to the dorsal thalamus. To address this issue we turned to a Pax6−/−↔Pax6+/+ chimera assay that directly tests whether Pax6 is required autonomously by cells to occupy their correct positions at a given developmental stage. The principle of this approach is as follows. If there is an absolute requirement for Pax6 for a tissue to develop, thenPax6−/− cells will not contribute to that tissue inPax6−/−↔Pax6+/+ chimeras, as occurs in the lens of the eye and in the nasal epithelium (Quinn et al., 1996). Alternatively, the requirement may be more subtle, as is seen in the distal part of the optic cup (Collinson et al., 2000), andPax6−/− cells either may make a reduced contribution to the tissue or be distributed abnormally within it. Pax6−/− cells would make a normal contribution if Pax6 is not required cell autonomously for the development of the tissue. Previous studies (Quinn et al., 1996; Collinson et al., 2000; J. C. Quinn and J. D. West, unpublished observations) have shown that the contribution of Tg+ cells to a particular tissue of aPax6+/+,Tg+↔Pax6+/+ chimera approximates the global Tg+ contribution, although the two are seldom identical (reflecting stochastic events during tissue construction); large deviations from the global Tg+ contribution inPax6−/−,Tg+↔Pax6+/+ chimeras are likely to be a consequence of the mutation. To provide a measure of inherent variation between individual tissues in our chimeras, we performed counts in several tissues in the control (Pax6+/+,Tg+↔Pax6+/+) balanced chimeras and measured the difference in each case from the global percentage chimerism and found an SD of 7.5%. APax6−/− contribution to aPax6−/−↔Pax6+/+ chimera of >15% (±2 SDs) above or below the global contribution measured by GPI isoform composition would therefore be evidence thatPax6−/− cells did not contribute normally to that tissue, indicating a cell autonomous requirement for Pax6 in that tissue.
We detect no abnormal distribution ofPax6−/−,Tg+cells in the medial part of the ventral telencephalon ofPax6−/−,Tg+↔Pax6+/+ chimeras.Tg+,Pax6+/+andTg+,Pax6−/−cells were equally well mixed with unlabeled cells inPax6+/+,Tg+↔Pax6+/+ (Fig.3b) andPax6−/−,Tg+↔Pax6+/+ (Fig. 3c) chimeras. Previous examination ofPax6−/−↔Pax6+/+ chimeras has shown thatPax6−/−,Tg+cells exhibit abnormal patterns of distribution in relation to their wild-type counterparts in all forebrain tissues that express Pax6 (Quinn et al., 1996; Collinson et al., 2000) (data from diencephalon in this study, see below; Quinn, unpublished observations). Furthermore, cells were able to contribute to the medial part of the ventral telencephalon regardless of their Pax6 genotype. There was no significant reduction inPax6−/− cell contribution to this region in the chimeras examined. In bothPax6−/−,Tg+↔Pax6+/+ andPax6+/+,Tg+↔Pax6+/+ chimeras, the Tg+ contribution to the medial part of the ventral telencephalon was similar to the global Tg+ contribution values defined by GPI isoform composition for each chimera (Fig.4). Therefore the loss of Pax6 function does not detectably impair the ability of cells to contribute to the medial part of the ventral telencephalon, implying that there is no cell-autonomous requirement for Pax6 for the development of this part of the brain. Taken together, the failure to detect abnormal distribution or reduced contribution ofPax6−/− cells in our chimeras provides compelling evidence that Pax6 does not participate directly in the genesis of the region of the ventral telencephalon that projects a transient tract to the dorsal thalamus.
Fig. 4.

Histogram showing quantification of chimerism inPax6−/−↔Pax6+/+ andPax6+/+↔Pax6+/+ chimeras. Global chimerism and chimerism for the medial part of the ventral telencephalon and the dorsal thalamus are shown for each chimera, with thenumbers above each bar indicating the percentages of Tg+ cells. Primary Tg+ signal/nuclei counts for the medial ventral telencephalon and the dorsal thalamus were as follows:Pax6+/+,Tg+↔Pax6+/+ chimeras: JC24, medial vtel = 2168/3721, dt = 2613/4390; JC58: medial vtel = 2135/3325, dt = 2427/3465;Pax6−/−,Tg+↔Pax6+/+ chimeras: JC56, medial vtel = 1412/3484, dt = 692/3179; JC61: medial vtel = 1513/3591, dt = 511/3633. These primary counts were divided by the tissue-specific correction factors (1.16 for the medial ventral telencephalon, 1.07 for the dorsal thalamus) to give the corrected hybridization index, which gives a true estimate of the percentage of Tg+ cells in the tissue for comparison with the global Tg+ contribution (see Materials and Methods). Large variation between tissue-specific and global Tg+ contribution, indicating a requirement for Pax6, is seen only in the dorsal thalamus ofPax6−/−↔Pax6+/+ chimeras (marked with ∗), where Pax6−/− cells are also abnormally distributed (Fig. 3). vtel, Ventral telencephalon.
In contrast, the ability of cells to contribute to the dorsal thalamus was affected dramatically by their Pax6 genotype.Tg+,Pax6+/+cells were evenly distributed in the dorsal thalamus ofPax6+/+,Tg+↔Pax6+/+ chimeras (Fig. 3e), butTg+,Pax6−/−cells were segregated from Pax6+/+ cells into radial stripes inPax6−/−,Tg+↔Pax6+/+ chimeras (Fig.3f,g). These stripes could be followed in a dorsoventral direction through many sections and extended into the ventral thalamus (data not shown). When Tg+ signals and nuclei were counted in 25-μm-wide strips running perpendicular to the ventricular surface, we observed that either nearly all or virtually none of the cells in these stripes were Pax6−/−(Fig. 3f,g), suggesting that stripes of mutant cells form perpendicular to the ventricular surface. The stripes of mutant cells in the dorsal thalamus varied considerably in width from one or two cells to tens of cells (Fig. 3, compare f,g). These stripes were not a consequence of clonal expansion during normal development, because they were not seen in the dorsal thalamus of Pax6+/+↔Pax6+/+ chimeras (Fig. 3e). Although the overall morphology of the dorsal thalamus in the balancedPax6−/−↔Pax6+/+ chimeras that were examined did not appear disturbed [and certainly did not recapitulate the distortions seen in Pax6−/−embryos (Stoykova et al., 1996; Warren and Price, 1997)], the ventricular surface formed a dramatic kink coinciding with particularly large patches of mutant cells (Fig. 3g). This indicated that the normal expansion of the diencephalic wall was retarded where large clumps of Pax6−/− cells were present. Analysis of broad areas of the dorsal thalamus showed that, on average, Pax6−/− cells were under-represented in the dorsal thalamus ofPax6−/−↔Pax6+/+ chimeras. AlthoughPax6+/+ cells contributed to the dorsal thalamus of Pax6+/+↔Pax6+/+ chimeras at levels comparable to the global contribution, the contribution ofPax6−/− cells to the dorsal thalamus of Pax6−/−↔Pax6+/+ chimeras was less than half the global contribution (Fig. 4). Taken together, the abnormal distribution and under-representation ofPax6−/− cells inPax6−/−↔Pax6+/+ chimeras provide strong evidence that Pax6 is required autonomously for cells to make a full contribution to the dorsal thalamus and to participate normally in its development.
Thalamocortical axons do not turn laterally into the telencephalon in Foxg1−/− embryos
In E12.5 Foxg1−/−embryos, axons labeled from the dorsal thalamus coursed dorsoventrally through the ventral thalamus (Fig.5a), but although a few axons approached the telencephalic/diencephalic boundary, hardly any entered the telencephalon and those that did appeared disorganized (Fig.5a–c). The telencephalon ofFoxg1−/− embryos is highly abnormal, and no retrogradely labeled cell bodies were detected in any telencephalic tissues (Fig. 5d). At neither E14.5 nor E15.5 were thalamocortical axons seen turning laterally into telencephalic structures (Fig. 5e–h). At E14.5 and E15.5, the tract penetrated farther than normal into the lateral hypothalamus (Fig. 5f–h, compare with Fig.1f–h).
Fig. 5.
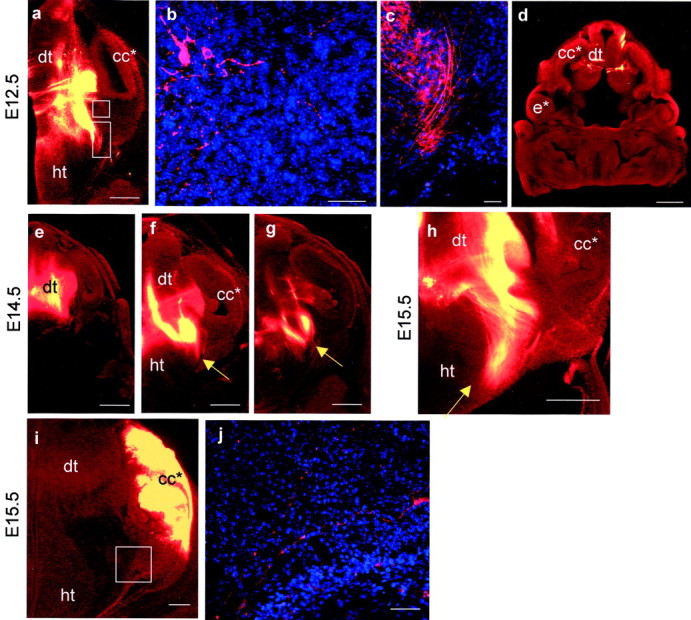
DiI labeling inFoxg1−/− embryos at E12.5 (a–d), E14.5 (e–g), and E15.5 (h–j).a–h, DiI injections into dorsal thalamus. a, A caudal E12.5 section showing the injection site and absence of DiI labeling from telencephalic structures (the Foxg1−/−telencephalon lacks recognizable ventral structures so is markedcc*). b, A higher magnification of thetop boxed area in a showing that the very few axons which approach the telencephalon are disorganized.c, A higher magnification of the bottom area boxed in a showing that labeled axons grow ventrally and medially rather than turning laterally into the telencephalon. d, A more rostral E12.5 section showing that no retrogradely labeled cells or axons can be detected in the rostral telencephalon. e–g, Caudal to rostral series of sections at E14.5 show the injection site and the trajectory of thalamic efferents. These do not penetrate the telencephalon; arrows mark lateral limit of the tract in each section. h, By E15.5 the tract has not penetrated the telencephalon but has continued ventrally within the thalamus toward the hypothalamus (marked with arrow).i, j, DiI injection into telencephalic structures at E15.5. i, Although no substantial tract leaves the telencephalon inFoxg1−/− embryos, higher magnification of the area boxed in jshows that a few disorganized axons are able to cross the telencephalic/diencephalic boundary. All sections were cut in the coronal plane. cc* and e* denoteFoxg1−/− correlates ofFoxg1+/+ cerebral cortex and eye;dt, dorsal thalamus; ht, hypothalamus. Scale bars: a, d,e–h, 500 μm; b,c, 50 μm.
Placement of DiI crystals in the telencephalon ofFoxg1−/− embryos labeled a few axons that had crossed the telencephalic/diencephalic boundary (Fig. 5i,j), although these appeared disorganized and did not extend to the dorsal thalamus. Similar injections in control embryos labeled a much larger number of axons that followed the trajectory of the thalamocortical tract and terminated in the dorsal thalamus (data not shown).
Most of the retinal axons navigate the thalamus appropriately inFoxg1−/− embryos but a subpopulation deviate into telencephalic structures
The optic tract forms at about the same time as the thalamocortical tract. Retinal axons navigating toward the dorsal thalamus encounter diencephalic and telencephalic tissues (Mason and Wang, 1997) that are implicated in the formation of the thalamocortical tract (Braisted et al., 1999; Tuttle et al., 1999, Pratt et al., 2000). We were therefore interested to see how retinal axons responded to the disrupted forebrain environment present inFoxg1−/− embryos. In E15.5 control embryos, DiI crystals placed in the optic cup labeled a tract leaving the retina (Fig. 6a), coursing over the ventral surface of the brain, and continuing dorsally and caudally to the lateral dorsal thalamus (Fig.6b,c). These axons were not seen entering telencephalic tissue (Fig. 6b–e). In E15.5Foxg1−/− embryos, DiI crystals placed in the optic cup label a tract in which the majority of axons followed the same trajectory as that seen in control embryos to reach the dorsal thalamus (Fig. 6f–j). Unlike in the wild type, a subpopulation of axons deviated into telencephalic structures (Fig. 6g,i).
Fig. 6.

DiI labeling of axons from the optic cup in Foxg1−/−(f–j) and control (a–e) embryos at E15.5.a–e, In control embryos, a rostral to caudal series shows the tract leaving the optic cup (a), growing over the lateral surface of the hypothalamus (b), and reaching the lateral aspect of the dorsal thalamus (c). Boxed areas in b and c are shown at higher magnification in d and e. d, Axons do not penetrate into telencephalic structures.e, Axons form a smooth tract running in a dorsoventral direction in the lateral dorsal thalamus.f–j, InFoxg1−/− embryos, a rostral to caudal series shows the tract leaving the optic cup (f), growing over the lateral surface of the hypothalamus (g) (unfilled yellow arrow in g shows axons penetrating the telencephalon), and reaching the lateral aspect of the dorsal thalamus (h). Boxed areas ing and h are shown at higher magnification in i and j. i, Some axons leave the main tract and penetrate into telencephalic structures (unfilled yellow arrow). j, Axons form a smooth tract running in a dorsoventral plane in the lateral dorsal thalamus. The main tract is indicated by yellow arrows in a–c andf–h. Note that short labeled fibers, marked with yellow arrowheads, sprouting medially from the main tract into the diencephalon are seen in both control (d, e) andFoxg1−/−(i, j) embryos. cc* ande* denoteFoxg1−/− correlates ofFoxg1+/+ cerebral cortex and eye;dt, dorsal thalamus; ht, hypothalamus;vtel, ventral telencephalon. All sections were cut in the coronal plane. Scale bars: a–c,f–h, 500 μm; d,e, i, j, 50 μm.
DISCUSSION
Disruption to thalamic afferents inPax6−/− embryos
Tract-tracing studies in several rodent species have identified a transient population of thalamic afferents that originate in the medial part of the ventral telencephalon before thalamocortical tract formation (Metin and Godement, 1996; Molnar et al., 1998; Braisted et al., 1999). The cell bodies of these projecting neurons map to ventral telencephalic domains expressing Nkx2.1 (transcription factor) and Netrin1 (secreted protein) mRNAs (Tuttle et al., 1999). In Mash1−/− embryos, these domains of gene expression are missing, the transient afferent tract does not form, and the thalamocortical tract does not penetrate the telencephalon. This led to the proposal that Mash1 is required in the medial part of the ventral telencephalon for the projection of this transient tract that guides thalamocortical afferents laterally into the ventral telencephalon (Tuttle et al., 1999).
Our observation that the ventral telencephalon does not project to the dorsal thalamus in Pax6−/−embryos could be explained by a primary defect in the projecting cells, in the environment through which their axons navigate, or both. Available evidence strongly suggests that the medial part of the ventral telencephalon develops normally inPax6−/− embryos. First, Pax6 is not expressed in this region at the time when its thalamic connections form (Stoykova et al., 1996, 2000; Hirata et al., 2002). Second, the expression patterns of Mash1 mRNA and protein,Nkx2.1 mRNA, and Netrin1 mRNA are not altered in this region in Pax6−/−embryos (Pratt et al., 2000; Stoykova et al., 2000; the present study). Third, Pax6−/− cells inPax6−/−↔Pax6+/+ chimeras show no abnormal distribution and contribute normally to this region. In the forebrain of Pax6−/−↔Pax6+/+ chimeras, abnormalities always manifest as segregation betweenPax6−/− andPax6+/+ cells rather than complete exclusion of Pax6−/− cells, even in areas with high persistent expression of Pax6 (Quinn et al., 1996; Collinson et al., 2000) (data from diencephalon in this study; Quinn, unpublished observations). We cannot rule out the formal possibility of a total exclusion of a subpopulation ofPax6−/− cells that would normally project to the thalamus if that subpopulation makes up <30% of the region analyzed (because this translates to a chimerism within 15% of the global chimerism of a balanced chimera, placing it within the limits of inherent variations between our control chimeric tissues; see Results). This is, however, unlikely because (1) the proportion of ventral telencephalic cells that project to the thalamus is likely to be high (see Results) and (2) a cell autonomous requirement for Pax6 is manifested as a segregation defect rather than exclusion in all other regions of the brain examined. The development of the thalamus is compromised in Pax6−/−embryos. First, Pax6−/−thalamic cells exhibit reduced proliferation (Warren and Price, 1997). Second, the Pax6−/− dorsal thalamus exhibits abnormal differentiation that is manifested as altered patterns of gene expression and projection of axons with altered navigation properties (Pratt et al., 2000). Third,Pax6−/− cells are abnormally distributed and under-represented in the dorsal thalamus ofPax6−/−↔Pax6+/+ chimeras (the present study). Taken together these findings provide compelling evidence that a primary defect in Pax6−/−thalamus is responsible for its failure to receive transient afferents from the ventral telencephalon. The lack of this afferent tract may be one of several factors contributing to the failure of subsequent thalamocortical development in Pax6−/−embryos (Kawano et al., 1999; Hevner at al., 2002). Other factors are likely to include cell-autonomous defects of thalamic cells and axons (Pratt et al., 2000) (chimera results of the present study).
Axons forming the tract of the post-optic commissure (TPOC) fail to reach the dorsal thalamus inPax6−/− embryos (Mastick et al., 1997). A recent preliminary report shows that TPOC axons can be rescued nonautonomously by transient expression of Pax6 within the thalamus at E10.5 (Mastick, 2001). This result is interesting in the context of this study because it suggests a more general Pax6-dependent role for the thalamus in attracting afferent axons.
Abnormal thalamic development inPax6−/− embryos
Previous work has demonstrated that proliferation is reduced in the Pax6−/− dorsal thalamus at E10.5 (Warren and Price, 1997). The under-representation ofPax6−/− cells in the dorsal thalamus of E12.5 Pax6−/−↔Pax6+/+ chimeras suggests a cell-autonomous proliferation defect that cannot be rescued by surrounding Pax6+/+ cells. Kinks in the ventricular zone coinciding with large areas consisting almost exclusively of Pax6−/− cells could reflect mechanical distortion imposed on the tissue by a patch of relatively slowly dividing mutant cells surrounded by more rapidly proliferating wild-type epithelium. That the tissue responds to these patches of mutant cells by forming a kink, rather than relieving the tension by allowing Pax6−/−and Pax6+/+ cells to mix laterally, provides a measure of the strong forces opposing their mixing.
Misexpression of cell surface molecules or secreted proteins in thePax6−/− thalamus is a likely mechanism for its failure to receive transient innervation from the ventral telencephalon. ThatPax6−/− andPax6+/+ cells did not intermingle in the thalamus of E12.5 Pax6−/−↔Pax6+/+ chimeras suggests a role for Pax6 in influencing cell–cell interactions in the developing thalamus by regulating the expression of cell surface molecules or secreted signaling proteins. This hypothesis is appealing because, in various developing systems, Pax6 directly or indirectly regulates the expression of many genes controlling cell–cell contact and signaling. These include genes encoding cell surface adhesion molecules L1, R-cadherin, β1 integrin, and trkB (Stoykova et al., 1997; Meech et al., 1999; Warren et al., 1999; Duncan et al., 2000) and secreted proteins Wnt7b, SFRP-2, and Netrin1 (Kim et al., 2001; Vitalis et al., 2001). We have previously made a preliminary report that, at E14.5,Pax6−/− andPax6+/+ thalamic cells exhibit different abilities to adhere to and grow on slices of wild-type forebrain in culture (Pratt et al., 2001), suggesting that the control of thalamic cell surface properties by Pax6 spans the period of thalamocortical tract formation.
Disruption of the ventral telencephalon prevents thalamocortical axons innervating the cerebral cortex inFoxg1−/− embryos
The thalamocortical phenotype ofMash1−/− embryos provides evidence for the importance of transient thalamic afferents from the ventral telencephalon in guiding thalamocortical axons (Tuttle et al., 1999), but the thalamus itself is also disrupted, making it hard to determine where the primary defects causing thalamocortical axon misrouting lie. In these mutants, most of the axons stall at the border between the dorsal and ventral thalamus (Tuttle et al., 1999), raising the possibility that the ventral telencephalon might be required to guide axons through the thalamus. To test this we examinedFoxg1−/− embryos in which development of the thalamus is normal but the telencephalon is severely disrupted and has no recognizable ventral structures (Xuan et al., 1995; Dou et al., 1999). Thalamic axons inFoxg1−/− embryos showed no clear pathfinding defects within the thalamus, indicating that a normal ventral telencephalon is not required for this segment of the thalamocortical tract to form. Thalamic axons did not, however, turn laterally into the telencephalon, indicating that this turn requires a normal ventral telencephalon. There is no evidence for a general physical barrier to axons at the diencephalic telencephalic boundary inFoxg1−/− embryos because our injections into the Foxg1−/−telencephalon or eye (see below) identified axons crossing this boundary. A striking feature of the lateral turn of thalamocortical axons into the ventral telencephalon is their avoidance of the hypothalamus. In vitro studies show that thalamocortical axons are repelled by molecules secreted by the hypothalamus (Braisted et al., 1999) and do not penetrate explants of hypothalamus (Pratt et al., 2000). Our observation that dorsal thalamic efferents inFoxg1−/− embryos penetrate farther ventrally than normal into the lateral part of the hypothalamus suggests that repulsion by the hypothalamus in vivo is not sufficient to propel thalamocortical axons laterally or to completely repel invasion by thalamocortical axons diverted from their normal course.
Optic tract formation in Foxg1−/−embryos
The optic tract grows from the retina to the dorsal thalamus and encounters tissues involved in guiding the thalamocortical tract (Mason and Wang, 1997). In Foxg1−/−embryos we labeled many axons that navigated from the retina to the lateral aspect of the dorsal thalamus along a similar trajectory to that seen in control embryos, indicating that diencephalic tissues are able to supply appropriate navigation cues to retinal axons in the absence of Foxg1. This result supports our premise, based on the absence of Foxg1 expression from the diencephalon and appropriate expression of diencephalic marker genes inFoxg1−/− embryos, that theFoxg1−/− diencephalon is normal (Xuan et al., 1995; Dou et al., 1999). We also observed a subset of axons that projected into the telencephalon, a feature never seen in control embryos. So although thalamocortical axons (which normally penetrate the telencephalon) are excluded in theFoxg1−/− mutant, some retinal axons (which are normally excluded from the ventral telencephalon) are able to penetrate in theFoxg1−/− mutant. It is tempting to speculate that the loss of Foxg1 from the developing telencephalon results in the simultaneous loss of attractive cues for thalamic axons and repulsive cues for some (but not all) retinal axons.Foxg1 is expressed in the nasal portion of the retina (Hatini et al., 1994; Xuan et al., 1995; Huh et al., 1999), so it is also possible that the pathfinding errors in theFoxg1−/− optic tract reflect an autonomous requirement for Foxg1 in a subset of retinal axons. These issues deserve examination in a future study.
Footnotes
This work was supported by the Medical Research Council, Biotechnology Biological Sciences Research Council, and Wellcome Trust (065035). We thank Jean Hebert and Susan McConnell for the Foxg1tm1(cre)skm mice, Robert Hill for helping lay the foundations for the chimera work, and Katy Gillies, Margaret Keighren, and Jean Flockhart for expert technical assistance.
Correspondence should be addressed to Dr. David J. Price, Genes and Development Group, Biomedical Sciences, University of Edinburgh, Hugh Robson Building, George Square, Edinburgh EH8 9XD, UK. E-mail:dprice@ed.ac.uk.
REFERENCES
- 1.Angevine JB., Jr Time of neuron origin in the diencephalon of the mouse. An autoradiographic study. J Comp Neurol. 1970;139:129–187. doi: 10.1002/cne.901390202. [DOI] [PubMed] [Google Scholar]
- 2.Auladell C, Perez-Sust P, Super H, Soriano E. The early development of thalamocortical and corticothalamic projections in the mouse. Anat Embryol (Berl) 2000;201:169–179. doi: 10.1007/pl00008238. [DOI] [PubMed] [Google Scholar]
- 3.Braisted JE, Tuttle R, O'Leary DD. Thalamocortical axons are influenced by chemorepellent and chemoattractant activities localized to decision points along their path. Dev Biol. 1999;208:430–440. doi: 10.1006/dbio.1999.9216. [DOI] [PubMed] [Google Scholar]
- 4.Braisted JE, Catalano SM, Stimac R, Kennedy TE, Tessier-Lavigne M, Shatz CJ, O'Leary DD. Netrin-1 promotes thalamic axon growth and is required for proper development of the thalamocortical projection. J Neurosci. 2000;20:5792–5801. doi: 10.1523/JNEUROSCI.20-15-05792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collinson JM, Hill RE, West JD. Different roles for Pax6 in the optic vesicle and facial epithelium mediate early morphogenesis of the murine eye. Development. 2000;127:945–956. doi: 10.1242/dev.127.5.945. [DOI] [PubMed] [Google Scholar]
- 6.Dou CL, Li S, Lai E. Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex. 1999;9:543–550. doi: 10.1093/cercor/9.6.543. [DOI] [PubMed] [Google Scholar]
- 7.Duncan MK, Cvekl A, Li X, Piatigorsky J. Truncated forms of Pax-6 disrupt lens morphology in transgenic mice. Invest Ophthalmol Vis Sci. 2000;41:464–473. [PubMed] [Google Scholar]
- 8.Favor J, Neuhauser-Klaus A, Erhling UH. The effect of dose fractionation on the frequency of erithrolnitrousurea induced dominant cataract and recessive specific locus mutations in germs cells of the mouse. Mutat Res. 1988;198:269–275. doi: 10.1016/0027-5107(88)90003-6. [DOI] [PubMed] [Google Scholar]
- 9.Garel S, Marin F, Grosschedl R, Charnay P. Ebf1 controls early cell differentiation in the embryonic striatum. Development. 1999;126:5285–5294. doi: 10.1242/dev.126.23.5285. [DOI] [PubMed] [Google Scholar]
- 10.Hatini V, Tao W, Lai E. Expression of winged helix genes, BF-1 and BF-2, define adjacent domains within the developing forebrain and retina. J Neurobiol. 1994;25:1293–1309. doi: 10.1002/neu.480251010. [DOI] [PubMed] [Google Scholar]
- 11.Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 12.Hevner RF, Miyashita-Lin E, Rubenstein JLR. Cortical and thalamic axon pathfinding defects in Tbr1, Gbx2, and Pax6 mutant mice: evidence that cortical and thalamic axons interact and guide each other. J Comp Neurol. 2002;447:8–17. doi: 10.1002/cne.10219. [DOI] [PubMed] [Google Scholar]
- 13.Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- 14.Hirata T, Nomura T, Takagi Y, Sato Y, Tomioka N, Fujisawa H, Osumi N. Mosaic development of the olfactory cortex with Pax6-dependent and –independent components. Dev Brain Res. 2002;136:17–26. doi: 10.1016/s0165-3806(02)00304-8. [DOI] [PubMed] [Google Scholar]
- 15.Huh S, Hatini V, Marcus RC, Li SC, Lai E. Dorsal-ventral patterning defects in the eye of BF-1-deficient mice associated with a restricted loss of shh expression. Dev Biol. 1999;211:53–63. doi: 10.1006/dbio.1999.9303. [DOI] [PubMed] [Google Scholar]
- 16.Kawano H, Fukuda T, Kubo K, Horie M, Uyemura K, Takeuchi K, Osumi N, Eto K, Kawamura K. Pax-6 is required for thalamocortical pathway formation in fetal rats. J Comp Neurol. 1999;408:147–160. doi: 10.1002/(sici)1096-9861(19990531)408:2<147::aid-cne1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 17.Keighren M, West JD. Analysis of cell ploidy in histological sections of mouse tissues by DNA-DNA in-situ hybridisation with digoxygenin labelled probes. Histochem J. 1993;25:30–44. doi: 10.1007/BF00161042. [DOI] [PubMed] [Google Scholar]
- 18. Kim AS, Anderson SA, Rubenstein JL, Lowenstein DH, Pleasure SJ. Pax-6 regulates expression of SFRP-2 and Wnt-7b in the developing CNS. J Neurosci 21 2001. RC132(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo C. Localisation of low abundance DNA sequences in tissue sections by in-situ hybridisation. J Cell Sci. 1986;81:143–162. doi: 10.1242/jcs.81.1.143. [DOI] [PubMed] [Google Scholar]
- 20.Mason CA, Wang LC. Growth cone form is behavior-specific and, consequently, position-specific along the retinal axon pathway. J Neurosci. 1997;17:1086–1100. doi: 10.1523/JNEUROSCI.17-03-01086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastick GS. Pioneer axon guidance errors in Pax6 mutant mouse embryos are rescued by electroporation-mediated gene transfer. Soc Neurosci Abstr. 2001;27:794.14. [Google Scholar]
- 22.Mastick GS, Davies NM, Andrew GL, Easter SS. Pax-6 functions in boundary formation and axon guidance in the embryonic mouse forebrain. Development. 1997;124:1985–1997. doi: 10.1242/dev.124.10.1985. [DOI] [PubMed] [Google Scholar]
- 23.Meech R, Kallunki P, Edelman GM, Jones FS. A binding site for homeodomain and Pax proteins is necessary for L1 cell adhesion molecule gene expression by Pax-6 and bone morphogenetic proteins. Proc Natl Acad Sci USA. 1999;96:2420–2425. doi: 10.1073/pnas.96.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metin C, Godement P. The ganglionic eminence may be an intermediate target for corticofugal and thalamocortical axons. J Neurosci. 1996;16:3219–3235. doi: 10.1523/JNEUROSCI.16-10-03219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molnar Z, Adams R, Blakemore C. Mechanisms underlying the early establishment of thalamocortical connections in the rat. J Neurosci. 1998;18:5723–5745. doi: 10.1523/JNEUROSCI.18-15-05723.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pratt T, Vitalis T, Warren N, Edgar JM, Mason JO, Price DJ. A role for Pax6 in the normal development of dorsal thalamus and its cortical connections. Development. 2000;127:5167–5178. doi: 10.1242/dev.127.23.5167. [DOI] [PubMed] [Google Scholar]
- 27.Pratt T, Price DJ, Mason JO. Pax6 and the development of thalamocortical projections. Soc Neurosci Abstr. 2001;27:797.8. [Google Scholar]
- 28.Quinn JC, West JD, Hill RE. Multiple functions for Pax6 in mouse eye and nasal development. Genes Dev. 1996;10:435–446. doi: 10.1101/gad.10.4.435. [DOI] [PubMed] [Google Scholar]
- 29.Quinn JC, West JD, Kaufman MH. Genetic background effects on dental and other craniofacial abnormalities in homozygous small eye (Pax6Sey/Pax6Sey) mice. Anat Embryol (Berl) 1997;196:311–321. doi: 10.1007/s004290050100. [DOI] [PubMed] [Google Scholar]
- 30.Skaliora I, Adams R, Blakemore C. Morphology and growth patterns of developing thalamocortical axons. J Neurosci. 2000;20:3650–3662. doi: 10.1523/JNEUROSCI.20-10-03650.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoykova A, Fritsch R, Walther C, Gruss P. Forebrain patterning defects in small eye mutant mice. Development. 1996;122:3453–3465. doi: 10.1242/dev.122.11.3453. [DOI] [PubMed] [Google Scholar]
- 32.Stoykova A, Gotz M, Gruss P, Price J. Pax6-dependent regulation of adhesive patterning, R-cadherin expression and boundary formation in developing forebrain. Development. 1997;124:3765–3777. doi: 10.1242/dev.124.19.3765. [DOI] [PubMed] [Google Scholar]
- 33.Stoykova A, Treichel D, Hallonet M, Gruss P. Pax6 modulates the dorsoventral patterning of the mammalian telencephalon. J Neurosci. 2000;20:8042–8050. doi: 10.1523/JNEUROSCI.20-21-08042.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuttle R, Nakagawa Y, Johnson JE, O'Leary DD. Defects in thalamocortical axon pathfinding correlate with altered cell domains in Mash-1-deficient mice. Development. 1999;126:1903–1916. doi: 10.1242/dev.126.9.1903. [DOI] [PubMed] [Google Scholar]
- 35.Vitalis T, Cases O, Engelkamp D, Verney C, Price DJ. Defect of tyrosine hydroxylase-immunoreactive neurons in the brains of mice lacking the transcription factor Pax6. J Neurosci. 2001;20:6501–6516. doi: 10.1523/JNEUROSCI.20-17-06501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warren N, Price DJ. Roles of Pax-6 in murine diencephalic development. Development. 1997;124:1573–1582. doi: 10.1242/dev.124.8.1573. [DOI] [PubMed] [Google Scholar]
- 37.Warren N, Caric D, Pratt T, Clausen JA, Asavaritikrai P, Mason JO, Hill RE, Price DJ. The transcription factor, Pax6, is required for cell proliferation and differentiation in the developing cerebral cortex. Cereb Cortex. 1999;9:627–635. doi: 10.1093/cercor/9.6.627. [DOI] [PubMed] [Google Scholar]
- 38.West JD, Flockhart JH. Genotypically unbalanced diploid↔ diploid foetal mouse chimaeras: possible relevance to human confined placental mosaicism. Genet Res. 1994;63:87–99. doi: 10.1017/s0016672300032195. [DOI] [PubMed] [Google Scholar]
- 39.Wilson SW, Rubenstein JL. Induction and dorsoventral patterning of the telencephalon. Neuron. 2000;28:641–651. doi: 10.1016/s0896-6273(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 40.Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]