

Abstract
The voltage-gated cardiac Na+ channel (Nav1.5), encoded by the SCN5A gene, conducts the inward depolarizing cardiac Na+ current (INa) and is vital for normal cardiac electrical activity. Inherited loss-of-function mutations in SCN5A lead to defects in the generation and conduction of the cardiac electrical impulse and are associated with various arrhythmia phenotypes1. Here we show that sirtuin 1 deacetylase (Sirt1) deacetylates Nav1.5 at lysine 1479 (K1479) and stimulates INa via lysine-deacetylation-mediated trafficking of Nav1.5 to the plasma membrane. Cardiac Sirt1 deficiency in mice induces hyperacetylation of K1479 in Nav1.5, decreases expression of Nav1.5 on the cardiomyocyte membrane, reduces INa and leads to cardiac conduction abnormalities and premature death owing to arrhythmia. The arrhythmic phenotype of cardiac-Sirt1-deficient mice recapitulated human cardiac arrhythmias resulting from loss of function of Nav1.5. Increased Sirt1 activity or expression results in decreased lysine acetylation of Nav1.5, which promotes the trafficking of Nav1.5 to the plasma membrane and stimulation of INa. As compared to wild-type Nav1.5, Nav1.5 with K1479 mutated to a nonacetylatable residue increases peak INa and is not regulated by Sirt1, whereas Nav1.5 with K1479 mutated to mimic acetylation decreases INa. Nav1.5 is hyperacetylated on K1479 in the hearts of patients with cardiomyopathy and clinical conduction disease. Thus, Sirt1, by deacetylating Nav1.5, plays an essential part in the regulation of INa and cardiac electrical activity.
Nav1.5, encoded by the SCN5A gene, has a critical role in the cardiac action potential2. Inherited mutations in SCN5A lead to cardiac-arrhythmia syndromes (Brugada syndrome, long-QT syndrome type 3 and inherited conduction disease)1, and pharmacologic agents that decrease cardiac sodium current can be proarrhythmic3. Post-translational modifications of Nav1.5 (phosphorylation, methylation, alanine-N-terminal acetylation, ubiquitination and palmitoylation) modulate its biophysical properties and its membrane localization4,5. Additionally, Nav1.5 is regulated by the cellular energetic state; NAD+/NADH alters membrane localization of the channel, at least in part, through protein kinase C (PKC)-mediated phosphorylation6,7. However, the role of dynamic lysine acetylation in governing Nav1.5 function is not known. Recognizing that Nav1.5 membrane localization is influenced by NAD+ (ref. 6), and that Sirt1 is an NAD+-dependent lysine deacetylase, we asked whether Sirt1 regulates the inward depolarizing sodium current (INa) and cardiac electrical activity by deacetylating Nav1.5.
We first tested whether Nav1.5 physically interacts with Sirt1. Native Nav1.5 from neonatal rat cardiomyocytes and adult mouse hearts coprecipitated with Sirt1 (Fig. 1a), and Sirt1 colocalized with Nav1.5 on the cell surface of mouse cardiomyocytes (Fig. 1b). We next examined the effect of Sirt1 on INa conducted by Nav1.5 in a heterologous expression system. In human embryonic kidney (HEK) 293 cells expressing Nav1.5, overexpression of Sirt1 increased INa, as assessed by whole-cell patch clamping, whereas the inhibition of Sirt1 by overexpression of catalytically inactive dominant-negative Sirt1 (H363Y) decreased INa (Fig. 1c). Similarly, overexpression of Sirt1 in neonatal rat cardiomyocytes increased endogenous INa (Fig. 1d) without changing Nav1.5 expression (Supplementary Fig. 1a, b), whereas treatment with Ex243, a pharmacologic Sirt1 inhibitor, decreased INa (Fig. 1d). Sirt1 overexpression in HEK293 cells resulted in a small (≈2-mV) hyperpolarizing shift in the voltage for half-maximal activation (V1/2) of INa from −39.8 mV to −41.9 mV (Supplementary Table 1). However, no substantial differences were observed in other kinetic parameters of Nav1.5, including steady-state inactivation, development of intermediate inactivation or recovery from inactivation (Supplementary Fig. 2 and Supplementary Table 1). To determine whether Nav1.5 is governed by endogenous Sirt1, we generated mice with cardiomyocytespecific deletion of Sirt1 (cSirt1−/−; Supplementary Fig. 1c, d) and measured INa in ventricular myocytes isolated from 6-7-month-old mice. INa was significantly lower in cSirt1−/− myocytes than in control littermate Sirt1fl/fl myocytes (Fig. 1e-g), despite an increase in total Nav1.5 protein in cSirt1−/− hearts (Supplementary Fig. 1e). There were no changes in the current-voltage relationship (Supplementary Table 2), steady-state inactivation, recovery from slow and fast inactivation, late sodium current (Supplementary Fig. 3a, b) or the time constants of the decay phase of INa (Supplementary Table 2) between cSirt1−/− and Sirt1fl/fl cardiomyocytes.
Figure 1.
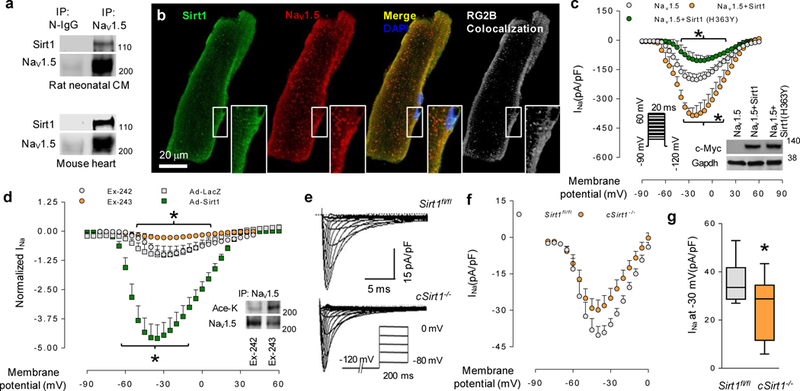
The interaction of Sirt1 with Nav1.5 and regulation of INa by Sirt1. (a) Co-immunoprecipitation (IP) of Sirt1 with Nav1.5 in rat neonatal cardiomyocytes (CM) and mouse heart. Nonimmune IgG (N-IgG) or Nav1.5 antibody was used for immunoprecipitation, and Sirt1 and Nav1.5 antibodies were used for immunoblotting. (b) Photomicrographs showing the localization of Sirt1 and Nav1.5 in cardiomyocytes. White, pixel colocalization analyzed using the RG2B plugin in ImageJ. Insets show higher-magnification views of the boxed areas. Scale bar, 20 μm. (c) Current-voltage (I/V) relationship for peak INa in HEK293 cells expressing full-length wild-type Nav1.5 without or with Sirt1 or dominant-negative Sirt1 (H363Y). n = 13 for Nav1.5, 12 for Nav1.5 + Sirt1 and n = 8 for Nav1.5 + Sirt1 (H363Y). Inset, immunoblot to measure the expression of c-Myc-tagged wild-type and mutant (H363Y) Sirt1. GAPDH was used as a loading control. *P < 0.05 Nav1.5 versus Nav1.5 + Sirt1; Nav1.5 versus Nav1.5 + Sirt1 (H363Y). (d) Normalized current-voltage relationships for isolated rat neonatal cardiomyocytes infected with Ad-Sirt1 or control Ad-LacZ and treated with 5-μM Sirt1 selective inhibitor Ex-243 or the inactive optical isomer Ex-242. *P < 0.05 Ad-Sirt1 versus Ad-LacZ; Ex-243 versus Ex-242. n = 6 cells for Ex-243 and AdSirt1, n = 7 cells for Ex-242 and Ad-LacZ. Insert, acetylation of endogenous Nav1.5, as measured by immunoblotting using antibody toward acetylated lysine epitope (Ace-K) following immunoprecipitation with Nav1.5 antibody. (e,f) INa in ventricular myocytes isolated from mice with conditional deletion of cardiomyocyte Sirt1 (cSirt1−/−) and littermate Sirt1fl/fl controls. Representative traces of INa with a pulse protocol (e) and the current-voltage relationship of INa (f) are shown. (g) INa density at −30 mV in ventricular myocytes from cSirt1−/− and Sirt1fl/fl mice. *P < 0.05, n = 11 in each group. Immunoblots are representative of three independent experiments. Uncropped blots are shown in Supplementary Figure 7. Independent sample t test was used. For XY scatter plots, data is shown as mean, and error bars represent s.e.m. For box-and-whisker plots, center line shows median, end of box shows interquartile range (IQR) and whiskers show minima and maxima within 1.5 IQR.
Next, we tested whether Nav1.5 is lysine acetylated and whether Sirt1 regulates this acetylation. Inhibition of Sirt1 with nicotinamide (NAM) stimulated the acetylation of full-length Nav1.5 expressed in HEK293 cells, whereas overexpression of Sirt1 or stimulation of Sirt1 activity with resveratrol decreased the levels of lysine acetylation of Nav1.5 (Fig. 2a). In addition, inhibition of Sirt1 in neonatal rat cardiomyocytes, either by adenoviral overexpression of dominant-negative Sirt1 (H363Y) or pharmacologically by a Sirt1 inhibitor (Ex-243), led to hyperacetylation of native Nav1.5 (Supplementary Fig. 4a and Fig. 1d). To identify the acetylated lysine residue(s) in Nav1.5 that are deacetylated by Sirt1, we focused on the lysine-rich intracellular region of Nav1.5-linking domains III and IV (III-IV interdomain linker), which is 61 amino acids in length. This interdomain linker is highly conserved among voltage-gated sodium channels and has an important role in the regulation of channel localization and kinetics4. Moreover, inherited mutations and deletions of lysine residues in this region are associated with clinical arrhythmia phenotypes8,9. We generated a recombinant fusion protein in which glutathione S-transferase (GST) is fused to the III-IV interdomain linker (GST-Nav1.5 (III-IV)), and found that this fusion protein interacted with Sirt1 using in vitro co-precipitation (Fig. 2b). Acetylation of GST-Nav1.5 (III-IV) by p300/CREB-binding protein (CBP) acetylase increased this interaction (Fig. 2b). Moreover, similarly to its effects on full length Nav1.5, manipulation of Sirt1 expression or activity altered the acetylation of GST-Nav1.5 (III-IV) expressed in HEK293 cells (Supplementary Fig. 4b). To identify the acetylated lysine(s) in the region of Nav1.5 that is deacetylated by Sirt1, we performed mass spectrometry (MS). We acetylated recombinant GST-Nav1.5 (III-IV) with p300/CBP and then treated the acetylated protein with Sirt1 in vitro (Supplementary Fig. 5a). After acetylation with p300/CBP, we identified the acetylated peptide kAcLGGQDIFmTEEQK by MS/MS analysis (Supplementary Fig. 5b,c). The acetylated lysine in this peptide corresponds to the highly-conserved lysine 1479 (K1479) in full-length Nav1.5. Liquid chromatography (LC)-MS/MS analysis failed to detect this acetylated peptide in untreated GST-Nav1.5 (III-IV) peptide, and the abundance of this acetylated peptide was markedly decreased when p300-acetylated GST-Nav1.5 (III-IV) was treated with Sirt1 (Supplementary Fig. 5c). To verify K1479 as a target of Sirt1, we mutated this residue in GST-Nav1.5 (III-IV) to a nonacetylatable arginine (K1479R) and assessed its acetylation when expressed in HEK293 cells. As compared to the WT GST-Nav1.5 (III- IV) peptide, acetylation of the K1479R mutant GST-Nav1.5 (III-IV) peptide was decreased; moreover, knockdown of Sirt1 increased acetylation of wild-type GST-Nav1.5 (III-IV) but not that of the K1479R mutant (Fig. 2c). To further assess Nav1.5 acetylation, we generated an antibody highly specific against the acetylated K1479 immunogen (Supplementary Fig. 5d-f). Using this antibody, we found that Sirt1 overexpression in HEK293 cells led to deacetylation of K1479 in GST-Nav1.5 (III-IV) (Fig. 2d). Similarly, K1479 in full-length Nav1.5 expressed in HEK293 cells was deacetylated when Sirt1 was overexpressed but hyperacetylated when Sirt1 activity was inhibited by either treatment with NAM or the expression of dominant-negative Sirt1 (H363Y) (Fig. 2e, f and Supplementary Fig. 4b). Moreover, K1479 was hyperacetylated in isolated cardiomyocytes and whole hearts of cSirt1−/− mice, as compared to those of littermate mice in which exon 4 of Sirt1 is flanked by LoxP (Sirt1fl/fl) (Fig. 2g-i). We also tested the role of other HDACs in the regulation of K1479 acetylation. Trichostatin A (TSA), an inhibitor of class I and II HDACs, does not inhibit class III HDACs such as Sirt1. Treatment of HEK293 cells expressing Nav1.5 with TSA also resulted in increased levels of acetylated K1479 (Supplementary Fig. 4c), which indicates that acetylated K1479 is also targeted by nonsirtuin deacetylases.
Figure 2.
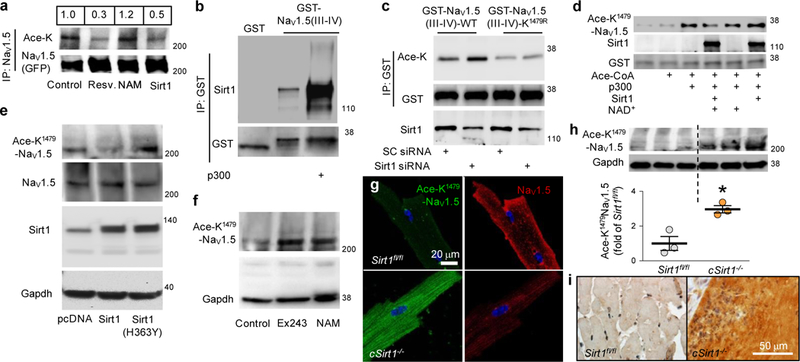
Regulation of Nav1.5 acetylation at K1479 by Sirt1. (a) Acetylation of Nav1.5 (GFP-tagged) expressed in HEK293 cells (control, no treatment) or treated with NAM (5 mM) or resveratrol (Resv., 100 μM), or in HEK293 cells overexpressing Sirt1. Quantification of the acetylated Nav1.5 to total Nav1.5 ratio is shown above the lanes. (b) Co-precipitation of recombinant Sirt1 with nonacetylated and acetylated recombinant GST-Nav1.5 (III-IV) peptide, as assessed by a GST pull-down experiment using lysates from expressing GST or the GST-Nav1.5 (III-IV) peptide. (c) Acetylation of the GST-NaV1.5 (III-IV) and K1479R GST-NaV1.5 (III-IV) peptides expressed in HEK293 cells treated with a scrambled siRNA (SC siRNA) or Sirt1 siRNA. (d) In vitro p300/CBP-mediated acetylation, and Sirt1-mediated deacetylation, of K1479 in recombinant GST-Nav1.5 (III-IV) peptide, using antibody toward Ace-K1479 epitope (Ace-K1479-Nav1.5). (e) K1479 acetylation in full-length Nav1.5 in HEK293 cells transfected with pcDNA, Sirt1 or a dominant-negative Sirt1 (H363Y). (f) K1479 acetylation in full-length Nav1.5 in HEK293 cells (control, vehicle) or treated with Ex-243 (5 μM) or NAM (5 mM). (g) Photomicrographs showing K1479 acetylation and plasma membrane expression of Nav1.5 in cardiomyocytes isolated from cSirt1−/− mice, as compared to Sirt1fl/fl mice. Scale bar, 20 μm. (h,i) Immunoblot (h) and immunostaining (i) showing K1479 acetylation in Nav1.5 in hearts from cSirt1−/− versus Sirt1fl/fl mice. n = 3 for each group. *P < 0.05 for cSirt1−/− versus Sirt1fl/fl. Immunoblots are representative of three independent experiments. Scale bar (i), 50 μm. Uncropped blots are shown in Supplementary Figure 7. Independent sample t test was used. Dot plot shows mean (line), and error bars represent s.e.m. Ace-K antibody directed against global acetylated lysine residues. Ace-K1479-Nav1.5 custom antibody directed specifically against acetylated K1479 of Nav1.5.
Next, we explored the importance of K1479 to a Sirt1-induced increase in INa. We generated full-length Nav1.5-mutant constructs and expressed them in HEK293 cells: a K1479Q construct was designed to mimic a constitutively acetylated channel and K1479R and K1479A constructs were designed to be nonacetylatable. The expression of acetylation-mimic Nav1.5 decreased peak INa as compared to wild-type Nav1.5, whereas the expression of nonacetylatable Nav1.5 increased INa (Fig. 3a, b). Moreover, in contrast to INa conducted by wild-type Nav1.5, Sirt1 did not stimulate INa in HEK293 cells expressing nonacetylatable Nav1.5 (compare Fig. 3c and Fig. 1c), and the expression of dominant-negative Sirt1 (H363Y) failed to inhibit INa conducted through nonacetylatable Nav1.5 (compare Fig. 3d and Fig. 1c). The kinetic properties of INa conducted by nonacetylatable Nav1.5 (K1479A and K1479R) were similar to those of wild-type Nav1.5; they showed no significant differences in steady-state activation, inactivation, development of intermediate inactivation or recovery from inactivation (Supplementary Tables 1 and 3 and Supplementary Fig. 2c, d). As compared to wild-type Nav1.5, the K1479Q acetylation mimic shifted the steady-state inactivation curve to more positive potentials (Supplementary Table 3 and Supplementary Fig. 2d), indicating that an increased fraction of channels is available for activation at normal resting membrane potential. However, despite this depolarizing shift in steady-state inactivation, INa conducted by K1479Q Nav1.5 was lower than that conducted by wild-type Nav1.5, which indicates that decreased current density is due to the reduced membrane localization of K1479Q Nav1.5.
Figure 3.
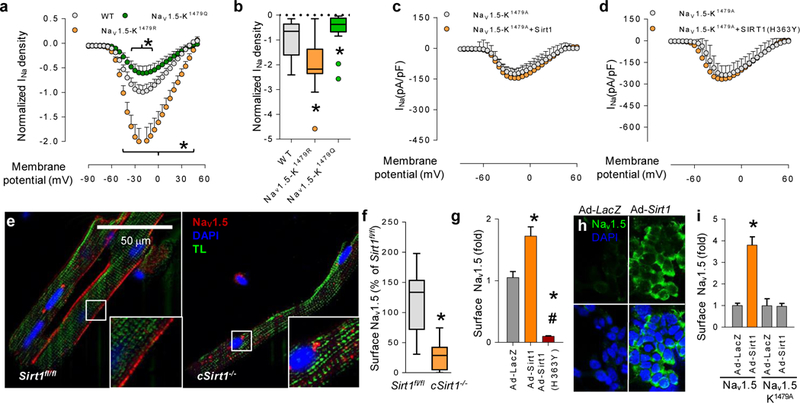
Regulation of INa and trafficking of NaV1.5 via Sirt1-mediated deacetylation of K1479. (a) INa in HEK293 cells expressing WT Nav1.5 or mutant Nav1.5 constructs mimicking acetylation (K1479Q) and deacetylation (K1479R). *P < 0.05 versus WT. n = 21 cells for WT, 20 cells for K1479Q and 11 cells for K1479R. (b) Sodium-current density at −40 mV. *P < 0.05 versus WT. (c,d) INa in HEK293 cells expressing nonacetylatable K1479A-Nav1.5 and expressing wild-type Sirt1 (c) or dominant-negative Sirt1 (H363Y) (d). n = 11 cells for Nav1.5-K1479A (c), 10 cells for Nav1.5-K1479A + Sirt1 (c), 10 cells for Nav1.5-K1479A (d) and 9 cells for Nav1.5-K1479A + Sirt1 (H363Y) (d). (e) Photomicrographs showing cell surface expression of Nav1.5 in cardiomyocytes from a cSirt1−/− mouse and a cSirt1fl/fl littermate control using an antibody against an extracellular Nav1.5 epitope in nonpermeabilized cells (magnification ×63, scale bar; 50 μm; TL, transmitted light). Insets show higher-magnification views of the boxed areas. (f) Quantification of surface Nav1.5 in cardiomyocytes isolated from cSirt1−/− mice and Sirt1fl/fl controls. n = 8 cells for Sirt1fl/fl and 7 cells for cSirt1−/−; *P < 0.05 versus Sirt1fl/fl. (g) Surface expression of Nav1.5, as assessed by a surface chemiluminescence assay, in HEK293 cells expressing Nav1.5 tagged with an extracellular FLAG epitope and subsequently transfected with the indicated adenoviral constructs for overexpression of Sirt1 or dominant-negative Sirt1 (H363Y). *P < 0.05 versus Ad-LacZ, #P < 0.05 versus Ad-Sirt1. n = 10 for Ad-LacZ and Ad-Sirt1, 8 for Ad-Sirt1 (H363Y). (h) Photomicrographs showing immunofluorescence staining of FLAG-Nav1.5 in the plasma membrane of HEK293 cells transfected with the indicated adenoviral constructs (magnification ×40). Scale bar, 10 μm. (i) Surface expression of Nav1.5 or nonacetylatable K1479A-Nav1.5 (both tagged with an extracellular FLAG epitope), as assessed by a surface chemiluminescence assay, following transfection with the indicated adenoviral constructs. *P < 0.05 versus Ad-LacZ. n = 9 for Nav1.5-Ad-LacZ, 11 for Nav1.5-Ad-Sirt1, 10 for Nav1.5-K1479A-Ad-LacZ and 8 for Nav1.5-K1479A-Ad-Sirt1. Independent sample t test was used. For XY scatter plots, data are shown as means, and error bars represent s.e.m. For box-and-whisker plots, whiskers show minima and maxima within 1.5 IQR. Outliers are shown as individual dots and are included in the statistical analysis. Ad-Sirt1 and Ad-Sirt1 (H363Y): adenoviruses expressing wild-type and dominant-negative Sirt1. Ad-LacZ: control virus.
Given the observations that Nav1.5 and Sirt1 colocalize at the cardiomyocyte membrane (Fig. 1b); that there is an appreciable decrease in the membrane localization of Nav1.5 in cSirt1−/− cardiomyocytes (Fig. 2g); and that Sirt1 affects the amplitude of INa but not its kinetics, we explored the role of Sirt1 in the trafficking of Nav1.5 to the plasma membrane. Activation of protein kinase A increases Nav1.5 trafficking to the plasma membrane10, whereas PKC activation results in decreased abundance of channels resident in the membrane11. Notably, a conserved serine residue (S1503), which resides in the III-IV interdomain linker, serves as a phospho-acceptor site that mediates the PKC-induced decrease in the cell surface expression of the channel7. To test the possibility that K1479 acetylation, similarly to S1503 phosphorylation, decreases cell surface expression of Nav1.5, we performed quantitative immunofluorescence of cardiomyocytes isolated from cSirt1−/− mice using an antibody that detects only an extracellular epitope of Nav1.5. cSirt1−/− cardiomyocytes showed a significant decrease in their expression of Nav1.5 at the cell membrane as compared to cSirt1fl/fl controls (Fig. 3e, f, Supplementary Fig. 1f and Supplementary Videos 1 and 2). Moreover, a quantitative cell surface luminescence assay using this antibody in HEK293 cells expressing Nav1.5 showed that expression of wild-type Sirt1 increased cell surface expression of the channel, whereas expression of dominant-negative Sirt1 (H363Y) decreased the abundance of Nav1.5 at the cell membrane (Fig. 3g-i). Notably, Sirt1 expression did not increase cell surface expression of nonacetylatable (K1479A) Nav1.5 (Fig. 3i), which underscores the requirement for K1479 acetylation in the regulation of Nav1.5 trafficking by Sirt1.
Next, we examined whether cSirt1−/− mice showed cardiac conduction abnormalities that are similar to those in Scn5a-heterozygousknockout mice and those observed in patients with loss-of-function SCN5A mutations12. Surface electrocardiograms (EKGs) performed on 4-5-month-old cSirt1−/− and Sirt1fl/fl mice showed no differences in baseline EKG intervals (Supplementary Table 4), which suggests that a lack of Sirt1 in cardiomyocytes has no adverse impact on cardiac electrical activity and conduction at this age. However, chronologic aging unmasked notable differences in cardiac conduction parameters between cSirt1−/− and Sirt1fl/fl mice. At approximately 9 months of age, cSirt1−/− mice exhibited prolongation of the QRS and QTc intervals as compared to aged-matched Sirt1fl/fl mice (P < 0.05 for each; Supplementary Table 4), indicating that the absence of Sirt1 in cardiomyocytes leads to age-dependent impairment of conductance of the electrical impulse and delayed ventricular repolarization, similar to prior reports on Scn5a+/− mice12. Expression of the gapjunction protein connexin 43 was not diminished in the hearts of cSirt1−/− mice (Supplementary Fig. 6a), which indicates that a defect in cell-to-cell electrical-impulse conduction likely does not contribute to the cardiac conduction abnormalities. To complement these studies, we intraperitoneally (i.p.) administered the Sirt1 inhibitor Ex-527 to ~10-11-month-old wild-type (Scn5a+/+) and Scn5a-heterozygousknockout (Scn5a+/−) mice. The efficacy of Ex-527 in inhibiting Sirt1 was verified by documenting increased acetylation of Nav1.5 on K1479 (Supplementary Fig. 6c) and activation of the unfolded protein response (Supplementary Fig. 6d) in the hearts of treated Scn5a+/+ mice. Scn5a+/+ mice treated with Ex-527 showed prolongation of the QTc and a strong trend toward QRS prolongation as compared to control mice (Supplementary Table 5). QRS prolongation reached significance in Scn5a+/− mice treated with Ex-527 (P = 0.05; Supplementary Table 5). Thus, pharmacologic inhibition of Sirt1 recapitulates the same conduction abnormalities observed in aged cSirt1−/− mice, and lack of one allele of Scn5a mice amplifies this phenotype. Also, similarly to cSirt1−/− mice, mice treated with Ex527 did not show a decrease in cardiac connexin 43 (Supplementary Fig. 6b), which indicates that Sirt1 likely does not regulate gap junction conduction.
The increase in QRS and QTc interval duration in cSirt1−/− mice prompted us to compare action-potential upstroke and duration in cSirt1fl/fl and cSirt1−/− cardiomyocytes. As compared to cSirt1fl/fl cardiomyocytes, cSirt1−/− cardiomyocytes showed decreased velocity of phase 0 of the action potential (Supplementary Fig. 3c)— consistent with a decrease in INa -prolonged action-potential duration (Supplementary Fig. 3d)—no change in resting membrane potential (Supplementary Fig. 3e) and no change in the amplitude of late INa (Supplementary Fig. 3b). These changes in action-potential duration and kinetics in cSirt1−/− cardiomyocytes are consistent with the observed EKG abnormalities.
To examine whether cardiac arrhythmias occur in cSirt1−/− mice, we performed continuous ambulatory telemetry of 6-9-month-old mice. Telemetry monitoring showed nonsustained ventricular tachycardia (VT) in 4 of 12 cSirt1−/− mice but in 0 of 10 Sirt1fl/fl mice (P < 0.05; Fig. 4a and Supplementary Table 6). In addition, although there were no differences in heart rate (maximum, minimum or mean), frequent sinus pauses were present in 6 of 12 cSirt1−/− mice, as compared to only 1 of 10 Sirt1fl/fl mice (P < 0.05), and there was a trend toward more episodes of two-degree atrioventricular (AV) block in the cSirt1−/− mice (Fig. 4b and Supplementary Table 6). Moreover, cSirt1−/− mice were at a markedly increased risk for premature death between the ages of 6 and 10 months (Fig. 4c), the period during which they manifested abnormalities in cardiac electrical conduction and arrhythmias. Although the left ventricular-ejection fraction was mildly decreased in cSirt1−/− as compared to cSirt1fl/fl mice, cardiac output was not depressed in 5-6-month-old cSirt1−/− mice, LV mass was not increased and no signs of clinical heart failure or pulmonary congestion were evident at this age (Supplementary Table 7). We followed four cSirt1−/− mice with long-term implanted telemetry monitors to identify the cause of death. Each of the mice developed marked bradyarrhythmias (sinus bradycardia with junctional-escape rhythms and/or AV block) within 12 h of death (Fig. 4d, e), which suggests that cSirt1−/− mice die of bradyarrhythmias.
Figure 4.
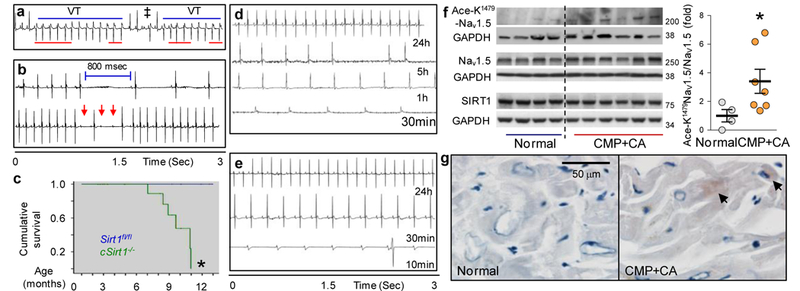
Bradyarrhythmias, tachyarrhythmias and conduction abnormalities related to increased K1479 acetylation of Nav1.5 in mice and humans. (a) A representative telemetry recording (n = 4) from a cSirt1−/− mouse showing nonsustained ventricular tachycardia (VT), a fusion beat (‡) and atrio-ventricular (A-V) dissociation (red lines). (b) Representative telemetry recordings (n = 6) from a cSirt1−/− mouse showing prolonged sinus pause (800 ms, top) and episodes of second-degree AV block (red arrows, bottom) followed by a junctional escape beat. (c) Kaplan-Meier survival curves for cSirt1−/− as compared to Sirt1fl/fl mice. n = 31 for Sirt1fl/fl and 27 for cSirt1−/−, *P < 0.05 versus Sirt1fl/fl. (d,e) Representative telemetry recordings (n = 4) from two cSirt1−/− mice showing onset of bradycardia in cSirt1−/− mice within 12 h of death. In d, recordings show normal sinus rhythm at 24 h before death (24 h), atrial fibrillation with a slow ventricular rate of ~120 beats/min at 5 h before death (5 h); sinus bradycardia at 160 beats/min at 1 h before death (1 h) and junctional escape rhythm at 120 beats/min at 30 min before death (30 min). In e, traces show normal sinus rhythm at 24 h before death (24 h), sinus bradycardia at 240 beats/min at 30 min before death (30 min) and complete heart block with a slow ventricular escape at 10 min before death (10 min). (f) Left, western blot showing expression of Nav1.5, Sirt1 and acetylated-K1479-Nav1.5 in ventricular tissue of hearts explanted from patients with dilated cardiomyopathy (CMP) as compared to hearts with normal function. GAPDH was used as loading control. Right, quantitation of Ace-K1479-Nav1.5 normalized to total Nav1.5 n = 4 normal and n = 7 CMP hearts, *P < 0.05 versus normal. (g) Photomicrographs showing acetyl-K1479-Nav1.5 immunostaining in the heart of a patient with CMP and conduction abnormalities (CA; QRS 176 ms, QTc 511 ms) as compared to that of a normal heart. Representative images from CMP heart (n = 4) and normal heart (n = 3). Arrows, Ace-K1479-Nav1.5. Scale bar, 50 μm. Independent sample t test was used for f and log-rank test was used for c. Uncropped blots are shown in Supplementary Figure 7. Dot plots show mean (line), and error bars represent s.e.m.
Finally, we explored the potential significance of K1479 acetylation in human cardiac conduction disease by determining whether lysine acetylation of Nav1.5 is associated with clinical conduction abnormalities. We obtained ventricular tissue from hearts explanted at the time of transplantation from patients with idiopathic dilated cardiomyopathy. Mean QRS and QTc were prolonged in these patients, as assessed by surface EKGs (Supplementary Table 8). We compared Nav1.5 expression and K1479 acetylation in these hearts to those of subjects who had no known heart disease, had normal left-ventricular function and had died of noncardiac-related causes. When compared to these normal human hearts, dysfunctional hearts with conduction abnormalities showed increased levels of Ace-K1479 Nav1.5, with no decrease in Sirt1 expression (Fig. 4f, g).
Several post-translational modifications govern trafficking of Nav1.5 (ref. 2). PKC-mediated phosphorylation of serine 1503 in the III-IV interdomain linker of the channel decreases its expression on the cell membrane7,11. Moreover, the energetic state of the cell also regulates Nav1.5 trafficking; NAD+ and NADH have opposing effects on the membrane expression of the channel6. Notably, NADH acts via PKC to decrease membrane expression of Nav1.5 (ref. 6). Our findings that Sirt1 plays an important part in regulating the trafficking of Nav1.5 to the cell membrane dovetails with these prior reports, and suggests that NAD+-dependent, Sirt1-mediated deacetylation of K1479 in the III-IV interdomain linker counteracts PKC-mediated phosphorylation of S1503 in the same region of the channel. Given that Nav1.5 trafficking is regulated by lysine ubiquitination13, as well as the role of Sirt1 in governing ubiquitination14, it is tempting to hypothesize a functional interaction between lysine acetylation and the ubiquitination of Nav1.5.
Inherited human SCN5A channelopathies are associated with a wide range of cardiac conduction abnormalities and arrhythmia phenotypes. SCN5A mutations that decrease INa can result in progressive cardiac conduction defects with widening of the QRS interval15-17, bundlebranch block16 and dysfunction of the sinoatrial pacemaker, resulting in sinus block and bradycardia15,17,18. In addition, reduced expression of Nav1.5 in Scn5a+/− mice leads to widespread conduction delay, as well as atrial and ventricular arrhythmias that worsen with age12,19. The heterogeneity of the conduction abnormalities and arrhythmias in cSirt1−/− mice, as well as their association with aging, are reminiscent of the phenotype of Scn5a+/− mice and suggest a global slowing of conduction, probably most prominently in the specialized conduction system and periatrioventricular nodal tissue. Age-related worsening of conduction disease is also present in patients with loss-of-function SCN5A mutations (as seen in inherited conduction disease20 and Brugada syndrome21), and suggests that the loss of Sirt1-dependent INa might add to the insults perpetrated by aging on the cardiac conduction system. Such insults include age-associated fibrosis22 that could compound the decrease in INa owing to lack of Sirt1. It is also possible that the decrease in INa caused by deficiency in Sirt1 contributes to cardiac fibrosis, as has been reported in Scn5a+/− mice12.
The III-IV interdomain linker of Nav1.5 is lysine-rich and is one of the most conserved regions of the protein. Several mutations or deletions involving lysines in this linker lead to abnormal Nav1.5 function and are associated with human arrhythmias. The ΔKPQ deletion, encompassing residues 1505-1507, disrupts fast inactivation and results in an autosomal-dominant form of long-QT syndrome23. It also leads to higher peak current and changes in current gating24. A loss of function ΔK1500 mutation results in reduced peak sodium current, but a larger sustained (late) sodium current, and leads to an overlapping phenotype of a prolonged QT interval, QRS widening (right bundle-branch block) and sinus pauses8. Most germane to our findings, in a large family with a malignant phenotype, a deletion mutation affecting K1479 (ΔK1479) was reported to cause Brugada syndrome, severe cardiac conduction disease, including prolonged sinus pauses, sudden cardiac death despite pacemaker placement, and sudden infant death syndrome25. When expressed in HEK293 cells, the Nav1.5 channel bearing the ΔK1479 deletion resulted in both a decrease in the peak current density and marked changes in channel kinetics9. By contrast, we found that replacement of K1479—the primary site for Nav1.5 acetylation—with a nonacetylatable residue alters membrane localization without inducing major changes in channel kinetics. The differences in the electrophysiological characteristics of Nav1.5 ΔK1479 and Nav1.5 with a nonacetylatable residue could be due to changes in protein folding caused by the deletion of K1479, but not by its replacement with a nonacetylatable residue, in this region of the protein, in which mutations of other amino acids are known to cause profound kinetic changes in the channel. Overall, these inherited human arrhythmia phenotypes, coupled with changes in Nav1.5 function and/or trafficking, underscore the important role of K1479 in regulating the cardiac sodium current.
Our findings on Sirt1-dependent regulation of Nav1.5 shed light on the role of K1479 not only in inherited forms of arrhythmia, but also in conditions that lead to acquired arrhythmias. Reduction of INa is typical in both experimental and human heart failure26-28. The finding that Nav1.5 is hyperacetylated at K1479 in patients with dilated cardiomyopathy and conduction disease suggests that the K1479- acetylation-mediated decline of INa might play a part in brady- and tachyarrhythmias associated with advanced human heart failure. It is important to note, however, that Sirt1 expression did not differ substantially between cardiomyopathic and normal hearts. Although this finding does not argue against the potential role of K1479 acetylation in human cardiac conduction disease, it does indicate that K1479 acetylation of Nav1.5 in this particular cohort of patients is due to mechanisms other than Sirt1 deficiency. Changes in the expression or activity of nonsirtuin HDACs or acetyltransferases could provide for such alternative mechanisms. It is also important to recognize that decreased expression or activity of cardiac Sirt1 could, via its effect on Nav1.5, lead indirectly to structural changes in the heart. Ion channel dysfunction can result in mechanical instability, ultimately leading to cardiac remodeling, as is evident in humans who harbor arrhythmogenic mutations in SCN5A and develop dilated cardiomyopathy29, and in Scn5a+/− mice, which develop cardiac hypertrophy and fibrosis as they age12.
Our findings do not exclude a role for other sirtuins or nonsirtuin HDACs in the modulation of cardiac electrophysiology. The finding that TSA alters K1479 acetylation suggests that K1479 acetylation is regulated by more than one HDAC, which is concordant with previous findings that many acetylated lysines targeted by Sirt1 are also deacetylated by nonsirtuin HDACs30. Deacetylation-mediated regulation of INa by HDACs other than Sirt1 might provide a redundant mechanism for K1479 deacetylation and explain why cSirt1−/− mice had only a modest reduction in INa and survived into adulthood; a compensatory increase in Nav1.5 protein expression is another explanation for the modest reduction of INa in cSirt1−/− mice. We also cannot rule out the possibility that Sirt1 might affect the trafficking and/or function of other ion channels. The QTc prolongation seen in cSirt1−/− mice could be the result of changes in the expression or function of K+ channels, Ca2+ channels or Ca2+-handling proteins.
In summary, these findings show that dynamic lysine acetylation, modulated by the Sirt1 deacetylase, is a functional post-translational modification of the cardiac sodium channel. By deacetylating Nav1.5 at K1479, Sirt1 stimulates its trafficking to the cell membrane and thus has an important role in the regulation of cardiac electrical activity. Consistent with its effect on INa, lack of cardiomyocyte Sirt1 in mice leads to cardiac conduction abnormalities and fatal arrhythmias. These findings suggest that pharmacological or genetic interventions that activate Sirt1 might be of benefit in treating cardiac arrhythmias that result from familial or acquired loss-of-function sodium channelopathies.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
ONLINE METHODS
Animals.
Sirt1 floxed mice (Sirt1fl/fl; Jackson Laboratories 008041, backcrossed into C57BL/6) were mated with mice overexpressing Cre-recombinase driven by the α-myosin heavy chain promoter (αMHC-Cre; Jackson Laboratories 011038, C57BL/6 background) to generate cSirt1−/− mice and littermate Sirt1fl/fl controls. 4-9-month-old male and female mice were used. All studies were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Iowa.
Reagents.
Nav1.5 (abcam-ab63288, Almone labs-ASC-005, MilliporeAB5493), Sirt1 (Thermo Scientific-PA5-23-063, Santa Cruz Biotechnologysc-15404), GAPDH (Trevigen-2275-PC), custom designed anti-acetyl-K1479 Nav1.5 (YenZym Antibodies) and acetyl-lysine (Cell signaling technology9681) antibodies were used. A custom designed anti-acetyl-K1479 Nav1.5 antibody was purified from sera from rabbits immunized with the peptide FNQQKK1479KAcLGGQDIC. The specificity of this antibody was determined by examining its ability to detect p300/CBP acetyltransferase-induced acetylation of III-IV interdomain linker of Nav1.5 and not the nonacetylated linker (Supplementary Fig. 6c). All other reagents were procured from Sigma-Aldrich, St. Louis, MO, unless specified. The SCN5A mutant constructs were generated by site-directed mutagenesis. Ex-243 and Ex-527 are specific inhibitors of Sirt1, whereas Ex-242 is an optical isomer of Ex-243 with no activity toward Sirt1.
Cell culture.
HEK293 cells were obtained from the American Type Culture Collection (ATCC) and were cultured in 10% FBS-supplemented DMEM media. Rat neonatal cardiomyocytes were freshly isolated and cultured in an equivolume mixture of DMEM and F-12 media containing 5% FBS.
Rat neonatal cardiomyocyte isolation.
Cardiomyocytes were isolated from newly delivered neonatal Sprague-Dawley rat pups using the neonatal cardiomyocyte isolation system (Worthington Biochemical Co, Lakewood, NJ).
Isolation of mouse ventricular myocytes.
Mouse ventricular myocytes were isolated from 6-7-month-old cSirt1−/− and Sirt1fl/fl control hearts by perfusing a Ca2+-free normal Tyrode solution containing collagenase (1 mg/ml, Type II, Worthington Biochemical, USA) and protease (0.1 mg/ml, Sigma-Aldrich, USA) on a Langendorff apparatus at 37 °C, as described31. Isolated ventricular myocytes were stored in high K+, low Cl− solution at room temperature (24 °C) and used within 18 h. Normal Tyrode solution contained (in mmol/liter) NaCl 143, KCl 5.4, MgCl2 0.5, CaCl2 1.8, NaH2PO4 0.5, glucose 11.1 and 5 HEPES (pH adjusted to 7.4 with NaOH). The high K+, low Cl− storage solution contained (in mmol/liter) K-glutamate 70, KCl 55, HEPES 10, MgCl2 3, taurine 20, KH2PO4 20, EGTA 0.5, adjusted pH to 7.3 with KOH.
Immunoprecipitation and immunoblotting.
Immunoprecipitation of Sirt1, Nav1.5 and GST-Nav1.5 (III-IV) was carried out by incubating 2 μg of the respective antibodies (Santa Cruz Biotechnology and Millipore) with 1 mg of cell lysate or tissue homogenate overnight, followed by 40 μl of protein A-Sepharose slurry (Amersham, Piscataway, NJ) for 4 h. After washing, immunoprecipitates were boiled in SDS-PAGE gel loading buffer, subjected to SDS-PAGE, transferred to nitrocellulose membrane and probed with 1:1,000 dilution of specified primary antibody and the appropriate peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). Western blotting of 50 μg of whole-cell lysates (WCL) was similarly performed by using appropriate primary and secondary antibodies. Chemiluminescent signal was developed by using SuperSignal West Pico/Femto substrate (Pierce, Rockford, IL), and blots were imaged with a GelDoc 2000 Chemi Doc system (Bio-Rad, Hercules, CA). Supplementary Figure 8 shows uncropped blots for Supplementary Figures.
Acetylation and deacetylation of Nav1.5.
For analysis of in vitro acetylation and deacetylation of Nav1.5 constructs, we followed established protocols32. Purified recombinant GST-Nav1.5 (III-IV) or GST-K1479A-NaV1.5 (III-IV) was desalted, and the buffer was exchanged with acetylation buffer (50 mM Tris·HCl, pH 8.0/137 mM NaCl/0.1 mM EDTA, pH 8.0/10% glycerol/1 mM DTT/0.1 mM PMSF/2 μM trichostatin A) using Zeba spin columns (Pierce). Five micrograms of Nav1.5 construct per acetylation reaction were incubated with acetyl-CoA (20 μM) and p300/CBP acetyltransferase (60 units, Active Motif) at 30 °C for 40 min with shaking. The acetylation reaction was followed immediately by the deacetylation reaction by adding deacetylation buffer (25 mM Tris·HCl, pH 8.0/137 mM NaCl/2.7 mM KCl/1 mM MgCl2/ 1 mg/ml BSA), 1 mM NAD+ and active recombinant Sirt1 (10 units; Biomol, Plymouth Meeting, PA), and incubated at 37 °C for 1 h with shaking. The reaction mixture was subjected to SDS-PAGE and immunoblotting. Acetylation of Nav1.5 and GST-Nav1.5 (III-IV) in cells was assessed by immunoprecipitation followed by immunoblotting with an acetyl-lysine-specific antibody (Cell Signaling). Acetylation of K1479 in mouse and human tissues was assessed using the custom acetyl-K1479 antibody.
Determination of cell surface expression of Nav1.5 in HEK293 cells.
A stable HEK293 cell line expressing full-length Nav1.5 tagged with an extracellular FLAG-myc epitope was created. Cells were plated in a 12-well dish at a density of 1.0 × 105 cells/well and placed on ice to stop cellular trafficking throughout experiment. Nonpermeabilized cells were then treated with primary α-FLAG-M2 antibody (1:50 in PBS/0.5% BSA) (Covance, New York, NY) for 90 min followed by washing with 1×-PBS/0.5% BSA (3×). Cells were then treated with a FITC conjugated-goat anti-mouse secondary antibody (1:1,000) (Sigma-Aldrich, St. Louis, MO) for 30 min in the dark. Cells were washed again with only 1×-PBS and treated with DAPI (0.5 μg/ml) for 1 min followed by a 5-min rinse in 1× PBS. Cells were imaged with a Leica TCS-SL confocal microscope and captured with Leica confocal software (Leica Microsystems, Mannheim, Germany).
Cell surface expression of wild-type Nav1.5 and K1479A-Nav1.5 was quantified by chemiluminescence. The wild-type and mutant constructs were expressed in HEK293 cells. Nonpermeabilized cells were labeled sequentially with an antibody that detects an extracellular domain of NaV1.5 (Abcam) followed by secondary secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG. All steps were performed at 4 °C to block Nav1.5 trafficking. The cells were then washed extensively, and HRP-labeled proteins were detected using SuperSignal West Femto chemiluminescent substrate (Pierce) and read in a TD20/20 luminometer (Turner, Sunnyvale, CA). Background chemiluminescence was established with HEK293 cells not expressing Nav1.5.
Adult mouse cardiomyocytes on laminin-coated coverslips were fixed, blocked and incubated with 1:100 anti-Nav1.5 antibody (Abcam, ab62388), which detects the extracellular domain of Nav1.5, overnight. The antigen-antibody complexes were probed with 1:1,000 AlexaFluor568 tagged anti-mouse secondary antibody and counterstained with DAPI. Images were acquired to detect fluorescence of AlexaFluor568, DAPI along with transmitted light imaging using a Zeiss confocal microscope (Model 710). The surface expression of Nav1.5 was determined by quantifying the fluorescence intensity of immunostaining for Nav1.5 on the cell boundary using ImageJ (1.49e) software. The cell boundary was determined by simultaneous transmitted light microscopy.
Immunohistochemistry.
Sections (5 μm) of formalin-fixed paraffin-embedded tissues were heated (95 °C, 20 min) in citrate buffer (10 mM) for antigen retrieval followed by incubation with primary antibody. Antigen-primary antibody complexes were incubated with a polyvalent biotinylated goat anti-rabbit secondary antibody and streptavidin peroxidase (STV-HRP) system to amplify the signals, followed by detection with diaminobenzidine (DAB) as a chromogen. Slides were counterstained with hematoxylin, dehydrated with alcohols and xylene and mounted in DPX. Images were captured by a charged coupled device (CCD) camera attached with the Olympus microscope (Model BX61).
Whole-cell patch clamping.
Conventional whole-cell patch clamp techniques were used to record Na+ currents from HEK293 cells constitutively expressing Nav1.5 and GFP, from rat neonatal cardiomyocytes, or from rodshaped, quiescent single isolated mouse ventricular myocytes. For HEK293 cells, the electrodes of 1-2 MΩ were filled with a pipette solution containing (in mmol/liter) NaF 10, CsF 110, CsCl 20, EGTA 10 and HEPES 10 (pH 7.35 with CsOH), and the bath solution contained (in mmol/liter) NaCl 40, 103 NMDG, KCl 4.5, CaCl2 1.5, MgCl2 1, and HEPES 10 (titrated to pH 7.35 with CsOH). For recordings from isolated rat neonatal myocytes, the pipette solution contained (in mmol/liter) NaF 10, CsF 120, CsCl 20, EGTA 10, HEPES 10 titrated to pH 7.4 with CsOH and the bath solution contained in (mmol/ liter) NaCl 25, NMDG 118.2, KCl 4, MgCl2 1, CaCl2 1.8, HEPES 10, glucose 10 and titrated to a pH of 7.4 with HCl. For recordings from mouse ventricular cardiomyocytes, the pipette solution contained (in mmol/liter) Cs-aspartate 90, CsCl 20, MgCl2 2, Mg- ATP 5, HEPES 10, Na2-creatine phosphate 2.5, tetraethylammonium chloride (TEA-Cl) 10, EGTA 5 with pH adjusted to 7.3 using CsOH and a modified Cs+-based low Na+ bath solution contained (in mmol/liter) CsCl; 130, NaCl; 5, MgCl2; 2.5, CaCl2; 0.2, HEPES; 20, and glucose 11, with added nifedipine (10 μM) and nickel chloride (100 μM) to block Ca2+ currents. In all recordings, the potentials were corrected for the liquid junction and 80% of the series resistance was compensated, yielding a maximum voltage error of ±1 mV. Low-pass filtered signals (5 KHz) from a patch-clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA) were digitized in an AD/DA converter (Digidata 1200, Molecular Devices, Sunnyvale, CA) at 20 KHz and were stored for later analysis. Na+ channel data were analyzed with pClamp 9.2 software (Molecular Devices). Cell capacitance was recorded directly from the patch amplifier after nullifying the transients following patch rupture. To prevent membrane-potential escape from the command voltage in voltage-clamp mode, access resistance was kept below 7 MΩ throughout the experiment, and relatively small cells were used. To minimize time-dependent drift in gating parameters, all protocols were initiated 5 min (10 min for mouse ventricular cardiomyocytes) after the whole-cell configuration was obtained. For generation of I-V curves, a 1-s prepulse to −120 mV was used to eliminate inactivation, and 200-ms test pulses between −80 and +30 mV were used to activate the channel. Intermediate inactivation was assessed using a two-pulse protocol. The first pulse to −20 mV from a holding potential of −120 mV ranged from 1 to 1,000 ms in duration to ensure that channels entered the intermediate inactivation state. This was followed by a brief recovery period to −120 mV for 20 ms to allow recovery from fast inactivation. The peak current during the second test pulse to −20 mV was normalized to the first pulse, and the points were fit using a single exponential function. Recovery from inactivation (after a 1,000-ms depolarization to −20 mV from a holding potential of −120 mV) was fit using a doubleexponential function, with τ fast representing recovery from fast inactivation and τ slow representing recovery from slow inactivation. Steady-state inactivation was measured by a two-pulse protocol with 1,000 ms conditioning pulses from −150 to −20 mV, followed by a 200-ms test pulse to −20 mV. To record membrane voltage from single mouse ventricular myocytes, we used a conventional whole-cell current clamp configuration. Action potentials (APs) were evoked by a depolarizing current (1.3 × threshold) for 1.5 ms at 3 Hz. Action potential duration (APD) or maximum velocity of upstroke of AP (Vmax) values were obtained from the average of five sequential APs selected from 41st to 45th beats evoked by 50 depolarizing current stimulus. The pipette solution contained (mmol/liter): K aspartate 120, HEPES 5, KCl 25, Na2ATP 4, MgCl2 1, Na2 phosphocreatine 2, NaGTP 2, CaCl2 1, and EGTA 10. The pH was adjusted to 7.4 with KOH; the bath solution contained (mmol/liter) NaCl 143, KCl 5.4, HEPES 5, MgCl2 0.5, NaH2PO4 0.5, CaCl2 1.8, glucose 10, adjusted pH to 7.4 with NaOH. Filtered signals (2 KHz) from the patchclamp amplifier were digitized at 10 KHz. All electrophysiological experiments were performed at room temperature (23-25 °C).
Surface EKGs.
Recordings were performed in cSirt1−/− and Sirt1fl/fl mice. Mice were anesthetized by isoflurane (1-2%) inhalation and anesthesia was maintained during the procedure by use of a nose cone. High-resolution multilead EKGs were performed using a Data Acquisition System (iWorx/IX-100B, Dover, NH) and bipolar electrodes in positions corresponding to human leads I (right to left shoulder), II (right shoulder to left thigh), III (left shoulder to left thigh) and modified chest V (back to left midclavicular line in the 5th interspace). Electrodes were placed subcutaneously by lightly puncturing through the skin. Data were collected, digitized and analyzed using LabScribe2 by an investigator blinded to genotype. QTc was calculated using a mouse-specific formula33.
Telemetry.
Radiotelemetry electrocardiographic monitors (model ETA-F20 (DSI, St. Paul, MN)) were implanted subcutaneously on the backs of cSirt1−/− and Sirt1fl/fl mice following an i.p. injection of ketamine 80 mg/kg and xylazine 8 mg/kg anesthesia, as previously described34. Mice were allowed to recover for at least 6 d. Implanted monitors were interfaced with PhysioTel Receiver RPC-1 model (DSI, St. Paul, MN). Data were sampled at 400-1,000 Hz and converted to digital format using the PowerLab data acquisition system and LabChart 7 software (AD Instruments, Colorado Springs, CO). Telemetry data (12 h/mouse) were scanned for atrial and ventricular arrhythmias by a cardiologist trained to read mouse electrocardiograms (BL) and blinded to genotype. At least 95% of the data for each mouse was suitable for analysis.
Transthoracic echocardiograms.
Echocardiograms were performed on mice anesthetized with midazolam using the Vevo 2700 VisualSonics System.
Mass spectrometry (MS) analysis.
For mass spectrometry analysis, we used a modification of the protocol described in Burgoyne et al.35. Briefly, the recombinant fusion protein in which GST is fused to the III-IV interdomain linker (GST-Nav1.5 (III-IV)) was purified and treated with either control solution or solution containing p300/CBP acetyltransferase for 16 h. Purified GSTNav1.5-(III-IV) was then resolved by SDS-PAGE and visualized with colloidal Coomassie stain. Bands corresponding to Nav1.5 (III-IV) were excised and diced into small pieces followed by trypsin (Promega, Madison, WI) ingel digestion using the ProteaseMax (Promega)-based system for enhanced extraction of peptides. Extracted peptides were then desalted via C18 ziptips (Millipore), and a small fraction was spotted along with α-cyano-4- hydroxycinnamic acid or 2,5-dihydroxybenzoic acid matrix onto a matrixassisted laser desorption/ionization (MALDI) target plate. An ABI 4800 (Applied Biosystems, Foster City, CA) MALDI time of flight (TOF) instrument or a Thermo LTQ-Orbitrap XL and a Waters Nano-Acquity LC system was used to acquire MS or MS/MS data from the samples. Peptide mass fingerprinting, MS/MS search and de novo sequencing were performed using mMass (Martin Strohalm, Prague, Czech Republic), Mascot (Matrix Science, Boston, MA) and Data Explorer (Applied Biosystems). Mascot searches were performed with a mass error of 50 p.p.m. for MALDI TOF data and 10 p.p.m. for Orbitrap against the SwissProt database version 57.15, including a maximum of three missed cleavages.
Human heart tissue.
Procurement of human myocardial tissue was performed under protocols approved by Institutional Review Board of the University of Pennsylvania. Informed consent was obtained from all subjects. Failing human hearts were procured at the time of orthotopic heart transplantation. Nonfailing hearts were obtained at the time of organ donation from cadaveric donors (Gift of Life Donor Program). In all cases, hearts were arrested in situ using ice-cold cardioplegia solution and transported on wet ice for cell isolation within 4 h of explant. As previously described, cardiomyocyte-enriched pellets were obtained by filtration and sedimentation36, followed immediately by freezing in liquid nitrogen and storage at −80 °C until assay.
Statistical analysis.
Data is presented as dot plot, XY scatter plot or box-andwhisker plot. Dot plot and XY scatter plot shows mean, and the error bars represent s.e.m. Box-and-whisker plots shows median (center line) and interquartile range (IQR, lower and upper end of box), and minima and maxima (whiskers) within 1.5 IQR. The outliers falling beyond 1.5 IQR are shown as individual dots and are included in the statistical analysis. To determine the group sizes, power analysis was done using preliminary data sets. Considering the independent sample t test and observed variability in pilot experiments, a group sample size of six was needed to achieve >95% power to reject null hypothesis at the significance level of 0.05, and therefore we used 6-12 mice per group. All data met assumptions of the statistical test and different groups had similar variance. In experiments with sample sizes smaller than five, individual data points were plotted. All statistical analysis was performed using SPSS (Version 17.0) unless specified. Significance of the difference between two groups was evaluated using independent sample t test. The significance of the difference in survival curves was determined by a log-rank test. Results were considered to be significant if P values were ≤0.05.
Supplementary Material
Acknowledgments:
This work was supported by NIH grant HL115955 to K.I. and B.L., and University of Iowa Endowed Professorship to K.I. M.K. was supported by a Cardiovascular Institutional Research Fellowship (T32 HL007121, PI: F. Abboud). Q.L. was supported by the Training Program in Hematology: Molecular & Cell Biology of Blood Cells (T32 HL007344, PI: S. Lentz). Procurement of human heart tissue was enabled by NIH grant HL105993 to K.B.M. Proteomics studies were supported by NIH grants HL068758 and HL104017 (PI: R.A. Cohen) and DK103750 to M.M.B. We acknowledge NIH support for services that we received under contract HHSN268201000031 (PI: C. Costello). We acknowledge J. Lee at the Dana-Farber Molecular Biology Core Facility for his support with the MALDI and Orbitrap MS instruments. Cardiomyocyte imaging data were acquired at the University of Iowa Central Microscopy Research Facility, with NIH 1S10RR02543901 funding for the shared Zeiss LSM 710 Confocal Microscope. Mouse echocardiograms were made possible with the NIH shared instrument grant 1S10ODO019941. We thank S. Dudley for the Nav1.5-expressing cell line, T. Kouzarides for the Sirt1 constructs and D. Roden for the Scn5a+/− mice. AUTHOR CONTRIBUTIONS K.I. and B.L. designed and conceived the project. A.V., C.M.L., J.-Y.Y., A.N., S.K., G.M.M., J.S.J., Q.L., Y.-R.K., M.K., J.L., M.G., A.K., H.M., X. Zhu, X.G., W.K., X. Zhang, S.D., S.-B.J., V.K. and M.M.B. carried out experimental work, analyzed data and participated in data interpretation. R.L.B. and D.S.M. helped with data interpretation. K.B.M. provided critical human heart tissue and data set. K.I., B.L. and A.V. wrote the manuscript. K.I. and B.L. supervised the research and interpreted the data.
Footnotes
Data availability.
The authors declare that the data supporting the findings of this study are available within the article and from the authors on reasonable request.
References:
- 1.Ruan Y, Liu N & Priori SG Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 6, 337–348 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Chen-Izu Y et al. Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. (Lond.) 593, 1347–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J. Med. 321, 406–412 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Marionneau C & Abriel H Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 82, 36–47 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Pei Z, Xiao Y, Meng J, Hudmon A & Cummins TR Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 7, 12035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu M et al. Cardiac Na+ current regulation by pyridine nucleotides. Circ. Res. 105, 737–745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valdivia CR, Ueda K, Ackerman MJ & Makielski JC GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am. J. Physiol. Heart Circ. Physiol. 297, H1446–H1452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant AO et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang ZS, Tranquillo J, Neplioueva V, Bursac N & Grant AO Sodium channel kinetic changes that produce Brugada syndrome or progressive cardiac conduction system disease. Am. J. Physiol. Heart Circ. Physiol. 292, H399–H407 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Hallaq H et al. Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovasc. Res. 72, 250–261 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Hallaq H et al. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am. J. Physiol. Heart Circ. Physiol. 302, H782–H789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Royer A et al. Mouse model of SCN5A-linked hereditary Lenègre’s disease: agerelated conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746 (2005). [DOI] [PubMed] [Google Scholar]
- 13.van Bemmelen MX et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4–2 mediated ubiquitination. Circ. Res. 95, 284–291 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Bosch-Presegué L et al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell 42, 210–223 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Benson DW et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schott JJ et al. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Tan HL et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Lei M, Zhang H, Grace AA & Huang CL SCN5A and sinoatrial node pacemaker function. Cardiovasc. Res. 74, 356–365 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Papadatos GA et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. USA 99, 6210–6215 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Probst V et al. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenègre disease. J. Am. Coll. Cardiol. 41, 643–652 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Yokokawa M et al. Comparison of long-term follow-up of electrocardiographic features in Brugada syndrome between the SCN5A-positive probands and the SCN5A-negative probands. Am. J. Cardiol. 100, 649–655 (2007). [DOI] [PubMed] [Google Scholar]
- 22.de Jong S, van Veen TA, van Rijen HV & de Bakker JM Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 57, 630–638 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Wang Q et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Chandra R, Starmer CF & Grant AO Multiple effects of KPQ deletion mutation on gating of human cardiac Na+ channels expressed in mammalian cells. Am. J. Physiol. 274, H1643–H1654 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Schulze-Bahr E et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum. Mutat. 21, 651–652 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Gao G et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation 124, 1124–1131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herren AW, Bers DM & Grandi E Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 305, H431–H445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valdivia CR et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 38, 475–483 (2005). [DOI] [PubMed] [Google Scholar]
- 29.McNair WP et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol. 57, 2160–2168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 26, 2782–2790 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Method references
- 31.Yoon JY et al. A novel Na+ channel agonist, dimethyl lithospermate B, slows Na+ current inactivation and increases action potential duration in isolated rat ventricular myocytes. Br. J. Pharmacol. 143, 765–773 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattagajasingh I et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 104, 14855–14860 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell GF, Jeron A & Koren G Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274, H747–H751 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Petric S et al. In vivo electrophysiological characterization of TASK-1 deficient mice. Cell. Physiol. Biochem. 30, 523–537 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Burgoyne JR et al. Oxidation of HRas cysteine thiols by metabolic stress prevents palmitoylation in vivo and contributes to endothelial cell apoptosis. FASEB J. 26, 832–841 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickey DM, Dries DL, Margulies KB & Potter LR Guanylyl cyclase (GC)-A and GC-B activities in ventricles and cardiomyocytes from failed and non-failed human hearts: GC-A is inactive in the failed cardiomyocyte. J. Mol. Cell. Cardiol. 52, 727–732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.