

Summary
Interferon α (IFNα) is a prompt and efficient orchestrator of host defense against nucleic acids but upon chronicity becomes a potent mediator of autoimmunity. Sustained IFNα signaling is linked to pathogenesis of systemic lupus erythematosus (SLE), an incurable autoimmune disease characterized by aberrant self-DNA sensing that culminates in anti-DNA autoantibody-mediated pathology. IFNα instructs monocytes differentiation into autoinflammatory dendritic cells (DCs) than potentiates the survival and expansion of autoreactive lymphocytes, but the molecular mechanism bridging sterile IFNα-danger alarm with adaptive response against self-DNA remains elusive. Herein, we demonstrate IFNα-mediated deregulation of mitochondrial metabolism and impairment of autophagic degradation, leading to cytosolic accumulation of mtDNA that is sensed via stimulator of interferon genes (STING) to promote induction of autoinflammatory DCs. Identification of mtDNA as a cell-autonomous enhancer of IFNα signaling underlines the significance of efficient mitochondrial recycling in the maintenance of peripheral tolerance. Antioxidant treatment and metabolic rescue of autolysosomal degradation emerge as drug targets in SLE and other IFNα-related pathologies.
Keywords: autoimmunity, SLE, lysosome, metabolism
Graphical Abstract
Highlights
-
•
IFNα obstructs autophagic flux in SLE monocytes through lysosomal alkalinization
-
•
IFNα signaling induces oxidative stress that affects lysosomal pH through mTOR
-
•
Impaired clearance of damaged mitochondria leads to cytosolic mtDNA accumulation
-
•
Autophagic escape of mtDNA is sensed by STING and primes monocytes autoimmunity
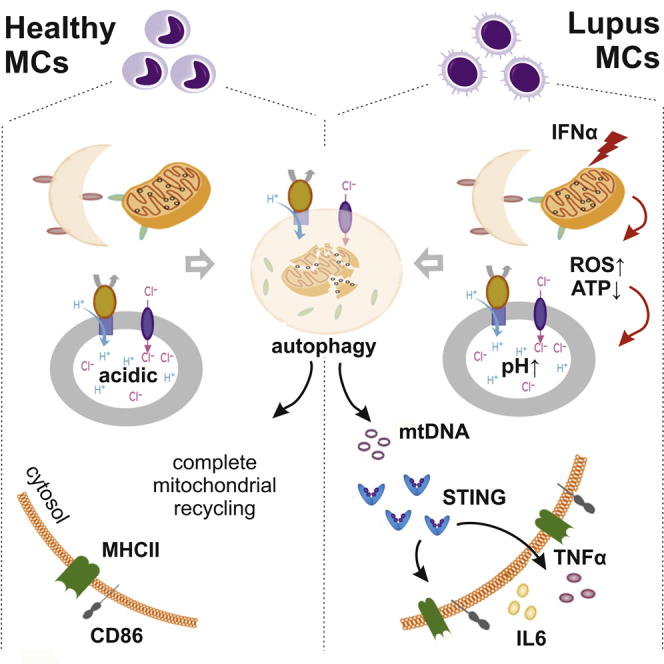
In lupus, sustained IFNα signaling leads to anti-DNA autoimmunity. Gkirtzimanaki et al. propose that excessive IFNα damages mitochondrial respiration, leading to oxidative stress that impairs lysosomal degradation and obstructs autophagic clearance. Undegraded mtDNA escapes in the cytoplasm and is sensed, priming monocytes cell-autonomously to initiate an anti-viral-like response against self-DNA.
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune inflammatory disease characterized by multiple pathophenotypes whereby interferon α (IFNα)-driven immunologic alterations culminate into persistent self-directed immune responses against autologous nucleic acids. Excessive and intractable tissue damage caused by autoantibodies, especially by anti-double-stranded DNA (dsDNA) immunoglobulin Gs (IgGs), or immune-complex depositions, affects various organs leading to increased morbidity and mortality (Weckerle et al., 2011). Genome-wide association studies (GWAS) studies reveal a genetic predisposition toward type I IFN activity in SLE with familial chilblain lupus considered as interferonopathy (Crow and Manel, 2015) and functional studies confirm its role in breakdown of peripheral tolerance and autoimmunity development (Crow, 2014, Liao et al., 2016). In support, hepatitis-C and leukemia patients treated with IFNα frequently develop a lupus-like syndrome with high titers of anti-dsDNA autoantibodies.
IFNα signaling and activation of nucleic acid-sensing and clearance pathways are intertwined (Shrivastav and Niewold, 2013), orchestrating antiviral immunity such as the differentiation of monocytes to potent antigen presenting dendritic cells (DCs) with increased capacity to drive T and B cell responses (Blanco et al., 2001, Ding et al., 2006, Jegerlehner et al., 2007, Ma et al., 2013, Pascual et al., 2006, Rönnblom and Pascual, 2008). In SLE, circulating monocytes are already differentiated into DCs with their function shaped by IFNα (Blanco et al., 2001, Rönnblom and Pascual, 2008). These IFNα-DCs exhibit increased HLA-DR, CD86, and BAFF expression, activate autoreactive T cells, and promote the expansion and differentiation of autoreactive B cells, therefore bridging innate danger sensing with adaptive autoimmune response (Gujer et al., 2011). However, the molecular mechanism(s) driving IFNα-mediated autoreactivity in SLE antigen-presenting cells (APCs) remain(s) elusive.
Autophagy is an evolutionarily conserved membrane-trafficking mechanism by which cells deliver cytoplasmic substrates to lysosomes for their recycling to maintain cellular metabolic equilibrium and homeostasis (Klionsky et al., 2011). Emerging literature implicates autophagy in intracellular danger sensing, antigen processing, and delivery for presentation in APCs (Alissafi et al., 2017, Rönnblom and Pascual, 2008). Moreover, it regulates the activation and differentiation of APCs through metabolic adaptation and maintenance of mitochondrial homeostasis (Ravi et al., 2014, Zhang et al., 2012). Sequestration and successful clearance of damaged mitochondria by autophagy (mitophagy) suppresses mitochondrial reactive oxygen species (mtROS) accumulation and prevents inflammation and generation of autoantigens by intracellular oxidation (Minton, 2016). Many antigens implicated in SLE pathology have undergone oxidative modification (dsDNA, HDL, β2GPI, Ro) that shows striking correlation with disease activity and organ damage (Perl, 2013). Additionally, failure of autophagic digestion of mitochondrial components such as mtDNA can lead to their cytosolic leakage and cause inflammation (Nakai et al., 2007). The highly immunogenic properties of mtDNA, an unmethylated dsDNA species of bacterial origin (Pisetsky, 2016, Zhang et al., 2010), when it is not properly recycled (Caielli et al., 2016, Oka et al., 2012), suggest that it can be perceived as a strong candidate to drive SLE autoreactivity.
In this study, we asked whether deregulated autophagy may be involved in the IFNα-mediated immunogenicity of monocytes in SLE and whether this is mediated by defects in their cell autonomous recycling that could result in activation of cytoplasmic DNA sensing. Our findings delineate that lupus characteristic IFNα signature in monocytes of active, untreated SLE patients is linked to aberrant mitochondrial metabolism, altered lysosomal pH, and defective autophagic degradation that results in cytoplasmic accumulation of mtDNA. Innate immune receptor stimulator of interferon genes (STING) senses it and drives differentiation of monocytes into autoreactive DCs to promote intracellular DNA-mediated, type I interferon-dependent immunity. Deciphering the role of IFNα in cell autonomous generation and sensing of self-antigens in lupus monocytes will provide us with important insights that may lead to better designed drug targeting to tame SLE autoimmunity.
Results
SLE Monocytes Display Increased Autophagosome Formation but Defective Autolysosomal Degradation
GWAS in SLE peripheral blood mononuclear cells (PBMCs) have identified ATG16L1, ATG5, ATG7, and IRGM autophagy genes as susceptibility loci (Zhang et al., 2015, Zhou et al., 2011). To understand the regulation of the autophagic flux in freshly isolated CD14+ monocytes of active, untreated SLE patients (Table S1), we monitored the expression and subcellular localization of indispensable autophagy components (Klionsky et al., 2016), namely the first ubiquitin-like conjugation system that is necessary for elongation of phagophore membrane (Atg5-Atg12), the second conjugation system that is required for autophagosome formation (PE-LC3B), the sequestrator of substrates to be cleared (SQSTM1/P62), and the acidic compartment for the digestion of autophagy substrates, the lysosome (LAMP1). As evidenced by the transcriptional upregulation of ATG5, the enhanced content in lipidated LC3B form and the presence of LC3+P62+LAMP1+ structures (Figures 1A, 1B, and 1D), autophagy is employed and autophagosome formation is enhanced in SLE monocytes from active patients. Notably, inefficient degradation, as evidenced by P62 protein accumulation in LC3+LAMP1+ vesicles in the absence of its transcriptional upregulation (Figures 1B–1D), suggests that proper autolysosomal resolution may be obstructed. Defective autolysosomal function can be either due to impaired autophagosome-to-lysosome fusion or ineffective acidification. Because LAMP1 and LC3B puncta were found to colocalize in SLE monocytes (Figure 1D), autophago-lysosomal fusion seems to be unaffected. Instead, lysosomal acidity was found to be compromised as Lysotracker Red-DND99 (LTR), a fluorophore linked to a weak base that is only partially protonated at neutral pH, exhibited significantly weaker intensity in SLE monocytes (Figure 1E). This alteration functionally affects lysosomal enzymes as indicated by the decreased cathepsin D activity (Figure 1F). Together, these results suggest that SLE monocytes harbor a pool of autophagosomes fused with lysosomes that are not properly acidified to digest damaged intracellular components, thus potentiating the accumulation of ineffectively cleared intracellular waste.
Figure 1.

Autophagic Flux Is Obstructed in SLE Monocytes
(A) Relative mRNA expression of ATG5 compared to GAPDH in CD14+ monocytes from healthy donors (n = 10) and SLE patients (n = 16).
(B) Western blot analysis for LC3B lipidation and SQSTM1/P62 protein levels in whole cell lysates of CD14+ monocytes from healthy donors (n = 8) and SLE patients (n = 8). One representative result is depicted. Relative intensities of LC3II to LC3I and SQSTM1/P62 to actin are shown.
(C) Relative quantity of SQSTM1/P62 mRNA levels compared to GAPDH in CD14+ monocytes from healthy donors (n = 8) and SLE patients (n = 8).
(D) Confocal microscopy for LC3B555, SQSTM1/P62488, LAMP-1633, and DAPI in CD14+ monocytes from healthy donors (n = 12) and SLE patients (n = 13). One representative result is depicted. % co-localization of LC3B and P62 puncta is shown.
(E) Confocal microscopy for Lysotracker DND99 in CD14+ monocytes from healthy donors (n = 7) and SLE patients (n = 7). One representative result is depicted. Averages of mean fluorescence intensities per cell are graphed.
(F) Relative cathepsin D activity in freshly isolated CD14+ monocytes from healthy donors (n = 9) and SLE patients (n = 9). Scale bar, 5 μM. Results are expressed as mean +SEM. ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005. All datasets were analyzed using non-parametric Mann-Whitney U test.
IFNα Signaling Impairs the Completion of Autophagy and Enhances the Immunogenic Potential of Monocytes
To dissect whether the observed defects on autophagy completion are inherent in SLE monocytes or mediated by soluble factors of inflammatory milieu, we cultured CD14+ monocytes from healthy donors with serum (10% v/v) from active SLE (SLEDAI ≥8), active rheumatoid arthritis (RA) patients, or healthy donors and monitored their autophagic flux. Treatment of healthy monocytes with SLE, but not RA serum, recapitulated the phenotype of SLE monocytes regarding autophagy induction and defective autolysosomal degradation (Figure S1; Tables S1 and S2). Previous work has shown that monocytes exposed to SLE serum acquire a DC-like phenotype with increased antigen presenting capacity (Blanco et al., 2001). To explore whether autolysosomal impairment is implicated in this process, we metabolically modulated the autophagic flux in healthy monocytes cultured in the presence of SLE serum with the use of rapamycin, an inhibitor of mechanistic target of rapamycin (mTOR) signaling and potent enhancer of autophagosome formation and autolysosomal degradation (Chen and Fang, 2002). Indeed, treatment with rapamycin prevented the SLE serum-induced upregulation of membrane HLADR and CD86 (Figure S1), suggesting that SLE serum contents can enhance antigen-presenting capacity of monocytes through autophagy modulation.
IFNα is a crucial cytokine that has been documented to drive the DC-like phenotype in SLE monocytes (Blanco et al., 2001). In line with this, Cxcl10 mRNA (Figure S1), a known IFNα-inducible gene, was upregulated in SLE serum-treated monocytes, and the extent of their SQSTM1/P62 accumulation correlated positively with serum IFNα concentrations (Figure S1). This prompted us to examine if type I IFN signaling directly affects the autophagic flux of healthy monocytes. We found that autophagy was induced (Figures 2A and 2B) as early as 4 hr, and the accumulation of SQSTM1/P62 followed and peaked at 18 hr, reaching the levels of accumulation seen upon lysosomal inhibition with hydroxychloroquine (CQ) (Figures 2B–2E and 2H). Lysosomal function was also compromised in IFNα-treated monocytes as evidenced by the significantly lower LTR intensity (Figure 2F) and reduced cathepsin D activity (Figure 2G), while rapamycin treatment restored lysosomal degradation upon IFNα signaling (Figures 2F–2H).
Figure 2.

Type I IFN Signaling Impairs Autophagolysosomal Degradation in Healthy CD14+ Monocytes
CD14+ monocytes from healthy donors were treated with recombinant IFNα (400 ng/mL) +/− CQ (46.1 μΜ), rapamycin (rapam.; 1 μΜ) as depicted.
(A) Relative mRNA expression of ATG5 compared to GAPDH (n = 9 healthy donors). Datasets were analyzed using non-parametric Mann-Whitney U test.
(B) Western blot analysis for LC3B lipidation and SQSTM1/P62 protein levels in whole cell lysates (n = 10 healthy donors). Relative intensities of LC3II to LC3I and SQSTM1/P62 to actin are shown. Datasets were analyzed using non-parametric Mann-Whitney U test.
(C) Relative quantity of SQSTM1/P62mRNA levels as in (A). Datasets were analyzed using non-parametric Mann-Whitney U test.
(D) Representative western blot analysis of autophagic flux upon IFNα signaling.
(E) Confocal microscopy for LC3B555, SQSTM1/P62488, LAMP-1633, and DAPI in CD14+ monocytes from healthy donors treated as indicated. One representative result is depicted. % co-localization of LC3B and P62 puncta is shown for monocytes cultured for 18 hr (n = 8).
(F) Confocal microscopy for Lysotracker DND99TM ± rapam. One representative result is depicted. Mean fluorescence intensity ratios are shown (n = 6). Datasets were analyzed using paired Student’s t test.
(G) Relative cathepsin D activity ± rapam. (n = 5). Datasets were analyzed using paired Student’s t test.
(H) Analysis of autophagic flux assessment with western blotting. Fold change of P62/actin intensity ratios are graphed (n = 4). Scale bar, 5 μM. Results are expressed as mean +SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005. Datasets were analyzed using paired Student’s t test.
To examine the functional importance of IFNα-dependent alterations of lysosomal environment that impair autophagy completion in IFNα-DCs, we assessed their MHCII antigen presenting capacity and pro-inflammatory cytokine secretion in the presence of rapamycin. It appears that addition of rapamycin decreased HLADR and CD86 membrane expression and significantly reduced TNFα and interleukin (IL)6 secretion (Figures 3A and 3B). Importantly, IFNα-treated CD14+ monocytes exposed to rapamycin demonstrated markedly reduced capacity to drive naive allogeneic CD4+ T cell proliferation in mixed lymphocyte reaction (MLR) ameliorating significantly their DC-like, inflammatory phenotype (Figure 3C). To address whether this effect is similarly significant for the immunoreactivity of SLE monocytes, we treated them ex vivo with rapamycin and performed MLR with naive allogeneic CD4+ T cells. In all cases, mTORC1 (the target of rapamycin inhibition) was found activated in freshly isolated cells from active SLE patients compared to healthy controls (Figure 3D), and addition of rapamycin reverted both their higher levels of HLADR and CD86 and their ability to prime T cells (Figure 3E) more efficiently than healthy monocytes (Figure S2).
Figure 3.

Impaired Autolysosomal Degradation Determines the Enhanced Immunogenic Potential of IFNα-Shaped and SLE Monocytes
CD14+ monocytes from healthy donors were cultured for 18 hr with IFNα (400 ng/mL) +/− rapam. (1 μΜ) as depicted.
(A) Levels of HLA-DR and CD86 membrane expression were measured by flow cytometry. A representative result is depicted. Geometric mean fluorescence intensity (GeoMFI) averages are plotted (n = 6).
(B) Concentrations of secreted IL6 and TNFα measured by ELISA in culture supernatants (n = 3).
(C) CFSE-labeled cord blood-naive CD4+ T cells were cultured for 6 days with allogeneic monocytes, previously treated for 18 hr with IFNα, +/− rapam., and analyzed for their proliferation. Proliferation index averages of 4 experiments are graphed. Right: histograms of CFSE dilution and size (FS) of CD4+ gated cells. GeoMFI of un-proliferated cells and FS counting are listed on right side of histograms. One representative result is depicted.
(D) p-4EBP1 and p-P70S6K expression in freshly isolated monocytes from healthy (n = 5) and SLE (n = 5) donors were analyzed by flow cytometry, and averages of their GeoMFIs are graphed.
(E) MLR between SLE monocytes ± rapam. (1 μM) for 1 hr, cocultured with allogeneic CFSE-labeled cord blood-naive CD4+ T cells for 6 days (n = 3). Averages of proliferation indexes as in (C) and averages of % HLADRhiCD86hi population after cocultures are graphed.
(F) Representative histogram of Lysosensor Green-189 intensity upon PLGA aNP addition in healthy monocytes cultured for 18 hr ± IFNα.
(G) Graphs of averages of the GeoMFIs of HLADR and CD86 as assessed by flow cytometry (n = 3). Representative histograms of HLADR and CD86 expression and a scatterplot showing the HLADRhiCD86hi populations.
(H) Concentrations of secreted IL6 and TNFα measured by ELISA in culture supernatants of the same experiments are depicted. Results are expressed as mean +SEM. ∗p < 0.05, ∗∗p < 0.005. All datasets were analyzed using paired Student’s t test.
Apart from optimizing lysosomal processing, modulation of mTORC1 activity is known to exert pleiotropic effects on DC differentiation and antigen presentation (Sukhbaatar et al., 2016). To specifically address that restoration of lysosomal pH can account for the alleviation of IFNα-dependent immunoreactivity of monocytes, we used poly(DL-lactide-co-glycolide) (PLGA) acidic nanoparticles (aNP) instead (Bourdenx et al., 2016). These biodegradable polymers are known to traffic to lysosomes and prevent lysosomal alkalinization even upon the presence of CQ. Indeed, co-treatment of healthy monocytes with aNPs and IFNα for 18 hr restored lysosomal acidification (Figure 3F) and significantly reduced the membrane expression of HLA-DR and CD86 (Figure 3G) and the secretion of IL6 and TNFα (Figure 3H).
Together, metabolic rescue of the IFNα-mediated autolysosomal deregulation inhibits the immunogenic potential of monocytes indicating the importance of efficient autophagic clearance for the amelioration of immunoreactivity upon danger signaling.
IFNα-Mediated Lysosomal Dysfunction Impedes Mitochondrial Clearance and Leads to Cell Autonomous mtDNA Accumulation
To decipher how impaired autophagic degradation can drive the immunogenicity of IFNα-shaped monocytes in the absence of an extracellular antigen, we examined the generation of an intracellular damage-associated molecular pattern (DAMP) that could originate from defective degradation of a cell autonomous autophagy substrate. A prominent cell intrinsic interferogenic DAMP that is physiologically recycled by autophagy is mtDNA (Krysko et al., 2011). We thus assessed mitochondrial homeostasis and clearance by autophagy (mitophagy) in SLE monocytes. Mitotracker (MTR) staining of monocytes from active, newly diagnosed SLE patients exhibited increased mean fluorescence intensity (MFI) and volume of MTR+ areas per cell compared to that observed in healthy monocytes (Figure 4A). Hyperpolarization of SLE mitochondria (increase in mitochondrial membrane potential [Δψ]) (Gergely et al., 2002a) can explain the increase in MTR MFI, but the observed increase in the volume of MTR+ areas per cell raised the possibility of alterations in mitochondrial recycling as well. Mitochondrial damage is recognized as indicated by PINK1 upregulation (Figure 4B) so mitochondrial recruitment to autophagosomes is induced. To test whether mitophagy is properly completed, we assessed mtDNA quantity; we quantified the relative amount of mtDNA (minArc region) copies calibrated with β2-microglobulin (β2 m) single copy gene of genomic DNA. We found accumulation of mtDNA in SLE monocytes compared to healthy controls (Figure 4C), suggesting either an increase in mitochondrial biogenesis or mtDNA escape from mitophagic degradation. Quantity assessment of the mitochondrial protein VDAC1—that is recycled by the proteasome as most of the outer mitochondrial membrane proteins (Geisler et al., 2010)—revealed no difference between healthy and SLE monocytes (Figure S3). This directed us toward the possibility of ineffective degradation of mitochondrial contents that are cleared by mitophagy. Notably, IFNα signaling induced mitochondrial hyperpolarization as indicated by the increase in intra-mitochondrial aggregates of JC1 dye, as early as 30 min post treatment, and this effect persisted until the 18 hr time point (Figure 4D). MTR staining, PINK1 expression, and quantification of nuclear-to-mitochondrial DNA m quantification cycle (Cq) ratios further confirmed that IFNα signaling recapitulated the phenotype of SLE monocytes concerning mitochondrial dynamics and mtDNA accumulation (Figures 4E–4G). Addition of rapamycin prevented mtDNA accumulation (Figure 4G), thus demonstrating that IFNα-dependent defects in the resolution of mitophagy impede mitochondrial clearance and lead to cell autonomous mtDNA accumulation.
Figure 4.

IFNα-Mediated Lysosomal Dysfunction Impedes Mitochondrial Clearance and Leads to mtDNA Accumulation
(Α) Confocal microscopy for Mitotracker CMXRos dye staining in freshly isolated CD14+ monocytes from healthy donors (n = 8) and SLE patients (n = 8). One representative result is depicted. Averages of MFI per cell are graphed. Datasets were analyzed using non-parametric Mann-Whitney U test.
(Β) TaqMan qPCR analysis for mtDNA content in freshly isolated monocytes of healthy donors (n = 5) and SLE patients (n = 5), expressed as a ratio of the Cq values of genomic DNA (gDNA) (β2 m)/mtDNA (mt minArc). Datasets were analyzed using non-parametric Mann-Whitney U test.
(C) Relative mRNA expression of PINK1 compared to GAPDH in CD14+ monocytes from healthy donors (n = 8) and SLE patients (n = 9). Datasets were analyzed using non-parametric Mann-Whitney U test.
(D) Confocal microscopy for JC-1 dye-staining in CD14+ monocytes from healthy donors ± IFNα (n = 4). One representative result is depicted. Average of MFI of J-aggregates per cell are shown. Datasets were analyzed using paired Student’s t test.
(E) Confocal Microscopy for Mitotracker CMXRos dye staining in freshly isolated CD14+ monocytes from healthy donors (n = 20) +/− IFN-α for 18h. One representative result is depicted. Αverages of MFI per cell are graphed.
(F) Relative mRNA expression of PINK1 compared to GAPDH in CD14+ monocytes from healthy donors (n = 10).
(G) Taqman QPCR analysis for mtDNA content in freshly isolated monocytes of healthy donors (n = 5) +/− IFN-α, rapam for 18 h expressed as a ratio of the Cq values of gDNA (β2m)/ mt DNA (mt minArc).
Scale bar, 5 μM. Results are expressed as mean + SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005. Datasets were analyzed using paired Student’s t test. As in (A) for CD14+ monocytes from healthy donors treated with IFN-α +/− rapam.
Type I IFN Signaling Affects Mitochondrial Metabolism Leading to Proinflammatory Oxidative Stress—mtROS and ATP Affect Lysosomal pH
To dissect the molecular mechanism governing SLE aberrancy in lysosomal degradation we performed next generation sequencing (RNA-seq) in the transcriptome of monocytes from active, untreated SLE patients (Tables S3 and S4). Pathway analysis revealed that type I IFN signature (representative genes IFI27, USP18, IFITM1, MX1), increase in antigen presentation and differentiation (MMP10, IL6, CSF3, MIR155HG, SIGLEC-1) coincide with response to oxidative stress and alterations in mitochondrial function and metabolism (ACOT4, CYP19A1, MOXD1, IFI6, GMPR) as well as in endocytic pH and ion channel regulation (IGLL5, CLIC5, ATP10A, KCNH3, CACNB4) (Figure 5A; Table S1). These results prompted us to formulate the hypothesis that IFNα-mediated deregulation of mitochondrial metabolism affects lysosomal pH, resulting in defective mitochondrial clearance. Lysosomal pH stability is affected by the localization and activity of mTORC1 subunit that functions to couple the cellular metabolic state to lysosomal degradation through ATP sensing (Cang et al., 2013). After confirming that mTORC1 is activated upon IFNα treatment (Matsumoto et al., 2009), we addressed whether aberrant OXPHOS, as evidenced by persistent mitochondrial hyperpolarization, can be coupled with IFNα-dependent lysosomal alkalinization through ATP sensing. We assessed the levels of both ATP and mtROS, in healthy monocytes cultured with IFNα. mtROS levels were increased as early as 2 hr post IFNα treatment whereas ATP levels were reduced, and this effect persisted until 18 hr as evidenced by MitoSOX staining and colorimetric assessment of intracellular ATP concentration (Figures 5B and 5C). Scavenging of mtROS with MitoTEMPO prevented the IFNα-mediated decrease in ATP formation and additionally enhanced its production compared to basal levels (Figure 5C) and restored lysosomal pH (Figure 5D). Addition of rapamycin reverted the aberrant OXPHOS product equilibrium, restoring mtROS production to physiological levels while also increasing ATP levels (Figures 5B and 5C). In support, oxygen consumption rate that was significantly lower upon IFNα signaling was restored with the addition of rapamycin or mtROS scavenging (Figure S6). Together, our results suggest the existence of a vicious cycle of defective mitochondrial respiration and oxidative stress that is generated and amplified upon IFNα signaling. Mitochondrial hyperpolarization, indicative of defective OXPHOS, increases the mtROS-to-ATP ratio and this impairs lysosomal degradation, which in turn impedes effective clearance of damaged mitochondria, thus numbing monocytes’ capacity to control oxidative stress.
Figure 5.

Type I IFN Signaling Affects Mitochondrial Metabolism Leading to Proinflammatory Oxidative Stress and Ionic Alterations of Lysosomal Compartments
(Α) Scatterplot (R/Bioconductor) of log2-transformed RNA-seq expression values of all human genes between active SLE (n = 7) and healthy control (n = 5) CD14+ monocytes. All non-differentially expressed genes are depicted as gray colored. Selected genes related to type I IFN signature, oxidative stress, metabolism, antigen presentation, and differentiation are noted in the up (red color) and down (blue color) differentially expressed genes.
(B) Relative mitochondrial O2− levels of healthy CD14+ monocytes ± IFNα, rapam. were assessed by flow cytometry using mitoSOX dye (n = 5) One representative histogram overlay is shown and gates indicating low and high mtROS levels are depicted. Averages of % of cells in the mitoSOX high population are graphed. Datasets were analyzed using paired Student’s t test.
(C) Concentration of intracellular ATP concentration ± IFNα, rapam., or mitoTEMPO in lysates of CD14+ monocytes (n = 5). Datasets were analyzed using paired Student’s t test.
(D) Confocal microscopy for Lysotracker DND99 in CD14+ monocytes from healthy donors ± IFNα, mitoTEMPO. One representative result is depicted. Averages of MFI per cell are graphed (n = 3). Datasets were analyzed using paired Student’s t test.
(E) Levels of HLA-DR and CD86 expression measured by flow cytometry in CD14+ monocytes upon IFNα signaling and mtROS scavenging. GeoMFI averages are plotted (n = 3). Datasets were analyzed using paired Student’s t test.
(F) Concentrations of secreted IL6 and TNFα measured by ELISA in culture supernatants of CD14+ monocytes (n = 7) cultured as in (D) are graphed. Scale bar, 5 μM. Results are expressed as mean +SEM. ∗p < 0.05, ∗∗p < 0.005.
To address whether IFNα-mediated effects in mitochondrial respiration drive monocytes inflammatory priming upon IFNα signaling, we scavenged mtROS and assessed their antigen presenting properties. Addition of MitoTEMPO prevented the IFNα-dependent increase of membrane expression of HLA-DR and to a lesser extent of CD86 (Figure 5E) and the concomitant IL6 and TNFα secretion (Figure 5F), thus ameliorating the effects of pro-inflammatory intracellular oxidative stress.
Autophagy Escape of mtDNA Is Sensed through STING and Primes Monocytes for Autoimmune Responses
Autophagic escape of mtDNA is a cell autonomous trigger that engages antiviral signaling to enhance the expression of a subset of IFN-stimulated genes and can induce inflammation and disease pathology (Oka et al., 2012). Unmethylated self DNA, whose cytoplasmic release is not related to programmed cell death, can be sensed by TLR9 in endosomes and by cytosolic sensors (DAI, IFI16/P202, DDX41, and cGAS) (de Jong et al., 2010, Klinman et al., 1996, West et al., 2015) that activate the STING adaptor molecule. As revealed by our RNA-seq data from SLE monocytes of active patients with strong IFNα signature, DNA cytosolic sensors are overexpressed (DAI, IFI16, ZBP1) whereas cGAS, that belongs to the family of IFN-stimulated genes (ISGs) (Ma et al., 2015), was not differentially expressed. To identify which pathway is engaged during mtDNA sensing upon IFNα signaling and translate it into increased immunogenicity, we silenced TLR9 and STING expression in healthy monocytes, cultured with GMCSF and IL4 or IFNα for 3 days, and measured their antigen presenting capacity. We found that IFNα-mediated increase in HLA−DR+CD86+ monocytes and CXCL10 secretion were significantly reduced upon STING knockdown, whereas no significant differences were found upon TLR9 silencing (Figures 6A and 6B). This effect was specific to IFNα, because STING knockdown did not affect HLA-DR and CD86 expression or CXCL10 secretion by monocytes treated with IL-4.
Figure 6.

Autophagic Escape of mtDNA Is Sensed through STING and Primes Monocytes for Autoimmune Responses
(A) CD14+ monocytes from healthy donors (n = 5) were electroporated with scramble, TLR9, and STING small interfering RNAs (siRNAs) and cultured for 3 days with GMCSF and either IL4 or IFNα. Levels of HLA-DR and CD86 expression were analyzed by flow cytometry. Averages of % of the HLADRhiCD86hi populations are graphed.
(B) CXCL10 secretion as a cumulative indicator of IFNα signaling upon scramble, STING, and TLR9 knockdown.
(C) IL6 and TNFα secretion in culture supernatants (n = 3).
(D) Relative STING mRNA levels compared to GAPDH in freshly isolated in CD14+ monocytes from (n = 5) healthy donors and (n = 6) active SLE patients. Results are expressed as mean +SEM. ∗∗p < 0.005.
(E) CD14+ monocytes from healthy donors (n = 4) transfected with functional or heat inactivated DNase I protein ± IFNα. Levels of HLA-DR and CD86 expression were measured by flow cytometry. Representative histograms of HLADR and CD86 counts between IFNα DNase I-treated and heat inactivated DNase I-treated cells are depicted. Averages of GeoMFIs are graphed. Results are expressed as mean +SEM. ∗p < 0.05, ∗∗p < 0.005. Datasets were analyzed using paired Student’s t test apart from (D) that was analyzed using non-parametric Mann-Whitney U test.
Interestingly, STING knockdown substantially reduced IFNα-mediated IL6 secretion while TNFα secretion was also reduced but did not reach statistical significance (p = 0.0626) (Figure 6C), suggesting that STING sensing can control type I interferon-mediated response to immunogenic nucleic acid presence in the cytoplasm. These results are in line with the upregulated expression of STING observed in SLE monocytes (Figure 6D) and recapitulate the effect of mTORC1 inhibition, lysosomal re-acidification, and mtROS scavenging upon IFNα signaling.
Finally, to confirm that the cytosolic escape of mtDNA from autolysosomes is sensed by STING upon IFNα signaling, we transfected monocytes with acidity-independent DNase I protein using DOTAP transfection reagent, a lipoplex that is internalized by the clathrin-mediated endocytosis pathway and targeted to the lysosomal compartment (Rejman et al., 2005). Transfection of DNase I reduced the autoinflammatory IFNα-mediated increase in antigen presenting capacity of monocytes based on HLADR and CD86 expression as compared to the delivery of heat inactivated DNase I (Figure 6E). These data confirm that ineffective degradation of a lysosomal nucleic acid substrate is being sensed and drives the response.
Discussion
A rational development of alternative, targeted therapies for a systemic autoimmune disease requires the comprehensive understanding of the pathogenic molecular mechanisms whose reversal may lead to restoration of homeostasis. Defective clearance and accumulation of biological waste together with self-DNA sensing and excessive IFNα production drive SLE pathogenesis (Crow, 2014, Niewold et al., 2007, Pascual et al., 2006, Tsokos, 2016). In this context, additional insight has been gained into our understanding of the innate immune pathways and sensors that are responsible for instigating self-DNA triggered autoinflammatory events in immune cells (Caielli et al., 2016, Oka et al., 2012, Shrivastav and Niewold, 2013). Nonetheless, whether and how sustained IFNα signaling may affect the continuous production of these self-DNA DAMPs to perpetuate the autoimmune response governing SLE remains elusive. Our work suggests a critical role for IFNα-mediated accumulation of mtDNA in SLE monocytes that escapes autophagic clearance and acts as a cell-autonomous cytoplasmic DAMP. Its sensing and activation of STING results in enhanced MHCII and CD86 expression and secretion of TNFα and IL6, enabling them to launch immunity to self-DNA and to promote the clonal expansion of autoreactive lymphocytes.
Our findings demonstrated that IFNα induces autophagy in naive CD14+ monocytes, an observation in line with the reported induction of autophagy by type I IFNs in several cancer cell lines (Schmeisser et al., 2013, Zhu et al., 2015). In silico analysis of the human LC3B promoter indicated the presence of Stat and IRF binding sites and LC3B gene transcripts were found upregulated in IFNα-treated monocytes (Figure S4), suggesting the possibility of a direct transcriptional regulation of autophagy components by IFNα. In addition to the induction of autophagy, IFNα impairs the completion of autophagic flux, monocytic lysosomes fail to properly acidify, leading to decreased digestion of lysosomal substrates and to accumulation of damaged autophagy substrates in the cytoplasm that are being sensed. Analysis of TFEB expression levels and nuclear localization, a transcription factor that controls the lysosomal-autophagic pathway (Settembre et al., 2011) and drives lysosomal biogenesis, together with the quantification of LAMP1 puncta per cell showed no difference in SLE and upon IFNα treatment (Figure S5), further confirming that deficiency in autophagic degradation is a result of deregulated lysosomal activity.
Our data dissect a metabolic pathway triggered by IFNα that results in deceleration of lysosomal digestion through changes of its pH regulation. In accordance with a previous study using SLE PBMCs (Gergely et al., 2002b), we observed that SLE monocytes harbor lower ATP and higher mtROS levels, indicative of high mitochondrial membrane potential that results from deregulated OXPHOS (Figure S6). Our RNA-seq data from SLE monocytes with high IFNα signature implicate disrupted mitochondrial metabolism, oxidative stress, and alterations in ionic channels as well as in the regulation of endosomal pH. In a state of mitochondrial hyperpolarization, as that seen in SLE PBMCs, H+ ions are extruded from the mitochondrial matrix within the electron transport chain (ETC)—and cytochromes become reduced, promoting production of ROS and generating oxidative stress, as indicated by the upregulation of AXL, FASLG, HERC5, ABTB2, ETS1, FBXO2, HERC6, IL1A, PARP15, PARP9, SPSB1, and TRIM69 genes in SLE monocytes.
Immunometabolism is an emerging frontier in immunology (Mathis and Shoelson, 2011). SLE T cell survival and hyperactivation is known to be supported by intensive OXPHOS (Wahl et al., 2012). Normalization of CD4+ T cell metabolism in lupus prone mice was proposed as a promising strategy to reverse lupus pathology (Yin et al., 2015). OXPHOS and its products, ATP and mtROS, are major determinants of intracellular oxidation and regulating cellular activation, differentiation, and survival. The same mitochondrial respiration byproducts modulate autophagy and its flux. mTOR is a mitochondrial membrane ΔΨm sensor that can be activated in a redox or ATP-dependent fashion, and its blockade has emerged as an effective approach to reverse T cell activation (Pearce et al., 2013, Sena et al., 2013, Yin et al., 2015). Residing on lysosomal membrane, mTORC1 subunit of mTOR possesses the capacity to regulate lysosomal pH and to determine its functionality (Korolchuk et al., 2011). Metabolic regulation of lysosomal function impacts on the resolution of autophagic degradation, affecting the fate of its substrates while modulating immune cell activation and differentiation (Chen et al., 2008). Our study provides a previously uncharacterized mechanism and unravels components affecting monocyte immunometabolism in human SLE, which could be exploited as an adjunct therapy to expedite remission and prevent disease flares. Acidic nanoparticles are used as tools to correct effects of aberrant immunometabolism, emerging as a more selective option for SLE treatment. PLGA aNPs, in particular, might also be degraded, their derivatives (lactic and glycolic acids) can support Δψmt., and their action remains unchanged upon hydroxychloroquine presence, a major drug for the treatment for lupus management.
Metabolic rescue of IFΝα-dependent deregulation of lysosomal acidification results in completion of autophagy and decreases the immunogenic potential of IFNα-induced monocytes. IFΝα signaling directly affects mitochondrial function and OXPHOS as indicated by the increased presence of Stat1 in mitochondria of IFNα-treated monocytes (Figure S6) that has been previously reported to affect Δψm. explaining—at least in part—the observed SLE mitochondrial hyperpolarization followed by increased generation of mtROS and reduced ATP. Oxidative stress is sensed as indicated by the upregulation of ROS-responding PINK1 gene, autophagy is induced, but IFNα-mediated lysosomal dysfunction impedes mitochondrial clearance leading to cell autonomous mtDNA accumulation that activates STING and prepares monocytes to defend against improperly localized bacterial-like nucleic acids. The observed functional restoration of lysosomal degradation and the resulting amelioration of their inflammatory phenotype suggest that mTORC1 inhibition can revert the inflammatory function of IFNα-shaped monocytes. Similar effects were attained by targeting any step of the proposed mechanism such as mtROS through scavenging, lysosomal pH through acidification by PLGA aNPs, DNA digestion through DNase I transfection into lysosomes, or STING knockdown in IFNα-shaped monocytes, thus hampering the IFNα-mediated differentiation of blood monocytes into DC-like APCs.
Inhibitors of mTOR, such as sirolimus and everolimus, are effective as part of triple immunosuppression regimens for the prevention of kidney transplant rejection—with their main advantages being the absence of nephrotoxicity and reduced occurrence of malignancies. The use of mTOR inhibitors in SLE (both human and murine) reduced severity of nephritis and general activity of the disease as well as antiphospholipid antibody production and vascular lesions associated with the antiphospholipid syndrome (Bride et al., 2016, Canaud et al., 2014, Oaks et al., 2016). Recent data from a multicenter trial suggest that rapamycin is effective in relapsed/refractory autoimmune cytopenias (Chen and Fang, 2002). In spite of these encouraging results, the role of mTOR inhibition in the treatment of SLE needs to be better documented. To date, their beneficial effects have been ascribed to their abrogating effects on mTOR activation in renal vascular endothelial cells and the normalization of T cell activation-induced calcium fluxing (Crow and Manel, 2015). Herein, we demonstrate an additional effect on lysosomal clearance in SLE monocytes—a critical event in both the initiation and relapse of the disease.
Our study established that mitochondrial hyperpolarization observed in SLE monocytes can be mediated by IFNα stimulation that activates a mechanism coupling cytokine signaling with OXPHOS. Reversal of IFΝα-dependent intracellular oxidation using scavenger of mtROS represents an additional target to be assessed for its efficacy in achieving durable remission, a major goal in SLE management. Antioxidant mtROS scavengers are currently being investigated in several diseases associated with mitochondrial dysfunction and oxidative stress such as antiphospholipid syndrome, multiple sclerosis, and vasculitis. Their efficiency in maintaining the homeostasis of neuronal and endothelial cells, while they simultaneously act as mild immunosuppressants, makes them attractive candidates to prevent or improve SLE pathology including renal, neuropsychiatric, and musculoskeletal manifestations.
Multi-targeted therapy is necessary to induce remission and prevent flares of SLE. Our data suggest unanticipated benefits of targeting IFNα in SLE and reveal additional downstream therapeutic targets that make break the “vicious” cycle of aberrant nucleic acid sensing in lupus monocytes, and thus may promote a deeper and a more durable remission in this disease. Development of sophisticated tools and approaches to in vivo regulate molecular aberrations that drive pathology can open the way to prevent and methodically treat complex human diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-LC3B | Novus Biologicals | Cat# NB100-2220; RRID: AB_10003146 |
| Mouse monoclonal anti-LC3B (clone 5F10) | Nanotools | Cat# 0231-100; RRID: AB_2722733 |
| Rabbit polyclonal anti-SQSTM1/P62 | MBL | Cat# PM045; RRID: AB_1279301 |
| Rabbit polyclonal anti-VDAC | Millipore | Cat# AB10527; RRID: AB_10806766 |
| Rat monoclomal anti-LAMP1 (clone 1D4B) | Abcam | Cat# ab25245; RRID: AB_449893 |
| anti-HLADR (clone L243) | Biolegend | Cat# 30762728 |
| anti-CD86 (clone IT2.2) | Biolegend | Cat# 30540506 |
| anti-CD80 (clone 2D10) | Biolegend | Cat# 305219 |
| anti-CD14 (clone HCD14) | Biolegend | Cat# 325605 |
| anti-pS6K PECy7 (clone CUPK43K) | eBioscience | Cat# 25900741 |
| anti-p4EBP1 PE (clone V3NTY24) | invitrogen | Cat# 12-9107-41 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Universal Type I interferon, human | PBL | Cat# 11200-1 |
| Recombinant human IL-4 | Peprotech | Cat# 200-04 |
| Recombinant human GMCSF | Peprotech | Cat# 300-03 |
| Resomer RG 503 H, Poly(D,L-lactide-co-glycolide) acid terminated, lactide:glycolide 50:50, Mw 24,000-38,000 | SIGMA | Cat#:719870 |
| Critical Commercial Assays | ||
| Cathepsin D Activity Assay Kit (fluorometric) | Abcam | Cat# ab65302 |
| ATP assay kit | Abcam | Cat# ab83355 |
| Amaxa Human Dendritic Cell Electroporation kit | Lonza | Cat# VPA-1004 |
| Mitochondrial Isolation Kit for cultured cells | Thermo Scientific | Cat# 89874 |
| VeriKine Human Interferon Alpha Multi-Subtype Serum ELISA Kit | PBL | Cat# 41110-1 |
| HumanTNFα Ready-Set-Go ELISA | eBioscience | Cat# 887346 |
| Human IL6 Ready-Set-Go ELISA | eBioscience | Cat# 887066 |
| Human CXCL10(IP-10) ELISA | eBioscience | Cat# BMS284INST |
| FITC AnnexinV apoptosis Detection kit | BD Bioscience | Cat# 556547 |
| Deposited Data | ||
| RNA-seq data, SRA Bioproject PRJNA392602 | NCBI’s Short Read Archive | https://www.ncbi.nlm.nih.gov/bioproject/PRJNA392602/ |
| Oligonucleotides | ||
| Stealth siRNA (Set of 3), TMEM173/STING Human, HSS139156,HSS139157,HSS139158 | Life Technologies | Cat# 1299003 |
| Stealth siRNAs (Set of 3), TLR9 Human, HSS147622, HSS147623, HSS147624 | Life Technologies | Cat# 1299003 |
| Stealth siRNA Negative Control Kit | Life Technologies | Cat# 12935-100 |
| Primers for IFNα, CXCL10, SQSTM1/P62, LC3, PINK1, Parkin, ATG5, huβ2Μ, mtMinArc (STAR Methods) | this paper | N/A |
| STING (TMEM173) primers and probe (FAM): Hs00736958_m1 assay | Life Technologies | Cat# 4331182 |
| GAPDH primers and probe (HEX) Hs03929097_g1 | Life Technologies | Cat# 4448489 |
| TLR9 primers and probe: Hs00152973_m1 (FAM) | Life Technologies | Cat# 4331182 |
| Software and Algorithms | ||
| GraphPad Prism | N/A | https://www.graphpad.com |
| Fiji | https://fiji.sc/ | |
| Macro for % colocalization for Fiji software | Daniel J. Shiwarski | N/A |
| Other | ||
| CD14 MicroBeads, human | Miltenyi | Cat# 130-050-201 |
| Monocyte Isolation Kit II (negative selection), human | Miltenyi | Cat# 130-091-153 |
| CD4+ CD25+ Regulatory T Cell Isolation Kit, human | Miltenyi | Cat# 130-091-301 |
| Lysosensor Green DND-189 | Life Technologies | Cat# L7535 |
| LysoTracker Red DND-99 | Life Technologies | Cat# L7528 |
| Mitotracker Red | Life Technologies | Cat# M7512 |
| CellTrace CFSE Cell Proliferation Kit | Life Technologies | Cat# C34554 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Life Technologies | Cat# M36008 |
| MitoTEMPO | SIGMA | Cat# SML0737 |
| Rapamycin | SIGMA | Cat# 553210 EMD Millipore |
| Plaquenil (hydroxychloroquine) | Sanofi Aventis | ATC code 8P01BA02 |
| DOTAP liposomal transfection reagent | Roche | REF: 11202375001 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katerina Gkirtzimanaki (kgirtzimanaki@gmail.com).
Experimental Model and Subject Details
Human Subjects
49 SLE patients followed by the Rheumatology Clinic of the University Hospital of Crete, diagnosed according to the American College of Rheumatology 1982 classification criteria were studied (median age = 42.5 years; range: 16 to 70yrs). Active SLE was defined as an SLE Disease Activity Index 2000 score (SLEDAI-2K) higher than 8 at the day of blood draw. Patients were abstained from their medications - including steroids and CQ - for at least 24 hours prior to blood sampling. Moreover, n = 26 patients were completely off therapy at the time of enrolment to the study. Healthy age- and sex-matched volunteers from the Department of Transfusion Medicine of the University Hospital of Heraklion served as controls. 6 patients followed by the Rheumatology Clinic of the University Hospital of Crete, diagnosed with Rheumatoid Arthritis according to the 1987 American College of Rheumatology (ACR) criteria, donated peripheral blood that served as control serum treatment (median age = 45.4 years). The study was approved by the Ethics Committee of the University Hospital of Heraklion and all subjects gave written informed consent prior to study (Protocol Number: 5944/14-6-2017). Sex/gender, age and clinical records of SLE patients are listed in Table S1 (excel file) and of RA patients in Table S2. Criteria of patient selection were disease activity (SLEDAI) and ability to abstain from therapy.
Methods Details
Peripheral blood monocyte isolation
PBMCs were isolated from heparinized human peripheral blood by Ficoll- Histopaque-1077 (GE-Healthcare) density centrifugation. Erythrocytes were eliminated by hypotonic lysis (1ml ddH2O for 25 s and 1ml 1.8% NaCl). Viability was measured 99% by trypan blue dye exclusion in every experiment. Monocytes were magnetically isolated (MACS, Miltenyi Biotec) and purity was evaluated by flow cytometry (CD14+ cells/total live cells). Preparations of ≥ 94% purity were used in all experiments.
Serum collection
Healthy, SLE and RA peripheral blood samples were collected in SST vacutainers and centrifuged at 2500 rpm for 15mins (RT). Serum was collected under sterile conditions and stored in aliquots at −80°C.
Cell culture
Magnetically isolated CD14+ monocytes were cultured in RPMI-1640/L- glutamine, supplemented with 10% heat-inactivated FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin for up to 24hours. When longer culture time was necessary, cells were cultured for 3 days and medium was further supplemented with GM-CSF (100 ng/ml, Peprotech) and IL-4 (20 ng/ml, Peprotech) or Universal Type I IFN (Hu-IFN-αA/D[Bg/II] - 400ng/ml, PBL) or heterologous healthy, SLE or RA serum (10% v/v in serum free medium).
Flow cytometry
Live cells (Annexin V Apoptosis kit, BD PharMingen) were stained with fluorochrome-conjugated antibodies (anti-CD14 clone HCD14, anti-CD4 clone Okt4, anti-CD86 clone IT2.2, anti-HLADR cloneL243, anti - p4EBP1 clone T36/45, anti-pP70S6K clone Cl 215247) for 20 min at 4°C in PBS/5% FBS. Mitochondrial ROS were measured by MitoSOX (Invitrogen) staining (1 mM for 15min at 37°C and 5% CO2). Mitochondrial membrane polarization was assessed by JC-1 (eBiosciences) staining (1 μg/ml for 10min at 37°C and 5% CO2) and lysosomal re-acidification by aNPs was assessed using.Lysosensor DND189 (Thermo Scientific) staining (1μΜ in medium for 20min at 37°C and 5% CO2). Samples were acquired on a FACSCalibur (BD Biosciences) and analyzed using the FlowJo software (Tree Star). For phospho-protein staining, cells were permeabilized and stained using the Intracellular Fixation and Permeabilization Buffer Set kit (eBioscience), according to manufacturer instructions.
Mixed Lymphocyte Reaction
Human cord blood (CB) was obtained from normal deliveries by the Ob/Gyn Dpt of the University Hospital of Heraklion. CB was collected in a heparinized tube following delivery of the infant and clamping and transecting of the umbilicus. CD4+CD25- T cells were magnetically isolated (Miltenyi, CD4+CD25+ Treg isolation kit 130-091-301, negative selection) by CB PBMCs (puriy ≥ 85%) and CFSE labeled (Thermo Fisher Scientific, Cell Trace C34554). Allogeneic monocytes (Miltenyi, Monocyte isolation kit II,130-091-153, negative selection) were isolated from unrelated adult PBMC donors (puriy ≥ 94%) 1 day prior to CB donation in the cases of healthy donors or the same day in the cases of SLE patients; HLA typing was not done. Healthy CD14+ PBMCs were pretreated for 18hrs with the following reagents, alone or in combination: Universal Type I IFN (IFNα), Rapamycin and CQ and washed before being added to the MLRs. SLE monocytes were pretreated for 1hr with rapamycin where indicated. 105 pretreated monocytes were plated in round-bottomed 96-well plates and 5x104 CFSE labeled T cells were added; MLRs were left in culture for 6 days in a humidified 37°C and 5% CO2 incubator. Every MLR culture was performed in triplicate. Proliferation index was calculated using FlowJo software platform.
DNase I Transfection
CD14+ PBMCs were transfected with 1.5 mg of DNase I (Roche) or Heat Inactivated DNase I (75°C for 10 min/U) using DOTAP transfection reagent (Roche) according to manufacturer’s instructions. After incubation with DOTAP-DNase I or DOTAP-HI-DNase I for 3 hours (transfection mix was added in culture medium), cells were washed to remove excess of transfection reagent and enzyme, stimulated with IFNα and incubated for 18hrs or GMCSF 100ng/ml and IFNα 400ng/ml or IL4 20ng/ml and incubated for 3 days. HLA-DR and CD86 markers were analyzed by flow cytometry.
Electroporation of primary monocytes with siRNAs
CD14+ PBMCs were isolated from blood specimens not older than 2hrs and incubated for 1hr in RPMI-1640 containing FBS 10%, streptomycin 100 μg/ml, penicillin 100 U/ml, Glutamine 2mM and sodium pyruvate 1mM, in a humidified 37°C/5% CO2 incubator before electroporation. 106 cells/sample were electroporated with 90nM (3 × 30nM) Stealth RNAi siRNA duplexes using Amaxa Human Dendritic Cell Nucleofector Kit. Cells were incubated with transfection mix in medium for 6hrs and were then washed and incubated for 3 days with GMCSF and IFNα or IL4. All transfection experiments were performed in triplicates (biological replicates) for all conditions. Transfection efficiency was assessed by flow cytometry for GFP in cells transfected with 5 μgs pmaxGFP® Vector (65%–75% efficiency).
RNA-sequencing library preparation
RNA was isolated from CD14+ PBMCs of 7 patients and 5 healthy controls. RNA-sequencing libraries were prepared with the TruSeq RNA kit using 1 μg total RNA according to Illumina’s protocols. All 12 samples were pooled together and mixed in equal amounts. Samples were sequenced in the Greek Genome Center (GGC) at BRFAA. Paired-end 40bp reads were generated with the NextSeq500.
RNA Sequencing library analysis
Raw sequencing data were aligned to the human genome GRCh37/hg19 with the use of tophat2 (version 2.0.9) (Kim et al., 2013) and «–b2-very-sensitive» parameter. Samtools (version 0.1.19) (Li et al., 2009) were used for data filtering and file format convertion while HT-seq count (version 0.6.1p1) (Anders et al., 2015) was utilized for assigning aligned fragments into exons using the « -s –no –m intersection –nonempty » command. Differential expression was performed with DESeq R package (Anders and Huber, 2010) and genes with fold change cut off 1.5 and p value ≤ 0.05 were considered to be differentially expressed (DEGs) between the 5 healhty and 7 patients (Table S3). Gene ontology and pathway analysis was performed in the DEGs with DAVID knowledgebase (Huang et al., 2009) Data have been deposited in the Short Read Archive (SRA) under the BioProject PRJNA392602.
Acidic nanoparticle (aNP) preparation
Resomer® RG 503H PLGA (Sigma-Aldrich, 719870) was used with a lactide-glycoside ratio of 50:50, Molecular weight 24000 to 38000. A stock solution of polymer was prepared by dissolving 31 mg of PLGA in 3.1 mL of THF (Sigma-Aldrich, 401757). 200 μL of this stock solution were added to 20 mL of deionized water under sonication. The suspension of PLGA-aNP was then slowly concentrated to provide a sample with a typical concentration of 0.18 mg/mL. For all experiments, PLGA-aNP solutions were used as freshly prepared.
Immunoblotting
Cells were lysed in RIPA buffer (50 mM Tris·HCl at pH 8, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 0.1% SDS) supplemented with protease and phosphatase inhibitors (Complete EDTA Free; Roche Applied Science) and equal amounts of proteins were subjected to SDS-PAGE on 12% gels and then transferred to Immobilon-Psq membrane (Millipore). Membranes were blocked with 5% skimmed milk, 1% BSA in TBST and then incubated with anti-LC3B, anti-TOMM20 and anti-Beclin1 by Novus Biologicals, anti-SQSTM-1 by MBL, anti-VDAC1 and anti-actin by Millipore, anti-Stat1 and anti-pStat1 by BD Biosciences, anti-pmTOR, anti-mTOR, anti-ULK1 and anti-pULK1 by Cell Signaling, anti-pBeclin1 from Abbiotec, anti-mouse HRP by Milipore and anti-rabbit HRP by Cell Signaling according to manufacturers suggestions. As control sample, protein extract from Neuro 2A cell line was used. The image was resolved by ECL system (Pierce and Millipore) and detected by ImageBlot (BIORAD). Relative intensity of bands was calculated with Fiji software.
Immunofluorescence (IF)
For IF microscopy cells were plated on poly-L-lysine coverslips (1,5-2 x105 cells /slide). When the appropriate treatments were finished, slides were washed once with PBS, fixed with 4% PFA for 20mins (RT) re-washed twice with PBS, refixed and permeabilized with 100% ice cold MetOH for 10mins (RT) and blocked with 2% BSA in 0,1% saponin (BS). Primary antibodies were left for 1hr at RT or O/N at 4°C. Slides were washed several times with BS and the secondary antibodies were incubated for 1hr at RT. Antibodies used were mouse anti-LC3B (1:20, 5F10 nanoTools), rat anti-Lamp-1 (1:400, 1D4B Abcam), rabbit anti-p62 (1:500, MBL), rabbit anti-TFEB (1:100, 954604 R&D Systems) followed by incubation with Alexa fluor® 555 anti-mouse IgG (1:500, Molecular Probes), Alexa fluor® 488 anti-rabbit IgG (1:500, Molecular Probes), Alexa fluor® 633 anti-rat IgG (1:250, Molecular Probes). For visualization of the nuclei DAPI (Sigma-Aldrich) was used. Samples were mounted with Mowiol and visualized using inverted confocal live cell imaging system Leica SP8.
For Mitotracker CMXRos and Lysotracker DND-99 (Molecular Probes) stainings, dyes were added in culture as suggested by the manufacturer, washed, fixed with 4% PFA for 20mins (RT), and mounted for immediate observation under the microscope.
Confocal Imaging Analysis
The quantification method of % colocalization used was based on an NIH funded macro, developed by Daniel J. Shiwarski, B.S., University of Pittsburgh and validated for the analysis of autophagic flux by Pampliega et al. (2013). This script (macro) was run at Fiji software and the numerical output expressed the extent of green and red puncta colocalization in RGB images.
TaqMan Assay
DNA was isolated with QIAamp DNA Micro kit (QIAGEN) following the manufacturer’s protocol. For the quantification of mitochondrial DNA copy number a multiplex RT- PCR assay was used according to Phillips et al. (2014). DNA samples (50ng/reaction) were used together with appropriate volumes of TaqMan Fast Universal PCR Master Mix (2x), No AmpErase UNG (Applied Biosystems)) at a CFX ConnectTM, Real-Time System. Total volume of each multiplex RT-PCR reaction was 25 μl. huβ2Μ primers - F: TTAACGTCCTTGGCTGGGTC, R: ACTGGAAGACAAAGGGCTCG, huβ2Μ probe: CAGATGCAGTCCAAACT-CTCACT, mtMinArc primers - F: CTGTTCCCCAACCTTTTCCT, R: CCATGATTGT-GAGGGGTAGG, mtMinArc probe: GACCCCCTAACAACCCCC STING (TMEM173) primers and probe: Hs00736958_m1 assay, GAPDH primers and probe: Hs02758991_g1 assay and TLR9 primers and probe: Hs00152973_m1 assay by Applied Biosystems.
Real time-PCR (RT-PCR)
Total RNA from monocytes was collected using the TRIzol (Invitrogen)extraction protocol with Turbo DNase (Ambion) treatment according to manufacturer’s instructions. cDNA was prepared using PrimeScript 1st strand cDNA Synthesis Kit (Takara) according to manufacturer’s protocol. Transcripts were quantified by incorporation of KAPA SYBR FAST qPCR Kit Master Mix (2X) (Kapa Biosystems) at a BIO-RAD CFX ConnectTM, Real-Time System. Expression was normalized to GAPDH and calculated by the change-in-threshold method [2ˆ(-ΔΔCT)].
Primer sequences used: IFNα–F: GGTGACAGAGACTCCCCTGA, R:CAGGCACAAGGGCTGTATTTCTT, CXCL10 – F:GTGGCATTCAAGGAGTACCTC, R:TGATGGCCTTCGATTCTGGATT, LC3B-F:CTGTTGGTGAACGGACACAG, R:CTGGGAGGCATAGACCATGT, ATG5-F:TGACGTTGGTAACTGACAAAGTG, R:AATGCCATTTCAGTGGTGTG, P62/SQSTM1– F:AGCAGATGAGGAAGATCGCC, R:CTGTAGACGGGTCCACTTCTT, PINK1–F:GGATATGGAGCAGTCA CTTACAG, R:GGCAGCACATCAGGGTAGTC, PARKIN- F:CGACCCTCAACTT GGCTACT R:TCTTTAATCAAGGAGTTGGGACA, TLR9 – F:CTGCCTTCCTACCCTGTGAG, R:GGATGCGGTTGGAGGACAA, GAPDH - F:CATGTTCCAATATGATTCCACC, R:GATGGG ATTTCCATTGATGAC.
ELISA
Detection of human IL-6, CXCL10, TNFα (eBioscience) and IFNα (VeriKine Human Interferon Alpha Multi-Subtype Serum ELISA Kit, PBL) in human sera and culture supernatants harvested at the indicated time points, were performed by sandwich ELISA following the manufacturer’s recommendations. Light absorbance was measured using an ELx800 Biotek plate reader. Background signal was subtracted. All samples were assessed in duplicates (technical replicates).
ATP Assay
CD14+ monocytes (106 cells /condition) were harvested, washed and lysed according to manufacturer’s protocol (Abcam) for the colorimetric assay. Light absorbance was measured using an ELx800 Biotek plate reader. ATP levels were calculated according to the formula given: [ATP] (nmol per μL or mM) = (Ts/Sv) ∗ D, Ts = ATP amount from standard curve (nmol or mM), Sv = sample volume added in sample wells (μL), D = sample dilution factor.
Isolation of mitochondria
Mitochondria were isolated from cultured cells with the Mitochondria Isolation Kit, Thermo Fisher Scientific (89874) according to manufacturer’s protocol. Briefly, cells were lysed using the reagent based method following the instructions to obtain a more purified fraction of mitochondria, with > 50% reduction of lysosomal and peroxisomal contaminants. For Western Blot analysis mitochondrial pellets were boiled with SDS-PAGE sample buffer: RIPA buffer supplemented with phosphatase and protease inhibitors (ThermoFisher Scientific) and additional SDS (20%) and β-mercaptoethanol (5%).
Cathepsin D activity detection
Cathepsin D activity was measured by the Fluorometric Assay Kit from Abcam (ab65302), according to manufacturer’s instructions. Briefly, 106 cells were harvested for each condition, lysed immediately and incubated with the preferred cathepsin-D substrate GKPILFFRLK(Dnp)-D-R-NH2, labeled with MCA. Fluorescence released upon substrate cleavage was measured in a FLx800 fluorescence microplate reader. For all experiments, cell lysates were assayed as freshly prepared.
Oxygen consumption rate measurements
Oxygen consumption rates were measured as described by Palikaras et.al. at Nature 2015, using a Clark-type electrode (Hansatech Instruments). 4x106 CD14+ monocytes treated as indicated were washed and collected in PBS buffer. The chamber was kept at 37°C and measurements were done for 10–15 min, depending on the oxygen consumption rate. The slope of the straight portions of the plots were used to derive the oxygen consumption rates. Cells were recovered after respiration measurements and collected for protein quantification. Rates were normalized to protein content as calculated by Bradford protein assay.
Reagents
Plaquenil (CQ: 46.1 μM) was purchased from Sanofi Aventis. Rapamycin (Rapam.:1 μM) was from SIGMA. Wortmanin (WM:1μΜ) was purchased from InvivoGen. Mitotracker CMXRos (MTR: 1 μM), MitoTEMPO (2nM), Lysotracker-DND99 (LTR: 1μΜ) and MitoSOX (5μΜ) were purchased from Molecular Probes – Thermo Fisher Scientific. JC-1 (2 μgs/ml) was purchased from eBiosciences. Recombinant DNase I and DOTAP liposomal transfection reagent were obtained from Roche. Human STING (TMEM173), TLR9 and Negative Stealth siRNA duplex oligonucleotides were purchased from Invitrogen.
Quantification and Statistical Analysis
Statistical Analysis
Results are presented as mean ± SEM. Statistical analysis was performed using paired Student’s t test or unpaired non-parametric Mann Whitney U test, as indicated in each Figure legend. All experiments were performed in the number of biological replicates mentioned as n in each Figure Legend. Briefly, experiments performed in different healthy donors (n corresponds to the number of healthy donors) were designed to assess the effect of different treatments assessing one donor / experiment. In this case, statistical analysis was performed using paired t test, while comparison of healthy with SLE samples was statistically analyzed using unpaired Student’s t test (non-parametric Mann Whitney). A P value < 0.05 was considered to indicate statistical significance (∗). All data were analyzed using GraphPad Prism software.
Data and Software Availability
The accession number for the RNA-sequencing data reported in this paper is NCBI Short Read Archive: PRJNA392602 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA392602/).
Acknowledgments
We thank all patients who contributed clinical samples, S. Georgakis for assisting with the experiments, A. Banos for assisting with sample preparation for RNA-seq, K. Palikaras for assisting with oxygen consumption analysis, and I. Gergiannaki for organizing the clinical records of SLE patients treated at the Rheumatology Clinic of the University Hospital of Crete. This work was implemented by the Greek General Secretariat of Research and Technology (co-funded by the European Social Fund [ESF] and National Resources) Aristeia I 2344, as well as the Foundation for Research in Rheumatology (FOREUM). K.G. was funded by IKY-Siemens (Post-Doc Fellowships of Excellence), and D.B. and P.V. were funded by European Union Project Innovative Medicine Initiative 6 (“BeTheCure” contract 115142-2) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 742390). This work was also supported by COST Actions Mye-EUNITER (BM1404, http://www.mye-euniter.eu) and TRANSAUTOPHAGY (CA15138, http://cost-transautophagy.eu) that are part of the European Union Framework Program Horizon 2020.
Author Contributions
Conceptualization and Methodology, K.G.; Investigation, K.G., E.K., D.N., A.P., and A.B.; Writing – Original Draft, K.G.; Writing – Review & Editing, G.B., D.T.B., and P.V.; Resources, A.M., P.S., G.B., D.T.B., and P.V.; Funding Acquisition, K.G., G.B., P.S., D.T.B., and P.V.; Supervision, D.T.B. and P.V.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2018
Footnotes
Supplemental Information includes six figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.001.
Contributor Information
Katerina Gkirtzimanaki, Email: kgirtzimanaki@gmail.com.
Panayotis Verginis, Email: pverginis@bioacademy.gr.
Supplemental Information
Table containing the 380 differentially expressed genes (DEGs) between patient and healthy group.
References
- Alissafi T., Banos A., Boon L., Sparwasser T., Ghigo A., Wing K., Vassilopoulos D., Boumpas D., Chavakis T., Cadwell K., Verginis P. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Invest. 2017;127:2789–2804. doi: 10.1172/JCI92079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco P., Palucka A.K., Gill M., Pascual V., Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- Bourdenx M., Daniel J., Genin E., Soria F.N., Blanchard-Desce M., Bezard E., Dehay B. Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy. 2016;12:472–483. doi: 10.1080/15548627.2015.1136769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bride K.L., Vincent T., Smith-Whitley K., Lambert M.P., Bleesing J.J., Seif A.E., Manno C.S., Casper J., Grupp S.A., Teachey D.T. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17–28. doi: 10.1182/blood-2015-07-657981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S., Athale S., Domic B., Murat E., Chandra M., Banchereau R., Baisch J., Phelps K., Clayton S., Gong M. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016;213:697–713. doi: 10.1084/jem.20151876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaud G., Bienaimé F., Tabarin F., Bataillon G., Seilhean D., Noël L.-H., Dragon-Durey M.-A., Snanoudj R., Friedlander G., Halbwachs-Mecarelli L. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N. Engl. J. Med. 2014;371:303–312. doi: 10.1056/NEJMoa1312890. [DOI] [PubMed] [Google Scholar]
- Cang C., Zhou Y., Navarro B., Seo Y.-J., Aranda K., Shi L., Battaglia-Hsu S., Nissim I., Clapham D.E., Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell. 2013;152:778–790. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Fang Y. A novel pathway regulating the mammalian target of rapamycin (mTOR) signaling. Biochem. Pharmacol. 2002;64:1071–1077. doi: 10.1016/s0006-2952(02)01263-7. [DOI] [PubMed] [Google Scholar]
- Chen C., Liu Y., Liu R., Ikenoue T., Guan K.-L., Liu Y., Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow M.K. Type I interferon in the pathogenesis of lupus. J. Immunol. 2014;192:5459–5468. doi: 10.4049/jimmunol.1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow Y.J., Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- de Jong S.D., Basha G., Wilson K.D., Kazem M., Cullis P., Jefferies W., Tam Y. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J. Immunol. 2010;184:6092–6102. doi: 10.4049/jimmunol.0802442. [DOI] [PubMed] [Google Scholar]
- Ding D., Mehta H., McCune W.J., Kaplan M.J. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J. Immunol. 2006;177:5878–5889. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Gergely P., Jr., Niland B., Gonchoroff N., Pullmann R., Jr., Phillips P.E., Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J. Immunol. 2002;169:1092–1101. doi: 10.4049/jimmunol.169.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergely P., Jr., Grossman C., Niland B., Puskas F., Neupane H., Allam F., Banki K., Phillips P.E., Perl A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:175–190. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujer C., Sandgren K.J., Douagi I., Adams W.C., Sundling C., Smed-Sörensen A., Seder R.A., Karlsson Hedestam G.B., Loré K. IFN-α produced by human plasmacytoid dendritic cells enhances T cell-dependent naïve B cell differentiation. J. Leukoc. Biol. 2011;89:811–821. doi: 10.1189/jlb.0810460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jegerlehner A., Maurer P., Bessa J., Hinton H.J., Kopf M., Bachmann M.F. TLR9 signaling in B cells determines class switch recombination to IgG2a. J. Immunol. 2007;178:2415–2420. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman D.M., Yi A.K., Beaucage S.L., Conover J., Krieg A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. USA. 1996;93:2879–2883. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Baehrecke E.H., Brumell J.H., Chu C.T., Codogno P., Cuervo A.M., Debnath J., Deretic V., Elazar Z., Eskelinen E.-L. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk V.I., Saiki S., Lichtenberg M., Siddiqi F.H., Roberts E.A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F.M. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011;13:453–460. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko D.V., Agostinis P., Krysko O., Garg A.D., Bachert C., Lambrecht B.N., Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–164. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X., Reihl A.M., Luo X.M. Breakdown of immune tolerance in systemic lupus erythematosus by dendritic cells. J. Immunol. Res. 2016;2016:6269157. doi: 10.1155/2016/6269157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Galluzzi L., Zitvogel L., Kroemer G. Autophagy and cellular immune responses. Immunity. 2013;39:211–227. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Ma Z., Jacobs S.R., West J.A., Stopford C., Zhang Z., Davis Z., Barber G.N., Glaunsinger B.A., Dittmer D.P., Damania B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA. 2015;112:E4306–E4315. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D., Shoelson S.E. Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 2011;11:81–83. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A., Ichikawa T., Nakao K., Miyaaki H., Hirano K., Fujimito M., Akiyama M., Miuma S., Ozawa E., Shibata H. Interferon-α-induced mTOR activation is an anti-hepatitis C virus signal via the phosphatidylinositol 3-kinase-Akt-independent pathway. J. Gastroenterol. 2009;44:856–863. doi: 10.1007/s00535-009-0075-1. [DOI] [PubMed] [Google Scholar]
- Minton K. Inflammasome: anti-inflammatory effect of mitophagy. Nat. Rev. Immunol. 2016;16 doi: 10.1038/nri.2016.33. 206–206. [DOI] [PubMed] [Google Scholar]
- Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Niewold T.B., Hua J., Lehman T.J.A., Harley J.B., Crow M.K. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaks Z., Winans T., Caza T., Fernandez D., Liu Y., Landas S.K., Banki K., Perl A. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol. 2016;68:2728–2739. doi: 10.1002/art.39791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O., Orhon I., Patel B., Sridhar S., Díaz-Carretero A., Beau I., Codogno P., Satir B.H., Satir P., Cuervo A.M. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual V., Farkas L., Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr. Opin. Immunol. 2006;18:676–682. doi: 10.1016/j.coi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Pearce E.L., Poffenberger M.C., Chang C.-H., Jones R.G. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013;9:674–686. doi: 10.1038/nrrheum.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips N.R., Sprouse M.L., Roby R.K. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: a multiplex real-time PCR assay. Sci. Rep. 2014;4:3887. doi: 10.1038/srep03887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisetsky D.S. The role of mitochondria in immune-mediated disease: the dangers of a split personality. Arthritis Res. Ther. 2016;18:169. doi: 10.1186/s13075-016-1063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S., Mitchell T., Kramer P., Chacko B., Darley-Usmar V.M. Mitochondria in monocytes and macrophages-implications for translational and basic research. Int. J. Biochem. Cell Biol. 2014;53:202–207. doi: 10.1016/j.biocel.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejman J., Bragonzi A., Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005;12:468–474. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- Rönnblom L., Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser H., Fey S.B., Horowitz J., Fischer E.R., Balinsky C.A., Miyake K., Bekisz J., Snow A.L., Zoon K.C. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy. 2013;9:683–696. doi: 10.4161/auto.23921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena L.A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D.A., Wang C.-R., Schumacker P.T., Licht J.D., Perlman H. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V.A., Arencibia M.G., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M., Niewold T.B. Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol. 2013;4:319. doi: 10.3389/fimmu.2013.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhbaatar N., Hengstschläger M., Weichhart T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol. 2016;37:778–789. doi: 10.1016/j.it.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos G.C. Systemic lupus erythematosus in 2015: cellular and metabolic requirements of effector T cells. Nat. Rev. Rheumatol. 2016;12:74–76. doi: 10.1038/nrrheum.2015.178. [DOI] [PubMed] [Google Scholar]
- Wahl D.R., Byersdorfer C.A., Ferrara J.L.M., Opipari A.W., Jr., Glick G.D. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol. Rev. 2012;249:104–115. doi: 10.1111/j.1600-065X.2012.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckerle C.E., Franek B.S., Kelly J.A., Kumabe M., Mikolaitis R.A., Green S.L., Utset T.O., Jolly M., James J.A., Harley J.B., Niewold T.B. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63:1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., Bestwick M., Duguay B.A., Raimundo N., MacDuff D.A. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Choi S.-C., Xu Z., Perry D.J., Seay H., Croker B.P., Sobel E.S., Brusko T.M., Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Morgan M.J., Chen K., Choksi S., Liu Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119:2895–2905. doi: 10.1182/blood-2011-08-372383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.M., Cheng F.J., Zhou X.J., Qi Y.Y., Hou P., Zhao M.H., Zhang H. Detecting Genetic Associations between ATG5 and Lupus Nephritis by trans -eQTL. J. Immunol. Res. 2015;2015:153132. doi: 10.1155/2015/153132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.J., Lu X.L., Lv J.C., Yang H.Z., Qin L.X., Zhao M.H., Su Y., Li Z.G., Zhang H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheum. Dis. 2011;70:1330–1337. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- Zhu S., Qiao Y., Wu J., Zang G., Liu Y.-J., Chen J. Human cancer cells respond to cytosolic nucleic acids via enhanced production of interferon-β and apoptosis. J. Immunother. Cancer. 2015;3:250. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table containing the 380 differentially expressed genes (DEGs) between patient and healthy group.