

Summary
Transdifferentiation of fibroblasts into induced neuronal cells (iNs) by the neuron-specific transcription factors Brn2, Myt1l, and Ascl1 is a paradigmatic example of inter-lineage conversion across epigenetically distant cells. Despite tremendous progress regarding the transcriptional hierarchy underlying transdifferentiation, the enablers of the concomitant epigenome resetting remain to be elucidated. Here, we investigated the role of KMT2A and KMT2B, two histone H3 lysine 4 methylases with cardinal roles in development, through individual and combined inactivation. We found that Kmt2b, whose human homolog’s mutations cause dystonia, is selectively required for iN conversion through suppression of the alternative myocyte program and induction of neuronal maturation genes. The identification of KMT2B-vulnerable targets allowed us, in turn, to expose, in a cohort of 225 patients, 45 unique variants in 39 KMT2B targets, which represent promising candidates to dissect the molecular bases of dystonia.
Keywords: KMT2B, histone H3 lysine 4 methylation, transdifferentiation, epigenetics, induced neuronal cells, mouse embryonic fibroblasts, myocytes, dystonia, cell fate conversion, MLL2
Graphical Abstract
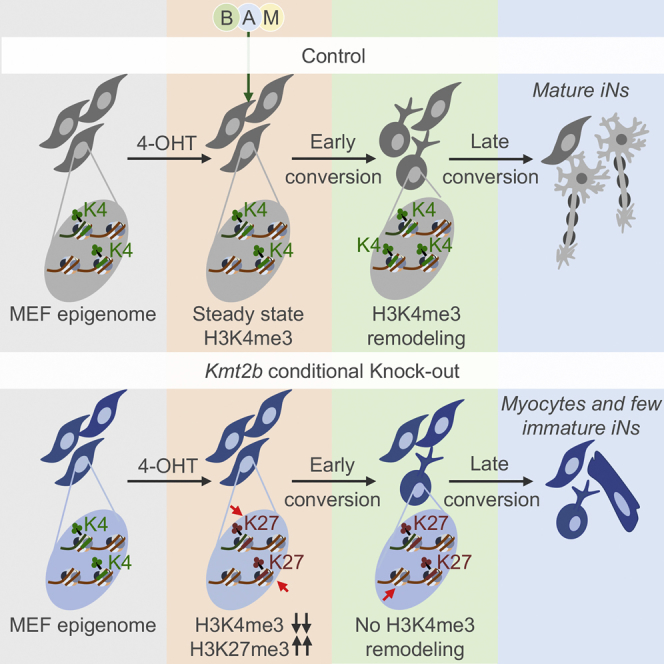
Highlights
-
•
KMT2B is critical for neuronal transdifferentiation, whereas KMT2A is dispensable
-
•
KMT2B is responsible for the activation of the neuronal maturation gene program
-
•
KMT2B represses the myocyte fate unleashed upon defective transdifferentiation
-
•
KMT2B dependence reveals candidate dystonia-causative genes
Barbagiovanni et al. demonstrate that KMT2B, in contrast to KMT2A, is fundamental for the epigenetic and transcriptomic resetting underlying transdifferentiation of fibroblasts into induced neuronal cells (iNs), acting both in the suppression of alternative fates and in the promotion of iN maturation. Transdifferentiation-specific KMT2B targets reveal dystonia-causative gene candidates.
Introduction
The conversion of murine embryonic fibroblasts (MEFs) into induced neuronal cells (iNs) through forced expression of the Brn2, Ascl1, and Myt1l transcription factors (TFs) (hereafter called BAM [Brn2, Ascl1, Mytl1] factors) defined the paradigm of transdifferentiation across germ layers (Vierbuchen et al., 2010). BAM factors synergistically cooperate in this process (∼20% efficiency), which does not require either proliferation or the transient reacquisition of pluripotency (Vierbuchen et al., 2010). ASCL1 instructs transdifferentiation as a pioneer TF, recognizing trivalent chromatin states marked by H3 lysine 4 monomethylation (H3K4me1), lysine 9 trimethylation (H3K9me3), and lysine 27 acetylation (H3K27ac) (Wapinski et al., 2013). BRN2 and MYT1L are, instead, critical for iN maturation and the suppression of alternative cell fates; upon Ascl1-only infection, MEFs that fail to become iNs deviate toward myocytes, an outcome that is prevented by co-transduction with Brn2 and Myt1l (Mall et al., 2017, Treutlein et al., 2016, Wapinski et al., 2013).
ASCL1 has been recently shown to effect massive remodeling of chromatin accessibility and nucleosome phasing, with open chromatin loci enriched for neuronal and muscle pathway genes (Wapinski et al., 2017). Moreover, MEF-to-iN conversion features a major chromatin reconfiguration in the first 5 days after Ascl1 induction, following which further chromatin transitions represent less than 20% of all changes underlying direct neuronal conversion (Wapinski et al., 2017). This suggests that these epigenetic transitions are not as gradual as during reprogramming to pluripotency (Cacchiarelli et al., 2015, Chronis et al., 2017), but the role of specific chromatin regulators in these accompanying waves of chromatin resetting remains elusive.
Here, we investigated the roles of KMT2A and KMT2B in MEF-to-iN conversion in light of their critical function in lineage decisions. KMT2A and KMT2B belong to Set1-Trithorax type H3K4 methylases, specifically responsible for the deposition of histone H3 lysine 4 trimethylation (H3K4me3) at gene promoters (Denissov et al., 2014).
Both associate with MENIN, which mediates their localization at specific loci, such as HOX genes (Glaser et al., 2006, Hughes et al., 2004, Lee et al., 2006). Despite their high homology, KMT2A and KMT2B are spatially and temporally non-redundant. Thus, knockout (KO) of either Kmt2a (Ernst et al., 2004) or Kmt2b (Glaser et al., 2006) is embryonic lethal at embryonic day 12.5 (E12.5) or E.10.5, respectively. Furthermore, KMT2A and KMT2B have specific roles during mammalian neuronal differentiation. During retinoic acid-based differentiation, the two enzymes regulate different HOX genes (Denissov et al., 2014), and Kmt2b−/− embryonic stem cells (ESCs) show a severe delay in in vitro differentiation toward the ectodermal lineage (Lubitz et al., 2007). Adult neurogenesis and function are also specifically affected. Indeed, Kmt2a−/− subventricular zone neural stem cells (SVZ NSCs) are impaired selectively in neuronal differentiation, whereas both KMT2A and KMT2B contribute to memory formation, albeit through distinct target pathways and with no apparent effects on brain and neuronal morphology (Kerimoglu et al., 2013, Kerimoglu et al., 2017, Lim et al., 2009). Finally, mutations in KMT2A and KMT2B cause two distinct brain disorders: Wiedemann-Steiner syndrome, featuring intellectual disability, and a newly recognized molecular subset of early-onset generalized dystonia, respectively (Meyer et al., 2017, Strom et al., 2014, Zech et al., 2016).
Results
Conditional Inactivation of Kmt2a and Kmt2b upon Transdifferentiation
To study the role of KMT2A and KMT2B during transdifferentiation, we employed conditional mouse strains carrying exon 2 of Kmt2a and/or Kmt2b flanked by LoxP sites, the knockin of the YFP-coding gene into one Rosa26 allele downstream of a LoxP-flanked transcription termination cassette (STOP cassette), and the gene coding for the tamoxifen-inducible version of Cre recombinase knocked into the second Rosa26 allele (Glaser et al., 2006, Kranz et al., 2010, Testa et al., 2004). Upon 4-hydroxytamoxifen (4-OHT) administration, Cre is expressed, generating a frameshift mutation in Kmt2a and/or Kmt2b (Figure 1A). MEFs were derived from Kmt2a (and/or Kmt2b)fl/fl Cre+ YFP+ embryos and from Kmt2a+/+Kmt2b+/+ Cre+ YFP+ or Kmt2afl/+ Cre+ YFP+ for Kmt2a conditional KO (cKO) as controls (Figure 1A). After 5 days of 4-OHT treatment followed by either 2 or 7 days in normal medium (Figure 1B), depletion of Kmt2a and/or Kmt2b was assessed by qRT-PCR (Figure 1C) and western blot (Figure 1D).
Figure 1.

Experimental System
(A) Schematic of Kmt2 cKO MEF derivation and 4-OHT treatment.
(B) Schematic of our experimental pipeline.
(C) Representative qPCR results for exon 2 copy number after 4-OHT treatment. Means ± SEM.
(D) Western blot analysis for KMT2B, with VINCULIN (VCL) as housekeeping, of MEFs treated 5 days with 4-OHT and left 7 days in culture without 4-OHT. The first lane was dedicated to ESCs.
Transdifferentiation was then implemented according to the original protocol (Vierbuchen et al., 2010; Figure 1B). The sustained expression of YFP (Figure S1A) along with permanent loss of Kmt2b exon 2 (Figure S1B) through the end of transdifferentiation excluded selection of Kmt2bfl/fl iNs that might have escaped recombination.
To decipher the role of the two KMT2 methylases, we integrated two complementary approaches to capture the acquisition of iN identity along transdifferentiation: morphological analysis through an automated and unbiased imaging method (ScanR) and cytofluorimetric analysis (fluorescence-activated cell sorting [FACS]) for the expression of polysialic acid-neural cell adhesion molecule (PSA-NCAM), a marker of iN induction (Figure 1B). Transcriptomic and epigenomic profiling was then performed at 5 and 13 days of transdifferentiation (Figure 1B). Day 5 was chosen as reference starting point because, at this time, MEFs are still equally competent to become iNs, having all undergone convergent transcriptional changes (Treutlein et al., 2016). Day 13 was selected as the endpoint because 8 days following transduction no more iNs are induced, and the 13-day iNs have been shown to be functionally mature (Vierbuchen et al., 2010). MEFs were also profiled at the epigenomic level at the onset of transdifferentiation to define their initial chromatin configuration (Figure 1B).
Kmt2b Ablation Impairs Transdifferentiation
We evaluated the transdifferentiation potential of Kmt2a−/− and/or Kmt2b−/− MEFs in terms of both efficiency of iN generation and degree of their maturation. To distinguish between the effect of their loss on cell mortality and transdifferentiation, we harnessed the observation that already 1 day after doxycycline administration (day 3), the vast majority of cells are post-mitotic (Vierbuchen et al., 2010). To confidently ascribe any change in cell number to cell mortality rather than to cell proliferation, we selected cKO and control samples starting transdifferentiation from the empirically validated same number of cells on day 3.
We found that loss of Kmt2b greatly impaired transdifferentiation efficiency in a manner independent of the mortality rate that accompanies transdifferentiation (Figures 2A and 2B; Table S1). In contrast, for the loss of Kmt2a, transdifferentiation efficiency was correlated with cell viability (Figures S1C and S1D; Table S1), pointing to the former as a by-product of the latter.
Figure 2.

KMT2B Is Essential for MEF-to-iN Transition
(A) Efficiency of transdifferentiation in ScanR experiments, calculated relating, for any chosen concentration, the number of Tuj1+ at 13 days to the number of DAPI 3 days after plating.
(B) Cell mortality in ScanR experiments, calculated as the ratio of DAPI 3 days (DAPI start) and 13 days (DAPI end) after plating.
(A and B) Kmt2a+/+Kmt2b+/+, n = 3; Kmt2bfl/fl, n = 2; Kmt2afl/flKmt2bfl/fl, n = 1.
(C) Efficiency of transdifferentiation in FACS experiments, calculated relating the number of PSA-NCAM+ cells at 13 days to the number of plated cells.
(D) Percentage of dead cells, calculated at 13 days, over the initial number of plated MEFs.
(C and D) The number of independent embryos per genotype corresponds to the one reported for time point 13 days of (E).
(E) Percentage of PSA-NCAM+ cells among transdifferentiating MEFs 5, 7, 9, 13, and 21 days after plating, assayed with FACS analysis. The number of independent embryos per time point per genotype is reported under the graph.
(F) Representative images of three different experiments, each reporting the specific cKO and its control. Kmt2afl/− (top left), Kmt2a+/+Kmt2b+/+ (top, center and right), Kmt2a−/− (bottom left), Kmt2b−/− (bottom center), and Kmt2a−/−Kmt2b−/− (bottom right) iNs at 13 days are reported (DAPI in blue and Tuj1 in red).
(G) Representative images of 21-day Kmt2a+/+Kmt2b+/+ (left), Kmt2a−/− (center), and Kmt2b−/− (right) iNs acquired with the bright-field microscope.
(H–J) Average neurite length per neuron, calculated with Neuritetracer on day 13 of transdifferentiation. In particular Kmt2a−/− iNs (H), Kmt2b−/− iNs (I), and Kmt2a−/− Kmt2b−/− iNs (J) were compared to controls.
In the case of Kmt2a ScanR experiments (F and H), Kmt2afl/− MEFs were used as a control. In (F), (H), and (J), MEFs were plated for BAM transduction 2 days after 4-OHT, whereas, in (A)–(E), (G), and (I), MEFs were plated 7 days after 4-OHT. (A)–(E) and (H)–(J), means ± SEM; ∗∗∗p < 0.0001; ∗p < 0.01; ns, not significant.
Next we sought to determine whether there is any redundancy between the two methylases by knocking out both enzymes. Kmt2a−/−Kmt2b−/− transdifferentiated MEFs showed both the highest cell death and the lowest conversion efficiency, indicating that KMT2A only partially compensates for the lack of KMT2B during transdifferentiation (Figures 2A and 2B; Table S1). Importantly, the combined loss of Kmt2a and Kmt2b did not affect MEF viability per se but was specifically triggered upon induction of transdifferentiation (Figures S1E and S1F).
We confirmed these observations through FACS-based quantitation of PSA-NCAM+ cells. Indeed, both Kmt2a−/− and Kmt2b−/− MEFs showed a lower efficiency of iN induction with respect to controls (Figure 2C), but although, for Kmt2a−/− MEFs, this was entirely accounted for by the higher rate of cell death (Figures 2D, S1G and S1H), for Kmt2b−/− MEFs, it was independent of cell death and, thus, imputable to a specific requirement for KMT2B in transdifferentiation (Figures 2D, S1I and S1J). Moreover, although cell mortality in Kmt2a−/− transdifferentiating MEFs was higher with respect to Kmt2b−/− and controls (Figure 2D), the percentage of Kmt2a−/− PSA-NCAM+ cells overlapped or was even higher than in controls along the entire process (Figure 2E). This indicates that PSA-NCAM+ and PSA-NCAM− fractions are at least equally vulnerable to cell death across Kmt2a−/− and Kmt2a+/+.
Finally, FACS analysis of the combined KO showed that, although the viability of Kmt2a−/−Kmt2b−/− MEFs was comparable with that of Kmt2a−/− and lower than that of controls (Figures 2D, S1K and S1L), their transdifferentiation efficiency was strongly reduced with respect both to control and the single cKO transdifferentiating MEFs (Figure 2C), underscoring that KMT2B is the main H3K4 trimethylase involved in direct cell conversion.
Then we analyzed the morphology of the generated iNs to probe the role of either methylase on iN maturation. To this aim, we investigated neurite elongation as a defining hallmark of complete iN conversion, finding that, in Kmt2b−/− iNs, it was severely reduced both at 13 (Figures 2F, 2I, S2A, and S2B) and 21 days (Figure 2G) of transdifferentiation. On the contrary, and consistent with the unaltered efficiency of iN conversion, neurite length in Kmt2a−/− iNs was fully comparable with that of Kmt2a+/− (Figures 2F, 2G, 2H, S2C, and S2D). Finally, the few double cKO iNs only showed minimal neurite elongation (Figures 2F, 2J, and S2E). This demonstrates that KMT2B, besides playing a fundamental role in MEF-to-iN conversion, has also a specific effect on iN maturation.
KMT2B Controls Transcriptome Resetting in MEF-to-iN Conversion
Given the role of KMT2B in H3K4me3 deposition at promoters (Denissov et al., 2014), its loss could impair the gene expression program underlying MEF-to-iN transition. To unveil the molecular basis of the dual phenotype observed upon loss of KMT2B (namely, less efficient iN conversion and defective iN maturation), we performed RNA sequencing (RNA-seq) on sorted PSA-NCAM+ and PSA-NCAM− Kmt2b−/− and control cells at day 13 of transdifferentiation. In particular, through the analysis of PSA-NCAM+ transcriptomes, we aimed to establish which genes responsible for transdifferentiation were differentially expressed in the absence of KMT2B and how Kmt2b−/− iNs differed from controls. Instead, through PSA-NCAM− transcriptomic profiling, we pursued mechanistic insight into the effect of Kmt2b loss on MEF-to-iN conversion.
The first component identified by principal-component analysis (PCA) clearly distinguished genotypes in both PSA-NCAM populations (Figures S3A and S3B), indicating that Kmt2b status represents the largest source of variation in the datasets. Specifically, through the analysis of PSA-NCAM+ cells, we found 1,508 differentially expressed genes (DEGs) between Kmt2b−/− and control iNs (at false discovery rate [FDR] < 0.01 and with at least a 50% fold change), the majority of which were downregulated in the latter, consistent with the gene-activating function of KMT2B (Figure 3A). To define which stage of transdifferentiation was most affected by loss of KMT2B, we clustered day 13 DEGs between Kmt2b−/− and control iNs using K-means clustering according to their established expression pattern throughout transdifferentiation (Wapinski et al., 2013; Figure 3B). Genes that are normally expressed on day 15 and downregulated on day 24 of transdifferentiation (clusters 2 and 3) were found to be already lowly expressed at 13 days in our Kmt2b−/− samples, further highlighting a defective MEF-to-iN transition. On the other hand, a high proportion of cluster 4, whose expression gradually increases throughout transdifferentiation, peaking at day 24, and that is enriched for synaptic genes and RE1-silencing transcription factor (REST) targets (4.79-fold enrichment, q-value [q] ∼ 1e−54 in neuronal progenitors), was downregulated in Kmt2b−/− iNs, further corroborating the defective neuronal fate acquisition in the absence of KMT2B.
Figure 3.

KMT2B Loss Impairs Transcriptomic Changes Underlying Transdifferentiation
(A) Volcano plot highlighting the top DEGs among Kmt2b−/− and control 13-day iNs.
(B) DEGs upon Kmt2b KO clustered on the basis of their expression pattern across stages of transdifferentiation (plotted are log-normalized fragments per kilobase pair per million reads sequenced [FPKM], scaled across the whole dataset).
(C) Heatmap showing the expression across genotypes in iNs sorted at 13 days of the transcriptional regulators belonging to the three subnetworks described by Treutlein et al. (2016). The significant DEGs between Kmt2b−/− and the control are indicated by red arrows. Each line represents a different embryo from which the MEFs were derived.
(D) Expression pattern during normal transdifferentiation (right) and across genotypes on day 13 (left) of the DEGs found in 13-day Kmt2b−/− PSA-NCAM− cells.
(E) GO enrichment analysis of genes upregulated in 13-day Kmt2b−/− PSA-NCAM− cells with respect to controls.
(F) MyoD1 normalized counts in control and Kmt2b−/− PSA-NCAM− cells. Means ± SD are reported. ∗, FDR < 0.01.
Recently, Treutlein et al. (2016) integrated the single-cell transcriptomes of Ascl1-only infected MEFs throughout transdifferentiation with those of 15-day BAM-infected iNs and identified the transcriptional regulators that most likely coordinate transdifferentiation progression, defining three subnetworks (i.e., MEF, initiation and maturation). We found that, in our PSA-NCAM+ dataset, the most disrupted subnetwork by loss of KMT2B was the maturation one, confirming at the molecular level the defective iN maturation phenotype we observed (Figure 3C). Indeed, the top 30 DEGs (Figures 3A and S3C) that have annotated function(s) included four key promoters of physiological neurite extension (Ppp1r9a, Myo16, Lrfn1, and Prka1b) and nine regulators of synapse formation, maturation, and function (Clstn3, Jph4, Syp, Sult4a1, Ptprn, Cadm3, Adcy1, Grin2b, and Ache). Together, these results show that the defective iN phenotype observed upon loss of KMT2B is determined by dysregulation of the transcriptional program sustaining iN maturation.
To gain deeper insight into this phenomenon, we next compared the transcriptomes of our 13-day iNs with those of single cells undergoing MEF-to-iN transition (Treutlein et al., 2016). Strikingly, Kmt2b−/− PSA-NCAM+ iNs showed a higher correlation with the transcriptomes of earlier stages of transdifferentiation, providing the molecular basis for our observation that the few Kmt2b−/− MEFs that manage to convert to iNs fail to undergo complete neuronal maturation (Figures S3D and S3E). Furthermore, these maturation-defective iNs also present an increased correlation with myocyte gene expression, suggesting a concomitant failure to fully suppress alternative fates (Figures S3D and S3E). Thus, to confirm whether Kmt2b−/− MEFs, which transdifferentiated less efficiently than controls, switched toward the myocyte fate, we compared Kmt2b−/− and control PSA-NCAM− 13-day transcriptomes and found that upregulated genes in Kmt2b−/− PSA-NCAM− cells were significantly enriched for targets bound by ASCL1 in the first phases of transdifferentiation (Wapinski et al., 2013; Figure 3D). Although these ASCL1-bound genes have been identified in MEFs transduced either with this TF alone (3.9-fold, p ∼ 2e−7) or with all BAM factors (3-fold, p ∼ 2e−10), a large fraction is then rapidly repressed by BRN2 and MYT1L in BAM-transduced MEFs (Figure 3D), preventing the shift toward myocytes (Treutlein et al., 2016). Therefore, the enrichment for such genes in Kmt2b−/− PSA-NCAM− cells indicates that loss of KMT2B prevents their expected silencing, rerouting conversion toward alternative fates.
Consistent with this observation, the top gene ontology (GO) enrichments of upregulated genes in Kmt2b−/− PSA-NCAM− cells were all related to myocyte fate acquisition and function (Figure 3E). Moreover, among these genes, we scored a 35-fold increase in MyoD1, a fundamental driver of myogenic differentiation (Pinney et al., 1988; Figure 3F).
Because we observed a very high correlation in fold changes between the transcriptomes (correlation [cor] ∼ 0.8, p ∼ 2e−16) of PSA-NCAM+ and PSA-NCAM− fractions (Figure 4A), we hypothesized that much of the impairment in Kmt2b−/− transcriptomic resetting occurred early during MEF-to-iN transition. Thus, we analyzed the genes that changed in the same direction in both PSA-NCAM fractions and whose level of expression is regulated during transdifferentiation (Wapinski et al., 2013). Indeed, most of the genes downregulated in the absence of KMT2B in both fractions are upregulated in the first phases of transdifferentiation (Figure 4B), and among them, we found the genes of the maturation subnetwork, Zfp612, Arnt2, and Lass4. In particular, Arnt2 and Zfp612 are upregulated or remain expressed only when MEFs are transduced with either Brn2 or Myt1l or all BAM factors but not with Ascl1 alone. Hence, in the absence of KMT2B, these genes could not be activated despite the presence of Brn2 and Myt1l. Furthermore, one of the main direct targets of ASCL1, the repressor ZFP238 (Wapinski et al., 2013), was deregulated both in PSA-NCAM+ and PSA-NCAM− Kmt2b−/− fractions with respect to controls (Figure 4B), underscoring that the loss of KMT2B disrupts the dynamic interplay among BAM factors on their targets.
Figure 4.

Early Dysregulation of MEF-to-iN Conversion in the Absence of KMT2B
(A) Correlation of fold changes in Kmt2b−/− across PSA-NCAM− and PSA-NCAM+ cells. All tested genes are plotted.
(B) Heatmap of expression across normal transdifferentiation (left) and on day 13 (right) of DEGs with genome-wide significance in both cell fractions and also significantly differentially expressed during normal transdifferentiation. Z scores were calculated separately for the left and right portions of the heatmap.
Together, the combined transcriptomic analysis of PSA-NCAM+ and PSA-NCAM− cKO and control cells demonstrates that, upon transdifferentiation, Kmt2b−/− MEFs undergo an early massive dysregulation of the transcriptional program enabling conversion. This, on one side, leads to a lower transdifferentiation efficiency and a substantial shift toward a myocytic fate; on the other, it prevents the few converting Kmt2b−/− cells from activating the full neuronal maturation program.
Elucidation of the KMT2B-Vulnerable Epigenome
Next, we proceeded to analyze how the epigenome resetting underlies MEF-to-iN conversion. To define how KMT2B loss affects the balance between H3K4me3 and histone H3 lysine 27 trimethylation (H3K27me3) deposition at specific loci (Austenaa et al., 2012), we performed chromatin immunoprecipitation coupled to deep sequencing (ChIP-seq) on MEFs 2 and 7 days after 4-OHT administration. We first observed that most of the genes that were downregulated in both cell fractions (i.e., PSA-NCAM+ and PSA-NCAM−) were H3K4me3-marked in control MEFs and lost this mark already 2 days after 4-OHT administration in Kmt2b−/− MEFs (Figure 5A). The loss of KMT2B had, instead, a milder effect, and on a smaller proportion of the transcription start site (TSS), for the genes downregulated in PSA-NCAM+ cells (Figures 5A and 5B). Moreover, the decrease in H3K4me3 was accompanied by an increase in H3K27me3 at these TSS, which, already in cKO MEFs before the onset of iN conversion, had a magnitude comparable with the increase observed at 5 days of transdifferentiation (Figure 5C). This suggests that the epigenome remodeling, which rapidly follows the loss of KMT2B, disfavors MEF-to-iN conversion and, more specifically, that the initial failure of Kmt2b−/− MEFs in keeping active a specific subset of genes (Figure 5C) has a major, very early effect on transdifferentiation efficiency, whereas the effect of Kmt2b loss on neuronal maturation likely occurs at later steps.
Figure 5.

The Histone Methylation Patterns of Kmt2b−/− MEFs Prefigure Gene Repression in Untransdifferentiated cKO Cells
(A) MEF H3K4me3 coverage (in reads per kilobase million [RPKM]) in Kmt2b−/−, control, and double-mutant MEFs 2 and 7 days after the last 4-OHT administration around the TSS of DEGs in Kmt2b−/− and control transdifferentiating cells.
(B) MEF H3K4me3 RPKM around the TSS of genes downregulated in both PSA-NCAM fractions (left) and in Kmt2b−/− PSA-NCAM+ cells only (right).
(C) H3K27me3 RPKM around the TSS of genes downregulated in Kmt2b−/− PSA-NCAM− cells, in MEFs, and at 5 days of transdifferentiation.
Thus, to specifically dissect the epigenome resetting underlying iN maturation, we performed ChIP-seq on control and Kmt2b−/− MEFs on days 5 and 13 of transdifferentiation, also including the day 5 Kmt2a−/− background.
The Kmt2a−/− sample clustered together with controls in the PCA, further demonstrating the lack of effect of the absence of KMT2A on transdifferentiation (Figure 6A).
Figure 6.

H3K4me3 Remodeling along Neuronal Transdifferentiation and Differentiation
(A) PCA across the union of H3K4me3 sites in Kmt2b−/− and control transdifferentiating MEFs at 5 and 13 days (in the latter after sorting for PSA-NCAM) and in MEFs prior to transdifferentiation.
(B) Quilt of the GO enrichment analysis regarding the cellular components of the genes both differentially H3K4 trimethylated and expressed (FDR < 0.01, fold change [FC] > 0.5) in Kmt2b−/− with respect to the control.
(C) Correlation of fold change in 13-day Kmt2b−/− iNs with fold change in Kmt2b−/− hippocampal neurons. Only genes significantly dysregulated in both datasets are plotted.
(D) Distribution of fold changes in H3K4me3 in in vivo Kmt2b−/− hippocampal neurons (Kerimoglu et al., 2017) of sites that lose H3K4me3 in transdifferentiating Kmt2b−/− versus control cells.
(E) Enrichments for biological processes among genes with decereased H3K4me3 in BAM-treated KO cells but not in CaMKII neurons, against the background of all H3K4me3-marked genes.
As expected by the ChIP-seq on MEFs, for the largest majority of differentially marked regions, the loss of H3K4me3 was maintained from day 5 to day 13, whereas, for a small subset of loci, the loss was reverted on day 13 (Figure S4A). The latter were particularly enriched for genes related to the regulation of nervous system development (2.4-fold enrichment, q ∼ 0.009) and intracellular ligand-gated ion channel activity (15-fold enrichment, q ∼ 0.007), suggesting delayed compensation on a subset of KMT2B targets enabling the acquisition, albeit severely compromised, of neuronal identity.
On the other hand, we identified 112 genes that presented lower H3K4me3 in Kmt2b−/− transdifferentiating MEFs already on day 5 and that showed a further decrease in the H3K4me3 level at 13 days. Among these, 47 were also downregulated in 13-day iNs (Figure S4B), indicating that KMT2B is necessary for the maintenance of the mark at these loci and for their sustained activation.
In light of the parallel changes in H3K4me3 on day 5 and 13 revealed by the PCA, we analyzed all samples together, adjusting for the differences linked to time points. Upon loss of Kmt2b, we found 994 sites, mapping at the TSS of 545 genes, showing decreased H3K4me3 and 95 sites, mapping at the TSS of 82 genes, presenting an increased H3K4me3. Among the 82 genes, only Runx1t1 was differentially expressed in Kmt2b−/− iNs with respect to controls (Figure S4C).
We found that, of the 545 genes showing a reduced H3K4me3, 143 were significantly differentially expressed, underscoring the critical importance of KMT2B-mediated H3K4me3 deposition as a source of differential expression (overlap, p ∼ 2e−59, hypergeometric test) while extending the observation that only subsets of KMT2B-dependent H3K4me3 targets are transcriptionally vulnerable (Hu et al., 2017). Some of the most significantly enriched GO categories among these genes were related to neuronal cell components (e.g., axon), pointing to a KMT2B-mediated gene expression program whose disruption underlies defective iN specification and maturation (Figure 6B).
Next, to investigate how the role of KMT2B in transdifferentiation relates to the physiological maintenance of the neuronal fate, we compared the transcriptomic and epigenomic data of Kmt2b−/− iNs with those of in vivo post-mitotic hippocampal neurons (transcriptomes from whole hippocampi and H3K4me3 profiles from hippocampal neurons, respectively), in which Kmt2b loss was driven by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent Cre expression (Kerimoglu et al., 2017). We observed that the 116 DEGs shared between the two datasets were dysregulated in the same direction (cor p ∼ 9e−7) (Figure 6C). Indeed, of the sites that lost H3K4me3 in our BAM-treated cKO cells, about 90% were detectable in some of the in vivo samples, and of these, 80% also showed a reduction in H3K4me3 in Kmt2b−/− hippocampal neurons (Figure 6D). Thus, loss of Kmt2b affects a core set of sites that lose H3K4me3 independently of the stage of neuronal determination, pointing to a fundamental convergence between the processes of iN conversion and the maintenance of physiologically acquired neuronal fate. Instead, the genes that lost the mark only in the transdifferentiation paradigm presented an enrichment for positive regulation of both neurogenesis and cell projection organization, concordant with the specific features of our impaired conversion process (Figure 6E).
Finally, to discriminate between the direct versus indirect KMT2B targets underlying the impaired transcriptomic remodeling of Kmt2b−/− MEF-to-iN transition, we set out to profile its genome-wide occupancy. To circumvent the lack of ChIP-grade commercial antibodies against KMT2B, we immunoprecipitated MENIN, the common subunit exclusive to KMT2A and KMT2B complexes, in both Kmt2a+/+Kmt2b+/+ and Kmt2b−/− transdifferentiating MEFs on day 5. We could thus ascribe MENIN targets in control samples to both methylases, whereas we defined by subtraction, as KMT2B-specific targets, the genes that lost MENIN binding in Kmt2b−/− with respect to control cells.
Although the low enrichment of the immunoprecipitation (IP) could only afford limited sensitivity, we could nevertheless identify candidate regions with reduced MENIN enrichment, which we intersected with promoters of genes downregulated and losing H3K4me3 in Kmt2b−/− iNs with respect to controls. This revealed a core of 12 candidate direct KMT2B target genes during iN transition (Figure S5). Importantly, these candidate direct targets included, again, Arnt2, a central gene of the maturation subnetwork (Treutlein et al., 2016) that, together with Gnao1, Chst10, Rtn1, and Gpr135, is related to neuronal specification, function, and metabolism.
Kmt2b Vulnerability Uncovers Candidate Genes Associated with Dystonia
It was recently shown that KMT2B mutations can cause early-onset dystonia (Meyer et al., 2017, Zech et al., 2016), a neurological disorder whose additional genetic causes remain still largely unknown. Indeed, the DEGs in our Kmt2b−/− samples were enriched for genes whose disruption in the mouse is associated with dystonia (Atp1a3, Tubb4a, Hpca, and GnaI) or myoclonus (Npas4, Scn8a, Nhlrc1, Cit, Kcnj11, Slc7a10, Bsn, Mfsd8, Kcna1, and Plaur) (5.7-fold, p ∼ 0.0027). Thus, we exploited the 216 candidate direct KMT2B targets predicted by the intersection of DEGs (at FDR < 0.05) and genes that show H3K4me3 reduction in Kmt2b−/− over controls. In particular, we combined these targets with an analysis of genetic variations detected through high-throughput sequencing of whole exome-captured DNA from 225 dystonia patients to define candidate dystonia-relevant gene variants.
First, the analysis of the exome aggregation consortium (ExAC) population data revealed that 45 of 216 targets (21%) exhibited probability of being loss-of-function-intolerant (pLI) scores of 0.9 or higher, 54 (25%) missense Z scores of 2.0 or higher, and 26 (12%) both pLI scores of 0.9 or higher and missense Z scores of 2.0 or higher (Lek et al., 2016; Table S2). Within all targets, 34% of KMT2B target genes were evolutionary constrained against at least one class of functional variation in ExAC (Lek et al., 2016), indicating that the KMT2B-sensitive gene set is enriched for a genomic sequence in which mutational changes are scarcely tolerated. For most of the KMT2B targets (79%), we found no known disease annotation (monogenic disease) in the OMIM database (Table S2).
Under an autosomal dominant disease model, whole-exome sequencing (WES) yielded 38 unique, predicted deleterious missense variant across 32 KMT2B targets, 1 each in 38 independent probands. Of these variants, 11 affected genes with a selective constraint against missense variation (missense Z scores ≥ 2.0). Six genes contained heterozygous missense variants in more than 1 proband (ALK, DOCK10, KDR, LRP2, SLC35F1, and SLC40A1). In addition, a total of 7 unique predicted loss-of-function variants affecting 7 KMT2B targets were identified among 7 separate probands. Of these variants, 1 resided in a gene with strong loss-of-function intolerance in the general population (NOL4, pLI score of 1.0). Four variants in 3 genes emerged as especially promising candidates for follow-up evaluation (Table 1). In a female proband with childhood-onset segmental isolated dystonia and a family history suggestive of dominant inheritance (case_224), we detected a c.457C > T (p.Arg153X) stop-gain variant in the NOL4 gene whose haploinsufficiency is predicted to be non-tolerable (pLI score of 1.0) (Lek et al., 2016); two male probands with a strikingly similar presentation of early adulthood-onset tremulous cervical isolated dystonia were each identified to have a distinct missense variant of the exact same codon 51 in SLC35F1 (case_112: c.152G > T [p.Arg51Met]; case_122: c.152G > C [p.Arg51Thr]), a gene constrained against missense variants (missense Z scores of 2.01) (Figure S6B); and a female proband manifesting late adulthood-onset cervical isolated dystonia (case_088) was found to harbor a missense variant at SLC40A1 codon 157 (c.469G > C [p.Asp157His]) (Figure S6C). Along with the observation that other missense mutations at this site have been implicated in hemochromatosis type 4 (MIM606069) (Callebaut et al., 2014, Hetet et al., 2003), a multisystem iron deposition disorder, this finding led us to reverse-phenotype this proband and uncover elevated serum ferritin levels as well as bilateral signal alterations on brain MRI in a pattern consistent with brain iron deposition (data not shown). Finally, to complete a comprehensive assessment of the mutational architecture in KMT2B targets among dystonia cases, we analyzed two additional murine datasets of KMT2B-vulnerable genes from brain neurons (Kerimoglu et al., 2013) and ESCs (Denissov et al., 2014), respectively (Table S3), scoring them against the same panel of 225 exomes from dystonia patients. We found that the deletion of Kmt2b in these different cellular systems led to the identification of different targets and that the percentage of candidate dystonia-causative genes was different in the three cellular types. Specifically, the percentage of dystonia candidate genes was significantly higher in the iN versus ESC set of KMT2B targets for pLi score (Table S4), suggesting that transdifferentiation uncovers greater vulnerability in the KMT2B-dependent pathways at play in dystonia.
Table 1.
Candidate variants in 4 probands with dystonia
| Disease model | Gene | pLI score (ExAC) | missense Z score (ExAC) | Known disease annotation (OMIM) | Variation nucleotidea | Variation amino acida | Variant type | Frequency ExAC; dbSNP142 | Frequency in-house exomesb | CADD score | Carrier # | Sex | Age at onset, y | Dystonia type | Family history | Comment on putative disease relevance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Autosomal-dominant | NOL4 | 1.0 | 2.7 | no | c.457C>T | p.Arg153X | stop-gain | not found; not found | not found | 37 | case_224 | F | 4 | segmental isolated dystonia (cervical dystonia, upper limb dystonic tremor) | positive | loss-of-function variant in gene with pLI≥0.9 |
| Autosomal-dominant | SLC35F1 | 0.39 | 2.01 | no | c.152G>T | p.Arg51Met | missense | not found; not found | not found | 30 | case_112 | M | 26 | focal isolated dystonia (tremulous cervical dystonia) | negative | mutational recurrence at codon-51 in gene with missense Z score ≥2.0 |
| Autosomal-dominant | SLC35F1 | 0.39 | 2.01 | no | c.152G>C | p.Arg51Thr | missense | not found; not found | not found | 24 | case_122 | M | 33 | focal isolated dystonia (tremulous cervical dystonia | negative | mutational recurrence at codon-51 in gene with missense Z score ≥2.0 |
| Autosomal-dominant | SLC40A1 | 0.98 | 1.77 | MIM606069 | c.469G>C | p.Asp157His | missense | not found; not found | not found | 31 | case_088 | F | 44 | focal isolated dystonia (cervical dystonia) | positive | mutational recurrence at codon-157, a site previously associated with hemochromatosis 4 |
Sex: M = male, F = female. ExAC = exome aggregation consortium, Cambridge, MA (http://exac.broadinstitute.org). pLI = probability of being loss-of-function intolerant. OMIM = Online Mendelian Inheritance in Man (https://omim.org/). CADD = Combined Annotation Dependent Depletion.
numbering according to NCBI accessions NM_003787.4 and NP_003778.2 for NOL4, NM_001029858.3 and NP_001025029.2 for SLC35F1, NM_014585.5 and NP_055400.1 for SLC40A1, and NM_006931.2 and NP_008862.1 for SLC2A3.
consisting of roughly 10,000 non-dystonia control exomes.
Discussion
Here, we showed that KMT2B is required for the BAM-mediated epigenome remodeling that underlies the MEF-to-iN conversion and that the molecular unravelling of this phenotype exposes dystonia candidate genes. In particular, we demonstrated, for KMT2B, a dual role in the promotion of iN conversion through suppression of the alternative myocytic fate as well as in iN maturation. In contrast, KMT2A proved dispensable and, despite being a paralog of KMT2B, was unable to replace it in transdifferentiation. Beyond identification of the first chromatin regulator essential for MEF-to-iN conversion, our findings support the following conclusions about the specificity of direct neuronal programming.
First, the requirement for KMT2B in iN generation only partially recapitulates its function during both in vitro and in vivo neuronal differentiation. In vitro, Kmt2b−/− ESCs present an impaired differentiation to neurons (Lubitz et al., 2007), whereas the deletion of Kmt2b in excitatory forebrain terminally differentiated neurons did not impinge brain morphology macroscopically but impaired memory formation and affected H3K4me3 deposition (Kerimoglu et al., 2013, Kerimoglu et al., 2017). This notwithstanding, we found high convergence in vulnerable H3K4me3 sites upon loss of KMT2B between in vivo matured neurons and transdifferentiated iNs. Conversely, the dispensability of KMT2A in MEF transdifferentiation stands in stark contrast with its essential role during neuronal differentiation, as observed both in murine SVZ (Lim et al., 2009) and in D. rerio (Huang et al., 2015). Together, these results highlight how different origins and/or transition paths to similar developmental endpoints (namely, transdifferentiation and differentiation) expose distinct epigenetic requirements. This, in turn, has major implications for the choice of programming or reprogramming paradigms in disease modeling and regeneration, especially when they are intended to model or remedy deficits in epigenetic regulation.
Second, we previously showed that the defective response to lipopolysaccharide (LPS) in Kmt2b−/− macrophages was imputable to a single downstream gene, Pigp, which aberrantly accumulated H3K27me3 (Austenaa et al., 2012). In MEF-to-iN transition, instead, we found that loss of KMT2B leads to massive transcriptional dysregulation and H3K4me3 redistribution that affects, starting already at the MEF level, the whole program of iN fate acquisition and maturation rather than specific master regulators, pointing to distinct logics of cell fate control downstream of the same chromatin modifier.
Third, the presence of H3K4me3 at lineage-specific genes has been recently proposed as a molecular roadblock to reprogramming in the nuclear transfer (NT) paradigm (Hörmanseder et al., 2017), leading to a model in which H3K4me3-dependent memory of transcriptional status in donor cells limits the reprogramming of their developmental potential. H3K4me3 inhibition in somatic cells thus favors their reacquisition of pluripotency but disables their ability to undergo inter-lineage cell conversion. Because the H3K4me3 inhibition upon NT was carried out by overexpressing the H3K4 demethylase Kdm5b, likely its global effects are neither directly comparable with nor symmetrical to our setting, in which the bulk H3K4me3 axis (operated by KMT2E and KMT2F) was left intact. It is worth noting, however, that inhibition of H3K4 demethylase LSD1 (KDM1A) promotes TF-induced reprogramming to pluripotency (Cacchiarelli et al., 2015). Thus, this symmetrical convergence with our findings points to a fundamental distinction in how NT- versus TF-based reprogramming handles H3K4me3 dynamics.
Finally, our work innovatively exploited transdifferentiation as a tool for the identification and prioritization of candidate causative variants. Because KMT2B loss-of-function mutations lead to dystonia (Meyer et al., 2017, Zech et al., 2016), we reasoned that the KMT2B-vulnerable targets exposed through the MEF-to-iN transition could guide our search for dystonia-relevant variants and genes in whole-exome sequences of dystonia patients. Indeed, we found, in 39 KMT2B targets, 45 unique protein-impactful alterations, including mutations in 3 promising candidates: NOL4, SLC35F1, and SLC40A1. NOL3, notable for its relationship to NOL4, has been previously associated with myoclonus (Russell et al., 2012), whereas for SLC35F1, we found a mutational recurrence at the same codon in 2 probands with very similar phenotypes. Finally, for SLC40A1, we scored a mutational recurrence at a codon previously associated with hemochromatosis type 4 and, therefore, with iron deposition. The proband was found to have elevated serum ferritin and possibly iron deposits on brain MRI (bilateral signal alterations of basal ganglia), in line with signal alterations of basal ganglia described in most KMT2B-mutated patients (Meyer et al., 2017) that had not yet been associated with iron deposits, raising the intriguing possibility that the brain signal alterations seen in KMT2B probands might be due to iron abnormalities induced by SLC40A1-mediated downstream effects. Together, these data offer a compendium of promising candidates that are now opened up for further validation, following the recent trend in human genomics in which mutations originally uncovered in single individuals are eventually consolidated in the definition of syndromes (Vissers et al., 2010, Gabriele et al., 2017).
Experimental Procedures
MEF Derivation
MEFs were derived from mice with cKO for Kmt2a (Kranz et al., 2010), Kmt2b (Glaser et al., 2006), or both methylases and housed and bred in a specific pathogen-free animal house. Control MEFs were derived from Kmt2+/+YFP+Cre+ embryos and, in the case of Kmt2a, also from Kmt2afl/+YFP+Cre+ ones. See the Supplemental Experimental Procedures for further details.
Mouse Experiments
Experiments involving animals were carried out in accordance with the Italian Law (D.lgs. 26/2014) that enforces Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010, on the protection of animals used for scientific purposes. The project started before the passing of D.lgs. 26/2014 and was thus notified to the Ministry of Health before the implementation of the current legislation. As the new legislation does not apply to projects approved before its enactment, for such projects only a notification of the experiments to the Ministry of Health was required (in accordance with the D.L.vo 116/92 [and following additions], which enforced EU 86/609 Directive), and this requirement was dutifully fulfilled. Following the coming into force of Italian Law (D.lgs. 26/2014) and the expiration of the previously notified project, we submitted for the continuation of the project a new application to the Ministry of Health that granted us the authorization under Project n 385/2016.
Immunofluorescence
MEFs were seeded with different cell densities in Nunc Lab-Tek Permanox chamber slides (Sigma-Aldrich) in duplicates. 3 and 13 days after plating for transduction, cells were stained and acquired with a BX61 upright microscope equipped with a motorized stage from Olympus. The software ScanR (Olympus) was used, and the objective utilized was 20× with a 0.75 numerical aperture. See the Supplemental Experimental Procedures for further details.
Cytofluorimetric Analysis
The PSA-NCAM antibody used is listed in Table S7. 3 min before each tube was acquired, 5–10 μL of propidium iodide (PI) (Sigma-Aldrich, P4170-1G) was added. Samples were acquired at BD FacsCalibur with the BD CellQuest Pro software and analyzed with the Flowjo software. Unpaired t test was executed. See the Supplemental Experimental Procedures for further details.
FACS
FACS was executed on 4-OHT-treated MEFs left in culture 7 days before transdifferentiation. MEFs were plated on Matrigel-coated dishes both for RNA-seq and the ChIP-seq on iNs at 13 days. Cells were sorted for PSA-NCAM positivity at MoFlo Astrios (Beckman Coulter) with the software Summit v6.2. The negative PSA-NCAM fraction was kept as well.
RNA-Seq
RNA was extracted with the RNeasy micro kit (QIAGEN). RNA quality was evaluated with the Agilent 2100 Bioanalyzer. Total RNA was depleted of rRNA with Ribo-Zero, and libraries were prepared with the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina) starting from 100 ng of RNA per sample and sequenced with the Illumina HiSeq machine at a read length of 100 bp, paired ends, and a coverage of 120 million reads. See the Supplemental Experimental Procedures for further details.
ChIP-Seq
ChIP-seq for MENIN was performed on bulk 5-day transdifferentiating MEFs, ChIP-seq for H3K4me3 on MEFs 2 and 7 days after the last 4-OHT administration, on the whole-cell population at 5 days, and on PSA-NCAM+ cells at 13 days. H3K27me3 ChIP-seq was performed on MEFs 2 and 7 days after the last 4-OHT administration and on the whole-cell population at 5 days. See the Supplemental Experimental Procedures for further details.
Exome Analysis
WES and analysis of all 225 dystonia subjects was performed as described previously (Zech et al., 2016, Zech et al., 2017). Assuming that causative mutations in the proband cohort are coding alleles of strong effect, we searched the full WES dataset for missense, in-frame insertion or deletion (indel), stop-gain and stop-loss, frameshift, and splice site variations in any of the KMT2B-sensitive genes (Figure S6A). Filtered variants were considered under both an autosomal dominant (1 heterozygous variant per gene per proband) and an autosomal recessive disease model (homozygous variant or 2 heterozygous variants per gene per proband). See the Supplemental Experimental Procedures for further details.
Ethics Approval
Recruitment of dystonia patients and genetic analyses were performed after approval by the local ethics review boards at each participating center. All dystonia patients provided written informed consent.
Acknowledgments
This work was supported by European Research Council (ERC) grant DISEASEAVATARS #616441 (to G.T.), the EPIGEN Flagship Project of the Italian National Research Council (CNR) (to G.T.), ERANET-Neuron grants from the Italian Ministry of Health (FoodForThought - F4T to G.T. and AUTSYN to P.-L.G.), Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG to G.T.), Regione Lombardia (Ricerca Indipendente 2012 to G.T.), the Umberto Veronesi Foundation (to P.L.-G. and S.A.), the Italian Ministry of Health (Ricerca Corrente grant to G.T.), Fondazione Italiana per la Ricerca sul Cancro (FIRC) (to P.L.R.), and the European Research Council (AdERC #340527 to V.B.). Recruitment of dystonia patients and genetic analyses were funded by a research grant from the Else Kröner-Fresenius-Stiftung as well as in-house institutional funding from Technische Universität München (Munich, Germany), Helmholtz Zentrum München (Munich, Germany), and Medizinische Universität Innsbruck (Innsbruck, Austria) as well as Czech Science Foundation grant GACR16-13323S and Charles University (Prague, Czech Republic) project Progres Q27/LF1.
Author Contributions
G.B. carried out all experiments, including animal crossing, MEF derivation and transdifferentiation, ScanR imaging analysis, FACS acquisition and analyses, qRT-PCR, and ChIP-seq. P.-L.G. performed RNA-seq and ChIP-seq analyses. M.Z. and J.W. analyzed KMT2B targets in exomes of dystonia patients. S.A. initiated the project, set up the transdifferentiation protocol, derived some MEFs of this study, and maintained colonies before G.B.’s arrival. G.B. and P.L.R. carried out BAM vector production. A.D.-C. imported the colonies and designed primers for exon 2 copy numbers. G.B., P.L.R., and E.T. carried out RNA-seq library preparation. S.B., B.H., and R.J. recruited dystonia patients. A.F.S. shared the mice and the antibody for KMT2B. M.C. and V.B. shared the transdifferentiation protocol. G.B., P.-L.G., P.L.R., and G.T. wrote the paper. G.T. conceived, designed, and supervised the study.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and eight tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.067.
Data and Software Availability
The accession number for the sequencing data reported in this paper is GEO: GSE120441.
Supplemental Information
? symbol indicates genes with no human counterpart.
? symbol indicates genes with no human counterpart.
References
- Austenaa L., Barozzi I., Chronowska A., Termanini A., Ostuni R., Prosperini E., Stewart A.F., Testa G., Natoli G. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012;36:572–585. doi: 10.1016/j.immuni.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Cacchiarelli D., Trapnell C., Ziller M.J., Soumillon M., Cesana M., Karnik R., Donaghey J., Smith Z.D., Ratanasirintrawoot S., Zhang X. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell. 2015;162:412–424. doi: 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I., Joubrel R., Pissard S., Kannengiesser C., Gérolami V., Ged C., Cadet E., Cartault F., Ka C., Gourlaouen I. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum. Mol. Genet. 2014;23:4479–4490. doi: 10.1093/hmg/ddu160. [DOI] [PubMed] [Google Scholar]
- Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017;168:442–459.e20. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., Anastassiadis K., Stunnenberg H.G., Stewart A.F. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141:526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- Ernst P., Fisher J.K., Avery W., Wade S., Foy D., Korsmeyer S.J. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell. 2004;6:437–443. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- Gabriele M., Vulto-van Silfhout A.T., Germain P.L., Vitriolo A., Kumar R., Douglas E., Haan E., Kosaki K., Takenouchi T., Rauch A. YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. Am. J. Hum. Genet. 2017;100:907–925. doi: 10.1016/j.ajhg.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser S., Schaft J., Lubitz S., Vintersten K., van der Hoeven F., Tufteland K.R., Aasland R., Anastassiadis K., Ang S.L., Stewart A.F. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133:1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- Hetet G., Devaux I., Soufir N., Grandchamp B., Beaumont C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood. 2003;102:1904–1910. doi: 10.1182/blood-2003-02-0439. [DOI] [PubMed] [Google Scholar]
- Hörmanseder E., Simeone A., Allen G.E., Bradshaw C.R., Figlmüller M., Gurdon J., Jullien J. H3K4 Methylation-Dependent Memory of Somatic Cell Identity Inhibits Reprogramming and Development of Nuclear Transfer Embryos. Cell Stem Cell. 2017;21:135–143.e6. doi: 10.1016/j.stem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Gao X., Cao K., Morgan M.A., Mas G., Smith E.R., Volk A.G., Bartom E.T., Crispino J.D., Di Croce L., Shilatifard A. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification. Mol. Cell. 2017;65:460–475.e6. doi: 10.1016/j.molcel.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.C., Shih H.Y., Lin S.J., Chiu C.C., Ma T.L., Yeh T.H., Cheng Y.C. The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev. Neurobiol. 2015;75:452–462. doi: 10.1002/dneu.22235. [DOI] [PubMed] [Google Scholar]
- Hughes C.M., Rozenblatt-Rosen O., Milne T.A., Copeland T.D., Levine S.S., Lee J.C., Hayes D.N., Shanmugam K.S., Bhattacharjee A., Biondi C.A. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Kerimoglu C., Agis-Balboa R.C., Kranz A., Stilling R., Bahari-Javan S., Benito-Garagorri E., Halder R., Burkhardt S., Stewart A.F., Fischer A. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J. Neurosci. 2013;33:3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimoglu C., Sakib M.S., Jain G., Benito E., Burkhardt S., Capece V., Kaurani L., Halder R., Agís-Balboa R.C., Stilling R. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017;20:538–548. doi: 10.1016/j.celrep.2017.06.072. [DOI] [PubMed] [Google Scholar]
- Kranz A., Fu J., Duerschke K., Weidlich S., Naumann R., Stewart A.F., Anastassiadis K. An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis. 2010;48:512–520. doi: 10.1002/dvg.20641. [DOI] [PubMed] [Google Scholar]
- Lee S., Lee D.K., Dou Y., Lee J., Lee B., Kwak E., Kong Y.Y., Lee S.K., Roeder R.G., Lee J.W. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc. Natl. Acad. Sci. USA. 2006;103:15392–15397. doi: 10.1073/pnas.0607313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.A., Huang Y.C., Swigut T., Mirick A.L., Garcia-Verdugo J.M., Wysocka J., Ernst P., Alvarez-Buylla A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458:529–533. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubitz S., Glaser S., Schaft J., Stewart A.F., Anastassiadis K. Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol. Biol. Cell. 2007;18:2356–2366. doi: 10.1091/mbc.E06-11-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M., Kareta M.S., Chanda S., Ahlenius H., Perotti N., Zhou B., Grieder S.D., Ge X., Drake S., Euong Ang C. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature. 2017;544:245–249. doi: 10.1038/nature21722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E., Carss K.J., Rankin J., Nichols J.M., Grozeva D., Joseph A.P., Mencacci N.E., Papandreou A., Ng J., Barral S., UK10K Consortium. Deciphering Developmental Disorders Study. NIHR BioResource Rare Diseases Consortium Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 2017;49:223–237. doi: 10.1038/ng.3740. [DOI] [PubMed] [Google Scholar]
- Pinney D.F., Pearson-White S.H., Konieczny S.F., Latham K.E., Emerson C.P., Jr. Myogenic lineage determination and differentiation: evidence for a regulatory gene pathway. Cell. 1988;53:781–793. doi: 10.1016/0092-8674(88)90095-5. [DOI] [PubMed] [Google Scholar]
- Russell J.F., Steckley J.L., Coppola G., Hahn A.F., Howard M.A., Kornberg Z., Huang A., Mirsattari S.M., Merriman B., Klein E. Familial cortical myoclonus with a mutation in NOL3. Ann. Neurol. 2012;72:175–183. doi: 10.1002/ana.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom S.P., Lozano R., Lee H., Dorrani N., Mann J., O’Lague P.F., Mans N., Deignan J.L., Vilain E., Nelson S.F. De Novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med. Genet. 2014;15:49. doi: 10.1186/1471-2350-15-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa G., Schaft J., van der Hoeven F., Glaser S., Anastassiadis K., Zhang Y., Hermann T., Stremmel W., Stewart A.F. A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis. 2004;38:151–158. doi: 10.1002/gene.20012. [DOI] [PubMed] [Google Scholar]
- Treutlein B., Lee Q.Y., Camp J.G., Mall M., Koh W., Shariati S.A., Sim S., Neff N.F., Skotheim J.M., Wernig M., Quake S.R. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature. 2016;534:391–395. doi: 10.1038/nature18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Südhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Wapinski O.L., Vierbuchen T., Qu K., Lee Q.Y., Chanda S., Fuentes D.R., Giresi P.G., Ng Y.H., Marro S., Neff N.F. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell. 2013;155:621–635. doi: 10.1016/j.cell.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O.L., Lee Q.Y., Chen A.C., Li R., Corces M.R., Ang C.E., Treutlein B., Xiang C., Baubet V., Suchy F.P. Rapid Chromatin Switch in the Direct Reprogramming of Fibroblasts to Neurons. Cell Rep. 2017;20:3236–3247. doi: 10.1016/j.celrep.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech M., Boesch S., Maier E.M., Borggraefe I., Vill K., Laccone F., Pilshofer V., Ceballos-Baumann A., Alhaddad B., Berutti R. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am. J. Hum. Genet. 2016;99:1377–1387. doi: 10.1016/j.ajhg.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech M., Boesch S., Jochim A., Weber S., Meindl T., Schormair B., Wieland T., Lunetta C., Sansone V., Messner M. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Mov. Disord. 2017;32:549–559. doi: 10.1002/mds.26808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
? symbol indicates genes with no human counterpart.
? symbol indicates genes with no human counterpart.