

Abstract
The study of adult stem cell populations provides insight into the mechanisms that regulate tissue maintenance in normal physiology and many disease states. With an impressive rate of epithelial renewal driven by a pool of multipotent stem cells, the intestine is a particularly advantageous model system for the study of adult stem cells. Until recently, the isolation and in vitro study of intestinal epithelial stem cells (IESCs) was not possible due to the lack of biomarkers and culture techniques. However, advances in molecular characterization and culture of IESCs have made in vitro studies on this cell type amenable to most laboratories. The methods described in this chapter will allow the investigator to adapt newly established techniques toward downstream analysis of IESCs in vitro.
Keywords: Intestinal epithelial stem cells, Intestinal epithelial isolation, Flow cytometry/fluorescence-activated cell sorting, In vitro stemness assay, Cell surface markers, RT-PCR
1. Introduction
The intestinal epithelium is one of the most proliferative tissues in the mammalian organism, undergoing a constant rate of physiological regeneration (1). This rapid tissue renewal is driven by a pool of multipotent intestinal epithelial stem cells (IESCs) that are critical in maintaining the absorptive function of the gut as well as the protective epithelial barrier. The intestinal epithelium is arranged in a monolayer along a crypt-villus axis containing well-defined cellular lineages. The crypts of the intestinal epithelium constitute the stem cell zone and the IESCs that reside in the crypts drive proliferation up the crypt-villus axis (2). As proliferating cells move from crypt base to villus tip, they undergo further division and differentiation until they are committed to one of the major post-mitotic lineages of the intestinal epithelium: Paneth cell, enteroendocrine cell, goblet cell (all secretory lineages), or enterocyte (absorptive lineage) (2–4) (Fig. 1). Other lineages, such as tuft cells, are less thoroughly defined and may exist as subtypes of the main four post-mitotic lineages (5). Clear anatomical distinctions between proliferating and differentiated compartments, as well as unidirectional cell migration along the crypt-villus axis, make the intestinal epithelium an attractive model to study stem cell maintenance and differentiation.
Fig. 1.
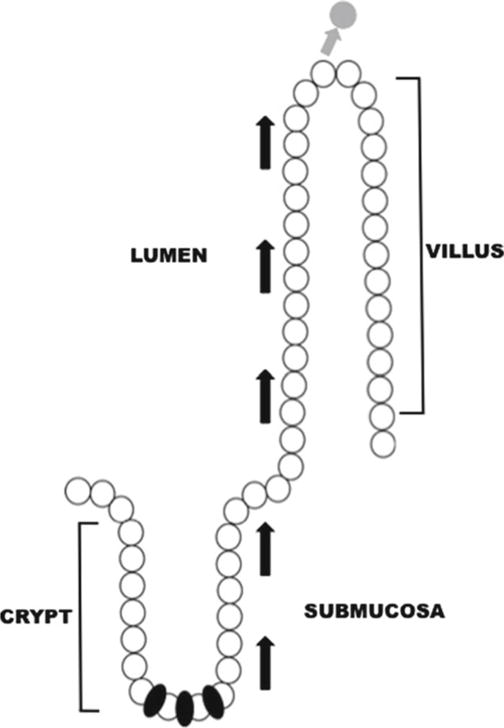
Stem cells and the intestinal crypt-villus axis. Cells in the intestinal epithelium are aligned along the crypt-villus axis. Under normal conditions, cells are polarized so that the basolateral epithelial membrane is in contact with the submucosa and the apical epithelial surface interacts with the intestinal lumen. Stem cells (indicated in black) reside at the base of the crypts and drive proliferation. The stem cell population also generates all four cellular lineages in the intestinal epithelium: enteroendocrine, Paneth, goblet, and enterocyte. Arrows indicate the direction of cell migration during proliferation and differentiation. Cells are eventually sloughed off of the villus tip into the lumen at the end of their life cycle (indicated by gray cell and arrow).
Though the anatomical division of proliferative and post-mitotic zones along the crypt-villus axis is quite striking, the crypt contains a heterogeneous cell population that presents a technical hurdle in the identification and isolation of IESCs. In addition to transit-amplifying progenitor cells, which reside toward the top of the crypts, Paneth cells at the base of the crypt are closely associated with crypt-based columnar IESCs (6, 7). The cellular heterogeneity of the crypts has driven decades of research into specific genetic markers for IESCs, which could facilitate their isolation from other crypt-based cell types. Until recently, the availability of IESC biomarkers has been extremely limited, but rapid technological advances over the past 5 years have identified a handful of biomarkers that can facilitate the isolation of IESCs in most research environments.
A landmark genetic lineage tracing study identified Lgr5 as a biomarker of stem cells in the small intestine and colonic epithelium (6). Currently, in vivo genetic lineage tracing remains the gold standard for de fining a gene as a stem cell biomarker. Briefly, transgenic mice are generated expressing tamoxifen-inducible creERT2, driven by expression of the gene of interest. These animals are then crossed to a Cre-reporter mouse, usually Rosa26-LacZ (floxed-stop), in which Cre-mediated recombination causes the irreversible expression of LacZ, allowing tissues to be analyzed for the presence of this reporter gene. Cells that are subsequently generated from the putative stem cell also express the LacZ reporter gene. Using this technique in mice where inducible Cre expression was driven by Lgr5, Barker et al. demonstrated that Lgr5 positive crypt-based columnar cells (CBC) were capable of giving rise to all post-mitotic lineages along the crypt-villus axis (6). Further studies used Lgr5EGFP reporter mice to demonstrate that “high” levels of Lgr5EGFP expression preferentially mark IESCs that are capable of forming cryptoid structures in vitro (8).
Using a novel culture system that will be discussed in more detail, the authors were able to drive single Lgr5High cells to generate the four major post-mitotic lineages of the intestinal epithelium in vitro (8). It is critical to note that (1) “high” expression of Lgr5EGFP is associated with IESCs, as Lgr5EGFP is not restricted only to CBCs but expressed at varying levels in a broad pattern throughout the crypts and (2) Lgr5Low cells do not produce cryptoids (Fig. 2a) (6, 8). The existence of a commercially available Lgr5EGFP mouse model facilitates the use of this marker for the identification and isolation of IESCs in broad research settings. A caveat, however, is that the Lgr5EGFP reporter exhibits mosaic expression of the EGFP transgene. This presents a technical hurdle in that Lgr5Negative populations cannot be used as a control group in experiments, as IESCs expressing high levels of Lgr5 protein, but not the EGFP transgene, will contaminate the Lgr5Negative population.
Fig. 2.
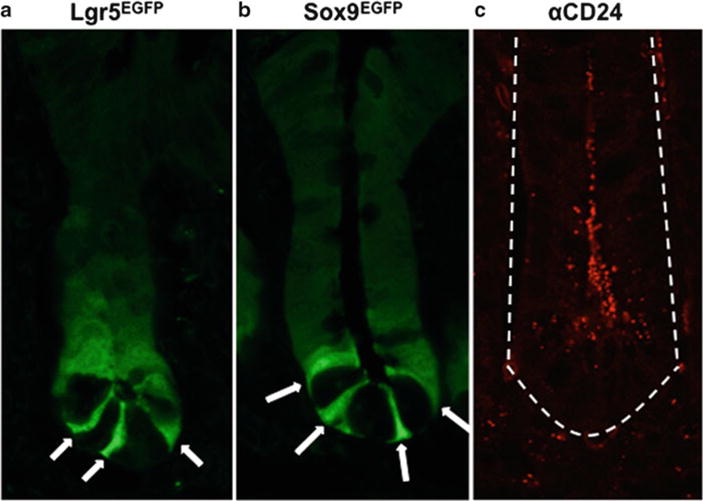
Lgr5, Sox9, and CD24 are expressed in the crypts of the small intestine. Lgr5 (a), Sox9 (b), and CD24 (c) are expressed broadly throughout the crypt base, but at varying levels. The difference in expression levels facilitates the isolation of or enrichment for IESCs. Arrows indicate Lgr5HIGH (a) and Sox9LOW (b) IESCs. Lgr5 and Sox9 expression is indicated by EGFP in commercially available transgenic mice (a, b), while CD24 expression is shown using an antibody (c). Dotted line indicates basolateral edge of crypt (c). All images are shown at ×1,260 original magnification.
Like Lgr5, Sox9 has been shown to be a marker of stem cells in the intestine, colon, pancreas, and liver by in vivo lineage tracing (9). Recent studies have demonstrated that distinct levels of Sox9 also mark IESCs. Sox9EGFP reporter mice were initially observed to demonstrate broad but variable expression of Sox9 throughout the crypt, similar to that seen in Lgr5EGFP mouse model (Fig. 2b) (10). Isolation of cell populations expressing distinct levels of Sox9EGFP by fluorescence-activated cell sorting (FACS) revealed that cells expressing “low” levels of Sox9EGFP are capable of forming cryptoid structures in the in vitro culture conditions originally described by Sato et al. (10, 11). Biochemical analysis of other Sox9EGFP populations (termed “sublow” and “high”) indicates that Sox9 also marks transit-amplifying progenitor cells (Sox9Sublow) and enteroendocrine cells (Sox9High) (10, 11). Like Lgr5, the EGFP reporter mouse for Sox9 is commercially available and readily adapted to the standard protocols discussed in this chapter.
While isolation of IESCs using Lgr5High and Sox9Low expression levels is a significant technological advance, it remains a time-consuming approach, as it requires the procurement and upkeep of transgenic reporter animals. To address this issue, a search was conducted for a cell surface antigen that could be used to enrich intestinal epithelial cell preparations for IESCs using FACS. Sox9Low expression was correlated with expression of candidate cluster-of-differentiation (CD) genes by flow cytometry to identify cell surface antigens that could facilitate IESC enrichment using commercially available antibodies. These studies demonstrated that “low” levels of CD24 could be used to enrich for IESCs from whole intestinal epithelium (11). Further characterization of the CD24Low population revealed that these cells are actively dividing and demonstrate upregulated levels of Lgr5, supporting an enrichment of IESCs (12). Additionally, when placed in culture conditions that support cryptoid development, cells from the CD24Low population demonstrate functional stem cell characteristics of multipotency and self-renewal (11, 12). CD24 also exhibits a broad expression pattern along the basolateral and apical membranes of many of the cells at the crypt base (Fig. 2c) (12). Though IESC enrichment is achievable through the use of commercially available antibodies against CD24, this approach is not as precise as the use of distinct levels of Lgr5 or Sox9 and yields a cell population with a higher degree of heterogeneity. Notably, two independent studies have shown that CD24 also labels post-mitotic Paneth cells (7, 12). However, in terms of cryptoid generation in vitro, the presence of contaminating Paneth cells in an IESC population appears to greatly increase stem cell survival and development of cryptoids (7). Paneth cells are believed to contribute to the IESC niche by providing EGF, Notch ligands, Wnt3a, and TGF-α (7).
Aside from genetic markers that facilitate the identification of IESCs, physical isolation of the intestinal epithelium from subepithelial components of the intestine (such as myofibroblasts, muscle, nerve, and lymphatic cells) is also an important technical consideration. To allow for analysis and isolation by flow cytometry and FACS, the intestinal epithelium must be dissociated to the single-cell level. However, the methods used to accomplish this level of dissociation have to be carried out without damaging critical cell surface proteins or compromising the plasma membrane. The single-cell isolation procedure is divided into two main phases, the first being to remove the semi-intact epithelial layer from the basement membrane and the second to dissociate the resultant epithelial sheets into single cells. The method described here utilizes the chelating agent ethylenediaminetetraacetic acid (EDTA) and reducing agent dithiothreitol (DTT) to release the epithelium from its subepithelial support tissues and a protease (dispase) to reduce the whole epithelium to single cells (Table 1). During an initial incubation of the whole intestinal tissue on ice, EDTA functions to sequester Ca2+ ions, inhibiting cadherins and separating the epithelium from the lamina propria (13). DTT is also added to the dissociation reagent at this time and reduces the disulfide bonds that confer viscosity to gastrointestinal mucus (14). The use of DTT as a mucolytic is critical for the easy recovery of single cells. A second incubation is then carried out at physiological temperature in EDTA, allowing for the full release of the epithelium from the lamina propria. The epithelium is recovered following mechanical disruption of the tissue by shaking, and the resultant epithelial sheets are then treated with dispase to cleave the proteins responsible for cell–cell bonds within the epithelium.
Table 1.
Dissociation reagents for intestinal epithelial prep
The development of a three-dimensional culture system supporting the growth of isolated IESCs into cryptoid structures represents a significant technological advance, as it provides a new tool for downstream analysis of IESCs. FACS can be used to sort single cells obtained through small intestinal epithelial preparations for downstream applications, including in vitro study of IESCs. The system, described in this chapter, combines murine extracellular matrix with a growth factor cocktail (Table 2) to support IESC proliferation without the presence of a supporting cell type or mesenchymal component (8). This culture system can be used as an assay for the properties of “stemness”—multipotency and self-renewal—in isolated cell populations. Multipotency is demonstrated through retrieval of developed cryptoids, which can then be sectioned and assayed by immunohistochemistry for markers of differentiated cell lineages. Retrieval, dissociation, and serial passaging of cryptoids provide evidence for self-renewal.
Table 2.
Growth factors and reagents for in vitro intestinal epithelial stem cell assay
| Reagent | Category | Function |
|---|---|---|
| Matrigel | Extracellular matrix | Structural support of IESCs through laminins (23) |
| R-spondin 1 (Rspo1) | Growth factor | Stimulates canonical Wnt signaling (24) |
| Jagged-1 peptide | Growth factor | Inhibits premature IESCs differentiation (25) |
| Noggin 1 | Growth factor | Known to cause crypt expansion in vivo (26) |
| Epidermal growth factor (EGF) | Growth factor | Stimulates intestinal growth in vivo (27) |
| Wnt3a | Growth factor | Provides mitogenic signals to IESCs (7) |
| B27 | Media supplement | Supplements for serum-free media (28) |
| N2 | Media supplement | Supplements for serum-free media (29) |
| HEPES | Media supplement | Maintains proper pH of IESC growth media |
| Y-27632 | Anoikis inhibitor | Inhibits apoptotic response to epithelial dissociation (30) |
Some caveats of the conventional IESC in vitro assay exist in the relative technical difficulty and cost of the system. Additionally, the assay is not truly clonogenic, as cell–cell interactions may occur within a single Matrigel scaffold. However, while the field of IESC biology is in need of a clonogenic assay amenable to high-throughput experiments, the conventional assay provides a test for “stemness” that avoids the cost and time commitment associated with the development of a transgenic animal for in vivo lineage tracing.
The protocols in this chapter have been standardized and are designed to walk the reader through the basic experimental design necessary to isolate single IESCs from whole murine intestine. Additionally, we outline the procedures for downstream analysis of putative IESCs and IESC-enriched populations by RT-PCR and in vitro assay for stemness. An overview of experimental work flow from intestinal epithelial isolation to downstream analysis is given in Fig. 3. Together, these protocols should facilitate the study of IESCs in a wide range of laboratories.
Fig. 3.
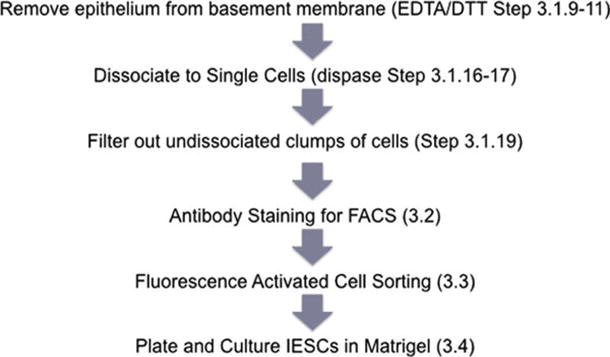
Workflow of the IESC isolation and culture.
2. Materials
2.1. Epithelial Isolation and Dissociation
Dulbecco’s Phosphate Buffered Saline (DPBS).
Hanks’ Balanced Salt Solution (HBSS).
Fetal Bovine Serum-Heat Inactivated (FBS).
Advanced Dulbecco’s Modified Eagle’s Medium/F12 (DMEM/F12, Gibco).
Dissociation reagent #1: 47 mL DPBS, 3 mL of 0.5 M (30 mM) EDTA (EDTA, Sigma), 75 μL of 1 M (1.5 mM) DTT (Sigma) (see Note 1).
Dissociation reagent #2: 47 mL DPBS, 3 mL of 0.5 M (30 mM) EDTA (see Note 1).
IESC media: DMEM/F12 supplemented with 1× N2 (invitrogen), 1× B27 w/o vitamin A (invitrogen), 10 mM HEPES, 2 mM l-glutamine, 100 μg/mL penicillin/streptomycin (see Note 2).
Dispase (Gibco).
DNAse (Roche, 10 mg/mL).
Tissue forceps.
Surgical scissors.
15-mL plastic conical tubes.
50-mL plastic conical tubes.
Water bath set to 37°C.
Petri dishes.
10-mL syringe.
40- and 70-μm filters (Becton Dickinson).
Y27632 (Sigma, 10 mM) (see Note 3).
Isoflurane.
70% ethanol.
Optional: paraformaldehyde (PFA).
2.2. Antibody Staining for Flow Cytometry or FACS
Staining media (same as IESC media listed in Subheading 2.1, see Note 2).
Polypropylene tubes (with caps if sorting, check manufacturer’s specifications to ensure the tubes are compatible with flow cytometry machines).
Optional: anti-CD45 (clone 30-F11, Biolegend) for exclusion of lymphocytes (15, 16).
Optional: anti-CD31 (clone 390, Biolegend) for exclusion of endothelial cells (17– 19).
Optional: anti-CD326 (EpCAM, clone 9C4, Biolegend) for positive selection of epithelial cells (see Note 4) (20).
Anti-CD24 (clone M1/69, Biolegend) for IESC enrichment (11, 12).
Propidium iodide (1 mg/mL).
2.3. Flow Cytometry or FACS
30-μm sterile filters to exclude large cell clumps prior to FACS.
Polypropylene collection tubes.
2.4. Matrigel Cultures
IESC media (same as listed in Subheading 2.1, see Note 2).
Growth factors (stock concentrations listed): Mouse R-spondin 1 (R&D Systems, 250 μg/mL) or Human R-spondin 1 (R&D Systems, 100 μg/mL), Noggin (PeproTech, 100 μg/mL), Epidermal Growth Factor (EGF, R&D Systems, 200 μg/mL) Jagged-1 peptide (AnaSpec 4.8 mM), and Wnt-3a (R&D Biosystems, 40 μg/mL) (see Note 5).
Matrigel, Growth Factor Reduced (BD Biosciences).
24-Well tissue culture plate (Corning). Other tissue culture plates may be incompatible with Matrigel due to the surface charge after plastic treatment. Corning’s plastic treatments are compatible with preserving the droplet of Matrigel and preventing contact with walls of the wells.
Sterile 1.7-mL snap-cap polypropylene tubes.
2.5. qRT-PCR
RT-PCR Micro Kit (The Ambion RNAqueous-Micro Kit is recommended for RNA isolation from 10 to 500,000 cells).
Taqman probes to assess the purity of sorted populations. See Table 3 (Applied Biosystems).
1.7-mL polypropylene tubes.
2× Taqman master mix (Applied Biosystems).
PCR-qualified water.
Table 3.
Gene list and corresponding Taqman probe numbers for RT-PCR analysis
| Gene | Lineage | Taqman catalog number |
|---|---|---|
| Lgr5 | Stem/progenitor | Mm00438890_m1 |
| Ascl2 | Stem | Mm01268891_g1 |
| Olfm4 | Stem | Mm01320260_m1 |
| Sox9 | Stem/progenitor | Mm00448840_m1 |
| Hes1 | Stem/progenitor | Mm00468601_m1 |
| Atoh1 | Stem/progenitor | Mm00476035_s1 |
| Neurog3 | Progenitor | Mm00437606_s1 |
| Sucrase isomaltase | Enterocyte | Mm01210305_m1 |
| Lactase | Enterocyte | Mm01285112_m1 |
| Tff | Goblet | Mm00495590_m1 |
| Mucin2 | Goblet | Mm00458299_m1 |
| Lysozyme | Paneth | Mm00727183_s1 |
| Chromogranin A | Enteroendocrine | Mm00514341_m1 |
| Substance P | Enteroendocrine | Mm00436880_m1 |
| 18s | Internal control | HS99999901_s1 |
3. Methods
3.1. Epithelial Isolation and Dissociation
- For one mouse, prepare:
- Two petri dishes containing 15 mL ice-cold DPBS.
- One 15-mL conical containing 10 mL of ice-cold dissociation reagent #1. Add Y27632 to final concentration of 10 μM just prior to use (see Note 3).
- One 15-mL conical containing 6 mL of dissociation reagent #2. Add Y27632 to final concentration of 10 μM just prior to use. Pre-warm tube at 37°C in a water bath.
Drop anesthetize mouse in isoflurane until respiration stops. Euthanize mouse by cervical dislocation.
Sterilize the ventral side of the mouse with 70% ethanol.
To allow access to the intestine, make a v-shaped incision through the peritoneal wall starting at the midline (just rostral to the genitals) to the rib cage. Cut the intestine at the pyloric sphincter. Grab the duodenal end with forceps and very carefully pull the intestine. This action will separate most of the mesentery from the intestine while preserving its integrity. Once the intestine has been mobilized, cut at the ileocecal junction. Place the intestine in the petri dish containing DPBS.
Flush fecal matter from proximal end of the intestine using a 10-mL syringe with ice-cold DPBS. A plastic micropipette tip can be cut to size to fit the syringe. The tip can then be placed directly into the lumen for flushing procedure. Remove and discard excess fat, mesentery, and pancreas from the intestinal tissue.
Optional: If downstream histological analysis of de-epithelialized tissue is required, mark the intestine by cutting a small slit at the proximal end.
Fillet open the intestine by cutting longitudinally through the lumen.
Place intestine in second petri dish containing ice-cold DPBS and lightly swirl intestine with forceps to remove any remaining fecal matter.
Place intestine in ice-cold dissociation reagent #1. Incubate on ice for 20 min.
Remove intestine from dissociation reagent #1 with forceps and place in dissociation reagent #2. Incubate at 37°C for 10 min.
Shake the tube containing intestine for 30 s to release epithelium from basement membrane (see Note 6 for important instructions on shaking procedure).
Remove remnant intestinal tissue (consisting of submucosa and muscularis). Optional: fix remnant intestinal tissue overnight in 4% PFA to assess efficiency of epithelial removal by immunostaining analysis (see Note 7).
Pellet cell solution at 1,000 × g for 5 min at 4°C.
Wash dissociated cells by removing supernatant and resuspending cells in 10 mL DPBS with 10% FBS. Optional: Remove a 25 μL aliquot of solution containing the resuspended pellet and place on a microscope slide. Intact crypts and villi and/or large sheets of intact epithelium should be observed (Fig. 4).
Pellet cell solution at 1,000 × g for 5 min at 4°C.
Remove supernatant and resuspend cells in 10 mL HBSS containing 8 mg dispase (see Note 8).
Incubate cell solution for 10 min at 37°C in a water bath. Shake conical tube with intestine vigorously every 2 min for a total of four times during incubation. Remove a 25 μL aliquot of solution containing the resuspended pellet and place on a microscope slide. The observed solution should consist primarily of single cells (Fig. 4) (see Notes 8 and 9). If cells are not sufficiently dissociated, continue incubating and shaking cell solution for up to four additional minutes as described above until solution is predominantly single cells. Do not exceed a total of 14 min at this step or significant cell lysis can occur.
Add 10% FBS and 50 μL of 10 mg/mL DNase to cell solution. Invert tube three to four times to thoroughly mix reagents.
Sequentially pass the solution through 70- and 40-μm filters to exclude large undissociated clumps of cells. Collect cell solution in a 50-mL conical tube. The filters can be gently tapped on the side of the tube to expedite filtration. Forceful pipetting of dissociated cells through the filters is not recommended.
Pellet cell solution at 1,000 × g for 5 min at 4°C.
Remove supernatant and resuspend cells in 10 mL HBSS containing 10% FBS to rinse dispase.
Pellet cell solution at 1,000 × g for 5 min at 4°C.
Remove supernatant and resuspend in 4 mL IESC media.
Fig. 4.
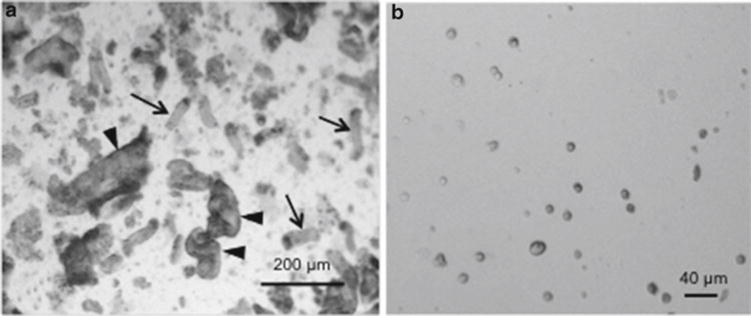
Intestinal epithelial cells after removal from basement membrane with EDTA/DTT protocol. (a) Intestinal epithelial cells in Subheading 3.1, step 14 with whole crypts intact. Arrows depict crypts while arrowheads indicate portions of villi. Images are ×40 original magnification. (b) Cell solution after dissociation to single cells in Subheading 3.1, step 17. Images are ×100 original magnification.
3.2. Antibody Staining for Flow Cytometry or FACS
The volumes below can be proportionally increased if a larger number of cells are to be analyzed or sorted.
Label polypropylene tubes and place on ice.
Dilute cells with IESC media containing Y27632 (10 μM) to obtain 5 × 106 cells in 500 μL of solution.
Add antibodies at manufacturers’ recommended concentrations to the tubes containing cell solution.
Incubate cell and antibody solution on ice for 60 min.
Add 2 mL of staining media to each tube to begin washing unbound antibody from cells.
Pellet cells at 500 × g for 5 min at 4°C.
Remove supernatant and resuspend cells in 2 mL of staining media. If dead cells are to be excluded from the flow cytometric analysis or FACS, add 2 μL of propidium iodide from a 1 mg/mL stock at least 10 min prior to analysis or sorting.
Pellet cells at 500 × g for 5 min at 4°C.
Remove supernatant and resuspend cells in 500 μL of staining media for flow cytometric analysis or FACS.
3.3. Flow Cytometry or FACS
All stages of cell preparation, antibody staining, and FACS should be incubated or stored on ice unless otherwise noted. The time between euthanasia and end of sort should be kept between 3 and 5 h. Significant increases in cell death (based on PI+ cells) and loss of RNA integrity (based on RT-PCR) is observed if isolation procedures go beyond 6 h.
Pass cell solution through a sterile 30-μm filter to prevent clogging of FACS instrument (see Note 10).
Apply cells to FACS. Use gating protocol shown on forward scatter (FCS) vs. side scatter (SSC) plot in Fig. 5a to exclude dead cells and debris. We have determined that approximately 95% of lymphocytes can be excluded by eliminating the dense population of smaller cells depicted in Fig. 5a. A more robust exclusion of non-epithelial cells can be accomplished by also excluding cells marked by CD31 (endothelial cells) and CD45 (lymphocytes).
Doublets or multimers can be excluded by gating on the diagonal pattern of events on a side-scatter-height vs. side-scatter-area bivariate histogram (Fig. 5b). If cells are properly dissociated, 50–75% of the cells should plot in a dense and narrow line of events falling diagonally along the side-scatter-height vs. side-scatter-area plot (Fig. 5b).
Collect IESCs by gating on Sox9LOW or CD24LOW populations (Fig. 5c, d). Sox9LOW cells typically comprise 0.5–1% of the single cells passing through doublet discrimination, while CD24LOW cells normally comprise 1.5–3% of the singlet fraction.
Fig. 5.
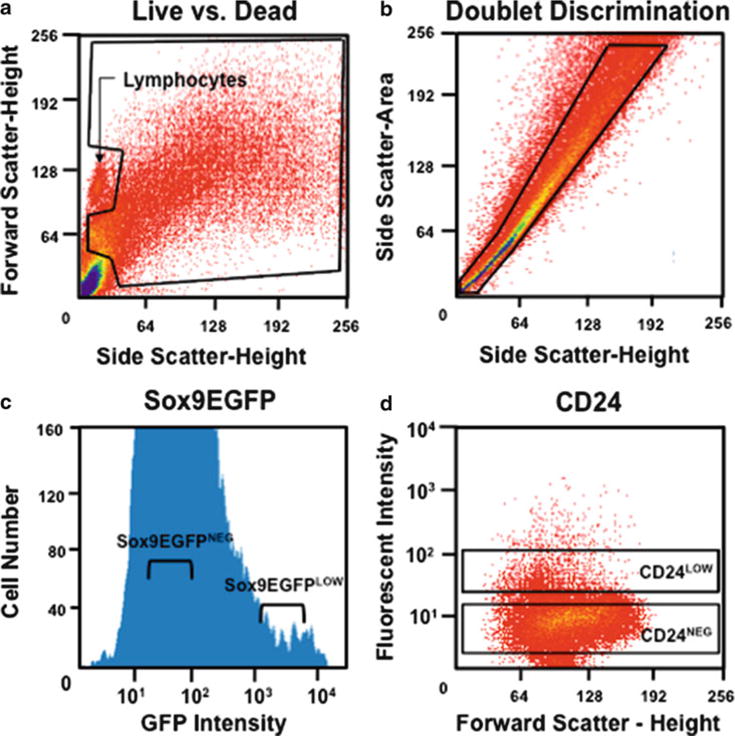
Cell sorting of IESCs using Sox9 EGFP and CD24. (a) Gating to exclude debris, lymphocytes, and dead cells. (b) Gating to include only single cells. (c) A univariate graph of Sox9 EGFP expression in cells passing through doublet discrimination. Cells within the “Sox9 EGFPLow” gate are IESCs. (d) FACS analysis of intestinal epithelial cells stained with anti-CD24 antibody. Cells within the “CD24Low” gate are enriched for IESCs.
3.4. Matrigel Cultures
Preparation: If culturing sorted cells is required, Matrigel must be thawed on ice overnight in a 4°C refrigerator or cold room. Matrigel quickly solidifies at room temperature; therefore, all tubes should be cooled on ice prior to addition of Matrigel. Pipette tips should be equilibrated to 4°C prior to use with Matrigel.
Preparation: Place culture plate in incubator to warm (see Note 11).
Pellet sorted cells at 1,000 × g for 5 min at 4°C.
While the cells are pelleting, add the following growth factors directly to the desired amount of Matrigel: Mouse or Human R-spondin 1 (1 μg/mL), Noggin (100 ng/mL), EGF (50 ng/mL), Jagged-1 peptide (1 μM), and Wnt-3a (2.5 ng/mL). Add all growth factors prior to mixing. To mix, rapidly pipette up and down 15 times paying careful attention not to introduce air bubbles into Matrigel. Store Matrigel on ice while preparing cells.
Transfer appropriate number of cells in suspension into an ice-cold 1.7-mL tube. Typical cell concentrations range from 2,000 to 20,000 cells per 50 μL of Matrigel (see Notes 12 and 13). If cell volume is less than 5% of final Matrigel volume, cells can be added directly to Matrigel. If the cell suspension volume is greater than 5% of Matrigel volume, pellet cells at 1,000 × g for 5 min, remove supernatant, and add IESC media to 5% of final Matrigel volume. Add cells to Matrigel and mix by rapidly pipetting up and down 15 times paying careful attention not to introduce air bubbles into Matrigel. This procedure is necessary to attain equal distribution of cells in the Matrigel.
Remove culture plate from the incubator. Use chilled pipette tips to plate 50 μL Matrigel droplets into the center of the well (Fig. 6). Avoid plating near the edge of well since localized plate charge will attract Matrigel to the sidewall making observation of cells under microscopy difficult. Matrigel is very fluid at this point so pay careful attention to not make rapid movements when handling the plate.
Place culture plate with Matrigel in the incubator at 37°C for 30 min to allow Matrigel to fully solidify (see Note 14).
Overlay Matrigel with 500 μL/well of IESC media containing 10 μM Y27632 (see Note 3).
Two days after plating, add the following growth factors to existing media: Mouse or Human R-spondin 1 (500 ng/mL), Noggin (100 ng/mL), and EGF (50 ng/mL). Existing media should be supplemented with growth factors on days 2, 6, 10, etc., after plating (Fig. 7a).
Four days after plating, replace IESC media (without Y27632) containing growth factors at the concentrations listed in step 9. Media containing growth factors should be changed at days 4, 8, 12, etc., after plating so that the cultures receive new growth factors (whether by supplementation (step 9) or in fresh media (step 10)) every other day and fresh IESC media every 4 days (Fig. 7a).
Monitor cryptoid formation and growth rate each day by microscopy (Fig. 7b).
Fig. 6.
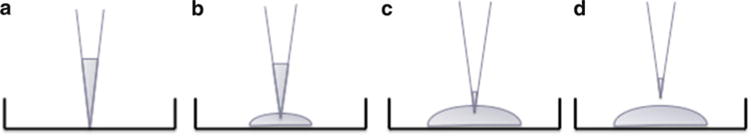
IESC plating technique in Matrigel. (a) Start with the tip of the pipette resting on the center of the well base. (b) While gently ejecting the Matrigel, slowly bring the pipette tip off the well base. (c, d) Leave a small amount of Matrigel in the pipette and remove the tip from Matrigel droplet to avoid introducing air bubbles.
Fig. 7.
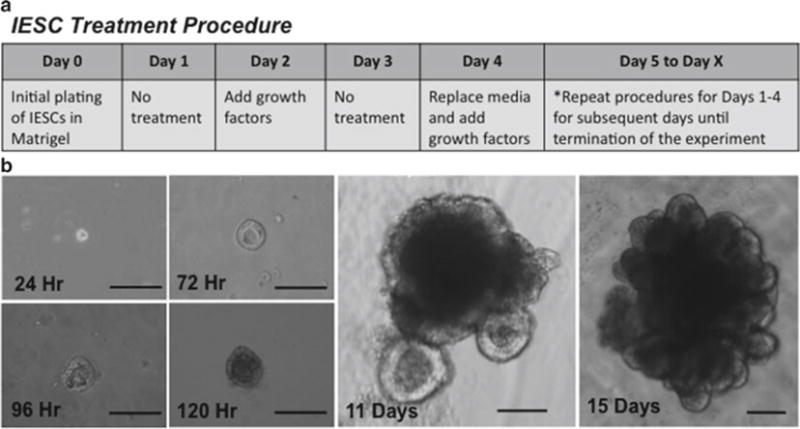
IESC treatment procedure and IESC development into Cryptoids. (a) Treatment schedule for IESC cultures. (b) Development of IESCs into cryptoids over time. Times listed are after initial plating. Note that crypt buds are well defined at day 11 and increase in number significantly by day 15. All scale bars are 100 μm.
3.5. RT-PCR Analysis
It is recommended that prior to collecting cells in RNA lysis buffer, the post-sort purity is assessed on at least 500 cells. If purity is greater than 90% based on the biomarker used for FACS, then cell populations can be further analyzed for purity using RT-PCR for lineage-specific biomarkers.
To assess purity of FACS isolated IESC populations, collect 10,000 cells of interest and control populations into separate tubes containing 500 μL of RNA lysis buffer. Control populations are de fined as (1) all cells that pass doublet discrimination (Fig. 5b) and/or (2) cells that fall into the negative population for the biomarker of interest (see Sox9 EGFP- or CD24-negative gates Fig. 5c, d).
Prepare RNA and treat with DNAse according to kit manufacturer’s protocols.
Prepare cDNA according to kit manufacturer’s protocols.
Use lineage-specific Taqman probes to assess gene expression in population of interest and compare to control populations (Table 3). Determine enrichment of IESC population by comparing non-IESC biomarker expression to progenitor/post-mitotic gene expression using the ΔΔCT method (11, 21).
4. Notes
Dissociation reagents #1 and #2 can be kept for up to 1 month at 4°C. 50 mL of dissociation reagent #1 is sufficient for epithelial isolation from 5 mice, 6–8 weeks of age. 50 mL of dissociation reagent #2 is sufficient for epithelial isolation from 8 mice, 6–8 weeks of age.
IESC media without growth factors can be stored for 1 week at 4°C.
Y27632 should only be added immediately before use.
Preliminary data demonstrate that when using EpCAM as a positive selection marker for epithelial cells, cryptoid formation decreases significantly. EpCAM should only be used if downstream applications do not include culturing of IESCs. For an alternative approach to enrich for epithelial cells, use CD31 (endothelial marker) and CD45 (lymphocyte marker) as negative selection biomarkers.
Experiments indicate that cryptoid formation efficiencies may be slightly higher with Human R-spondin than Mouse R-spondin; however, the difference between these efficiencies is negligible for most applications (unpublished data). Both Human and Mouse R-spondin should be tested for each individual application.
Holding the conical tube relatively parallel to the ground, shake the tube with enough force to provide accelerations of approximately 2.5–3.5 g. (The Apple iPhone has a built-in accelerometer that can be used to practice. The accelerometer data can be viewed in real time and uploaded using a free application for the iPhone called Context Logger which is available online). Shake the tube at 2.5–3.0 shake cycles per second for at least 30 s for a total of 80–90 shake cycles. One shake cycle is a back and forth motion. The solution should be cloudy with cells and tissue, and the remnant intestinal tissue should float toward the middle of the solution when the entire epithelium has been removed.
To quantify the amount of epithelial tissue removed from the basement membrane, the intestinal remnant tissue can be fixed for histologic analysis. After soaking the tissue in PFA overnight, transfer the remnant tissue to 30% sucrose for at least 1 and up to 3 days. “Swiss roll” the intestine and embed in OCT compound. Section tissue, stain with H&E, and image to determine the level of tissue removal.
Eight milligram of dispase per 10 mL of HBSS is the manufacturer’s recommended concentration. For a more complete dissociation to single cells, the concentration of dispase can be increased. Additionally, tissue can be transferred to 50-mL conical tube prior to shaking. The increased volume of the tube aids in increasing shear force on the cells.
Shaking should be more forceful and faster (3–3.5 shake cycles per second) than previous shaking step. Vigorous shaking does not result in decreased cell viability.
FACS instrument should be fitted with a 100-μm nozzle for epithelial cells.
A slightly warm plate allows the Matrigel to polymerize more quickly and preserves the dimensions of the Matrigel droplet.
A significant volume (~5%) of Matrigel is lost during pipetting. Prepare an excess volume of Matrigel to compensate for this loss. Use tips refrigerated at 4°C to limit volume loss and avoid premature polymerization of the Matrigel in the pipette tip. Cells should be plated in Matrigel at a range of 2,000–20,000 cells per 50 μL of Matrigel (50 μL is the amount of Matrigel used in each well of a 24-well plate. For a 96-well plate, use 5 μL of Matrigel). Cell densities outside of this range may be used, but previous experiments show the most efficient cryptoid formation within the suggested range.
The introduction of air bubbles causes the Matrigel to polymerize incorrectly and will negatively affect culture results.
Matrigel must be allowed to fully polymerize prior to the addition of the media to avoid dilution and degradation.
Acknowledgments
We would like to thank Victoria Bali, Ph.D., for critical reading of the document. Additionally, we would like to acknowledge Jill Carrington, Ph.D., who to our knowledge was the first to coin the term “cryptoid.” The original work described in this chapter was funded by the National Institutes of Health, 1-K01-DK080181-01, the American Gastroenterological Association Research Scholar Award, the North Carolina Biotechnology Center Grant, and the UNC-Chapel Hill the Center for Gastrointestinal Biology and Disease, 5P30DK034987 (S.T. Magness).
References
- 1.Wright N, Allison M. The biology of epithelial cell populations. Clarindon; Oxford: 1984. [Google Scholar]
- 2.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian theory of the origin of the four epithelial cell types. Am J Anat. 1974;141(4):537–561. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 3.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. I. Columnar cell. Am J Anat. 1974;141(4):461–479. doi: 10.1002/aja.1001410403. [DOI] [PubMed] [Google Scholar]
- 4.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. III. Entero-endocrine cells. Am J Anat. 1974;141(4):503–519. doi: 10.1002/aja.1001410405. [DOI] [PubMed] [Google Scholar]
- 5.Isomaki AM. A new cell type (tuft cell) in the gastrointestinal mucosa of the rat. A transmission and scanning electron microscopic study. Acta Pathol Microbiol Scand A suppl. 1973;240:1. [PubMed] [Google Scholar]
- 6.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 9.Furuyama K, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43(1):34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 10.Formeister EJ, et al. Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am J Physiol Gastrointest Liver Physiol. 2009;296(5):G1108–G1118. doi: 10.1152/ajpgi.00004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gracz AD, Ramalingam S, Magness ST. Sox9-expression marks a subset of CD24-expressing small intestine epithelial stem cells that form organoids in vitro. Am J Physiol Gastrointest Liver Physiol. 2010;298(5):G590–G600. doi: 10.1152/ajpgi.00470.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Furstenberg RJ, et al. Sorting mouse jejunal epithelial cells with CD24 yields a population with characteristics of intestinal stem cells. Am J Physiol Gastrointest Liver Physiol. 2010;300(3):G409–G417. doi: 10.1152/ajpgi.00453.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Obrink B. Epithelial cell adhesion molecules. Exp Cell Res. 1986;163(1):1–21. doi: 10.1016/0014-4827(86)90554-9. [DOI] [PubMed] [Google Scholar]
- 14.Holden KG, et al. Gel electrophoresis of mucous glycoproteins. II. Effect of physical deaggregation and disulfide-bond cleavage. Biochemistry. 1971;10(16):3110–3113. doi: 10.1021/bi00792a020. [DOI] [PubMed] [Google Scholar]
- 15.Trowbridge IS, Ralph P, Bevan MJ. Differences in the surface proteins of mouse B and T cells. Proc Natl Acad Sci U S A. 1975;72(1):157–161. doi: 10.1073/pnas.72.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabre JW, Williams AF. Quantitative serological analysis of a rabbit anti-rat lymphocyte serum and preliminary biochemical characterisation of the major antigen recognised. Transplantation. 1977;23(4):349–359. doi: 10.1097/00007890-197704000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Muller WA, et al. A human endothelial cell-restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J Exp Med. 1989;170(2):399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohto H, et al. A novel leukocyte differentiation antigen: two monoclonal antibodies TM2 and TM3 define a 120-kd molecule present on neutrophils, monocytes, platelets, and activated lymphoblasts. Blood. 1985;66(4):873–881. [PubMed] [Google Scholar]
- 19.Ashman LK, et al. Different epitopes of the CD31 antigen identified by monoclonal antibodies: cell type-specific patterns of expression. Tissue Antigens. 1991;38(5):199–207. doi: 10.1111/j.1399-0039.1991.tb01898.x. [DOI] [PubMed] [Google Scholar]
- 20.Bergsagel PL, et al. A murine cDNA encodes a pan-epithelial glycoprotein that is also expressed on plasma cells. J Immunol. 1992;148(2):590–596. [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffin PJ, Fogarty WM. Some properties of a protease from Bacillus polymyxa. Biochem J. 1971;125(4):109P. doi: 10.1042/bj1250109pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasaki T, et al. Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp Cell Res. 2002;275(2):185–199. doi: 10.1006/excr.2002.5499. [DOI] [PubMed] [Google Scholar]
- 24.Kim KA, et al. Mitogenic in fluence of human R-spondin1 on the intestinal epithelium. Science. 2005;309(5738):1256–1259. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 25.Li L, et al. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity. 1998;8(1):43–55. doi: 10.1016/s1074-7613(00)80457-4. [DOI] [PubMed] [Google Scholar]
- 26.Haramis AP, et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science. 2004;303(5664):1684–1686. doi: 10.1126/science.1093587. [DOI] [PubMed] [Google Scholar]
- 27.Dignass AU, Sturm A. Peptide growth factors in the intestine. Eur J Gastroenterol Hepatol. 2001;13(7):763–770. doi: 10.1097/00042737-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Brewer GJ, Cotman CW. Survival and growth of hippocampal neurons in de fined medium at low density: advantages of a sandwich culture technique or low oxygen. Brain Res. 1989;494(1):65–74. doi: 10.1016/0006-8993(89)90144-3. [DOI] [PubMed] [Google Scholar]
- 29.Johe KK, et al. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996;10(24):3129–3140. doi: 10.1101/gad.10.24.3129. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25(6):681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]