

Abstract
Mice with disruption of Lrrk1 and patients with nonfunctional mutant Lrrk1 exhibit severe osteopetrosis phenotypes because of osteoclast cytoskeletal dysfunction. To understand how Lrrk1 regulates osteoclast function by modulating cytoskeleton rearrangement, we examined the proteins that are differentially phosphorylated in wild‐type mice and Lrrk1‐deficient osteoclasts by metal affinity purification coupled liquid chromatography/mass spectrometry (LC/MS) analyses. One of the candidates that we identified by LC/MS is L‐plastin, an actin bundling protein. We found that phosphorylation of L‐plastin at serine (Ser) residues 5 was present in wild‐type osteoclasts but not in Lrrk1‐deficient cells. Western blot analyses with antibodies specific for Ser5 phosphorylated L‐plastin confirmed the reduced L‐plastin Ser5 phosphorylation in Lrrk1 knockout (KO) osteoclasts. micro computed tomography (Micro‐CT) analyses revealed that the trabecular bone volume of the distal femur was increased by 27% in the 16 to 21‐week‐old L‐plastin KO females as compared with the wild‐type control mice. The ratio of bone volume to tissue volume and connectivity density were increased by 44% and 47% (both P < 0.05), respectively, in L‐plastin KO mice. Our data suggest that targeted disruption of L‐plastin increases trabecular bone volume, and phosphorylation of Ser5 in L‐plastin in the Lrrk1 signaling pathway may in part contribute to actin assembly in mature osteoclasts.
Keywords: L‐plastin, Lrrk1, osteoclast, protein kinase, serine phosphorylation
1. INTRODUCTION
Osteoporosis is one of the most physically debilitating conditions associated with aging. There are two major known causes of osteoporosis: low peak bone mineral density, which typically appears around the age of 30, and a high bone loss rate that occurs particularly after menopause and during the natural aging process. Bone loss occurs with age in part because the rate of bone resorption (BR) surpasses the rate of bone formation (BF). Effective inhibitors of BR, such as bisphosphonates, have been widely used in the clinic to treat high turnover bone diseases. However, treatment with bisphosphonates results in suppression of BF besides BR and blunts the anabolic actions of parathyroid hormone (PTH).1 Because bisphosphonates are incorporated within the bone matrix with high affinity, long term treatment with these drugs may impair fracture healing, cause jaw osteonecrosis, and increase the risk for atypical fractures of the femur.2, 3, 4, 5, 6 Although cathepsin K inhibitor has shown promise in large animal and human clinical studies, treatment with cathepsin K inhibitor causes periosteal BF due to decreased growth hormone degradation.7 Discontinuation of cathepsin K inhibitor results in reversal of treatment effects.8 Therefore, novel antiresorptives that avoid antianabolic actions are needed to enhance endosteal BF and improve fracture repair.
In our previous studies, we have demonstrated that mice with disruption of the Lrrk1 gene displayed a severe osteopetrosis phenotype resulting from the dysfunction of mature osteoclasts (OC). OCs lacking LRRK1 failed to form peripheral sealing zones and ruffled borders on bone slices due to defects in receptor activator of nuclear factor‐ κB ligand (RANKL)‐induced cytoskeletal rearrangement.9 In contrast to bisphosphonate‐treated monocytes, precursors derived from Lrrk1 knockout (KO) mice differentiate normally into mature OCs but fail to resorb bone. While BR in KO mice is reduced dramatically, BF is not significantly affected. Lrrk1 KO mice have normal teeth, are healthy through 1 year of age, and respond to anabolic PTH treatment, but are resistant to ovariectomy‐induced bone loss.9 More recently, mutations in the Lrrk1 gene have been identified in human patients.10 The clinical features of affected patients resulting from the dysfunction of Lrrk1 in OC were very similar to the skeletal phenotypes observed in the Lrrk1 KO mice.9, 10 These studies suggest that Lrrk1 could be an antagonist drug target for treatment of osteoporosis.
Lrrk1 contains a protein kinase C (PKC)‐like serine/threonine protein kinase domain and a guanosine triphosphate (GTP) binding Roc domain to which GTP binding stimulates Lrrk1 kinase activity in vitro. Our structure‐function studies revealed that ANK, ROC, and the kinase domains of Lrrk1 were essential for the Lrrk1 mediated BR function in OCs.11 We have also demonstrated that Tyr‐527 phosphorylation of c‐Src was altered in Lrrk1 deficient OCs.9 Phosphorylation and activation of Rac1/Cdc42 were also reduced in Lrrk1 KO cells.11 Because mice with Lrrk1 disruption exhibit a more severe osteopetrosis phenotype than c‐Src KO mice and Rac1/Rac2 double KO mice, it can be predicted that Lrrk1 signaling may target other signaling molecules besides the c‐Src/Rac1/Cdc42 small GTPase signaling pathways via posttranslational modification.12 To understand how Lrrk1 regulates formation of F‐actin rings and podosomes in OCs, we have examined proteins that are differentially phosphorylated in the wild type (WT) and Lrrk1 deficient OCs by LC/MS. In this study, we show that L‐plastin is differentially phosphorylated in Lrrk1 deficient OCs and that disruption of the L‐plastin gene in mice causes an osteopetrosis phenotype.
2. MATERIALS AND METHODS
2.1. Recombinant proteins and antibodies
Recombinant macrophage colony‐stimulating factor (M‐CSF) and RANKL were ordered from R&D Systems (#3588; Minneapolis, MN). A pan antibody specific to L‐plastin was obtained from the Cell Signaling Technology (Danvers, MA) and an antibody specific to phospho‐L‐plastin at residue serine 5 was obtained from Signalway Antibody (#12455; Baltimore, MD). A polyclonal antibody against β‐actin was purchased from Sigma (#122M4782; St. Louis, MO). A High‐SelectTM TiO2 Phosphopeptide Enrichment Kit was obtained from Thermo Fisher Scientific (#A32993; Waltham, MA).
2.2. In vitro osteoclast culture and differentiation
Primary OC precursors were isolated from the spleen of 5‐week‐old Lrrk1 KO and WT mice as described previously.9 The isolated precursors were maintained in α‐MEM supplemented with 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 µg/mL), and M‐CSF (20 ng/mL) at 37°C in 5% CO2 for 3 days to stimulate monocyte proliferation. The cells were then induced to differentiate in a medium containing 20 ng/ml M‐CSF and 30 ng/mL RANKL for 5‐6 days. Differentiated OCs were retreated with fresh 20 ng/mL M‐CSF and 30 ng/mL RANKL for 30 minutes before harvesting and lysing for Western blot or mass spectrometry analyses.
2.3. Mass spectrometry (MS) and Western blot analyses
Mature OCs derived from the spleen of Lrrk1 KO and WT control mice were lysed in a denaturing lysis buffer containing 4 M guanidine hydrochloride, 50 mM Tris‐HCl (pH 8.0), 150 mM NaCl, 10 mM MgCl2, and 5 mM TCEP‐HCl for 20 minutes at room temperature followed by centrifugation at 12 000 rpm for 10 minutes. The cellular lysates were reduced, precipitated twice with four volumes of acetone, and digested with trypsin (0.1 mg/mL in 50 mM NH4HCO3) at 37°C overnight. Digested peptide lysates were desalted with a Sep‐Pac C18 cartridge and dried. The phospho‐peptides were enriched with spherical porous titanium dioxide (TiO2) resin spin tips from the High‐ SelectTM TiO2 Phospho‐peptide Enrichment Kit according to the manufacturer's instructions. The enriched phospho‐peptides were then dried, resuspended in 0.1% formic acid, and directly subjected to LS/MS at the University of California, Riverside, MS Core Facility. An aliquot of total cellular lysate (30 μg) was separated by 10% NuPAGE for Western blot analysis as described previously.11
2.4. Micro CT analyses of formalin‐fixed long bones
Dr. Sharon Celeste Morley from the Washington University School of Medicine, St. Louis, MO, USA provided 10% formalin fixed femurs from 16 to 21‐week‐old WT and L‐plastin KO female mice.13 Bone micro‐architectures were assessed by µ‐CT (Scanco Invivo CT40, Switzerland) as described previously.14, 15 The bone in PBS was scanned by X‐ray at 55 kVp volts (trabecular bone) or at 75 Kvp volts (cortical bone) at a resolution of 10.5 µm/slice. A scout view of the whole bone was used to measure the bone length and to select the sampling sites. The region of the trabecular bone was analyzed from the distal femur between 0.315 and 1.365 mm (second spongiosa) above the distal femur growth plate. For femoral cortical bone analysis, 0.525‐mm slices on each side of the center (total 1.05 mm, 100 slides) were analyzed. Parameters such as bone volume (BV; mm^3), the ratio of BV to tissue volume (BV/TV, %), trabecular number (Tb.N, mm−1), trabecular thickness (Tb.Th., mm), trabecular space (Tb.Sp., mm), and connectivity density (Conn.D., 1/mm^3) were compared between L‐plastin KO females and WT control mice. All procedures were performed with the approval of the Institutional Animal Care and Use Committee of VALLHCS and Washington University School of Medicine, St. Louis, MO, USA.
2.5. Statistical analysis
Data are presented as mean ± SEM and analyzed by the Student t test or two‐way analysis of variance as appropriate.
3. RESULTS
3.1. Phosphorylation of L‐plastin at serine (Ser) residues 5 was reduced in Lrrk1‐deficient cells
Lrrk1 consists of ankyrin repeats, leucine rich repeats, a GTPase‐like domain of Roc, a COR domain, a PKC‐like serine/threonine kinase domain, and WD40 repeats. It is believed that kinase activity is required for Lrrk1 function in OCs.11 To identify the proteins that are differentially phosphorylated in the WT and Lrrk1 deficient OCs, total cellular proteins were extracted from mature OCs derived from WT and Lrrk1 KO mice, and the trypsin‐digested peptides were then examined for the serine/threonine residues phosphorylation by LS/MS analyses. One of the identified protein was L‐plastin, which was phosphorylated at Ser5 in WT but not Lrrk1 KO OCs (Figure 1). To confirm this finding, Western blot analyses were carried out with an antibody specific for phosphoserine, Ser5 phosphorylated L‐plastin, and a pan antibody specific to total L‐plastin. Figure 2A shows Coomassie blue staining of WT and KO OC lysate to show that similar amounts of total protein had been loaded. Immunoblotting analyses with an antiphosphoserine antibody revealed reduced phosphorylated protein with a molecular weight of 65 kDa in Lrrk1 deficient OC lysates (Figure 2B). A parallel blot with anti‐Ser5‐L‐plastin antibody recognized the same 65 kDa phosphorylated protein in WT cells, and reduced phosphorylated L‐plastin in Lrrk1 KO OCs (Figure 2C). The expression level of either total L‐Plastin or β‐actin was unaffected in Lrrk1 KO OCs, as expected.
Figure 1.
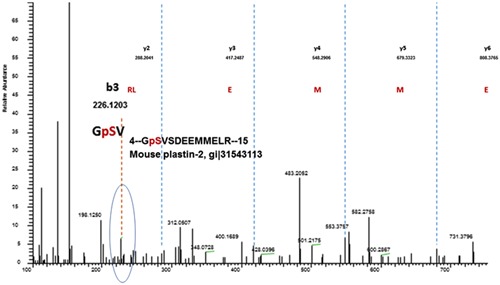
Phosphorylation of L‐plastin at serine (Ser) residues 5 was absent in Lrrk1 deficient cells. OC precursors derived from Lrrk1 knockout and control wild‐type mice were differentiated in the presence of RANKL and M‐CSF into mature OCs and lysed for protein extraction. Cellular proteins were digested with trypsin and the phospho‐peptides were enriched with spherical porous titanium dioxide (TiO2) resin spin tips for LS/MS analyses. Phosphorylation of L‐plastin, an actin bundling protein, at Ser5 was only present in WT mature OCs but not in Lrrk1 deficient cells as detected by LC/MS. LC/MS, liquid chromatography/mass spectrometry; M‐CSF, macrophage colony‐stimulating factor; OC, osteoclasts; RANKL, receptor activator of nuclear factor‐ κB ligan; WT, wild type
Figure 2.
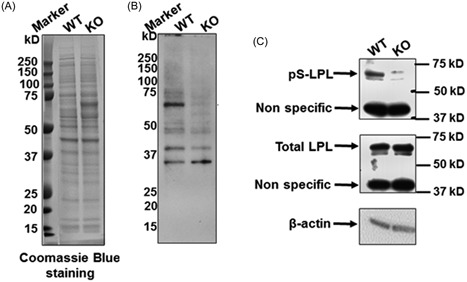
Reduced L‐plastin Ser5 phosphorylation in Lrrk1 KO OCs by Western Blot. OC precursors derived from WT and Lrrk1 KO mice were differentiated in the presence of RANKL and M‐CSF, and total cellular protein was extracted for Western blot analyses with specific antibodies. A, Coomassie blue staining of total protein from WT and KO OCs. B, Western blot detects differential levels of serine‐phosphorylated proteins between WT and Lrrk1 KO cells. C, Western blot shows that Ser5 is phosphorylated but does not show the that the level of L‐Plastin (LPL) was affected in LRRK1 KO OCs. The anti‐phospho‐L‐plastin antibody was obtained from Signalway Antibody (#12455; Baltimore, MD). KO, knockout; OC, osteoclasts; WT, wild type
3.2. L‐plastin KO mice exhibit mild osteopetrosis in long bones
To determine the role of L‐plastin in regulating bone mass, we examined the micro‐architecture of long bones derived from 16 to 21‐week‐old female mice by micro‐CT. There was no significant difference in the length of the femurs from WT and KO mice (15.45 ± 0.08 vs 15.1 ± 0.16 mm, P > 0.05). Micro‐CT analyses revealed that trabecular BV of the distal femur was increased by 27% in the 16 to 21‐week‐old L‐plastin KO females compared with the gender‐matched WT control mice (P > 0.05). However, the BV to tissue volume ratio (BV/TV) and connectivity density were significantly increased by 44% and 47%, respectively, in L‐plastin KO mice (Figure 3 A‐C & Figure 3G). There were no significant differences in the trabecular number, thickness, or separation between KO and WT femurs (Figures 3D‐F). Cortical bone phenotypes were not significantly affected by the lack of L‐plastin. The BV, TV, and BV/TV of the cortical bone samples were comparable between the femurs derived from WT and KO mice (data not shown).
Figure 3.
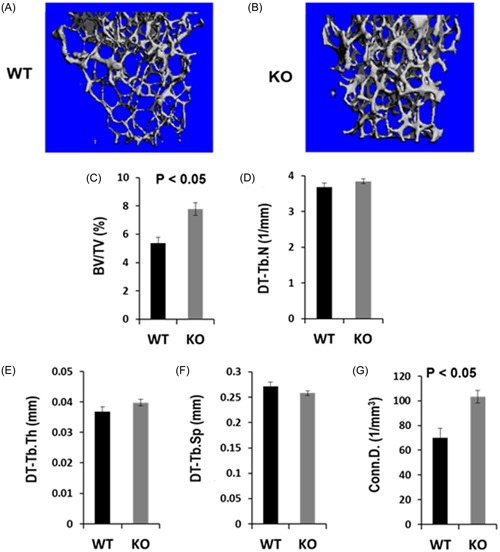
L‐Plastin KO mice exhibit mild osteopetrosis in long bones. Femurs isolated from 16 to 21‐week‐old WT and L‐plastin KO female mice were scanned by micro‐CT images. A & B, Representative micro‐CT images show an increased bone volume in the distal metaphysis of the femur in L‐plastin KO female as compared to WT control mouse. C‐H, Quantitative data of the trabecular bone parameters of the distal femoral metaphysis in WT control (N = 6) and L‐Plastin KO (N = 8) mice show increases in the ratio of BV to tissue volume (BV/TV) and connectivity density. P < 0.05 indicates statistical significance. BV, bone volume; KO, knockout; OC, osteoclasts; TV, tissue volume; WT, wild type
4. DISCUSSION
In our previous studies, we demonstrated that Lrrk1 deficient monocytes can differentiate into larger, TRAP‐positive multinuclear OCs. However, these mature OCs either showed diffused F‐actin in the cytoplasm and failed to assemble sealing rings on bone slices, or form giant flat cells on the bone surface that are not associated with resorption pits.9 The dysfunction of Lrrk1 deficient OCs contributed to the disruption of cytoskeletal rearrangement, sealing ring formation, and podosome assembly in OCs. However, the mechanisms by which Lrrk1 influenced cytoskeletal organization remained unknown. In the current study, we demonstrated, by proteomic approaches, that phosphorylation of L‐plastin at serine residues 5 was present in WT mature OCs, but was absent in differentiated Lrrk1 deficient OCs, and that deficiency of L‐plastin in female mice caused an increase in trabecular bone volume leading to mild osteopetrotic phenotypes. Our micro‐CT scanning also found that lack of L‐plastin in OCs did not change the cortical bone volume or the femur length of the long bones.
In previous studies, it was found that L‐plastin deficient OCs exhibited normal differentiation and peripheral podosomes, but these cells showed significant decreases in the formation of the adhesive contact with the bone surface.16 It has also been demonstrated that phosphorylation of L‐plastin at serine residue 5, after a variety of stimuli, leads to increased stability of actin‐based OC sealing rings, and podosomes that are critical for osteoclast adhesion, and the early process of BR.16 Our findings together with other laboratories suggest that phosphorylation of Ser5 of L‐plastin is involved in the Lrrk1 signal pathways, and may in part contribute to the disrupted cytoskeletal rearrangement in Lrrk1 deficient OCs, and the reduced BR observed Lrrk1 KO mice.
L‐plastin is phosphorylated on serine 5 and serine 7 in hematopoietic cells in vivo in response to signals triggering the activation of the immune response, cell migration, and proliferation.16, 17, 18 Although in vivo and in vitro phosphorylation of serine 5 by cyclic adenosine monophosphate (cAMP)‐dependent protein kinase A is well documented, it is still unknown which kinase(s) phosphorylate serine 7.17, 18 Lommel et al19recently provided evidence that the extracellular signal‐regulated kinase/mitogen‐activated protein kinase (ERK/MAPK) pathway was involved in L‐plastin serine 5 phosphorylation in breast cancers, with ribosomal S6 kinase 1 and 2 able to directly phosphorylate the L‐plastin serine 5 residue. Other potential candidate kinases that are involved in L‐plastin phosphorylation are PKC and PI3K.20, 21, 22 Interestingly, the N‐terminal sequence of L‐plastin proteins (RGSVS) does bear the consensus substrate motifs (R/KxS) for PKC protein kinases.23 Because Lrrk1 is an MAPKKK that contains a PKC‐like serine/threonine protein kinase domain, it is possible that Lrrk1 directly phosphorylates the serine residues of PKC substrate motifs. Although we demonstrated in this study that serine 5 phosphorylation of L‐plastin was present in WT OCs, we do not have evidence that L‐plastin is a direct biological substance of Lrrk1. Further studies are needed to determine if Lrrk1 directly or indirectly modifies L‐plastin serine residues in OCs.
We found that the magnitude of increase in trabecular bone mass was much smaller in L‐plastin KO as compared with the severe osteopetrosis phenotype observed in the Lrrk1 KO female mice of similar ages.9 We also noted that the integrin αvβ3, c‐Src, and Rac/cdc42 small GTPase proteins are key regulators of the OC cytoskeleton and activity.24, 25, 26, 27 Deletion of any of these signaling molecules compromises the capacity of OCs to remodel the cytoskeleton and resorb bone, leading to osteopetrosis phenotypes. Interestingly, mice with disruption of integrin, Src or RAC/Cdc42 proteins exhibited a less severe bone phenotype than Lrrk1 KO mice, suggesting Lrrk1 may phosphorylate multiple biological substrates and be involved in multiple signaling pathways in OCs, and L‐plastin might be one of the Lrrk1 targets in the Lrrk1 signaling pathways.
The limitations of our study are as follows. First, the role of L‐plastin downstream of Lrrk1 such as actin bundling, F‐actin ring formation, and other potential L‐plastin target membrane receptors (eg RANK and M‐CSF receptor) and/or other membrane proteins in OCs was not studied becuase it has been reported that L‐plastin phosphorylation facilitates the surface transport of activation‐induced receptors and immune synapse formation in T cells.28, 29 Second, histomorphometric analyses were not performed to confirm that reduced BR is the cause for increased trabecular bone mass in the KO mice. Third, the skeletal phenotype was evaluated only in L‐plastin KO females but not males. Further studies are needed to confirm the skeletal phenotype of L‐plastin KO in males and elucidate the molecular mechanisms by which Lrrk1 regulates L‐plastin phosphorylation.
In conclusion, we have demonstrated that targeted disruption of L‐plastin increases trabecular bone volume but had no effect on cortical bone. The magnitude of increase in trabecular bone mass, however, is much smaller in L‐plastin KO versus Lrrk1 KO mice of similar ages. Lrrk1 phosphorylation of Ser5 of L‐Plastin may in part contribute to actin assembly in mature OCs.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health grants 1R21 AR066831‐01, 1R21AR072831‐01 (to WX), and ASBMR GAP grant (to WX). Subburaman Mohan (SM) is a recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs. SCM is supported by the NIH grant R01AI104732. The Department of Veterans Affairs in Loma Linda, California provided facilities to carry out this study.
Si M, Goodluck H, Zeng C, et al. LRRK1 regulation of actin assembly in osteoclasts involves serine 5 phosphorylation of L‐plastin. J Cell Biochem. 2018;119:10351‐10357. 10.1002/jcb.27377
References
REFERENCE
- 1. Chavassieux PM, Arlot ME, Reda C, Wei L, Yates AJ, Meunier PJ. Histomorphometric assessment of the long‐term effects of alendronate on bone quality and remodeling in patients with osteoporosis. J Clin Invest. 1997;100(6):1475‐1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kidd LJ, Cowling NR, Wu ACK, Kelly WL, Forwood MR. Bisphosphonate treatment delays stress fracture remodeling in the rat ulna. J Orthop Res. 2011;29(12):1827‐1833. [DOI] [PubMed] [Google Scholar]
- 3. Nase JB, Suzuki JB. Osteonecrosis of the jaw and oral bisphosphonate treatment. J Am Dent Assoc. 2006;137(8):1115‐1119. ; quiz 69‐70 [DOI] [PubMed] [Google Scholar]
- 4. Taylor KH, Middlefell LS, Mizen KD. Osteonecrosis of the jaws induced by anti‐RANK ligand therapy. Br J Oral Maxillofac Surg. 2010;48(3):221‐223. [DOI] [PubMed] [Google Scholar]
- 5. Schilcher J, Michaëlsson K, Aspenberg P. Bisphosphonate use and atypical fractures of the femoral shaft. N Engl J Med. 2011;364(18):1728‐1737. [DOI] [PubMed] [Google Scholar]
- 6. Sellmeyer DE. Atypical fractures as a potential complication of long‐term bisphosphonate therapy. JAMA. 2010;304(13):1480‐1484. [DOI] [PubMed] [Google Scholar]
- 7. Boonen S, Rosenberg E, Claessens F, Vanderschueren D, Papapoulos S. Inhibition of cathepsin K for treatment of osteoporosis. Curr Osteoporos Rep. 2012;10(1):73‐79. [DOI] [PubMed] [Google Scholar]
- 8. Langdahl B, Binkley N, Bone H, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res. 2012;27(11):2251‐2258. [DOI] [PubMed] [Google Scholar]
- 9. Xing W, Liu J, Cheng S, Vogel P, Mohan S, Brommage R. Targeted disruption of leucine‐rich repeat kinase 1 but not leucine‐rich repeat kinase 2 in mice causes severe osteopetrosis. J Bone Miner Res. 2013;28(9):1962‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iida A, Xing W, Docx MKF, et al. Identification of biallelic LRRK1 mutations in osteosclerotic metaphyseal dysplasia and evidence for locus heterogeneity. J Med Genet. 2016;53(8):568‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeng C, Goodluck H, Qin X, Liu B, Mohan S, Xing W. Leucine rich repeat kinase 1 regulates osteoclast function by modulating RAC1/Cdc42 small GTPase phosphorylation and activation. Am J Physiol Endocrinol Metab. 2016;311:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xing WR, Goodluck H, Zeng C, Mohan S. Role and mechanism of action of leucine‐rich repeat kinase 1 in bone. Bone Res. 2017;5:17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morley SC, Wang C, Lo WL, et al. The actin‐bundling protein L‐plastin dissociates CCR7 proximal signaling from CCR7‐induced motility. J Immunol. 2010;184(7):3628‐3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xing W, Pourteymoor S, Mohan S. Ascorbic acid regulates osterix expression in osteoblasts by activation of prolyl hydroxylase and ubiquitination‐mediated proteosomal degradation pathway. Physiol Genomics. 2011;43(12):749‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xing W, Kim J, Wergedal J, Chen ST, Mohan S. Ephrin B1 regulates bone marrow stromal cell differentiation and bone formation by influencing TAZ transactivation via complex formation with NHERF1. Mol Cell Biol. 2010;30(3):711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma T, Sadashivaiah K, Chellaiah MA. Regulation of sealing ring formation by L‐plastin and cortactin in osteoclasts. J Biol Chem. 2010;285(39):29911‐29924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janji B, Giganti A, De Corte V, et al. Phosphorylation on Ser5 increases the F‐actin‐binding activity of L‐plastin and promotes its targeting to sites of actin assembly in cells. J Cell Sci. 2006;119(Pt 9):1947‐1960. [DOI] [PubMed] [Google Scholar]
- 18. Lin CS, Lau A, Lue TF. Analysis and mapping of plastin phosphorylation. DNA Cell Biol. 1998;17(12):1041‐1046. [DOI] [PubMed] [Google Scholar]
- 19. Lommel MJ, Trairatphisan P, Gäbler K, et al. L‐plastin Ser5 phosphorylation in breast cancer cells and in vitro is mediated by RSK downstream of the ERK/MAPK pathway. FASEB J. 2016;30(3):1218‐1233. [DOI] [PubMed] [Google Scholar]
- 20. Jones SL, Wang J, Turck CW, Brown EJ. A role for the actin‐bundling protein L‐plastin in the regulation of leukocyte integrin function. Proc Natl Acad Sci U S A. 1998;95(16):9331‐9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paclet MH, Davis C, Kotsonis P, Godovac‐Zimmermann J, Segal AW, Dekker LV. N‐Formyl peptide receptor subtypes in human neutrophils activate L‐plastin phosphorylation through different signal transduction intermediates. Biochem J. 2004;377(Pt 2):469‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pazdrak K, Young TW, Straub C, Stafford S, Kurosky A. Priming of eosinophils by GM‐CSF is mediated by protein kinase CbetaII‐phosphorylated L‐plastin. J Immunol. 2011;186(11):6485‐6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang JH, Toita R, Kim CW, Katayama Y. Protein kinase C (PKC) isozyme‐specific substrates and their design. Biotech Adv. 2012;30(6):1662‐1672. [DOI] [PubMed] [Google Scholar]
- 24. McHugh KP, Hodivala‐Dilke K, Zheng MH, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105(4):433‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c‐src proto‐oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693‐702. [DOI] [PubMed] [Google Scholar]
- 26. Croke M, Ross FP, Korhonen M, Williams DA, Zou W, Teitelbaum SL. Rac deletion in osteoclasts causes severe osteopetrosis. J Cell Sci. 2011;124(Pt 22):3811‐3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu M, Sun B, Saar K, et al. Deletion of Rac in mature osteoclasts causes osteopetrosis, an age‐dependent change in osteoclast number and a reduced number of osteoblasts in vivo. J Bone Miner Res. 2016;31(4):864‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wabnitz GH, Köcher T, Lohneis P, et al. Costimulation induced phosphorylation of L‐plastin facilitates surface transport of the T cell activation molecules CD69 and CD25. Eur J Immunol. 2007;37(3):649‐662. [DOI] [PubMed] [Google Scholar]
- 29. Wabnitz GH, Michalke F, Stober C, et al. L‐plastin phosphorylation: a novel target for the immunosuppressive drug dexamethasone in primary human T cells. Eur J Immunol. 2011;41(11):3157‐3169. [DOI] [PubMed] [Google Scholar]