

Abstract
We report a first-in-patient study of vamorolone, a first-in-class dissociative steroidal anti-inflammatory drug, in Duchenne muscular dystrophy. This 2-week, open-label Phase IIa multiple ascending dose study (0.25, 0.75, 2.0, and 6.0 mg/kg/day) enrolled 48 boys with Duchenne muscular dystrophy (4 to <7 years), with outcomes including clinical safety, pharmacokinetics and pharmacodynamic biomarkers. The study design included pharmacodynamic biomarkers in three contexts of use: 1. Secondary outcomes for pharmacodynamic safety (insulin resistance, adrenal suppression, bone turnover); 2. Exploratory outcomes for drug mechanism of action; 3. Exploratory outcomes for expanded pharmacodynamic safety. Vamorolone was safe and well-tolerated through the highest dose tested (6.0 mg/kg/day) and pharmacokinetics of vamorolone were similar to prednisolone. Using pharmacodynamic biomarkers, the study demonstrated improved safety of vamorolone versus glucocorticoids as shown by reduction of insulin resistance, beneficial changes in bone turnover (loss of increased bone resorption and decreased bone formation only at the highest dose level), and a reduction in adrenal suppression. Exploratory biomarkers of pharmacodynamic efficacy showed an anti-inflammatory mechanism of action and a beneficial effect on plasma membrane stability, as demonstrated by a dose-responsive decrease in serum creatine kinase activity. With an array of pre-selected biomarkers in multiple contexts of use, we demonstrate the development of the first dissociative steroid that preserves anti-inflammatory efficacy and decreases steroid-associated safety concerns. Ongoing extension studies offer the potential to bridge exploratory efficacy biomarkers to clinical outcomes.
Graphical abstract
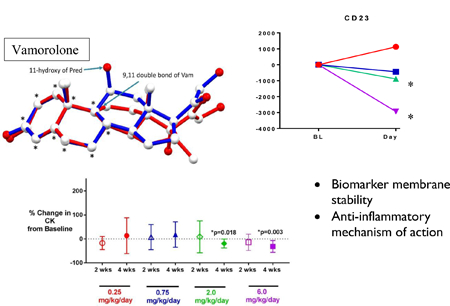
1. Introduction
Duchenne muscular dystrophy (DMD) is a progressive muscle disease caused by the genetic loss of dystrophin protein at the myofiber plasma membrane. The resulting membrane instability and chronic bouts of myofiber degeneration and regeneration trigger pathological activation of nuclear factor of kappa B (NF-κB) inflammatory pathways. Pathological increases in NF-κB activation is detected in dystrophin-deficient muscle at birth, years before obvious clinical symptoms of DMD, and this chronic inflammatory state most likely exacerbates membrane damage and leads to disease progression (1, 2). In the majority of patients with DMD, treatment with glucocorticoid anti-inflammatory drugs (i.e. prednisone, deflazacort) delays disease progression and is standard of care (3, 4). However, despite their clinical benefits, glucocorticoids have serious adverse effects that diminish patient quality of life, including increased bone fragility and frequent vertebral fractures, stunting of growth, delayed puberty, metabolic disturbance and weight gain, adrenal suppression, adverse effects on mood and behavior, and others (5). The concerns of parents and physicians regarding adverse effects lead to variable use of glucocorticoids in the management of patients with DMD, such that about half of patients in the United States and Europe do not receive this standard of care (6, 7).
Vamorolone (formerly VBP15) is a first-in-class steroidal drug that shares a high degree of chemical and 3-dimensional structural homology to glucocorticoids, and also binds the glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) with similar affinity (8–11). Whereas glucocorticoid/GR complexes dimerize and bind to the glucocorticoid response element (GRE), vamorolone/GR complexes do not dimerize and appear to confer decreased gene transcriptional activity of vamorolone compared to glucocorticoids (10). This leads to retention of drug activities associated with receptor/ligand monomers, but loss of drug activities associated with receptor/ligand dimers. Receptor/ligand monomers of the GR (gene NC3R1) have recently been shown to mediate the anti-inflammatory effects of glucocorticoids by blocking access of NF-κB transcriptional complexes to the promoter elements of pro-inflammatory genes (12). On the other hand, the pervasive gene transcriptional activity of receptor/ligand dimeric complexes appear increasingly responsible for the severe side effect profiles of glucocorticoid drugs, and these adverse effects are highly attenuated with vamorolone in animal models and Phase I human clinical studies (9, 10, 13–15). The dissociation of monomeric vs. dimeric vamorolone/ GR complexes is predicted to retain efficacy and reduce safety concerns, hence, a dissociative steroidal drug. If vamorolone is shown to be safe and effective, it could replace glucocorticoids in the treatment of many conditions where chronic use leads to debilitating side effects, such as in DMD.
As the primary biochemical defect in DMD is loss of the dystrophin protein at the myofiber cell membrane, and subsequent susceptibility to physical membrane damage, agents able to impart greater physical stability to the myofiber plasma membrane may ameliorate this primary cellular defect. The very high levels of serum creatine kinase (CK) observed in patients with DMD, beginning at birth and extending throughout life with progressive attenuation due to progressive muscle atrophy, reflect the loss of muscle fiber membrane integrity due to dystrophin deficiency. Glucocorticoids destabilize muscle fiber membranes; serum CK levels increase rapidly with intravenous and subcutaneous administration of glucocorticoids in patients with muscular dystrophy (16, 17). In addition to inhibiting NF-ĸB, vamorolone was developed to have a superior membrane-stabilizing activity compared to prednisolone. In preclinical studies, vamorolone demonstrated potent membrane stabilizing properties contrasting with membrane de-stabilizing properties of prednisolone (10, 16).
This Phase IIa trial of vamorolone (VBP15–002) was designed as a 2-week open-label, sequential multiple ascending dose trial with a 2-week washout period conducted in ambulant boys with DMD (ages 4 to <7 years), naïve to previous treatment with glucocorticoids.Well-established pharmacodynamic biomarkers predictive of later clinical safety concerns of glucocorticoids were used as secondary outcome measures. These clinically-bridged biomarkers are insulin resistance (fasting glucose and insulin), adrenal suppression (first-in-morning cortisol), and changes in bone turnover (osteocalcin and procollagen type 1 pro-peptide [P1NP] [bone formation], and -terminal telopeptide [CTX] [bone resorption]). As these biomarkers are known to be acutely responsive to glucocorticoids, we could accurately measure them in the 2-week treatment period and determine dose-responsiveness in vamorolone-treated DMD boys (18, 19). We also included exploratory pharmacodynamic biomarkers that were previously defined as responsive to glucocorticoids. These included 7 pro-inflammatory serum proteins which previously showed consistent suppression after glucocorticoid treatment in pediatric pro-inflammatory disease (DMD and inflammatory bowel disease [IBD]), and 6 serum biomarkers significantly increased by glucocorticoids, and were thus postulated as markers for additional safety concerns (20, 21).
Materials and Methods
2.1. Identity of Investigational Medication
ReveraGen BioPharma, Inc., through a designated central pharmacy (Almac Clinical Services, Durham. NC; Craigavon, UK), provided vamorolone in bulk to the registered pharmacist or designated staff at each study site. Vamorolone active pharmaceutical ingredient was manufactured by Olon Ricerca Biosciences (Concord, OH). Vamrolone drug product was manufactured by Velesco Pharmaceutical Service (Kalamazoo, MI) and supplied as a cherry-flavored suspension (4% by weight). Bulk vamorolone supply was stored at refrigerated temperature (2–8°C; 36–46°F). Excursions to ambient temperature were allowed.
2.2. Laboratories
This study utilized a central clinical laboratory (ACM Medical Laboratory, Inc., Rochester, NY; York, UK; Singapore). In addition to clinical safety parameters, this central laboratory also performed analysis on the following biomarkers: osteocalcin, P1NP, CTX, fasting glucose, fasting insulin. Analysis of morning cortisol was performed by PRA Health Sciences (Groningen, the Netherlands) via immunoassay. Analysis of glutamate dehydrogenase (GLDH) was performed by Randox Laboratories Ltd. (Antrim Co. Antrim, UK) via immunoassay. Exploratory biomarker analyses were performed by SomaLogic Inc. (Boulder, CO) via their proprietary SOMAscan™ aptamer profiling assay.
2.3. Study Design and Conduct
The trial design was a multi-center, open-label, sequential multiple ascending dose study evaluating the safety, PK, PD, and exploratory efficacy following oral administration of vamorolone 0.25 , 0.75 , 2.0, and 6.0 mg/kg/day for two weeks, with two-week follow-up off drug, in boys ages 4-<7 years of age with DMD. The study was approved by the Institutional Review Board at each participating site. The legal guardian for each study participant signed informed consent prior to any study procedures. The primary objective of this study was to evaluate the safety of multiple ascending oral doses of vamorolone in ambulant boys ages 4-< 7 years of age with DMD. The secondary objectives were to investigate the single-dose and multiple-dose PK of oral vamorolone at multiple dose levels, and to investigate the effects of single and multiple oral doses of vamorolone on serum PD biomarkers. Secondary objectives included evaluation of biomarkers bridged to clinical outcomes. The exploratory objectives were to evaluate biomarkers of safety and efficacy that were not yet bridged to clinical outcomes. Main criteria for inclusion were male subjects, 4 - <7 years of age at study entry, diagnosed with DMD by confirmed dystrophin deficiency, who were able to complete the Time to Stand Test without assistance at the study Screening and Baseline Visits. The study was conducted by the Cooperative International Neuromuscular Research Group (CINRG) at 11 United States (US) and non-US study sites. Twelve subjects were assigned to each of the 4 dose cohorts and received an oral dose of vamorolone once daily with 8 ounces of whole milk (or equivalent high fat food portion). Following completion of this study, participants had the option to enroll in a 6-month open label extension study (VBP15–003), and then a 24-month, open-label long-term extension study (VBP15-LTE).
2.4. Sample Size
A sample size of 12 subjects in each cohort was determined to be sufficient, with the following considerations: a sample size of 12 subjects is large enough to have a confidence interval of no wider than 50% for the proportion of adverse events assessed or any other event outcome. For continuous outcomes such as safety laboratory markers or PD biomarkers, a two-sided 95.0% confidence interval for the mean will be no wider than 0.8 standard deviations (SD) from the observed mean, with 90.0% coverage probability, based on the t statistic; thus, the total confidence interval will be approximately 1.6 SDs wide for any continuous parameter if its underlying distribution is approximately normal.
2.5. Analysis Populations
Two analysis populations were defined for this study. The Safety Population consisted of all subjects who received at least one dose of study medication. The Safety Population was used for safety and PD analyses. The PK Population consisted of all subjects who received at least one dose of study medication and had sufficient quantifiable plasma concentration data for PK analysis.
2.6. Safety
Safety was assessed by clinical laboratory analyses, treatment, and post-treatment; physical examinations with weight, vital sign measurements, and 12-lead ECG were performed during the pre-treatment, treatment, and post-treatment periods. Adverse events (AEs) were monitored throughout the course of the study. The liver-selective enzymes glutamate dehydrogenase (GLDH) and gamma-glutamyl transferase (GGT) were studied as exploratory biomarkers in VBP15–002 to monitor potential vamorolone-induced liver injury (22–24).
2.7. Pharmacokinetic Methods
Analysis of plasma concentrations of vamorolone was performed using a validated LC-MS/MS method (PRA Health Sciences, Lenexa, KS). All PK parameters were calculated using non-compartmental analysis (NCA). For the pharmacokinetic analysis, negative plasma concentrations that occurred at pre-dose time (0 hr) were treated as zero (0). All other concentrations were included in the PK analysis. Nominal/Relative times were identical for all Data used in this study. The NCA was performed using Phoenix® WinNonlin® (Certara). All concentration-time graphs were prepared using Matlab® R2016b for Windows® . The maximum plasma concentration (Cmax) and time to Cmax (tmax) were taken directly from the data. For the calculation of the AUClast and AUCinf, the option linear up-log down trapezoidal method was chosen with uniform weighting of the data. The elimination rate constant, kel, was calculated as the negative of the slope of the terminal log-linear segment of the plasma concentration-time curve. For the cases where the subjects showed a clear washout phase (by visual inspection), at least 3 terminal plasma concentration time points were selected for determination of kel. For subjects not showing such clear elimination, kel was assumed to be equal to the average kel of the rest of the subjects who received the same dose. The AUCinf was calculated as: AUCinf= AUClast + Clast/kel.where Clast is the final concentration. Elimination half-life (t½) was calculated according to: t1/2 = ln (2)/kel. Clearance (CL), uncorrected for bioavailability, was calculated as CL = Dose/ AUCinf.
2.8. Pharmacokinetic Analysis
The primary objectives of the PK analysis were:1) to investigate the first-and multiple-dose PK of oral vamorolone, and 2) to explore linearity of the different doses administered in boys ages 4 to <7 years with DMD. The secondary objective of the PK analysis was to compare the PK of vamorolone in boys with DMD and healthy adult male subjects who participated in the Phase I Randomized, Placebo-Controlled, Double-Blind, Single Ascending Dose and Multiple Ascending Dose Study (9). All subjects who received at least one dose of vamorolone study medication and had sufficient data for PK analysis were included in the PK Analysis Population. Twelve subjects were assigned to each of the 4 cohorts and received an oral dose of vamorolone Once daily with 8 ounces of whole milk (or equivalent high fat food portion). In the Phase VBP15–001 study in healthy adult volunteers, vamorolone showed enhanced bioavailability with a fat meal, as has also been observed with other hydrophobic steroidal drugs (9). Blood sample times for the determination of vamorolone plasma concentrations were as follows: Day 1:0 (pre-dose), 1, 2, 4, 6, 8 hours; day 14:0 (pre-dose), 1, 2, 4, 6, 8 hours. Forty-seven subjects had evaluable PK data and were included in the PK Analysis Population.
2.9. Pharmacodynamics
To address the safety concerns associated with glucocorticoids, pharmacodynamic (PD) biomarkers that are bridged to later clinical safety concerns of glucocorticoids were studied as secondary outcomes. These comprised PD biomarkers of insulin resistance (fasting glucose, fasting insulin), low bone mineral density (PD biomarkers of bone turnover [serum osteocalcin, P1NP, and CTX]), and adrenal axis suppression (first-in-morning cortisol). Serum osteocalcin, P1NP, and NTX (N-terminal telopeptide, a comparable bone resorption biomarker) were previously shown to respond to glucocorticoid treatment within the 2-week treatment time frame that was incorporated in the VBP15–002 study (18, 19).
Fasting serum glucose and insulin were measured at Baseline (0.5 hour pre-dose Day 1) and Week 2 (0.5 hour pre-dose Day 14). Insulin was measured quantitatively using the ADVIA Centaur® Insulin assay, a two-site sandwich immunoassay using direct chemiluminescent technology (Siemens Healthcare Diagnostics, Tarrytown, NY USA). Osteocalcin, P1NP, and CTX, were measured at the screening visit (blood drawn in the afternoon) and Week 2 (blood drawn 6 hours post morning dose); these times were chosen to minimize confounding diurnal variations. Osteocalcin was quantified using LIASON® Osteocalcin, a chemiluminescence immunoassay (DiaSorin, Stillwater, MN). CTX was quantified by the β-CrossLaps electrochemiluminescence immunoassay (Roche Diagnostics, Indianapolis, IN). P1NP was quantified using the Total P1NP electrochemiluminescence assay (Roche Diagnostics, Indianapolis, IN). All tests were designed for quantitative determination in human serum.
Glucocorticoids impact the hypothalamic-pituitary-adrenal (HPA) axis through negative feedback loops, and pharmacological glucocorticoids cause transient but significant reductions in the secretion of cortisol by the adrenal cortex within hours of a single, oral dose (18). The measurement and interpretation of adrenal hormones is also affected by circadian variation with a peak serum cortisol level pre-awakening followed by a rapid fall in serum cortisol post-awakening, hence the importance of timing of blood collection relative to time of day.Prolonged dosing of patients with pharmacological glucocorticoids leads to adrenal suppression - which is caused by chronic suppression of the HPA axis- and, in turn, a chronic decrease in endogenous cortisol production (25, 26). First-in-morning cortisol can be used as a screening tool for chronic adrenal suppression, and in patients with normal circadian regulation, first-in-morning serum cortisol levels <3.6 μg/dL (100 nmol/L) suggest chronic adrenal suppression (27, 28). First-in-morning blood cortisol levels were obtained ~24 hours after the previous dose of vamorolone at the 2-week visit. Cortisol was measured using the ADVIA Centaur® cortisol immunoassay, designed for quantitative determination in serum (Siemens Healthcare Diagnostics, Tarrytown, NY USA).
Serum creatine kinase (CK), an abundant enzyme in muscle fibers, was pre-specified as an exploratory pharmacodynamic efficacy biomarker in the Phase IIa protocol. Serum CK is elevated in the setting of muscle membrane damage caused by muscular dystrophy. To test if vamorolone improves membrane stabilization, serum CK activity was measured at Baseline, after two weeks of treatment with vamorolone, and after two weeks of washout following the last dose of vamorolone. The CK activity was measured by ADVIA® Chemistry Systems (Siemens Healthcare Diagnostics, Tarrytown, NY USA), an assay developed for human serum/plasma.
2.10. SOMAscan Assays
Serum from a subset of 39 subjects was accessioned for SOMAscan™ aptamer profiling. The number of subjects evaluated at each dose level was 7 at 6.0 mg/kg, 10 at 2.0 mg/kg, 11 at 0.75 mg/kg and 11 at 0.25 mg/kg. Number of subject per dose level varied because Somalogic™ ended performance of contractual assays, and therefore only samples accessed by December 1, 2017 were available for testing. Two serum samples were tested for each subject; Baseline (pre-treatment), and after two weeks of daily treatment with vamorolone at the indicated dose level. The Baseline sample was obtained during a Screening Visit prior to Study Day −1, in the afternoon. The sample following two weeks of treatment was obtained 6 hours after the morning dose. The SOMAscan™ assay measures 1,310 serum proteins, over a 108 dynamic range, using modified nucleic acid probes that are specific and sensitive for each of the proteins (aptamers). The aptamers generally bind to a larger epitope than do antibodies, and thus can be both more specific and also identify three dimensional structures (e.g. multimeric protein complexes). Because the detection reagents are nucleic acids, the method enables amplification of signals using PCR, and addressing of detection aptamer reagents using sequence tags. Serial dilutions of 70 µL of sera are used to assay each protein in the center of its standard curve. Data for each subject studied was received for all 1,310 proteins from SomaLogic™ with global normalization of all relative protein unit values. A data filter was then applied so that only data from the pre-specified 7 efficacy and 6 safety biomarkers were analyzed (Figure 2, Figure 3).
Figure 2: Line graphs of exploratory safety biomarkers demonstrating absolute change in Relative Fluorescence Units (RFU) from Baseline to Day 14 in VBP15–002.
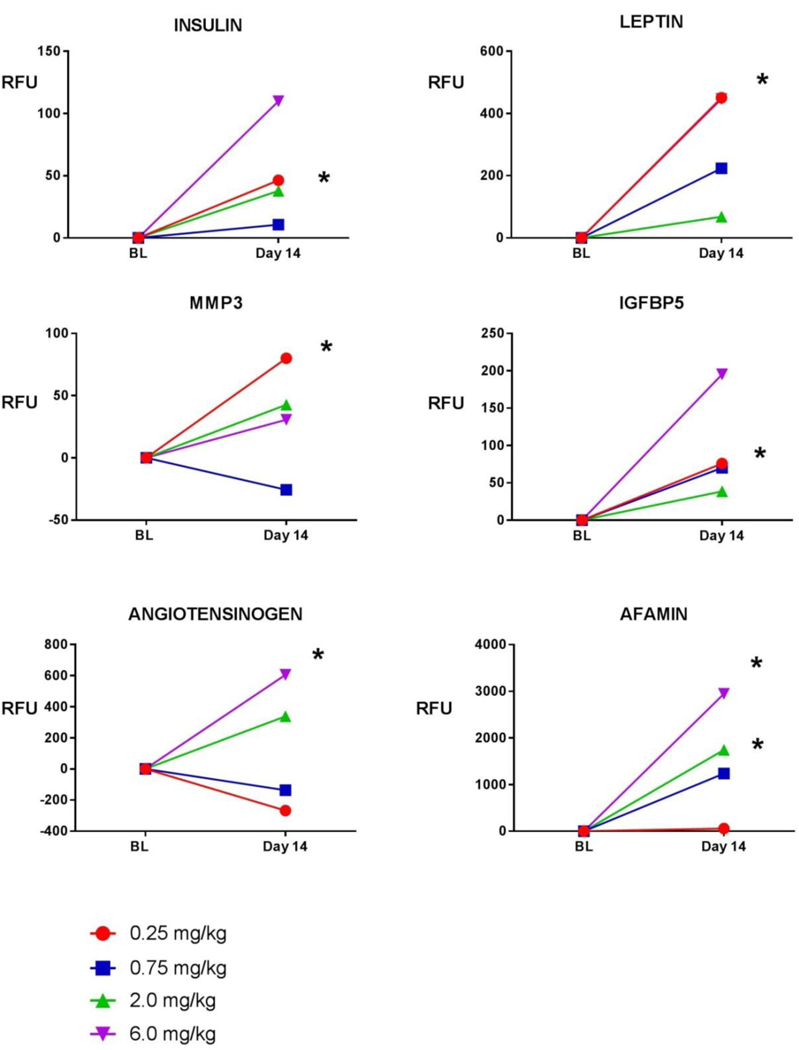
Shown are 4 dose levels. Baseline values were compared with Day 14 values using a two-tailed paired t-test. Asterisks (*) indicate significant increases from baseline (pre-dose) to following 2 weeks of treatment (post-dose) for a given dose level (p<0.05). Abbreviations: BL= baseline; RFU = Relative Fluorescence Units; MMP3 = matrix metalloproteinase 3; IGFBP5 = insulin-like growth factor binding protein-5.
Figure 3:
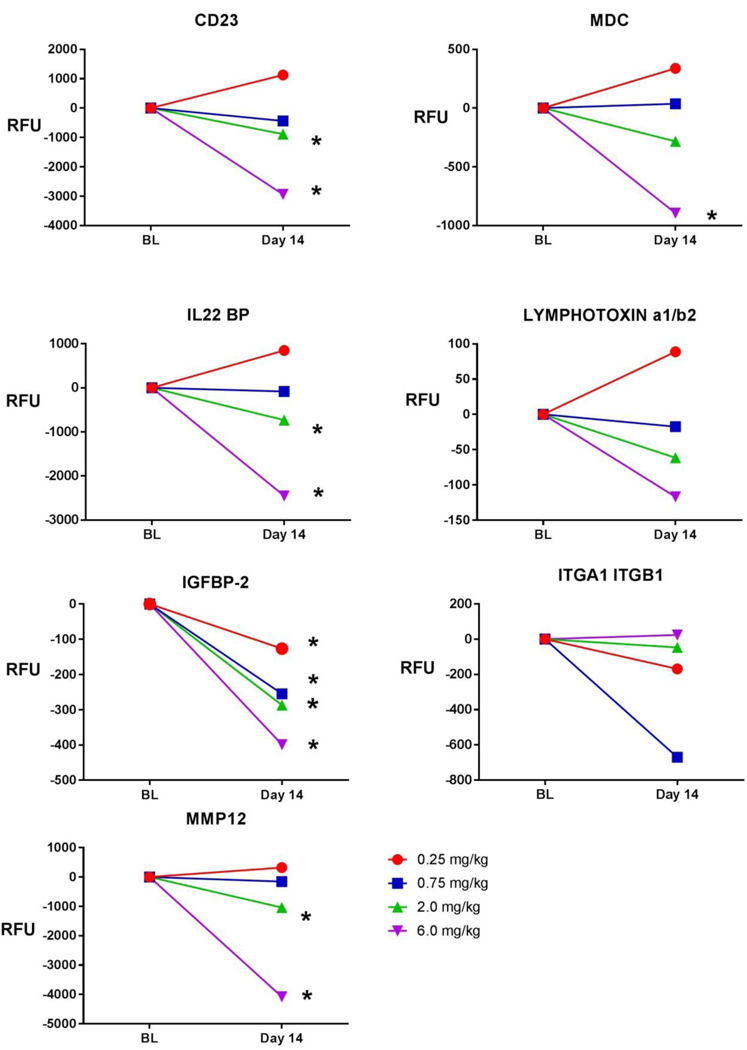
Line graphs of exploratory efficacy biomarkers demonstrating absolute change in Relative Fluorescence Units (RFU) from Baseline to Day 14 in VBP15–002. Shown are 4 dose levels. Baseline values were compared with Day 14 values using a two-tailed paired t-test. Asterisks (*) indicate significant increases from baseline (pre-dose) to following 2 weeks of treatment (post-dose) for a given dose level (p<0.05). Abbreviations: BL= baseline; RFU = Relative Fluorescence Units; MDC = macrophage-derived chemokine; IL22 BP = interleukin-22 binding protein; IGFBP2 = insulin-like growth factor binding protein 2; ITGA1 ITGB1 = integrin a1b1; MMP 12 = matrix metalloproteinase 12
2.11. Statistical Analysis
All statistical tests were two-sided and a resultant p-value of less than or equal to 0.05 was considered significant. Descriptive summaries of variables were provided where appropriate. In general, for continuous variables, the number of non-missing values (n) and the mean, standard deviation (SD), median, minimum, and maximum were tabulated. Unless otherwise noted, baseline was defined as the last measurement taken prior to first exposure to study drug, including Day 1 pre-dose measurements. Analyses were performed using Statistical Analysis System (SAS) (release 9.4) for Windows™. No adjustments for multiplicity on inferential statistical testing were performed in this study. Outliers were not removed. Analyses of the SOMAscan™ biomarkers were performed using GraphPad Prism™. Assay results are reported in Relative Fluorescence Units (RFUs). Mean percentage change from baseline in serum RFUs are summarized by dose level. For each biomarker, data were log transformed, and a parametric 2-tailed, paired-sample t- test was conducted to compare pre-treatment to post-treatment. A time-line graph was created presenting mean and subject-level change from baseline response, by biomarker and dose level.
2. Results
VBP15–002 was a 4-week, Phase IIa multiple ascending dose trial, comprised of two weeks of daily treatment with vamorolone followed by a two-week washout period in steroid-naïve, 4 to <7 year old boys with DMD. Twelve patients were recruited in each of four dose groups (0.25, 0.75, 2.0, and 6.0 mg/kg/day) (48 patients total). Clinical safety through adverse event reporting was collected throughout the study. The demographic and clinical information for the subjects enrolled are reported in Table 1.
Table 1:
Demographics and Clinical Information from 48 Study Participants. Abbreviations: W= white; B= black; A= Asian; Unk= unknown; Non H/L= not Hispanic or Latino; H/L= Hispanic or Latino; del= deletion; dupl= duplication; n/a = not available; TTSTAND=time to stand test from the floor; m/s=meters per second
| Dose Level (mg/ kg) |
Age at Enrollment (years) |
Race | Ethnicity | Mutation type | Starting exon |
Baseline TTSTAND Velocity (m/s) |
Baseline TTSTAND Seconds |
|---|---|---|---|---|---|---|---|
| 0.25 | 6 | W | Non H/L | Multiple Exon Del | 8 | 0.13 | 7.85 |
| 4 | W | Non H/L | Multiple Exon Del | 8 | 0.18 | 5.53 | |
| 4 | W | Non H/L | Multiple Exon Del | 47 | 0.26 | 3.9 | |
| 6 | B | Non H/L | Multiple Exon Del | 51 | 0.13 | 7.59 | |
| 6 | W | Non H/L | No large del/dupl | n/a | 0.14 | 7.3 | |
| 6 | W | Non H/L | Single Exon Del | 45 | 0.2 | 5 | |
| 6 | W | Non H/L | No large del/dupl | n/a | 0.1 | 10.2 | |
| 6 | W | Non H/L | No large del/dupl | n/a | 0.27 | 3.65 | |
| 4 | W | Non H/L | Multiple Exon Del | 8 | 0.16 | 6.44 | |
| 4 | W | Non H/L | Multiple Exon Del | 45 | 0.13 | 7.8 | |
| 4 | W | Non H/L | Multiple Exon Del | 10 | 0.29 | 3.4 | |
| 6 | W | Non H/L | Multiple Exon Del | 52 | 0.22 | 4.5 | |
| 0.75 | 6 | A | Non H/L | Multiple Exon Del | 8 | 0.14 | 7.14 |
| 4 | W | Non H/L | Single Exon Del | 44 | 0.25 | 4.02 | |
| 4 | W | Non H/L | Multiple Exon Del | 46 | 0.12 | 8.4 | |
| 5 | Unk | Hisp or Lat | Single Exon Del | 45 | 0.13 | 7.8 | |
| 5 | W | Non H/L | Multiple Exon Del | 45 | 0.13 | 7.97 | |
| 4 | W | Non H/L | Multiple Exon Del | 5 | 0.33 | 3.06 | |
| 4 | W | Non H/L | Multiple Exon Del | 51 | 0.33 | 3 | |
| 4 | W | Non H/L | Multiple Exon Del | 16 | 0.23 | 4.31 | |
| 6 | W | Non H/L | Multiple Exon Del | 48 | 0.27 | 3.7 | |
| 5 | W | Non H/L | Multiple Exon Del | 49 | 0.31 | 3.2 | |
| 6 | W | Non H/L | Multiple Exon Del | 49 | 0.38 | 2.8 | |
| 5 | W | Non H/L | Single Exon Del | 51 | 0.27 | 3.7 | |
| 2 | 4 | W | Non H/L | Multiple Exon Del | 22 | 0.28 | 3.6 |
| 4 | W | Non H/L | No large del/dupl | n/a | 0.18 | 5.7 | |
| 4 | W | Non H/L | Multiple Exon Del | 33 | 0.17 | 6.03 | |
| 4 | W | Non H/L | Single Exon Del | 50 | 0.12 | 8.5 | |
| 4 | W | Non H/L | Multiple Exon Del | 53 | 0.16 | 6.1 | |
| 6 | W | Non H/L | Multiple Exon Del | 3 | 0.26 | 3.8 | |
| 4 | W | Non H/L | Multiple Exon Del | 45 | 0.11 | 9.2 | |
| 5 | W | Non H/L | Multiple Exon Del | 48 | 0.26 | 3.9 | |
| 6 | W | Non H/L | Multiple Exon Del | 3 | 0.21 | 4.7 | |
| 5 | W | Non H/L | No large del/dupl | n/a | 0.29 | 3.5 | |
| 6 | W | Non H/L | Multiple Exon Del | 8 | 0.26 | 3.9 | |
| 4 | W | Non H/L | Single Exon Del | 8 | 0.4 | 2.5 | |
| 6 | 5 | W | Hisp or Lat | Multiple Exon Del | 48 | 0.15 | 6.5 |
| 5 | W | Hisp or Lat | Single Exon Del | 45 | 0.16 | 6.4 | |
| 5 | W | Non H/L | Multiple Exon Del | 44 | 0.14 | 7 | |
| 5 | W | Non H/L | Multiple Exon Del | 48 | 0.25 | 4 | |
| 6 | W | Non H/L | Single Exon Del | 51 | 0.24 | 4.1 | |
| 4 | W | Non H/L | Single Exon Del | 52 | 0.07 | 14.06 | |
| 4 | W | Non H/L | Single Exon Dupl | 12 | 0.26 | 3.81 | |
| 4 | W | Non H/L | Single Exon Del | 7 | 0.21 | 4.7 | |
| 4 | W | Non H/L | Multiple Exon Del | 48 | 0.16 | 6.15 | |
| 4 | W | Non H/L | Single Exon Del | 44 | 0.23 | 4.31 | |
| 6 | W | Non H/L | No large del/dupl | n/a | 0.2 | 5 | |
| 5 | W | Non H/L | Single Exon Del | 45 | 0.21 | 4.8 |
All study participants completed the 4-week VBP15–002 Phase IIa study and were invited to enroll in a 24-week Phase II extension study (VBP15–003) at the same dose level, and in the long term extension study that followed (VBP15-LTE).
3.1. Clinical Safety Assessment
There were no clinically significant safety concerns during the study, as assessed by clinical laboratory analyses, physical examinations with body mass index (BMI), vital sign measurements, and 12-lead electrocardiogram (ECG) performed during the pre-treatment (Baseline), treatment (2 weeks), and post-treatment washout periods (2 weeks). During the trial, there were a total of 46 treatment-emergent adverse events (TEAEs) among the 48 subjects. The TEAEs with the highest incidence were viral upper respiratory tract infection (12.5%), diarrhea (8.3%), pain in extremity (6.3%) and vomiting (6.3%). Twenty subjects (41.7%) had TEAEs that were considered by the investigator to be remotely related or unrelated to study drug, and 8 subjects (16.7%) had TEAEs that were considered by the investigator to be possibly or probably related to study drug. No subject had a TEAE that was assessed as definitely related to study drug. Twenty-six subjects (54.2%) had TEAEs considered by the investigator to be mild in severity, and two subjects (4.2%) had TEAEs considered by the investigator to be moderate in severity. No subject had a TEAE considered to be severe, or a TEAE that resulted in discontinuation from the study. There were no serious adverse events (SAEs). There were no clinically significant changes from baseline with respect to BMI, ECG findings, clinical laboratory values, or vital signs.
Drug-induced liver injury is typically monitored by alanine aminotransferase (ALT) and aspartate aminotransferase (AST). However, as expected, all DMD participants showed muscle disease-related elevation of serum ALT and AST at baseline, prior to any drug treatment. The liver-selective enzymes GLDH and GGT were studied as exploratory clinical laboratory tests in VBP15–002 to monitor potential vamorolone-induced liver injury (20–22). No dose-related persistent elevations of GLDH or GGT were seen, suggesting that vamorolone does not confer acute or subacute hepatic toxicity.
3.2. Pharmacokinetic Assessment
In boys with DMD, oral vamorolone was absorbed at a moderate rate with peak concentrations typically occurring at 2 or 4 hours on both Days 1 and 14. The drug half-life across all doses was similar on both days, averaging about 2 hours. Peak concentrations and AUCinf values were generally comparable on Days 1 and 14 for each of the dose levels. The mean ratios on Day 14 to Day 1 were 1.7 with a SD of 1.0 for Cmax and 2.5 with a SD of 5.1 for AUCinf. The large mean and SD of the AUCinf ratio is mainly driven by a single participant (Cohort 4 – 6 mg/kg) who on Day 1 had a very low AUCinf, probably due to low bioavailability of vamorolone. The mean AUCinf ratio for Day 14/Day 1 excluding this participant is 1.7 with SD 1.3. A summary of the observed (Cmax and tmax) and calculated (AUCinf, t1/2, and CL) pharmacokinetic parameters for all doses for Day 1 and Day 14 is provided in Table 2.
Table 2:
Summary of pharmacokinetic parameters for vamorolone after oral administration of 0.25, 0.75, 2.0 and 6.0 mg/kg doses once daily. Values are mean (SD) and for tmax mean [median] (SD). Abbreviations: Cmax = maximum peak concentration; tmax = time at which Cmax is observed; AUC = area under the curve; t1/2 = half-life; CL= clearance.
| Dose, mg/kg/day |
Day 1 | Day 14 | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.25 | 0.75 | 2.0 | 6.0 | 0.25 | 0.75 | 2.0 | 6.0 | |
| Cmax [ng/ml] | 22.9 (13.4) |
75.9 (25.9) |
199 (111) |
855.6 (471) |
32.2 (15.2) |
124.7 (42.5) |
252.5 (96) |
970 (270) |
| tmax [hr] | 3.6[4] (1.2) |
4.6[4] (2.1) |
2.5[2] (1.3) |
2.7[2] (1.3) |
3.8[4] (1.8) |
3.8[4] (2.2) |
2.8[2] (1) |
2.3[2] (0.86) |
|
AUCinf [hr∙ng/ml] |
118 (48) |
379 (117) |
761 (352) |
3279 (1693) |
164 (61) |
544 (155) |
1138 (467) |
3606 (897) |
| t1/2 [hr] | 2.1 (0.85) |
1.8 (0.43) |
1.9 (0.79) |
1.9 (0.95) |
1.9 (0.96) |
2.1 (0.8) |
1.9 (1.02) |
1.4 (0.35) |
| CL [ml/hr/kg] | 2459 (897) |
2285 (1103) |
2697 (1285) |
2320 (1375) |
1828 (919) |
1509 (482) |
2047 (771) |
1777 (476) |
Clearance was consistent across the Day 1 dose levels as well as for the Day 14 dose levels, but the latter tended to average slightly lower (about 2400 mL/hr/kg on Day 1 and 1900 mL/hr/kg on Day 14). Overall, higher doses resulted in higher plasma concentrations of vamorolone, as expected. In particular, plasma drug concentrations versus time showed a rapid to moderate rate of absorption, a maximum (Cmax) at a tmax of 2 or 4 hours, and a decline phase with a typical t1/2 of 2 hours. For all doses investigated, the mean PK profiles on Day 1 and Day 14 appear similar; no drug accumulation between doses was observed, consistent with the short half-life and daily dose schedule. In conclusion, the PK of vamorolone was similar to that of glucocorticoids such as prednisolone.
3.3. Serum Pharmacodynamic Safety Biomarker Measures Bridged to Clinical Outcomes
3.3.1. Insulin resistance measured by fasting insulin and glucose
Mean fasting glucose and mean fasting insulin levels were compared between Baseline (0.5 hour pre-dose Day 1) and Week 2 (0.5 hour pre-dose Day 14). At both Baseline and Week 2, mean levels of fasting glucose were within the normal range (60–99 mg/dL) for this age group for all vamorolone dose level groups. The mean changes from Baseline to Week 2 in fasting glucose were clinically unremarkable and not significant for any dose level group. There were no appreciable changes in mean fasting insulin levels at the 0.25, 0.75, or 2.0 mg/kg dose levels. At the 6.0 mg/kg dose, the mean (± SD) fasting insulin increased from 3.96 ± 2 µIU/mL to 6.73 ± 4.6 µIU/mL (paired t-test, p-value 0.063). All data for fasting glucose and insulin by dose level group are shown in Supplemental Table S1.
3.3.2. Bone turnover markers
After 2 weeks of daily treatment with vamorolone at 2.0 and 6.0 mg/kg doses, there was a significant decrease in CTX (bone resorption) correlated with increasing dose level (intragroup paired t test: 2.0 mg/kg, p = 0.006; 6.0 mg/kg, p = <0.001 (Table 3). Osteocalcin (bone formation) decreased significantly only at the 6.0 mg/kg dose level (intragroup paired t test; p = <0.001). At vamorolone doses of 2.0 and 6.0 mg/kg, significant decreases in P1NP (bone formation) were also observed (intragroup paired t test: 2.0 mg/kg, p = <0.001; 6.0 mg/kg, p =20<0.001 (Table 2). However, there was also a significant decrease observed at 0.25 mg/kg, demonstrating a pattern of unclear dose-responsiveness of P1NP (paired t test; p = 0.01).
Table 3:
Change in bone turnover biomarkers pre and post 14 days of vamorolone therapy. Abbreviations: CTX = C- terminal telopeptide; P1NP = procollagen type 1 pro-peptide
| Vamorolone dose |
Biomarker |
Visit |
N | Mean | SD |
Paired t-test p-value |
| 0.25 mg/kg/day |
Osteocalcin (ng/mL) |
Baseline | 12 | 37.9 |
11.6 | ns |
| Week 2 | 12 | 38.5 |
12 | |||
| CTX (pg/mL) |
Baseline | 11 | 871 |
161 | ns | |
| Week 2 | 12 | 964 |
158 | |||
| P1NP (ng/mL) |
Baseline | 12 | 556 |
185 | 0.01 | |
| Week 2 | 12 | 444 |
93.9 | |||
| 0.75 mg/kg/day |
Osteocalcin (ng/mL) | Baseline | 8 | 34.8 |
7.7 | ns |
| Week 2 | 10 | 34.8 | 9.1 | |||
| CTX (pg/mL) | Baseline | 8 | 915 | 281 | ns | |
| Week 2 | 10 | 913 | 276 | |||
| P1NP (ng/mL) | Baseline | 8 | 472 | 121 | ns | |
| Week 2 | 10 | 417 | 98.5 | |||
| 2.0 mg/kg/day |
Osteocalcin (ng/mL) | Baseline | 10 | 40.8 | 6.13 | ns |
| Week 2 | 10 | 36.5 | 9.16 | |||
| CTX (pg/mL) | Baseline | 10 | 905 | 265 | 0.006 | |
| Week 2 | 10 | 675 | 172 | |||
| P1NP (ng/mL) | Baseline | 10 | 519 | 96 | <0.001 | |
| Week 2 | 10 | 341 | 72.9 | |||
| 6.0 mg/kg/day |
Osteocalcin (ng/mL) | Baseline | 10 | 44.8 | 6.33 | <0.001 |
| Week 2 | 10 | 29.9 | 4.69 | |||
| CTX (pg/mL) | Baseline | 10 | 904 | 203 | <0.001 | |
| Week 2 | 10 | 649 | 205 | |||
| P1NP (ng/mL) | Baseline | 10 | 516 | 116 | <0.001 | |
| Week 2 | 10 | 310 | 59 |
3.3.3. Adrenal suppression measured by first-in-morning cortisol
Mean values of the 2-week first -in-morning cortisol 24 hours after the last drug dose were 10.43 for 0.25 mg/kg, 9.76 for 0.75 mg/kg, 7.32 for 2.0 mg/kg, and 3.01 µg/dL for 6.0 mg/kg. After 2 weeks of daily treatment, a depressed morning cortisol (<3.6 μg/dL [100 nmol/L]) consistent with adrenal suppression was observed in 0 of 11 tested participants who received 0.25 mg/kg, 0 of 11 tested participants who received 0.75 mg/kg, 2 of 11 (18.2%) tested participants who received 2 mg/kg, and 6 of 10 (60.0%) tested participants who received vamorolone 6 mg/kg (Figure 1). Findings in boys with DMD were similar to those previously reported in healthy adult volunteers receiving a daily dose of vamorolone for 2 weeks, where 0% of adults treated with 1 mg/kg, 0% of adults treated with 3 mg/kg, 50% of adults treated with 9 mg/kg, and 100% of adults treated with 20 mg/kg showed adrenal suppression (9).
Figure 1: Pharmacokinetic (Cmax)/Pharmacodynamic (First-in-Morning Cortisol Level) Analysis after 14 Days of Daily Vamorolone Therapy.
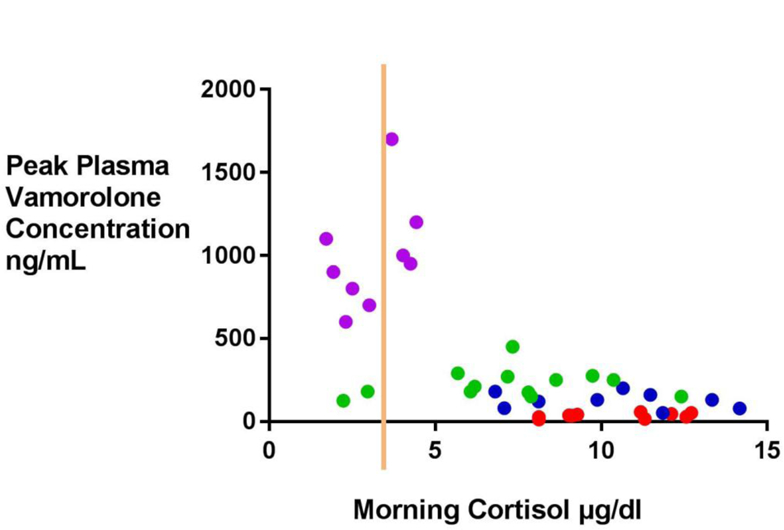
Blood samples were drawn in the morning pre-dose after 2 weeks of daily vamorolone therapy. Each dot represents an individual patient. Adrenal suppression is defined as a first-morning cortisol of <3.6 μg/dL [100 nmol/L] (represented by the orange line). Dot colors depict dose levels (purple = 6.0; green= 2.0; dark blue = 0.75; red= 0.25 mg/kg).
3.4. Exploratory Pharmacodynamic Biomarkers
Six exploratory safety biomarkers defined in glucocorticoid-treated DMD and IBD children (20) were tested in the VBP15–002 trial. wo (angiotensinogen [AGT] and afamin [AFM]) showed treatment-associated increases, with higher doses of vamorolone leading to greater mean increases in protein levels, in a pattern similar to glucocorticoids. The additional four proteins did not increase in a pattern that correlated with dose level (MMP-3, insulin, leptin, IGFBP-5) (Figure 2). A summary of means data by biomarker and dose level can be found in Supplemental Table S2. A summary of paired analysis data (parametric, two-tailed paired t-test) can be found in Supplemental Table S3.
Seven pre-specified exploratory efficacy biomarkers (pro-inflammatory proteins) responsive to glucocorticoid treatment in DMD and IBD were tested(20).Six(CD23,MDC,IL-22BP, IGFBP-2, LTa1/b2, and MMP-12) showed dose-responsive decreases with increasing doses of vamorolone, with the greatest decreases from baseline occurring at the highest doses (Figure 3). Only ITGA1/ITGA2 (integrin complex) showed no pattern of dose response, and this biomarker also showed lack of validation in IBD in the original discovery/validation report (20). These data provide strong support for the anti-inflammatory mechanism of action of vamorolone. A summary of means data by biomarker and dose level can be found in Supplemental Table 4. A summary of paired analysis data (parametric, two-tailed paired t-test) can be found in Supplemental Table S5.
Serum CK, a biomarker of muscle disease activity, was tested at baseline, two weeks treatment, and after two weeks wash-out (Figure 4). After two weeks washout (Week 4), mean (±SD) decreases in CK from baseline were dose-dependent and ranged from −7074 ± 5624 U/L (6.0 mg/kg) to 484 ± 14365 U/L (0.25 mg/kg) U/L across the four dose level groups. The decreases in mean CK values at Week 4 compared to baseline were significant for the 2.0 mg/kg (p=0.018) and 6.0 mg/kg (p=0.003) daily dose level groups.
Figure 4:
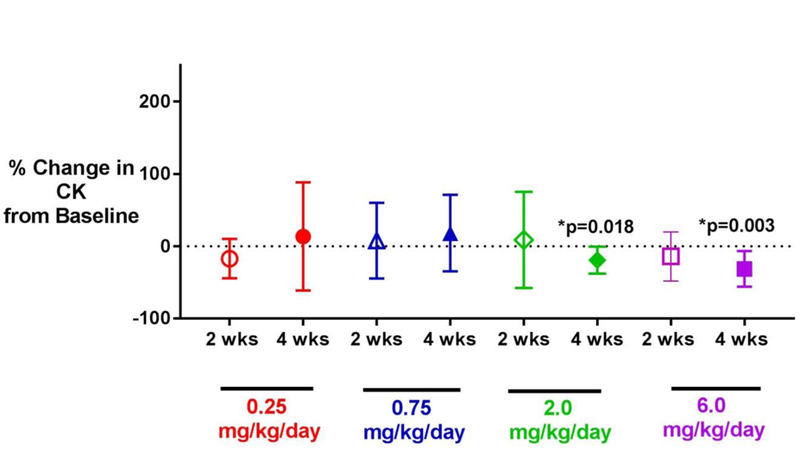
Change in CK activity from baseline to 2 weeks of daily vamorolone treatment, and from baseline to 4 weeks (following 2 weeks of daily vamorolone dosing, and 2 weeks washout) in VBP15–002. Shown is the mean percent change ± standard deviation. Baseline values were compared using a two-tailed paired t-test; there was no adjustment for multiple comparisons. Asterisks (*) indicate significant p-value <0.05). There was a significant decrease in CK activity from baseline to 4 weeks after daily dosing with 2.0 and 6.0 mg/kg of vamorolone. Abbreviations: CK = creatine kinase.
4. Discussion
We describe the first-in-patient clinical trial of vamorolone in boys with DMD. The overall design of the trial was a multiple-ascending dose study; safety and pharmacokinetics were measured as key outcomes. Our findings demonstrated that vamorolone had an acceptable safety and tolerability profile through the maximum daily dose tested (6.0 mg/kg) in patients with DMD. The half-life of vamorolone is quite short (2 hours) and similar to glucocorticoids such as prednisolone, with no evidence of drug accumulation between daily doses. The pharmacokinetics of vamorolone in 4 to <7 year old boys with DMD were essentially dose-proportional (linear) and similar to that in healthy adult volunteers (9).
Pharmacodynamic safety biomarkers bridged to clinical outcomes suggest improved safety of vamorolone compared to glucocorticoids in DMD boys. Glucocorticoids cause increased fasting insulin and glucose at doses of 0.2 mg/kg/day or lower, following 1–2 weeks of treatment (18). Our data suggest that vamorolone does not share safety concerns of insulin resistance with glucocorticoids following 2 weeks of treatment, although at the 6.0 mg/kg dose, increasing insulin levels suggest possible early development of insulin resistance. Long-term treatment with vamorolone and measures of glycosylated hemoglobin will better address clinical significance. In the Phase I VBP15–001 study of vamorolone in healthy adult volunteers, there was no evidence of insulin or glucose changes in a similar two-week multiple ascending dose design (1.0, 3.0, 9.0 and 20.0 mg/kg/day dose groups) (9).
Treatment with glucocorticoids leads to both acute and chronic suppression of the adrenal axis. Treatment with vamorolone led to some adrenal suppression in both boys with DMD (this study) and in healthy adult volunteers (9). Dose-response data for adrenal suppression in DMD patients treated with glucocorticoids is not available for comparison, but in adult volunteers we found about a 10-fold improvement (dose/dose) compared to effects of prednisone (9, 18).
Glucocorticoids cause decreases in osteocalcin (biomarker for bone formation) and increases in CTX (biomarker for bone resorption), and these changes can be correlated with bone morbidities. As shown in Table 3, boys with DMD treated with daily vamorolone showed dose-related decreases in both bone formation (osteocalcin; 6.0 mg/kg) and bone resorption (CTX; 2.0 and 6.0 mg/kg) biomarkers, the latter in contrast to glucocorticoids (18). Children undergo linear growth and bone modeling, which correlates with an increase in bone turnover markers. As with adrenal suppression, dose-response data regarding bone biomarker changes in children with DMD treated with prednisone are not available. However, studies have demonstrated decreased bone turnover in boys with DMD treated with glucocorticoids (29, 30). Another study showed that in ambulatory boys with DMD, there are elevations in bone resorption above ranges seen in healthy children, independent of treatment with glucocorticoids (31). Serum osteocalcin is reduced in both deflazacort and prednisone-treated DMD boys and in other subjects without DMD (29, 31–34). Comparing published data of DMD boys treated with glucocorticoids and DMD boys treated with vamorolone, vamorolone significantly reduced osteocalcin at a daily dose of 6.0 mg/kg and above, whereas prednisone induced similar reductions in osteocalcin at a daily dose of 0.75 mg/kg (~10-fold difference). The reduced potency of vamorolone in decreasing bone formation markers relative to prednisone, coupled with the opposite effect of the two drugs on bone resorption markers, and the lack of any changes of these markers in adult volunteers through 20 mg/kg of daily dosing (9) suggests the potential for improvement in bone safety with vamorolone compared to traditional glucocorticoids. This reduction in an important safety concern was also demonstrated in preclinical studies(10).
Exploratory efficacy biomarkers showed compelling dose-responsive anti-inflammatory effects (Figure 3). Serum CD23, also known as a low affinity immunoglobulin epsilon Fc receptor (FCER2) is expressed on B cells and antigen-presenting cells (35). ross-linking of CD23 mediates the activation of human macrophage inflammatory reaction and pro-inflammatory cytokine production (36). Vamorolone led to a decrease in CD23 at the 2.0 mg/kg and 6.0 mg/kg daily dose in boys with DMD, in a dose-responsive pattern, after 2 weeks of treatment (Figure 3). Macrophage-derived chemokine (MDC), also known as Chemokine (C-C) motif chemokine 22 (CCL22), plays a role in the trafficking of activated lymphocytes to inflammatory sites (37). Two weeks of daily vamorolone treatment led to significantly decreased MDC at the 6.0 mg/kg dose in boys with DMD (Figure 3). IL22 Binding Protein (IL22BP), also known as IL22RA, is the soluble receptor of Interleukin 22 (IL-22). IL-22 is a pro-inflammatory cytokine produced by cells in both innate and acquired immune responses. Elevated circulating levels of IL22BP can be seen in inflammatory diseases such as psoriasis, rheumatoid arthritis, and interstitial lung diseases (38). Vamorolone decreased IL-22BP in a dose-dependent manner, with statistical significance at the 2.0 and 6.0 mg/kg dose levels (Figure 3). Taken together, the exploratory biomarkers for efficacy provide compelling evidence of the anti-inflammatory mechanism of action of vamorolone. These anti-inflammatory effects would be expected to benefit muscle pathology in DMD (2), as well as increase dystrophin levels in Becker muscular dystrophy (39, 40).
An additional exploratory biomarker of efficacy tested was serum CK, a marker of muscle disease activity. Dose-dependent decreases in CK were seen at 2 weeks following the last of 14 daily doses of vamorolone in the two highest dose groups (Figure 4). We hypothesize that them embrane-stabilizing effects of vamorolone, or anti-inflammatory activity, or both could lead to reductions in disease activity and observed reductions in serum CK that persist for two weeks after completion of two weeks of daily drug dosing.
4.1. Conclusions
Biomarker data indicate that vamorolone has an improved safety profile compared with prednisone. Biomarkers demonstrate proof-of-concept that vamorolone has an anti-inflammatory mechanism of action, and also offers the potential to stabilize the myofiber membrane. In addition, bone turnover markers indicate an opposite, decreased effect on bone resorption that could reflect the novel mineralocorticoid receptor antagonism of vamorolone.
Supplementary Material
Acknowledgments:
We wish to acknowledge the contributions of the Cooperative International Neuromuscular Research Group, patients, and families.
Funding: This work was funded by a National Institutes of Health grant from the National Institute for Neurological Diseases and Stroke, (R44NS095423) (EPH, PRC), a National Institutes of Health grant from the National Institute for Arthritis and Musculoskeletal and Skin Diseases (1U34AR068616) (PRC), a National Institutes of Health grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (5U54HD090254) (JvdA, EPH, LSC, YH), a Department of Defense grant W81XWH-15–1-0265 (YH), and a Muscular Dystrophy Association grant MDA353094 (YH). Support for the vamorolone development program was provided by the Muscular Dystrophy Association (USA), Foundation to Eradicate Duchenne (USA), Parent Project Muscular Dystrophy (USA), Duchenne UK [UK],Joining Jack [UK],Duchenne Children’s Trust [UK], Duchenne Research Fund (UK), Save Our Sons (Aus), Michael’s Cause (USA), Pietro’s Fight (USA), Alex’s Wish (UK), Ryan’s Quest (USA), CureDuchenne (USA). Vamorolone was developed through a partnership with the National Institutes of Health NCATS TRND program (Therapeutics for Rare and Neglected Disease), with support for drug production, formulation, and toxicology studies.
LSC, JMD, JMMcC, JvdA, KN, MRC, and EPH are employees of ReveraGen BioPharma. JMMcC, KN, and EPH are co-founders of ReveraGen and own founder shares. LSC, JMD, JvdA own stock options of ReveraGen. MJ and PS are employees of Summit Analytical, a biostatistics clinical research organization. BDS and LJMG own Camden Group, LLC, a clinical research organization. ALS, LPM, AA, and MS are employees of TRiNDS LLC, a clinical trials management organization. WJJ, BDS, LJMG, MJ and PS received payment from ReveraGen for statistical analysis and consultancy. Patents awarded relevant to the results include: WO2017004205 (A1), US2016060289 (A1), US2015011519 (A1), US9649320 (B2); US2017027959 (A1).
The authors take full responsibility for the contents of this manuscript, which do not represent the views of the Department of Veterans Affairs or the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest Statement:
References
- 1.Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 65:826–834 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg AS, Puig M, Nagaraju K,Hoffman SA.R Villalta Rao A, Wakefield LM, Woodcock J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med 7:299rv4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, Clemens P, Hoffman EP, Cnaan A, Gordish-Dressman H; CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391: 451–461 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, DR Weber; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17(3): 251–267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma J, McMillan HJ, Karaguzel G, Goodin C, Wasson J,Matzinger A, DesClouds P,Cram D, Page M, Konji VN, Lentle B, Ward L The time and determinants of first fractures in boys with Duchenne muscular dystrophy. Osteoporosis Int 28: 597–608 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Vry J, Gramsch K, Rodger S, Thompson R, Steffensen BF, Rahbek J, Doerken S, Tassoni A, Beytía ML, Guergueltcheva V, Chamova T, Tournev I, Kostera-Pruszczyk A, Kaminska A, Lusakowska A, Mrazova L, Pavlovska L, Strenkova J, Vondráček P, Garami M, Karcagi V, Herczegfalvi A, Bushby K, Lochmüller H, Kirschner J. European Cross-Sectional Survey of Current Care ractices for Duchenne Muscular Dystrophy Reveals Regional and Age-Dependent Differences. J. Neuromuscul. Dis 3(4):517–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griggs RC, Miller JP, Greenberg CR, Fehlings DL, Pestronk A, Mendell JR, Moxley RT 3rd, King W, Kissel JT, Kwik V, Vanasse M, Florence JM, Pandya S, Dubow JS, Meyer JM. Efficacy and safety of deflazacort vs. prednisone and placebo for Duchenne muscular dystrophy. Neurology 87(20): 2123–2131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. .Reeves KM, Hoffman EP, Nagaraju K, Damsker JM, McCall JM. VBP15: preclinical characterization of a novel anti-inflammatory delta 9,11 steroid. Bioorg, Med. Chem 21(8):2241–2249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, Nagaraju K, Gordish-Dressman H, Damsker JM, McCall JM. Phase 1 trial of vamorolone, a 33 first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 134: 43–52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, Sali A, Miller BK, Phadke A, Scheffer L, Quinn J, Tatem K, Jordan S, Dadgar S, Rodriguez OC, Albanese C, Calhoun M, Gordish-Dressman H, Jaiswal JK, Connor EM, McCall JM, Hoffman EP, Reeves EK, Nagaraju K. VBP15, a novel anti-inflammatory and membranestabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med 5: 1569–1585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baudy AR, Reeves EK, Damsker JM, Heier C, Garvin LM, Dillingham BC, McCall J, Rayavarapu S, Wang Z, Vandermeulen JH, Sali A, Jahnke V, Duguez S, DuBois D, Rose MC, Nagaraju K, Hoffman EP. −9,11 modification of glucocorticoids dissociates nuclear factor-ĸB inhibitory efficacy from glucocorticoid response element-associated side effects. J Pharmacol Exp Ther 343(1); 225–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson WH, Vera IMS, Nwachukwu JC, Weikum E, Herbst AG, Yang Q, Bain DL, Nettles KW, Kojetin DJ, Ortlund EA. Cryptic glucocorticoid receptor-binding sites pervade genomic NF-κB response elements. Nat. Commun 9(1):1337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dillingham BC, Knoblach SM, Many GM, Harmon B, Mullen AM, Heier CR, Bello L, McCall JM, Hoffman EP, Connor EM, Nagaraju K, Reeves EKM, Damsker JM. VBP15, a novel anti-inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis. Cell Mol. Neurobiol 35: 377–387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damsker JM, Dillingham BC, Rose MC,Heier Balsley C.R., Watson AM, Stemmy EJ, Jurjus RA,Huynh T,Tatem K, Uaesoontrachoon K, Berry DM, Benton AS, Freishtat RJ, Hoffman EP, McCall JM, Gordish-Dressman H, Constant SL, Reeves EK, Nagaraju K. VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice. PLoS One 8: e63871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damsker JM, Conklin LS, Sadri S, Dillingham BC, Panchapakesan K, Heier CR, McCall JM, Sandler AD. VB 15, a novel dissociative steroid compound, reduces NFkB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis. Inflamm. Res 65: 737–743 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Sreetama S, Chandra G, Van der Meulen JH, Ahmad MM, Suzuki P, Bhuvanendran S, Nagaraju K, . . Jaiswal Hoffman, J.K.. Membrane stabilization by modified steroid offers a potential therapy for muscular dystrophy due to dysferlin deficit. Molecular Therapy, August 16 pii: S1525–0016(18)30368-X. 10.1016/j.ymthe.2018.07.021. [Epub ahead of print] PMID: 30166241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi K, Oimomi M, Shinko T, Shutta K, Matsuo B, Takai T, Imura H. Response of serum creatine phosphokinase to steroid hormone. Arch. Neurol 32: 89–91 (1975). [DOI] [PubMed] [Google Scholar]
- 18.Kauh E, Mixson L, Malice MP, Mesens S, Ramael S, Burke J, Reynders T, Van Dyck K, Beals C, Rosenberg E, Ruddy M. Prednisone affects inflammation, glucose tolerance, and bone turnover within hours of treatment in healthy individuals. Eur. J. Endocrinol 166: 45967 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Fleishaker DL, Mukherjee A, Whaley FS, Daniel S, Zeiher BG. Safety and pharmacodynamic dose response of short-term prednisone in healthy adult subjects: a dose ranging, randomized, placebo-controlled, crossover study. BMCM usculoskelet.Disord 17:293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hathout Y, Conklin LS, Seol H, Gordish-Dressman H, Brown KJ, Morgenroth LP, Nagaraju K, Heier CR, Damsker JM, van den Anker JN, Henricson E, Clemens P, Mah JK, McDonald C, Hoffman EP. Serum pharmacodynamic biomarkers for chronic corticosteroid treatment of children. Sci. Rep 6:31727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heier CR, Fiorillo AA, Chaisson E, Gordish-Dressman H, Hathout Y, Damsker JM, Hoffman EP, Conklin LS. Identification of pathway-specific serum biomarkers of response to glucocorticoid and infliximab treatment in children with inflammatory bowel disease. Clin. Transl. Gastroenterol 7(9): e192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Church RJ, Watkins PB. The transformation in biomarker detection and management of drug-induced liver injury. Liver Int 37:1582–1590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanigan KM, Voit T, Rosales XQ, Servais L, Kraus JE, Wardell C, et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne musculardy strophy:resultsof a double-blind randomized clinical trial. Neuromuscul. Disord 24:16–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosales XQ, Chu ML, Shilling C, Wall C, Pastores GM, Mendell JR. Fidelity of gamma-glutamyl transferase (GGT) in differentiating skeletal muscle from liver damage. J. Child. Neurol 23:748–51 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Rensen N, Gemke RJ, van Dalen EC, Rotteveel J, Kaspers GJ. Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. Cochrane Database Syst Rev 11:CD008727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmet A, Brienza V, Tran A, Lemieux J, Aglipay M, Barrowman N, Duffy C, Roth J, Jurencak R. Frequency and duration of adrenal suppression following glucocorticoid therapy in children with rheumatic diseases. Arthritis Care Res (Hoboken) 69:1224–30 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Maguire AM, Biesheuvel CJ, Ambler GR, Moore B, McLean M, Cowell CT. Evaluation of adrenal function using the human corticotrophin-releasing hormone test, low dose Synacthen test and 9am cortisol level in children and adolescents with central adrenal insufficiency. Clin Endocrinol 68:683–91 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Le Roux CW, Meeran K, Alaghband-Zadeh J. Is a 0900-h serum cortisol useful prior to a short synacthen test in outpatient assessment? Ann Clin Biochem 39:148–50 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Söderpalm AC, Magnusson P, Ahlander AC, Karlsson J, Kroksmark AK, Tulunius M, Swolin-Eide D. Low bone mineral density and decreased bone turnover in Duchenne muscular dystrophy. Neuromuscul. Disord 17: 919–28 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Misof BM, Roschger P, McMillan HJ, Ma J, Klaushofer K, Rauch F, Ward LM. Histomorphometry and bone matrix mineralization before and after bisphosphonate treatment in boys with Duchenne muscular dystrophy: a paired transiliac biopsy study. J. Bone Miner. Res 31: 1060–1069 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Bianchi ML, Mazzanti A, Galbiati E, Saraifoger S, Dubini A, Cornelio F, Moradi L.Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos Int 14: 761–767 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Babadjanova G, Allolio B, Vollmer M, Reincke M, Schulte HM. Comparison of the pharmacodynamic effects of deflazacort and prednisolone in healthy subjects. Eur J Clin Pharmacol 51(1): 53–7 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Saviola G, Abdi AL, Shams Eddin S, Coppini A, Cavalieri F, Campostrini L, Sacco S, Bucci M, Cirino G G, Rossini M. Compared clinical efficacy and bone metabolic effects of low-dose deflazacort and methyl prednisolone in male inflammatory arthropathies: a 12-month open randomized pilot study. Rheumatology (Oxford). 46: 994–8 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Rufo A, Del Fattore A, Capulli M, Carvello F, De Pasquale L,. Pierroz Ferrari, D., Morandi L,DeSimone M,Rucci N,Bertini E, Bianchi ML, De Benedetti F, Teti A.Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J Bone Miner Res 26: 1891–903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beavil AJ, Edmeades RL, Gould HJ, Sutton BJ. Alpha-helical coiled-coil stalks in the low-affinity receptor for IgE (Fc epsilon RII/CD23) and related C-type lectins. Proc. Natl. Acad. Sci. U S A 89: 753–757 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rambert J, Mamani-Matsuda M, Moynet D, Dubus P, Desplat V, Kauss T, Dehais J, Schaeverbeke T, Ezzedine K, Malvy D, Vincendeau P, Mossalayi MD. Molecular blocking of CD23 supports its role in the pathogenesis of arthritis. PLoS One 4: e4824 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cronshaw G, Kouroumalis A, Parry R, Webb A, Brown Z, Ward SG SG. Evidence that phospholipase-C-dependent, calcium-independent mechanisms are required for directional migration of T-lymphocytes in response to the CCR4 ligands CCL17 and CCL22. J. Leukoc. Biol 79: 1369–80 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Shabgah AG, Navashenaq JG, Shabgah OG, Mohammadi H, Sahebkar A. Interleukin-in human inflammatory diseases and viral infections. Autoimmun. Rev 16: 1209–1218 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Fiorillo AA, Heier CR, Novak JS, Tully CB, Brown KJ, Uaesoontrachoon K, Vila MC, Ngheim PP, Bello L, Kornegay JN, Angelini C, Partridge TA, Nagaraju K, Hoffman EP. TNF-α-induced microRNAs control dystrophin expression in Becker muscular dystrophy. Cell Rep 12(10): 1678–1690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiorillo AA, Tully CB, Damsker JM, Nagaraju K, Hoffman EP, Heier CR. Muscle miRNAome shows suppression of chronic inflammatory miRNAs with both prednisone and vamorolone. Phys Genomics June 8 10.1152/physiolgenomics.00134.2017. [Epub ahead of print] (2018). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.