

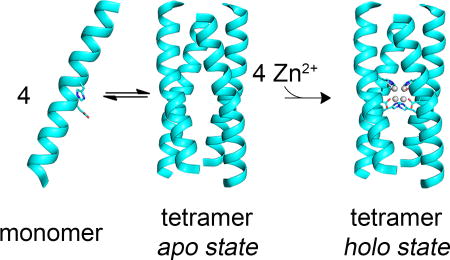
Abstract
De novo design provides an attractive approach, which allows one to test and refine the principles guiding metalloproteins in defining the geometry and reactivity of their metal ion cofactors. Although impressive progress has been made in designing proteins that bind transition metal ions including iron-sulphur clusters, the design of tetranuclear clusters with oxygen-rich environments remains in its infancy. In previous work, we described the design of homotetrameric four-helix bundles that bind tetra-Zn2+ clusters. The crystal structures of the helical proteins were in good agreement with the overall design, and the metal-binding and conformational properties of the helical bundles in solution were consistent with the crystal structures. However, the corresponding apo-proteins were not fully folded in solution. In this work, we design three peptides, based on the crystal structure of the original bundles. One of the peptides forms tetramers in aqueous solution in the absence of metal ions as assessed by CD and NMR. It also binds Zn2+ in the intended stoichiometry. These studies strongly suggest that the desired structure has been achieved in the apo state, providing evidence that the peptide is able to actively impart the designed geometry to the metal cluster.
Keywords: de novo protein design, multinuclear transition metal ion clusters, coiled coils, four-helix bundles, spectroscopic characterization
Graphical abstract
Introduction
De novo design of metalloproteins is an important tool for understanding how metal binding affects protein folding and for building novel structural and enzymatic functions in unnatural protein scaffolds.1–7 In the last years, there have been considerable successful de novo designs of non-heme metalloproteins utilizing helical bundles.8–10 Indeed, design of helical bundles is advantageous as sequences of the constituting helices are usually based on heptad or other repeats, and the residues in the core of the bundles are generally converted to metal-binding sites.11–13 Mononuclear metal-binding sites have been successfully incorporated into de novo designed three-helix bundles resulting in different functions, such as zinc-mediated hydrolysis and copper redox catalysis.10,14 Di-metal sites have been built in de novo designed four-helix bundles with diverse catalytic functions.9,15–17 Recently, we have redesigned this scaffold to stabilize the radical semiquinone form of cathecols within the dizinc analogue,18 or to direct N-hydroxylation of arylamines or quinone trimerization by neat four-electron reduction of dioxygen at the diferric site.19–22 Meanwhile, lanthanide ion binding sites have been built in the cores of three-helix bundles and have potential as MRI contrast regents.23 It is noteworthy that uranyl ions can be extracted from seawater by binding to de novo designed binding site in a three-helix bundle.24 Multinuclear iron-sulfur clusters as well as Cys-rich tetra-cadmium sites have been installed in de novo designed three-helix and four-helix bundles with programmable redox properties.25–28 Recently, we have de novo designed the first oxygen-liganded tetranuclear zinc clusters in four-helix bundles.29
De novo design of multinuclear metal clusters in helical bundles has proven to be a difficult task, because the large size of multinuclear clusters affects the folding behavior of the helical bundles. Breathing and dissociative mechanisms have been shown to be crucial mechanisms of metal binding in helical bundles.30 However, multinuclear metal cofactor binding inside helical bundles is complicated by many factors. Firstly, the helix association state and the nuclearity/geometry of the metal complex are structurally and thermodynamically coupled; secondly, structural rigidity of the helical bundles renders it difficult to predict the effects of the ligand geometry on the global fold; thirdly, how the different shells of interactions around the multinuclear metal coordination site influences folding of the helical bundles.28 In our recent de novo design of tetranuclear zinc clusters in four-helix bundles, we showed by crystal structure analysis that two tetrameric bundles, 4DH1 and 4DH2, accommodate a tetranuclear zinc cluster in their core.29 However, they exhibit different folding features in solution: the signatures of helical structure inferred from far UV circular dichroism spectroscopy (CD) of 4DH2 increases after addition of zinc ions, while 4DH1 appears to be largely pre-organized and hence insensitive to zinc ions. Moreover, 4DH1 displays different oligomerization state by analytical ultracentrifugation from the other designed helical bundles.29
Based on these findings, in this work we design three new 4DH analogues with the aim of examining the features of this class of metalloproteins in solution. Unexpectedly, only one out of three, namely 4DH3, showed the expected folding and metal-binding ability as assessed by CD and Nuclear Magnetic Resonance (NMR) spectroscopies. We discuss the two unsuccessful designs in terms of helix-helix interfaces in coiled coils, suggesting the reasons for misfolding.
The solution study of apo-4DH3 showed a monomer/tetramer equilibrium, which demonstrates the hydrogen bond network, observed by X-ray crystallography in our previous work, is established even in the absence of the metal cofactor. Moreover, the tetramer is stable in solution in the absence of metal ions, and hence ready to host the tetranuclear zinc cluster. Thus, the peptide is able to actively impart the designed geometry to the metal cluster.
Methods
Protein design
The starting backbone was generated by Coiled Coil Crick Parametrization program (CCCP, arteni.cs.dartmouth.edu/cccp/). 4DH1 crystallographic structure (PDB: 5WLL)29 was used as template in the fitting of backbone coordinates. Metal binding residues, second/third shell residues and hydrophobic core (a and d positions) residues were kept accordingly. Peripheral residues were first screened for aromatic (Trp and Tyr) residues insertion by manual screening through Rosetta fixed-backbone (fixbb) symmetry design module, and the best Trp/Tyr pairs were chosen according to their attractive and repulsive van der Waals terms.31 Subsequently, a full design run was accomplished excluding Cys and Met residues for b, c, e, f and g positions between residues 1–10 and 19–26 and b, e, f and g positions between residues 12–18.
Peptide Synthesis
The 26 amino acid peptides were synthesized by automatic solid-phase synthesis using an ABI 433A peptide synthesizer (Applied Biosystem, Foster City, CA, USA) with Fastmoc-protocols on a 0.2 mmol scale. The resin was a H-Rink amide ChemMatrix (Sigma), with a substitution level of 0.4 mmol/g. Fmoc group was removed with two 20% piperidine solution washings (3 and 7 minutes), and conditional washings were added upon conductivity measurements. Coupling was performed with a 5:5:10 equivalents of Fmoc-amino acid:HCTU:DIPEA solution in NMP for 40 minutes. Capping was performed after each coupling step with Ac2O/HOBt/DIPEA solution in NMP. Peptide cleavage was carried out with a 95:2.5:2.5 (v/v/v 20 mL) solution of TFA:TIS:H2O for two hours. Resin was finally washed three times with pure TFA and the eluates concentrated by rotary evaporation. The peptides were precipitated by adding 5 volumes of cold diethyl ether and separated by centrifugation. Purification was accomplished by preparative reverse-phase HPLC on a Vydac C4 column (22×250 mm) with water and acetonitrile (0.1 % TFA) as eluents. Identity and purity of the peptides was assessed by analytical HPLC coupled to a Shimadzu ESI IT-TOF mass spectrometer.
Circular Dichroism
CD measurements were performed using a J-815 spectropolarimeter equipped with a thermostated cell holder (JASCO, Easton, MD, USA). CD spectra were collected at 25°C, from 260 to 200 nm at 0.2 nm intervals with a 20 nm min−1 scan speed, at 2.5 nm band width and at 16 s response. Cells of 0.5, 0.1 and 0.01 cm path length were used in the measurements. Mean residue ellipticities θ were calculated using the equation θ = θobs/(10·l·C·n), in which θobs is the ellipticity measured in millidegrees, l is the path length of the cell in centimeters, C is the concentration in moles per liter, and n is the number of residues in the protein. Protein (monomer) concentration was determined by UV-Vis spectroscopy, using ε280 6990 M−1cm−1.32 Protein stock solutions were freshly prepared in H2O at 1 mM concentration (pH 3.5). Upon dilution, the pH was adjusted to 3.5 with trifluoroacetic acid. To conduct oligomerization studies, the peptide concentration ranged from 1 µM to 177 µM (as monomer) in 20 mM acetate buffer at pH 4.6, and prepared by dilution from a 0.5 mM stock solution. Mean residue ellipticity was fit as a function of peptide concentration using the program OriginPro. The equilibrium constant Kdiss is defined as [M]n/[T], in which [T] is the concentration of the oligomeric species, [M] is the concentration of monomer, and n is the association stoichiometry. Thus, Kdiss = n(αn)(Cn−1)/(1 − α), where α is the fraction of monomer [(θobsd − θassoc)/(θmon − θassoc)] and C is the total peptide concentration. The values of θassoc and θmon were varied during fitting.
NMR
All solution for 1D-NMR analysis were prepared in H2O/ D2O (90:10 v/v). The apo form of the three analogues were analyzed at 1 mM concentration (pH 3.5). 1D-NMR spectra were acquired at 25 °C, on a Bruker Avance 600 spectrometer equipped with a triple resonance cryo-probe. Suppression of the solvent signal was accomplished by excitation sculpting sequence. 2D-DOSY spectrum of the 4DH3 analogue (0.5 mM) was obtained in D2O. Exponential fitting of the signal decay was made with the Bruker proprietary software TopSpin. 128 scans were recorded for each FID, with total acquisition time 4h for the experiment. The theoretical Stoke’s radius for holo-protein was calculated with the program HYDROPRO,33 using default parameters except for: radius of the atomic probe 3.2 Å, temperature 298 K, solvent viscosity 0.012 poise.34
Size-exclusion Chromatography
Analytical size-exclusion chromatography (SEC) was carried out for the 4DH3 peptide on an AKTA FPLC (GE) fitted with a Superdex 75 column (GE; cutoff 3000–70000) and eluted at 10 °C with MES (20 mM, pH 6.5) / NaCl (200 mM) buffer solution, at a 0.5 mL/min flow rate. To analyze the association state in the holo form, the buffer solution contained also 50 µM ZnCl2. Peptide samples were prepared at 200 µM and 200 µL were injected. Four size standards were employed for calibration: blue dextran (2,000 kDa), Carbonic Anhydrase (29 kDa), Ribonuclease A (13.7 kDa), Aprotinin (6.5 kDa). Standard curve (Figure S1) and elution parameters (Table S1) are reported in the supporting information.
Results
Protein design
In this study, three new sequences were designed based on the 4DH series. Basically, four helices were arranged with D2 symmetry, using Crick parameters from our previous designs.29 The 26-mer helix bears 4 heptad repeats if we consider acetyl and amide capping at N- and C- termini, respectively. The central heptad repeat is occupied by the DXXH metal binding motif, where Asp and His occupy a and d positions, respectively (Figure 1a–b). Previous structural analysis showed that this binding motif forms a cuboidal zinc metal cluster of the form [Zn2+4(Nδ•His)4(µ3-η1:η3-O2C•Asp)4] (Figure 1c). The tetranuclear metal ion site is surrounded by the hydrophobic core composed by Leu and Ile residues in a and d positions.35,36 In our previous paper,29 we were interested in the design of residues at positions c and g, which form extensive hydrogen-bonding networks, acting as second and third-shell ligands to the primary ligands in the active site (Figure 1d). It is also known that the residues at the b and e positions of coiled coils play significant roles in stabilizing coiled coils.37 While these residues were primarily apolar in the region adjacent to the active site in our previously designed peptides 4DH1 and 4DH2 (Figure 1e), in this work we explored the introduction of polar and electrostatic interactions. Therefore, polar residues were screened at several b and e positions, as well as c and g positions distant to the active site, using the Rosetta symmetric fixbb module.31 Finally, aromatic residues, included to facilitate peptide concentration measurements, were allowed to vary at c, f and g positions. Three sequences were chosen based on the rank order of their Rosetta-computed energies (Figure 1e). Here, we present the solution properties of these variants.
Figure 1.

Design of tetranuclear clusters in helix bundles. a) D2-symmetrical 4DH analogues, bearing the DXXH metal-binding motif, have four Asp and four His residues coordinating 4 ions at the core. Second-shell Asp residues at the c–g interface (shown in red) are included to interact with the first-shell His ligands. The positions of the three orthogonal two-fold axes are also indicated. b) Crystal structure of 4DH1 (PDB ID 5WLL): the main chain is presented as cartoon, zinc ions as spheres, and coordinating residues as sticks. c) Designed tetranuclear zinc cluster, showing the relative orientations among the carboxylates. d) Model structure of 4DH analogues, including the first-shell ligands Asp12 and His16, the second-shell Asp11, and the third-shell Arg14. e) Sequence alignment of 4DH analogues. Coordinating residues are in bold, while b and e positions in proximity of the metal-binding site are underlined.
Initial screening of the designed peptides
Circular dichroism spectroscopy reveals that only 4DH3 and 4DH4 fold into helical structures
CD spectroscopy was adopted to verify the helical global folding of the three analogues. The peptides were screened in the apo-state at pH 3.5, which allows protonation of the Asp and His residues and improves solubility. Only 4DH3 has a significant α-helical content at this pH as determined by mean residue ellipticities (θ) at 192, 208 and 222 nm (Table 1 and Figure 2a, black). 4DH4 shows a lower helical content with respect to 4DH3 at 10 µM concentration (Figure 2a, red), whereas 4DH5 has random coil structure in these experimental conditions (Figure 2a, blue). Preliminary analysis of the three peptides at higher concentrations (0.1, 1 mM), reveals that only 4DH3 preserves its helical character, with concentration-dependent increase in helical content, as expected for coiled coil formation (Figure 2b–c, black). 4DH4 shows deepening of the minima centered at 205 and 225 nm as the concentration increases, characteristic of helical aggregates (Figure 2b–c, red).38 4DH5 exists as a random coil in these experimental conditions (Figure 2b–c, blue).
Table 1.
Far-UV region CD parameters for 4DH3, 4DH4 and 4DH5 at 10 µM concentration in H2O.
| θ192a | θ208a | θ222a | θratiob | |
|---|---|---|---|---|
| 4DH3 | 26.57 | −14.72 | −13.30 | 0.90 |
| 4DH4 | 14.49 | −11.60 | −9.24 | 0.80 |
| 4DH5 | −2.36 | −8.53 | −3.96 | 0.46 |
The values of θ are reported in kdeg cm2 dmol−1 res−1.
θratio is the ratio θ222/θ208.
Figure 2.

CD characterization of the three analogues as a function of peptide concentration. a) Comparison of far UV CD spectra of 4DH3 (black), 4DH4 (red) and 4DH5 (blue) at 10 µM concentration. b) Comparison of far UV CD spectra of 4DH3 (black), 4DH4 (red) and 4DH5 (blue) at 100 µM concentration. c) Comparison of far UV CD spectra of 4DH3 (black), 4DH4 (red) and 4DH5 (blue) at 1 mM concentration. All experiments were performed in H2O at pH 3.5.
1H-NMR screening suggests the equilibrium between different 4DH3 oligomeric states in water
NMR spectra of the three analogues were acquired in water (pH 3.5) at 1 mM concentration. Spectra analysis substantially confirms the results obtained by the preliminary CD analysis.
4DH5 shows sharp signals in the NH region, but they are not dispersed in the NH and Cα region, as would be expected for well-folded globular proteins (Figure 3, blue). These data are consistent with the hypothesis of a random coil structure for this analogue. 4DH4 shows a poorly defined and dispersed spectrum that suggests the presence of an oligomeric aggregate (Figure 3, red).
Figure 3.

NMR characterization of the three analogues, 1 mM in H2O/ D2O (90:10 v/v), pH 3.5. a) The 1D 1H NMR spectra in the NH region of 4DH3 (top in black), 4DH4 (middle in red) and 4DH5 (bottom in blue). b) The 1D 1H NMR spectra in the αCH region of 4DH3 (top in black), 4DH4 (middle in red) and 4DH5 (bottom in blue).
High field shifts for methyl protons hint the formation of a well-defined hydrophobic core in 4DH3 (Figure 3b, black). Amide signals appear well-defined and well-dispersed from 6 to 9 ppm (Figure 3a, black). These findings indicate the formation of a globular fold that consists of a defined number of associated helices, sequestering hydrophobic residues from the solvent.39
Solution analysis of 4DH3
Apo-4DH3 is in equilibrium between the monomer and the tetramer
Based on the above reported CD and NMR results, further analyses were carried out only on 4DH3. CD spectroscopy was adopted to evaluate the number of helices involved in the association, and to determine the thermodynamic driving force. At 1 mM concentration, the CD spectrum is consistent with nearly complete helix formation, whereas at lower concentrations, the spectra are consistent with a combination of α-helix and random coil conformations (Figure 2, black).
Fitting of the concentration dependence of the θ at 222 nm, with a simple tetramerization model, gave a Kdiss of 3.8 × 10−16 M3 in 50 mM buffer acetate at pH 4.6, consistent with previously characterized tetrameric designed coiled coils (Figure 4)40,41 Fitting of the experimental data for oligomeric states other than four gave worse R2 values.
Figure 4.

Dependence of the mean residue molar ellipticity at 222 nm as a function of 4DH3 concentration in 50 mM buffer acetate at pH 4.6. The error bars indicate the standard deviation of three independent experiments. The smoothed curve represents the best fit for the oligomerization equation from monomer to tetramer, as reported in the methods section.
As expected, when the same experiment was performed in 10 mM MES buffer at pH 6.5, only slight changes in the θ222 values were observed upon concentration increasing (data not shown). This finding reflects the behavior of the DF class of proteins, in which the thermodynamic cost of hindering four negatively charged residues is too high in the absence of the metal cofactor.42
To confirm the hypothesis that 4DH3 is in equilibrium between the monomer and the tetramer states, DOSY-NMR analysis was performed to evaluate the diffusional properties of the associated oligomer. As shown in Figure 5, a single species can be observed in a 0.5 mM 4DH3 solution (in D2O, pD = 3.91), corresponding to a diffusion coefficient of 1.2±0.1 10−10 m2/s. Based on the hydrodynamic radius of the glycerol, used as an internal standard, the observed radius for 4DH3 is 16±2 Å.21 Experimental value has been compared to the hydrodynamic radius as calculated directly from the designed model of the tetramer. The theoretical radius of the holo-form has been calculated to be 19 Å from the designed model.33 Discrepancy between apo and holo forms is expected for this class of compounds, which reach the optimal fold upon metal binding at neutral pH.
Figure 5.

2D 1H DOSY spectrum of 500 µM 4DH3 in D2O, in presence of glycerol as internal standard.
4DH3 tetramer binds up to four zinc ions
The metal binding properties of 4DH3 were analyzed by following the increase of helical content upon addition of zinc.42 The minimum at 222 nm of a 40 uM solution of 4DH3, in a 10 mM MES buffer at pH 6.5, decreased smoothly upon addition of ZnSO4, resembling a saturation mechanism of binding (Figure 6a). Thus, the CD titration data were fit by linear regression of the initial and final linear regions of the curves before and after binding saturation (Figure 6b). The intercept of the two lines gives the peptide:metal expected ratio of 1:1 (corresponding to four zinc ions for each tetramer). Protein aggregation and precipitation at higher concentrations, however, prevented investigation of the oligomeric state by NMR diffusion-oriented experiments. The association state of the designed peptide 4DH3 both in the apo and holo forms was then investigated by SEC. Figure 7 summarizes the SEC results with and without ZnCl2. in the elution buffer. 4DH3 produced two well-defined chromatographic peaks when eluted at pH 6.5 in the absence of zinc ion. The first, less intense, eluted peak (elution volume 15.6 mL), corresponds to an apparent molecular weight approximately 2-fold higher than the monomer molecular weight (apparent Mw was 6000 Da). The second peak, 5-fold more intense than the previous, corresponds to the unfolded monomer eluting with the column volume, as its weight is close to the lower cutoff (3000–70000 Da), confirming the CD data under the same pH conditions. In the presence of zinc, 4DH3 produces a very broad, tailing peak, attributable to dissociation equilibria taking place into the column. Multi-peak fitting analysis was performed to elucidate the intermediate species. Three Gaussian curves were needed to fit the observed peak, corresponding to oligomerization state n = 3.4, 2.0, 0.9, respectively.
Figure 6.

Zn2+-binding properties of 4DH3. a) CD spectral changes of a 40 μM 4DH3 solution (as monomer concentration) upon addition of ZnSO4 (0.2 eq each) in 10 mM MES buffer pH 6.5. The black arrow indicates the observed changes by increasing the metal:peptide ratio up to 2. b) Dependence of the mean residue molar ellipticity at 222 nm as a function of metal:peptide ratio. The dashed line intersects at the 1:1 ratio.
Figure 7.

Size-exclusion chromatography of 4DH3 at pH 6.5 (MES 20 mM, NaCl 200 mM), both in the presence (black line) and in the absence (blue line) of 50 µM ZnCl2. In the apo state, two single well-resolved peaks are observed, indicative of a momomer-dimer equilibrium. In the presence of zinc, a single poorly defined peak is produced. Multi-peak fitting (green lines and red dashed lines for the sum) led identification of the aggregation state of the three species under the main peak, resulting in apparent molecular weights corresponding to oligomerization state n = 3.4; 2.0; 0.9, respectively.
Discussion
In this work, we designed three new analogues of the 4DH series. We have previously shown that this class of peptides is able to fold into a homotetrameric four-helix bundle, and to host, in their core, an abiological metal cofactor composed by four Zn2+ ions bridged by four aspartates residues. In our two previous designs, solution characterization was limited by the lack of defined structure in the apo form. Here, we designed three new sequences by increasing the designable space of the two non-equivalent interfaces of the antiparallel four-helix bundle. This necessarily increases the number of sequences that could erroneously satisfy the desired fold, thus raising the risk of failure in such a difficult design task. In fact, only two out of three were found to be helical in the apo form, and only one was stable as a tetramer without aggregation by CD and NMR spectroscopies.
We therefore analyzed the designed models, by direct comparison of the bundle interfaces (Figure 8). Interfaces between residues at b–e positions are closely related in the three analogues; however, both 4DH4 and 4DH5 would expose a Val-Ala patch on both extremities of the bundle, which could account for the aggregation observed for 4DH4. 4DH5, instead, shows a great divergence in the c-g interface with respect to the other two analogues. The main difference lies for the pair of residues 4–21. Both 4DH3 and 4DH4 present two hydrophobic/aromatic residues (Tyr-Trp for 4DH3; Leu-Trp for 4DH4), whereas 4DH5 does not show any hydrophobic residue in g position. 4DH series is composed by only 4 heptad repeats, and one of them is dedicated to metal binding, thus limiting the thermodynamic driving force for folding given by the Leu/Ile hydrophobic core. The lack of hydrophobic residues for 4DH5 in the c–g interface is therefore not completely counterbalanced by the designed ion pairs (only −0.6 kcal/mol each).41
Figure 8.

Structural differences among the designed models of 4DH3, 4DH4 and 4DH5. The main chain is presented as cartoon, residues in the b and e (on the left) and c and g (on the right) positions as sticks. Hydrophobic, polar, negative- and positively-charged residues are coloured in green, magenta, red and blue, respectively. Helices 2 and 3 (on the left) and helices 3 and 4 (on the right) are transparent to allow a clearer view of the interfaces.
We thus calculated the free energy of tetramerization of apo-4DH3 at 25 °C, and we found it to be −21 kcal/mol, comparable to previously designed antiparallel tetrameric coiled coils.36,40,41 This finding highlights that the apo form of this class of proteins is highly stable under acidic conditions and pre-organized for metal binding. We expect that a dense network of hydrogen bonds could stabilize the protonated aspartates also in the apo form, as already outlined in our previous structural study and in other brilliant examples.43,44 At neutral pH, more than two oligomerization states are in equilibrium, as the metal-binding titration clearly shifted this equilibrium towards a well folded oligomeric state, with a saturation mechanism of binding. Interestingly, SEC analysis clearly suggests that the apo form is in equilibrium with a dimer intermediate at pH 6.5, which further moves to the tetrameric state upon zinc binding. Full conversion to the 4DH3 tetramer holo state was not observed under the explored conditions mostly because of dilution in the SEC column and successive dissociation equilibria. Protein precipitation prevented injection at higher concentration, and future designs will focus at increasing solubility of the holo state at neutral pH.
Conclusion
The primary goal of this work was to design new 4DH analogues, which would allow elucidating the behavior of this class of metalloproteins in solution. One of the three newly designed sequences, 4DH3, adopts the expected folding in aqueous solution. In fact, 4DH3 is stable and tetrameric both in the apo and the holo forms, as assessed by CD and NMR spectroscopies. We also demonstrated that for this class of proteins zinc binds the tetramer in solution with the desired stoichiometry, thus recovering one of the main features of Photosystem II, which is the ability to release and recover the metal cofactor of the OEC, during turnovers. This finding is crucial for the simple and sustainable development of a bioinspired catalyst for solar energy uptake,45 as well as the binding of abiological cofactors within de novo designed proteins.46–48
Supplementary Material
Acknowledgments
The authors thank Dr. Fabiana Paragliola for recording zinc titration data, and Fabrizia Sibillo for technical assistance. This work was primarily supported by NIH grant R35GM122603 to W.F.D., with additional support from the NSF (CHE1413295), the Research Department of Campania Region (STRAIN Project, POR FSE 2007/2013, grant number B25B0900000000 for a postdoctoral fellowship to M. C.), and University of Napoli “Federico II” for mobility grants to W.F.D. and M.C.
Dedication
We dedicate this work to Vincenzo Pavone, friend, mentor and inspiring scientist. His work in the areas of bioinorganic chemistry, peptide design and protein design has inspired multiple generations of chemists and will be valued for years to come. He pioneered the use of β-substituted amino acids in foldamer design long before it became fashionable, built a wide range of bioactive peptides, and contributed greatly to the first de novo design of catalytically active peptides and metalloproteins. We are deeply indebted to Vincenzo for his pioneering vision.
References
- 1.Chino M, Leone L, Zambrano G, Pirro F, D'Alonzo D, Firpo V, Aref D, Lista L, Maglio O, Nastri F, Lombardi A. Biopolymers. 2018 doi: 10.1002/bip.23107. [DOI] [PubMed] [Google Scholar]
- 2.Hecht MH, Zarzhitsky S, Karas C, Chari S. Curr. Opin. Struct. Biol. 2018;48:124. doi: 10.1016/j.sbi.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Kohler V, Lewis JC, Ward TR. Chem. Rev. 2018;118:142. doi: 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- 4.Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y, Lombardi A. Chem. Soc. Rev. 2016;45:5020. doi: 10.1039/c5cs00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Chem. Rev. 2014;114:3495. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins DW, Armstrong CT, Ross Anderson JL. Curr. Opin. Chem. Biol. 2014;19:90. doi: 10.1016/j.cbpa.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Nastri F, Bruni R, Maglio O, Lombardi A. Coordination Chemistry in Protein Cages. Wiley; Hoboken, NJ: 2013. p. 43. [Google Scholar]
- 8.Peacock AF. Curr. Opin. Chem. Biol. 2016;31:160. doi: 10.1016/j.cbpa.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Chino M, Maglio O, Nastri F, Pavone V, DeGrado WF, Lombardi A. Eur. J. Inorg. Chem. 2015;2015:3371. doi: 10.1002/ejic.201500470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tegoni M. Eur. J. Inorg. Chem. 2014;2014:2177. [Google Scholar]
- 11.Maglio O, Nastri F, Lombardi A. Ionic Interactions in Natural and Synthetic Macromolecules. Wiley: Hoboken, NJ; 2012. p. 361. [Google Scholar]
- 12.Lombardi A. Nat. Chem. Biol. 2015;11:760. doi: 10.1038/nchembio.1918. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt NW, Grigoryan G, DeGrado WF. Protein Sci. 2017;26:414. doi: 10.1002/pro.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tebo AG, Pecoraro VL. Curr. Opin. Chem. Biol. 2015;25:65. doi: 10.1016/j.cbpa.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maglio O, Nastri F, Martin de Rosales RT, Faiella M, Pavone V, DeGrado WF, Lombardi A. C.R. Chimie. 2007;10:703. [Google Scholar]
- 16.Shiga D, Funahashi Y, Masuda H, Kikuchi A, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Kamei M, Tanaka T. Biochemistry. 2012;51:7901. doi: 10.1021/bi3007884. [DOI] [PubMed] [Google Scholar]
- 17.Olson TL, Espiritu E, Edwardraja S, Canarie E, Flores M, Williams JC, Ghirlanda G, Allen JP. Biochim. Biophys. Acta, Bioenerg. 2017;1858:945. doi: 10.1016/j.bbabio.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Ulas G, Lemmin T, Wu Y, Gassner GT, DeGrado WF. Nat. Chem. 2016;8:354. doi: 10.1038/nchem.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, DeGrado WF. Nat. Chem. 2012;4:900. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snyder RA, Butch SE, Reig AJ, DeGrado WF, Solomon EI. J. Am. Chem. Soc. 2015;137:9302. doi: 10.1021/jacs.5b03524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chino M, Leone L, Maglio O, D'Alonzo D, Pirro F, Pavone V, Nastri F, Lombardi A. Angew. Chem. Int. Ed. 2017;56:15580. doi: 10.1002/anie.201707637. [DOI] [PubMed] [Google Scholar]
- 22.Chino M, Leone L, Maglio O, Lombardi A. Methods Enzymol. 2016;580:471. doi: 10.1016/bs.mie.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 23.Slope LN, Peacock AFA. Chem. - Asian J. 2016;11:660. doi: 10.1002/asia.201501173. [DOI] [PubMed] [Google Scholar]
- 24.Zhou L, Bosscher M, Zhang C, Özçubukçu S, Zhang L, Zhang W, Li CJ, Liu J, Jensen MP, Lai L, He C. Nat. Chem. 2014;6:236. doi: 10.1038/nchem.1856. [DOI] [PubMed] [Google Scholar]
- 25.Laplaza CE, Holm RH. J. Am. Chem. Soc. 2001;123:10255. doi: 10.1021/ja010851m. [DOI] [PubMed] [Google Scholar]
- 26.Sommer DJ, Roy A, Astashkin A, Ghirlanda G. Biopolymers. 2015;104:412. doi: 10.1002/bip.22635. [DOI] [PubMed] [Google Scholar]
- 27.Nanda V, Senn S, Pike DH, Rodriguez-Granillo A, Hansen WA, Khare SD, Noy D. Biochim. Biophys. Acta, Bioenerg. 2016;1857:531. doi: 10.1016/j.bbabio.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaytsev DV, Morozov VA, Fan J, Zhu X, Mukherjee M, Ni S, Kennedy MA, Ogawa MY. J. Inorg. Biochem. 2013;119:1. doi: 10.1016/j.jinorgbio.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Zhang SQ, Chino M, Liu L, Tang Y, Hu X, DeGrado WF, Lombardi A. J. Am. Chem. Soc. 2018;140:1294. doi: 10.1021/jacs.7b08261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh D, Pecoraro VL. Inorg. Chem. 2004;43:7902. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 31.DiMaio F, Leaver-Fay A, Bradley P, Baker D, André I. PLoS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Science. 1995;4:2411. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortega A, Amorós D, García de la Torre J. Biophys. J. 2011;101:892. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardy RC, Cottington RL. J. Res. Natl. Bur. Stand. 1949;42:573. [Google Scholar]
- 35.Fletcher JM, Boyle AL, Bruning M, Bartlett GJ, Vincent TL, Zaccai NR, Armstrong CT, Bromley EH, Booth PJ, Brady RL, Thomson AR, Woolfson DN. ACS Synth. Biol. 2012;1:240. doi: 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- 36.Thomas F, Boyle AL, Burton AJ, Woolfson DN. J. Am. Chem. Soc. 2013;135:5161. doi: 10.1021/ja312310g. [DOI] [PubMed] [Google Scholar]
- 37.Yadav MK, Leman LJ, Price DJ, Brooks CL, 3rd, Stout CD, Ghadiri MR. Biochemistry. 2006;45:4463. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly SM, Jess TJ, Price NC. Biochim. Biophys. Acta, Proteins Proteomics. 2005;1751:119. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Faiella M, Andreozzi C, Martin de Rosales RT, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat. Chem. Biol. 2009;5:882. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Betz SF, DeGrado WF. Biochemistry. 1996;35:6955. doi: 10.1021/bi960095a. [DOI] [PubMed] [Google Scholar]
- 41.Fairman R, Chao H-G, Lavoie TB, Villafranca JJ, Matsueda GR, Novotny J. Biochemistry. 1996;35:2824. doi: 10.1021/bi952784c. [DOI] [PubMed] [Google Scholar]
- 42.Martin de Rosales RT, Faiella M, Farquhar E, Que L, Jr, Andreozzi C, Pavone V, Maglio O, Nastri F, Lombardi A. J. Biol. Inorg. Chem. 2010;15:717. doi: 10.1007/s00775-010-0639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, Gilmore JM, Xu C, DiMaio F, Pereira JH, Sankaran B, Seelig G, Zwart PH, Baker D. Science. 2016;352:680. doi: 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burton AJ, Thomson AR, Dawson WM, Brady RL, Woolfson DN. Nat. Chem. 2016;8:837. doi: 10.1038/nchem.2555. [DOI] [PubMed] [Google Scholar]
- 45.Blakemore JD, Crabtree RH, Brudvig GW. Chem. Rev. 2015;115:12974. doi: 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- 46.Roxbury D, Zhang SQ, Mittal J, DeGrado WF, Jagota A. J. Phys. Chem. C Nanomater. Interfaces. 2013;117:26255. doi: 10.1021/jp405618p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim KH, Ko DK, Kim YT, Kim NH, Paul J, Zhang SQ, Murray CB, Acharya R, DeGrado WF, Kim YH, Grigoryan G. Nat. Commun. 2016;7:11429. doi: 10.1038/ncomms11429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polizzi NF, Wu Y, Lemmin T, Maxwell AM, Zhang SQ, Rawson J, Beratan DN, Therien MJ, DeGrado WF. Nat. Chem. 2017;9:1157. doi: 10.1038/nchem.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.