

Abstract
Prostate cancer (PCa) often recurs as incurable castration‐resistant prostate cancer (CRPC) after the failure of androgen deprivation therapy. CRPC development relies on androgen receptor (AR) signaling. The IL6/STAT3 pathway is also a key driver of CRPC. The crosstalk between IL6/STAT3 and the AR pathways provides opportunities to explore next‐generation agents to treat PCa. Through screening of around 600 natural compounds in our newly established prostate tumorigenesis model, potential STAT3 signaling inhibitors were found and additionally examined for effects on AR signaling. The small molecular compound 154 exhibited dual effects on IL6/STAT3 and AR pathways. We show here that compound 154 inhibits AR and STAT3 transcriptional activity, reduces the expression of phosphorylation of STAT3 (Y705) and downregulates the mRNA levels of AR target genes. Compound 154 also inhibits protein expression of AR and AR splice variants (ARv567es and AR‐V7) without altering AR mRNA levels. Compound 154 binds to AR directly, but not to STAT3 and is identified as an antagonist of the AR amino‐terminal domain (NTD) by disrupting protein‐protein interactions between STAT3 and the AR NTD. Moreover, compound 154 does not reduce AR nuclear translocation. Compound 154 possesses the potential to become a leading compound in novel therapies against CRPC.
Keywords: amino‐terminal domain, androgen receptor, drug development, prostate cancer, STAT3
Abbreviations
- Abi
abiraterone
- ADT
androgen deprivation therapy
- AR
androgen receptor
- ARE
androgen receptor response element
- AR‐FL
full‐length AR
- ATCC
American Type Culture Collection
- CHX
cycloheximide
- Cinobufagin‐3‐acetate
compound 154 or 154
- CRPC
castration‐resistant prostate cancer
- Crypt
cryptotanshinone
- CTCs
circulating tumor cells
- DARTS
drug affinity responsive target stability
- DBD
DNA binding domain
- DMEM
Dulbecco's Modified Eagle's Medium
- ENZ
enzalutamide
- ER
estrogen receptor
- FSK
forskolin
- GR
glucocorticoid receptor
- HEK
human embryonal kidney
- LBD
ligand binding domain
- mCRPC
metastatic castration‐resistant prostate cancer
- NTD
amino‐terminal domain
- PCa
prostate cancer
- PR
progesterone receptor
- PSA
prostate‐specific antigen
1. INTRODUCTION
Prostate cancer (PCa) has the highest incidence of male cancer in Europe and the second highest worldwide.1 Androgen deprivation therapy (ADT), including drug administration and surgery, has been routines to treat PCa. PCa patients initially are sensitive to ADT. However, in some patients, cancer relapses and progression to even more aggressive castration‐resistant prostate cancer (CRPC) eventually occurs.2 Studies show that most CRPC patients possess high level of androgen receptor (AR) and AR target gene expression, including prostate‐specific antigen (PSA). Mutated forms of constitutively active AR splice variants and activated proliferative pathways such as mitogen‐activated protein kinase and STAT3 signaling are common.3, 4
AR protein, which drives CRPC development, consists of several distinct functional domains: a ligand binding domain (LBD), a short‐hinge region, a DNA binding domain (DBD), and an amino‐terminal domain (NTD).5 Several AR inhibitors are used clinically or entering clinical phase studies. Abiraterone (Abi) blocks androgen synthesis as a CYP17A1 inhibitor. VT‐464 and galeterone (TOK‐001) act as lyase inhibitors. Other AR antagonists target the AR LBD, such as enzalutamide (ENZ), ARN‐509.6 However, these AR inhibitors predominantly fail because of mutations in the LBD which create drug resistance.7
The AR LBD is dispensable for transcriptional activity of AR because its deletion results in the constitutive activation of AR transcription.8, 9 It is reported that knockdown of full‐length AR (AR‐FL) in the 22Rv1 and CWR‐R1 models does not change the expression of AR target genes or androgen‐independent growth. Interestingly, knockdown of truncated AR variants inhibits both.8, 10, 11 Circulating tumor cells (CTCs) in metastatic CRPC (mCRPC) following ENZ and Abi resistance harbor high AR NTD expression and AR LBD loss according to the Epic Science platform.12 The strategy to target the AR NTD has potential to overcome resistance to current AR inhibitors that occur due to truncation (AR splice variants) or point mutations of the AR LBD.6
Constitutively active AR splice variants with mutations in the AR LBD, such as ARv567 and AR‐V7, have been found in CRPC patients. Studies show that ENZ and Abi resistance associates with increasing levels of AR‐V7 and ARv567es mutants, in contrast to AR‐FL.13 ARv567es is a constitutively active receptor and contributes to mCRPC progression.14 As one of the most abundant and best characterized variants among more than 20 already clarified AR splice variants,14 AR‐V7 is suggested to be a predictive marker in primary tumors and as a prognostic factor in CRPC patients.9 Higher expression of constitutively active AR‐V7 was found in CTCs from CRPC patients.9, 15, 16
The STAT3 pathway is essential to drive PCa progression to mCRPC, integrates with other signaling pathways to reactivate the AR pathway, and regulates interactions between tumor cells and the microenvironment.17, 18 reported that constitutively active STAT3 induced resistance to ENZ, and the JAK2 inhibitor AG490 could reverse ENZ resistance in LNCaP cells. Thus, it is promising to study STAT3 inhibitors as a therapeutic target for CRPC progression.
Here, we report the small molecule cinobufagin‐3‐acetate (compound 154 or 154), a natural product isolated from Traditional Chinese Medicine Chan′su (being made of the skin secretions of giant toads, including Bufo gargarizans and B. melanostictus), as a dual inhibitor of IL6/STAT3 and AR pathways, which (a) reduces AR and STAT3 transcriptional activity; (b) inhibits AR splice variants ARv567es and AR‐V7; (c) targets at the AR NTD rather than the AR LBD and affects the association between STAT3 and AR NTD.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
All the cell lines used in this study are described in Table S1. Human PCa cell lines LNCaP and 22Rv1 were purchased from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium with 10% FCS. VCaP cells were from ATCC and maintained in Dulbecco's Modified Eagle's Medium (DMEM) medium containing 10% FCS. MCF7 and DU145 cells (ATCC) were cultured in DMEM medium with 10% FCS. EPT3‐M1‐STAT3 and EPT3‐PT1‐AR (6.3)19 were cultured in Ham's F‐12 medium with 10% FCS. EPT2D5‐miR146a cells were kept in MCDB medium (Biological Industries, Beit Haemek Ltd, Rehovot, Israel) supplemented with 200 nmol/L hydrocortisone, 1% minimum essential media nonessential amino acids solution, 5 μg/mL transferrin, 10 nmol/L triiodothyronine, 5 ng/mL EGF, 5 μg/mL insulin, 5 μg/mL sodium selenite, 100 ng/mL testosterone (Sigma Aldrich, St. Louis, MI), 50 μg/mL bovine pituitary extract (Invitrogen Life Sciences, Waltham, MA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 5% FCS. MG‐132, cycloheximide (CHX), Cryptotanshinone (Crypt), S3I‐201, ENZ, Abi, IL6, and AG490 were purchased from (Sigma Aldrich).
Our newly established prostate tumorigenesis model has become attractive in drug discovery. The stepwise tumorigenesis model consists of the prostate primary epithelial EP156T cells, mesenchymal nontransformed EPT1, premalignant EPT2, primary tumor derived EPT3‐PT1 and metastasis‐derived EPT3‐M1 cells. The STAT3‐activated EPT3‐M1 cells were isolated by FACS and formed tumors with much higher efficiency than the larger STAT3 negative cell population, demonstrating that STAT3‐activated cells are tumor initiating cells.19
2.2. MTS assay
LNCaP, 22Rv1, EPT3‐M1‐STAT3, EPT2‐D5‐miR146a cells were seeded in 96‐well plates for 24 hours followed by treatment with different doses of 154. MTS assay was done after 3 days by adding 10 μL/well CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI) for 4 hours and the absorbance at 490 nm recorded using a Biotek machine.
2.3. Plasmids and transfection
LNCaP and human embryonal kidney (HEK) 293T cells were transiently transfected with Cignal AR Reporter (CCS‐1019L) (SABioscience, Qiagen, Venlo, Netherlands) plus or minus an AR expressor pLENTI7.3/V5‐DEST AR vector, or STAT3 reporter (SABioscience, Qiagen), or ARv567es construct (from Dr. Stephen Plymate, University of Washington, Seattle, USA14) using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Waltham, MA) for 24 hours, following treatment with 1 nmol/L R1881 and the indicated concentrations of 154 or ENZ.
MCF7 cells were cultured in phenol red‐free DME medium with 10% charcoal‐stripped FCS in one 96‐well plate for 24 hours. Cells were transfected with 100 ng/well Cignal GRE Reporter (CCS‐006L) or Cignal ERE Reporter (CCS‐005L) (SABioscience, Qiagen) for 16 hours, then treated with different doses of 154 or ENZ with or without 100 nmol/L dexamethasone, 10 nmol/L 17β‐estradiol (E2), respectively, for 6 hours. Alternatively, MCF7 cells were transfected with 100 ng/well Cignal PR Reporter (CCS‐6043L) (SABioscience, Qiagen) for 16 hours, then treated with different doses of 154 or ENZ plus or minus 10 nmol/L progesterone for 18 hours.
Luciferase activity was measured by the Dual Luciferase Assay Kit (Promega) using a Luminescence Microplate Reader (BioTek, Winooski, VT). Values were normalized to Renilla luciferase activity of DMSO vehicle. FLAG‐AR‐NTD plasmid, AR‐1‐558‐Gal4DBD and 5xGal4UAS‐TATA‐luciferase vectors were kind gifts from Dr. Marianne Sadar (University of British Columbia, Vancouver, Canada7) and FLAG‐tagged STAT3 plasmid from Dr. Saïd M. Sebti (H. Lee Moffitt Cancer Center and Research Institute, FL, USA20) and were transfected into LNCaP cells as described above.
Our lab has established a fast track androgen receptor response element (ARE) reporter system with an internal normalization system of ARE driven mCherry fluorescent signals.21 The virus particles were generated by transfecting CS‐GS241B‐mCHER‐Lv207‐01 and HIV packaging mix in HEK 293T lentiviral packaging cell line (GeneCopoeia™, Richmond, CA, Cat. No. HPK‐LvTR‐20). The lentiviral particles in the supernatant were harvested 48 and 72 hours post transfection and treated with Lenti‐Pac™Lentivirus Concentration Solution (GeneCopoeia™, Cat. No. LPR‐LCS‐01). LNCaP cells were infected with harvested lentiviral particles combined with polybrene at a final concentration of 8 μg/mL. Cells were incubated with fresh medium at 37°C for 48 hours. The infected cells were grown with 12.5 μg/mL hygromycin and stably transduced LNCaP‐207‐01 cells were selected.
2.4. PSA level detection
The supernatant of LNCaP cells were centrifuged at 12 000g for 1 minute at room temperature, then 0.5 mL of supernatants were quantified via the Elecsys total PSA assay (04641655160, Roche Diagnostics, Rotkreuz, Switzerland) in a Cobas analyzer (Roche, Basel, Switzerland) at the accredited laboratory of Clinical Biochemistry (LKB) Haukeland University Hospital, Bergen, Norway. The linear range of PSA detection is from 0.003 to 100 ng/mL in this assay.
2.5. Multi‐plate fluorescence reader
LNCaP‐207‐1 cells were cultured in RPMI 1640 medium with 10% charcoal‐stripped FCS. At 90% confluency, cells were stimulated with 1 nmol/L R1881. Simultaneously different doses of 154 compound, ENZ or Abi were added for 24 hours, respectively. Then cells were analyzed for fluorescence signals by Synergy H1 Hybrid Reader (BioTek) using Gen5 software (BioTek). The excitation was set at 584 nm and emission at 610 nm for mCherry signals, and 470 nm and emission at 509 nm for GFP signals.
2.6. Flowcytometry
LNCaP‐207‐1 cells were grown with RPMI 1640 containing 10% charcoal‐stripped FCS. Treatment started on the second day and after 24 hours the cells were harvested, washed twice with PBS and mixed with 400 μL PBS for further detection. Then the samples were immediately analyzed on the LSRFortessa cytometer (BD Biosciences, Heidelberg, Germany). All analyses were performed with FlowJo software (Tree Star, Ashland, OR). 1% events were accepted as false‐positive in the negative controls for all the experiments.
2.7. DARTS assay
LNCaP cell were lysed with cold M‐PER buffer (Thermo Fisher Scientific, Pierce cat. no. 78501) containing protease inhibitors (Roche, cat. no. 11836153001) and phosphatase inhibitors (Pierce cat. no. 78420) and centrifuged (18 000g for 10 minutes at 4°C). Lysates were diluted to the same final volume and proteolyzed in TNC buffer (50 mmol/L Tris HCl [pH 8.0], 50 mmol/L NaCl, 10 mmol/L CaCl2). 500 μmol/L 154 or the same volume of DMSO were added and incubated for 1 hour at RT. Pronase solution (1.25 mg/mL) was diluted serially by mixing with 1X TNC buffer to create 1:300, 1:1000, 1:3000, and 1:10 000 Pronase stock aliquots. Pronase was added into both DMSO and drug groups and incubated for 30 minutes at RT. Digestion was stopped by adding 4X loading buffer and heating to 90°C for 10 minutes immediately prior to the western blot assay according to publications.22, 23
2.8. DNA microarray
This method has been described previously.19 Total RNA was isolated and tested for RNA integrity by 1% agarose gel electrophoresis, then converted to Cy3‐labeled cRNA targets and hybridized to Agilent Whole Human Genome 44k Microarrays (Cat.no. G4845A, Agilent Technologies, Santa Clara, CA). Samples were analyzed by the Agilent Microarray Scanner Bundle. Microarray data were analyzed by J‐Express software.24 We used mean spot signals as intensity measure, normalized the expression data over the entire arrays and log2‐transformed and considered genes changed more than 2.0 fold with FDR value <5% as differentially expressed genes. The ArrayExpress ID for the cell lines is E‐MTAB‐5102.
2.9. Immunofluorescence staining
EPT3‐PT1‐AR (6.3) cells were stably transfected with a reporter vector where mCherry expression was driven by an ARE‐containing promoter region and seeded in Ham's F‐12 medium with 10% FCS for 24 hours. EPT3‐PT1‐AR (6.3) or VCaP cells were treated with DMSO, 154 (1 μmol/L), ENZ (10 μmol/L) in the presence or absence of R1881 (1 nmol/L) for 24 hours. Cells were fixed with 4% paraformaldehyde, and mounted onto Millipore microscope slides with 7 μL of ProLong® Gold Antifade Mountant (Thermo Fisher Science, Waltham, MA) with DAPI. Images were examined using the LeicaDMRBE microscope or Cytation5 Cell Imaging Multi‐Mode Reader.
2.10. Reverse transcriptase and qPCR gene expression analysis
LNCaP cells were seeded in six‐well plates. 1 μmol/L 154, 10 μmol/L ENZ plus, or minus 1 nmol/L R1881 were added into the relevant wells for 24 hours. Then 700 μL of Trizol were added into each well and RNA was extracted using the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. Reverse transcription and real‐time quantitative PCR (qPCR) were done as described.19 The TaqMan assays used for qPCR of human AR (Hs00985639_m1), PSA (Hs00169842_m1), TMPRSS2, and β‐actin (Hs99999903_m1) were obtained from Applied Biosystems (Thermo Fisher Scientific).
2.11. Western blot analysis
The levels of expression of phosphorylated STAT3 (Y705), AR, and PSA proteins were determined by western blotting following the procedures described.25 The following antibodies from Abcam, Cambridge, UK, were used in western blotting: anti‐phospho‐STAT3 (Tyr705) (ab76315); total STAT3 (ab119352); anti‐phospho‐STAT1 (Tyr701) (ab30645); total STAT1 (ab119352); β‐actin (ab8226); GAPDH (ab181602); FLAG (ab1162); AR V7 (ab198394); TATA binding protein TBP (ab51841); AR (ab133273); PSA (ab53774); and AR N20 (Santa Cruz Biotech, Santa Cruz, CA, sc‐816).
2.12. Co‐immunoprecipitation
LNCaP cells were transfected with 200 μg/T75 flask FLAG‐STAT3 and 200 μg/T150 flask FLAG‐AR‐NTD plasmids for 24 hours, followed by 1 μmol/L 154, 10 μmol/L Crypt with or without 50 ng/mL IL6 for 6 hours. LNCaP cells were collected in the medium and centrifuged at 800g for 5 minutes, then washed with cold PBS twice before adding into the cells soft RIPA buffer (PBS, 1% sodium deoxycholate, 20 mmol/L sodium molybdate, 50 mmol/L NaF, 25 mmol/L β‐glycerophosphate [Sigma Aldrich], 1 mmol/L EDTA, 1% Nonidet P‐40, and protease and phosphatase inhibitor cocktail [Roche, 11836153001]). Cell lysates were passed several times through a 27 1/2‐gauge needle to disrupt nuclei. Extracts were precleared with 2 μL anti‐mouse IgG (Sc‐2025) and 30 μL Protein G beads (Life Technologies, Waltham, MA, 10004D) to reduce the amount of the IgG chain, supernatant were collect with magnet and immunoprecipitated (IP) with 2 μL STAT3 (Abcam ab119352, 1 μL/mg protein) and 80 μL Protein G beads overnight at 4°C on a rotator. Beads were boiled at 95°C for 5 minutes and supernants were collected with magnet and then subjected to immunoblotting.
2.13. Compliance with design and statistical analysis requirements
All experiments were conducted for at least three times. Significance in groups (n = at least 3) was determined by two‐tailed Student's t test or statistical analysis of microarrays for microarray data and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 were considered significant.
The density of each protein band corresponding to its signal intensity was normalized to β‐actin or GAPDH and quantified by using the software ImageJ (NIH, Bethesda, MD).
3. RESULTS
3.1. Compound 154 is a dual modulator of the IL6/STAT3 and AR pathways
In a screen of 600 natural compounds, cinobufagin‐3‐acetate (compound 154) was found to efficiently inhibit the proliferation of STAT3‐activated cells (EPT3M1‐STAT3) and human prostate tumor cells (LNCaP, 22Rv1, and EPT2‐D5‐miR146a) in a dose‐dependent manner after treatment for 24 hours (Figure 1A, Figure S1). Here we sought to determine whether 154 was able to suppress STAT3 phosphorylation and activation. To confirm the inhibitory effect of 154 on STAT3 activity, the luciferase reporter assay was carried out using a STAT3‐luciferase vector in HEK 293T cells, following treatment with 154 or AG490 (a JAK2 inhibitor) in the presence of IL6 for 24 hours (Figure 1B). 154 reduced STAT3 transcriptional activity dose‐dependently with IL6. Then we tested the effect of 154 on STAT3 in the STAT3 activated cells EPT3‐M1‐STAT3, a cell line known to possess high basal level of STAT3.19 As shown in Figure 1C, treatment with 154 for 24 hours inhibited the phosphorylation of STAT3 on Tyr705. Figure 1D shows that 154 could selectively lead to a time‐dependent reduction of STAT3 activity. These findings suggest that 154 is a small molecule inhibitor of STAT3 signaling.
Figure 1.
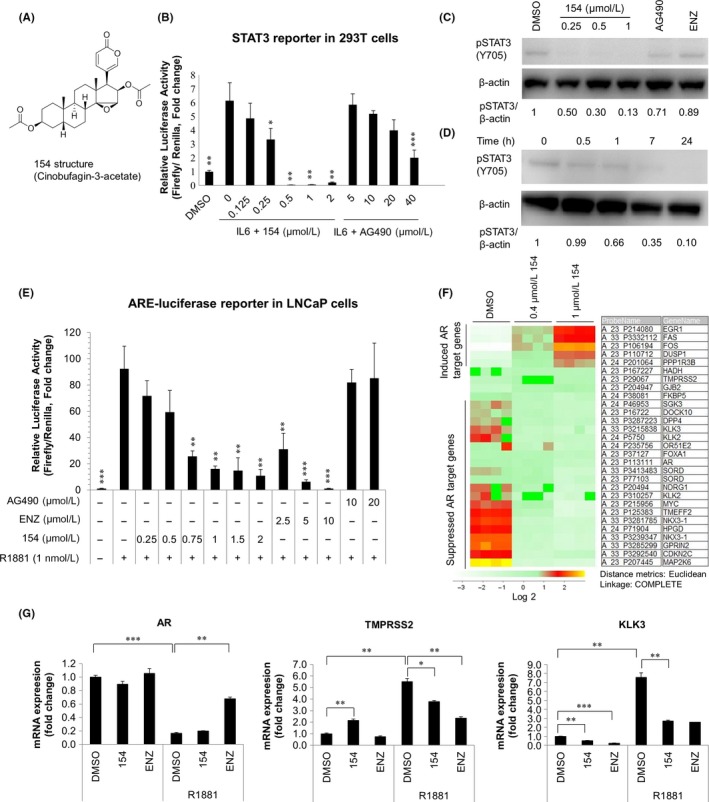
Compound 154 co‐targets IL6/STAT3 and AR pathway. A, Chemical structure of 154 (cinobufagin‐3‐acetate; CAS No. 4026‐97‐5; C28H36O7; molecular weight 484.25). B, 293T cells transfected with STAT3 promoter luciferase reporter were treated with indicated doses of compound 154 or AG490 with 10 ng/mL IL6 for 24 h. Luciferase assays were conducted to test if 154 affects STAT3 transcriptional activity. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Each group was compared with the group of IL6 treatment. C, EPT3M1‐STAT3 cells were treated in the presence or absence of 154 (0.25, 0.5, 1 μmol/L), AG490 (10 μmol/L) or enzalutamide (10 μmol/L) for 24 h. D, EPT3M1‐STAT3 cells were treated with 1 μmol/L 154 for the indicated times. Signal intensity was quantified with ImageJ (NIH). pSTAT3 level was normalized to β‐actin and fold change relative to control was calculated by using pSTAT3/β‐actin ratio. E, LNCaP cells were transfected with pLENTI7.3/V5‐DEST‐AR vector and Cignal ARE Reporter for 24 h, then treated with 154, AG490 or enzalutamide in the absence or presence of R1881 for 24 h. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. **P ≤ 0.01, ***P ≤ 0.001. Each group was compared to the group of R1881 (1 nmol/L) treatment. F, Agilent microarray gene expression data. LNCaP cells were treated with 0.4 or 1 μmol/L 154 for 24 h. The lower rows show suppressed genes, and the upper rows show genes induced by 154. DMSO treatment was used as a control. G, LNCaP cells were treated with either 1 μmol/L 154 or enzalutamide (10 μmol/L) in the presence of 1 nmol/L R1881 for 24 h. Real‐time qPCR was utilized to analyze the effect of 154 on the AR target genes. DSMO treated cells were used as the calibrator. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001
To examine the ability of 154 to affect transcriptional activity of the AR, LNCaP cells were cultured in charcoal‐stripped FCS RPMI‐1640 medium for 48 hours and cotransfected with an AR expressor pLENTI7.3/V5‐DEST AR vector and ARE reporter.21 The ARE luciferase activity in LNCaP cells was significantly activated by the synthetic androgen R1881 and could be blocked by compound 154 with similar effect as ENZ. The JAK2 inhibitor AG490 did not change ARE luciferase activity (Figure 1E). To test if 154 could affect the AR target gene expression, we treated LNCaP cells with 154 followed by real‐time quantitative PCR (qPCR) and microarray assays. Compound 154 at 0.4 and 1 μmol/L repressed several AR target genes, such as NKX3.1, KLK3, and FKBP5 according to Agilent microarray assays (Figure 1F). Compound 154 downregulated mRNA levels of androgen‐induced KLK3 (PSA) and TMPRSS2 according to quantitative real‐time PCR (qPCR) (Figure 1G). The results suggest that 154 may be an AR pathway modulator and inhibitor.
To determine whether 154 is a selective AR pathway inhibitor, we examined the effect of compound 154 on three other nuclear luciferase reporters, progesterone receptor (PR), estrogen receptor (ER), and glucocorticoid receptor (GR). Here we chose MCF7 cells to determine the selectivity of compound 154 on PR, ER, and GR, because it is reported that MCF7 cells are ERα+/PR+/AR+ cells with weak expression of GR.26 Figure 2A‐C shows that 154 is also a PR pathway modulator, but does not affect ER or GR pathways in MCF7 cells, indicating that 154 is a selective modulator. In order to test the effect of 154 on secreted PSA levels in LNCaP cells, supernatants were collected and tested after treatment with 154 or ENZ for 24 hours. In Figure 2D, 154 dramatically reduced the level of PSA in LNCaP cells compared to ENZ, even at the very low concentration 0.125 μmol/L. The results confirm the AR inhibitory activity of compound 154.
Figure 2.
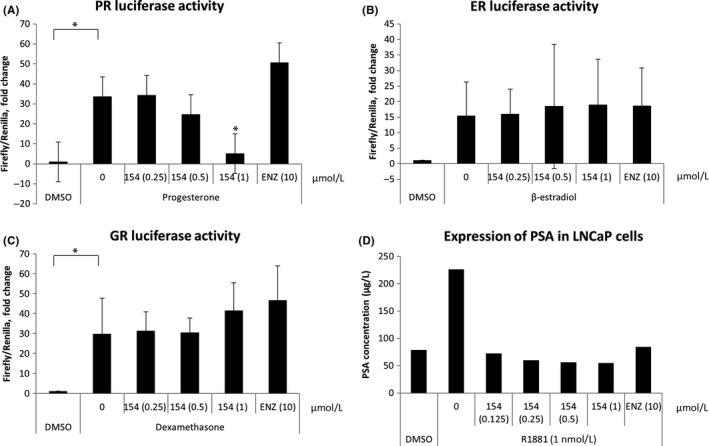
Compounds 154 is a selective AR inhibitor. MCF7 cells were cultured in 96‐well plates. A, MCF7 cells were transfected with 100 ng/well Cignal PR Reporter for 16 h, then treated with different doses of 154 or 10 μmol/L enzalutamide without (DMSO vehicle) or with 10 nmol/L progesterone for 18 h. B, MCF7 cells were transfected with 100 ng/well Cignal ERE Reporter or (C) Cignal GRE Reporter for 16 h, then treated with different doses of 154 or enzalutamide with or without 100 nmol/L dexamethasone, 10 nmol/L 17β‐estradiol (E2), respectively, for 6 h. D, LNCaP cells were cultured for 48 h, then treated with different doses of 154 or enzalutamide for 24 h, supernatants were collected for PSA measurements. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05
3.2. 154 reduces AR protein expression without altering AR mRNA level
AR protein remains crucial during PCa progression. AR mutations contribute to PCa development and lead to CRPC. First, we tested the effect of 154 on AR protein levels. Figure 3A shows that 154 reduced AR protein expression in a dose‐dependent manner in LNCaP cells. 154 decreased AR in the absence or presence of 1 nmol/L R1881 at 24 hours. A time‐course of treatment with 1 μmol/L 154 in LNCaP (Figure 3B) and 22Rv1 (Figure 3C) cells showed that downregulation of AR protein started at 8 hours. Figure 3D shows that 154 treatment did not affect AR mRNA levels in 22Rv1 cells.
Figure 3.
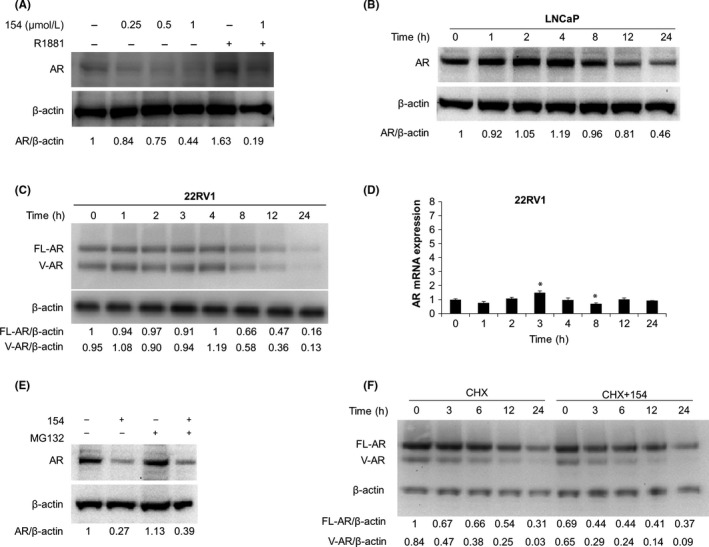
Compound 154 lowers AR protein levels. A, LNCaP cells treated with 0.25‐1 μmol/L 154 or 1 nmol/L R1881 for 24 h. B, LNCaP cells treated with 1 μmol/L 154 for the indicated times. C, 22Rv1 cells treated with 1 μmol/L 154 for the indicated times. D, 22Rv1 cells treated with 1 μmol/L 154 for the indicated time and then subjected to real‐time qPCR. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05. Each group was compare to the first group (Time = 0). E, LNCaP cells treated with 1 μmol/L 154 for 20 h, then co‐incubated with 10 μmol/L MG132 for 4 h. F, 22Rv1 cells treated with 1 μmol/L 154 with or without 50 μg/mL cycloheximide (CHX) at the same time and incubated for the indicated times. Signal intensity was quantified with ImageJ (NIH). AR or FL‐AR level was normalized to β‐actin and fold change relative to control was calculated by using AR/β‐actin or FL‐AR/β‐actin ratio
Next, we investigated whether the mechanism of AR downregulation by 154 was via an increase in the rate of AR degradation. The MG132 proteasome inhibitor did not prevent protein loss (Figure 3E), indicating that the molecular basis for this decline was not increased proteasome‐mediated AR degradation. To directly address whether 154 increased the rate of AR degradation in 22Rv1 cells, we used CHX to block new protein synthesis in a pulse‐chase assay. However, AR protein half‐life in 22Rv1 cells, was not decreased by 154, demonstrating that 154 did not directly enhance AR degradation (Figure 3F). The data suggest that 154 reduced the rate of AR synthesis.
Constitutively active AR splice variants include ARv567es and AR‐V7 which are important to CRPC development. We examined whether 154 changed the level of these two AR active splice variants in LNCaP or 22Rv1 cells. Figure 4A shows that 154 significantly reduced AR transcriptional activity in the presence or absence of ARv567es in LNCaP cells, while ENZ only works in the absence of ARv567es (Figure 4A left). Western blot analysis using an antibody against the AR NTD confirmed the approximate 1:1 ratio of FL‐AR to ARv567es in whole cell lysates of LNCaP cells treated with 154, ENZ and Abi (Figure 4B). As shown in Figure 4C, 154 decreased AR‐V7 expression in 22Rv1 cells in contrast to ENZ. Thus, 154 selectively inhibits constitutively active AR splice variants ARv567es and AR‐V7.
Figure 4.
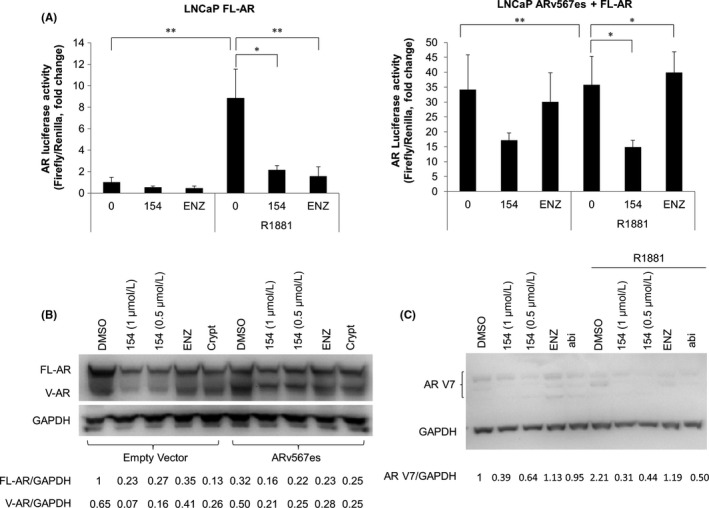
154 inhibits constitutively active AR splice variants ARv567es and AR‐V7. A, ARE‐luciferase activity in LNCaP cells with endogenous FL‐AR (left) or with both FL‐AR and transfected ARv567es (right) in the presence of ARE construct with or without 10 μmol/L enzalutamide or 1 μmol/L 154. B, LNCaP cells transfected with empty vector (left) or ARv567es (right) were treated with 10 μmol/L enzalutamide, cryptotanshinone or 1 μmol/L 154 for 24 h. Proteins were detected using the AR‐N20 antibody. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05, **P ≤ 0.01. Signal intensity was quantified with ImageJ (NIH). FL‐AR level was normalized to GAPDH and fold change relative to control was calculated by using FL‐AR/GAPDH ratio. C, 22Rv1 cells were treated with 10 μmol/L enzalutamide, abiraterone, cryptotanshinone or 1 μmol/L 154 in the presence or absence of 1 nmol/L R1881 for 24 h. Proteins were detected using AR‐V7 antibody. Signal intensity was quantified with ImageJ (NIH). ARV7 level was normalized to GAPDH and fold change relative to control was calculated by using AR V7/GAPDH ratio
3.3. 154 does not reduce AR nuclear translocation
AR translocation from cytoplasm to nucleus is a key step of AR pathway regulation.27 To examine if 154 reduces AR nuclear translocation and thereby affects the binding of AR to ARE, EPT3‐PT1‐AR (6.3) cells were established and treated with ENZ or 154 with R1881 for 24 hours. ENZ treatment impaired AR nuclear translocation that was induced by R1881 (Figure 5A). 154 did not remarkably alter the ratio of cytoplasmic and nuclear AR (Figure 5A).Western blot further confirmed that 154 did not reduce AR nuclear translocation that was triggered by R1881 in LNCaP cells (Figure 5B), which is different from AR antagonists like ENZ and ARN509.
Figure 5.
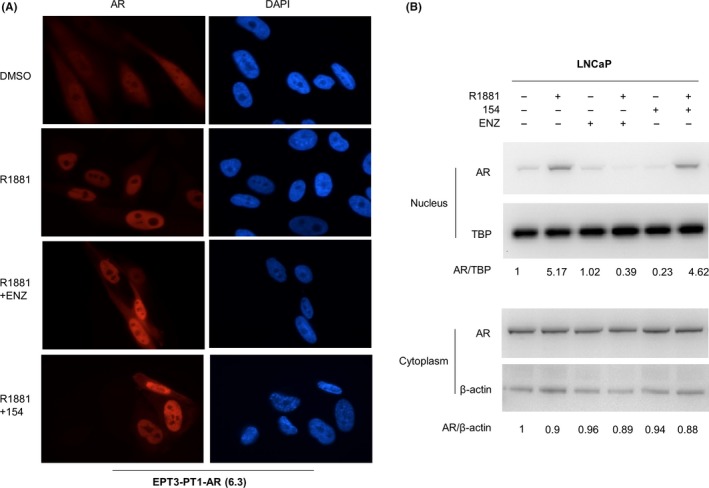
154 does not prevent AR nuclear translocation. A, Fluorescence microscopy representative results. EPT3‐PT1‐AR (6.3) cells were treated with 1 μmol/L 154 or 10 μmol/L enzalutamide plus 1 nmol/L R1881 or DMSO control for 24 h. B, LNCaP cells were pretreated for 4 h with 1 μmol/L 154 or 10 μmol/L enzalutamide before addition of 1 nmol/L R1881 or DMSO control for 1 hr. Signal intensity was quantified with ImageJ (NIH). AR or PSA level was normalized to β‐actin and fold change relative to control was calculated by using AR/β‐actin ratio
3.4. 154 as an antagonist of the AR NTD
EPI 001 was the first compound that targeted the disordered domain of the AF‐1 region of the AR NTD and inhibited AR transcriptional activity regardless of other AR domains.7, 28 ENZ binds to the AR LBD to compete with androgens like R1881.29 In order to test whether 154 could compete with R1881 as an AR LBD inhibitor, we tested the effect of increasing doses of R1881 in the presence of 1 μmol/L 154 on AR transcriptional activity and AR and PSA protein expression. In Figure 6A, the ability of ENZ to block AR activity was dramatically reduced at high concentrations of R1881. However, 154 inhibited AR activity consistently, regardless of increasing levels of R1881 in both LNCaP (Figure 6A, left) and 293T (Figure 6A, right) cells. As exemplified in Figure 6B, it was further confirmed that 154 still reduced both AR and PSA protein levels even at 50 nmol/L R1881, while ENZ did not. These data support that 154 does not compete for binding to the AR LBD.
Figure 6.
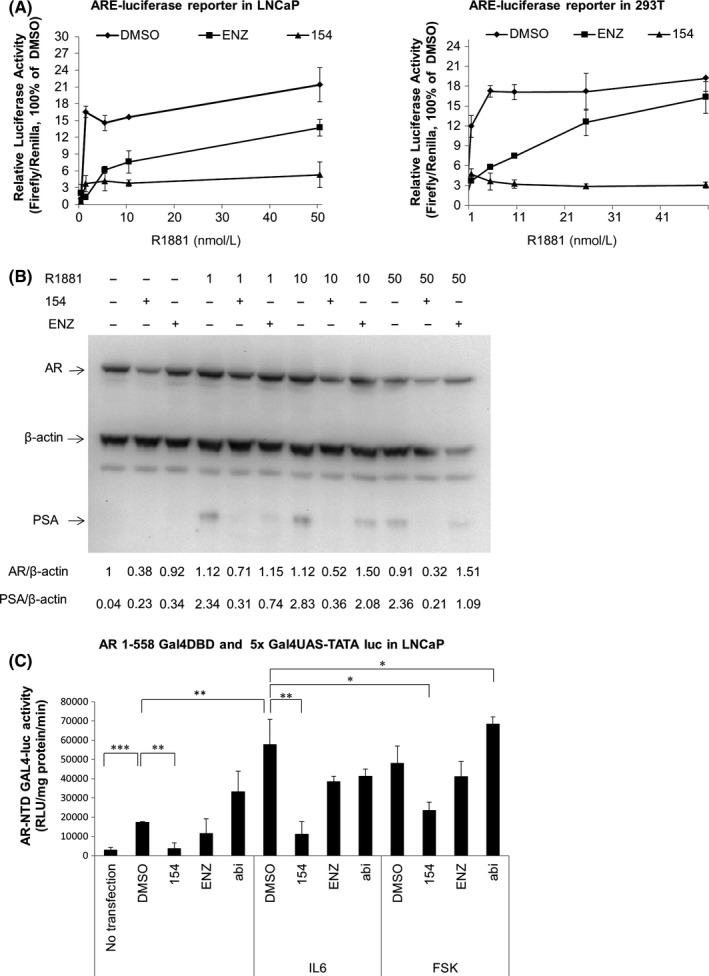
Compound 154 is an AR NTD inhibitor in LNCaP cells. A, LNCaP (left) and 293 (right) cells were co‐transfected with pLENTI7.3/V5‐DEST‐AR vector and ARE luciferase reporter for 24 h, then treated with 1 μmol/L 154, 10 μmol/L enzalutamide with or without increasing concentrations of R1881 for 24 h. B, LNCaP cells were treated with 1 μmol/L 154, 10 μmol/L enzalutamide with or without increasing concentrations of R1881 for 24 h. Signal intensity was quantified with ImageJ (NIH). AR level was normalized to TBP or β‐actin and fold change relative to control was calculated by using AR/TBP or AR/β‐actin ratio. C, AR NTD transactivation assay of LNCaP cells, transfected with 100 ng/well 5xGal4UAS‐TATA‐luciferase and 2 μg/well AR‐(1‐558)‐Gal4 DBD plasmid for 24 h, followed by incubation with 50 ng/mL IL6 or 50 μmol/L forskolin (FSK) or 1 μmol/L 154 for 24 h. All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001
The ability of 154 to inhibit transactivation of the AR NTD was tested in LNCaP cells cotransfected with an expression vector of a chimeric protein encoding amino acids 1‐558 of the human AR NTD fused to the Gal4DBD with a reporter gene containing the Gal4‐binding site. Androgen and the antiandrogen ENZ have no effect on this chimera due to lack of the AR LBD in the Gal4DBD‐AR1‐558 chimera. 154 reduced both IL6‐induced and forskolin (FSK, Figure 6C) induced transactivation of the AR NTD to baseline levels. Thus, 154 inhibits AR activity stimulated by both IL6 and FSK related pathways.
Hormone sensitive VCaP, 22Rv1, and LNCaP cells were found to harbor abundant AR NTD expression compared to no expression in the hormone insensitive cell line DU145 and very low expression in the EPT3‐M1 cells with high STAT3 level (Figure 7A). In both 22Rv1 (Figure 7A, middle) and VCaP (Figure 7A, right) cells, 154 reduced the expression of AR NTD. Interestingly, ENZ enhanced the level of AR variants in 22Rv1 cells, but reduced the AR NTD in VCaP cells. This has been illustrated previously13 and might be associated with drug resistance in mCRPC. Figure 7B shows that 154 treatment lowered the AR NTD signal activity in VCaP cells compared to ENZ, Crypt, or vehicle treatment, consistent with Western blot data. Taken together, 154 is an AR NTD inhibitor.
Figure 7.
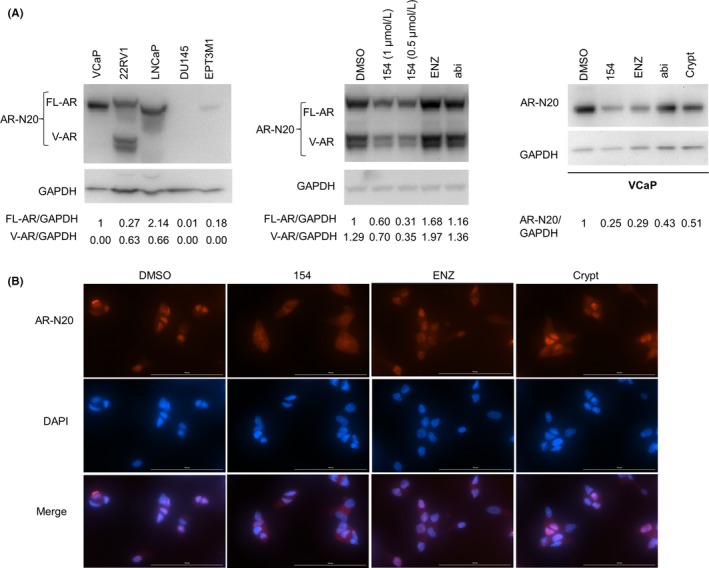
Compound 154 is an antagonist of the AR NTD in other prostate cancer cell lines. A, AR NTD protein expression analyzed by AR‐N20 antibody in different cell lines (left). 22Rv1 (middle) and VCaP cells (right) were treated with 0.5, 1 μmol/L 154, 10 μmol/L enzalutamide, 10 μmol/L abiraterone, 10 μmol/L cryptotanshinone, or vehicle for 24 h. Signal intensity was quantified with ImageJ (NIH). FL‐AR (left, middle) or AR‐N20 (right) level was normalized to GAPDH and fold change relative to control was calculated by using FL‐AR/GAPDH or AR‐N20/GAPDH ratio. B, Indirect immunofluorescence assay of VCaP cells treated with 1 μmol/L 154, 10 μmol/L enzalutamide, 10 μmol/L cryptotanshinone or vehicle for 24 h using the anti‐AR‐N20 antibody
3.5. 154 disrupts the association between STAT3 and AR NTD
To directly address if 154 targets AR and STAT3 protein, drug affinity responsive target stability (DARTS) assay was used for target identification which is based on the principle that the targeted protein bound by drugs will be more resistant against proteases. Western blotting showed protection of the target protein AR, whereas digestion of the nontarget proteins STAT3 and β‐actin was unchanged by 154. This suggests that 154 targets AR directly (Figure 8A).
Figure 8.
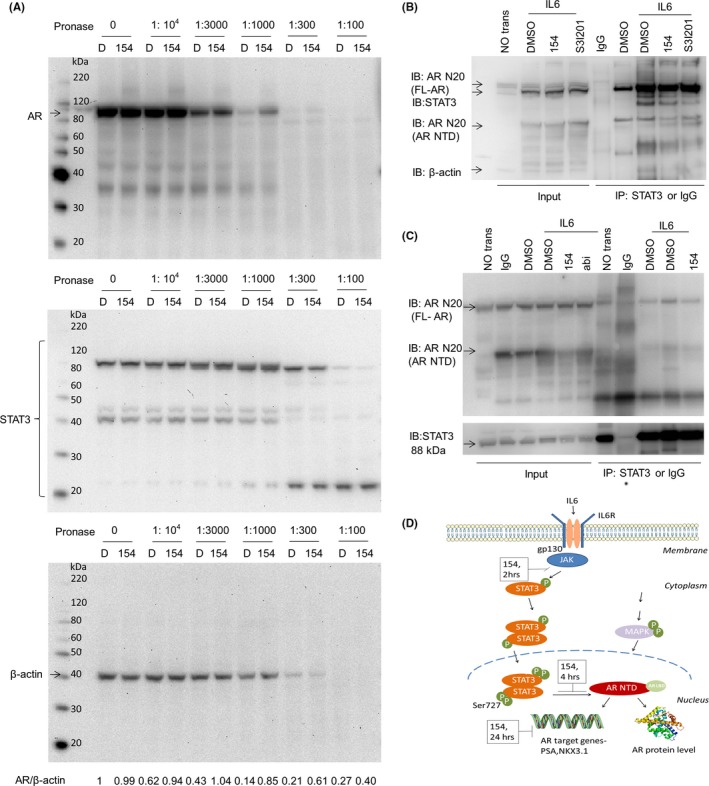
154 impairs the association between STAT3 and AR NTD. A, Left panels show DARTS. LNCaP cell lysates were prepared as described in,23 incubated with 500 μmol/L 154 or an equivalent amount of vehicle (DMSO) for 1 hr, followed by digestion with Pronase with protein ratios of 1:10 000, 1:3000, 1:1000, 1:300, and 1:100 for 30 min. Then the samples were subjected to western blotting with antibodies against AR (ab133273, 1:1000), STAT3 (ab119352, 1:2500), or β‐actin (ab8226, 1:2000), D = DMSO. Signal intensity was quantified with ImageJ (NIH). AR level was normalized to β‐actin and fold change relative to control was calculated by using AR/β‐actin ratio. B and C, LNCaP cells were transfected with FLAG‐STAT3 and FLAG‐AR‐NTD plasmids and were treated with 1 μmol/L 154 or 10 μmol/L abiraterone in the presence or absence of IL6 (50 ng/mL) for 4 h (B) or 6 h (C). Extracts were immunoprecipitated (IP) with STAT3‐antibody (ab119352) or negative control IgG and immunoblotting (IB) was done with the anti‐AR N20 antibody (Sc‐816, 1:1000) and anti‐STAT3 (ab119352, 1:5000). D, The proposed mechanism of compound 154 on dual STAT3 and AR pathway signaling17
It is reported that STAT3 interacts with AR NTD at amino acids 234‐558 in LNCaP cells after exposure to IL6 for 6 hours.30 According to our study, 154 inhibited both the IL6/STAT3 and AR pathways. Based on this, we proposed the hypothesis that the series of changes caused by 154 in the AR pathway mainly relies on the association between IL6‐activated STAT3 and AR NTD. In order to investigate this possibility, we transiently transfected LNCaP cells with FLAG‐STAT3 plasmid and FLAG‐AR‐NTD plasmid and next stimulated with IL6 for 4 hours (Figure 8B) and 6 hours (Figure 8C), respectively, and then immunoprecipated with anti‐STAT3 antibody to pull down STAT3 associated proteins, which were subjected to the western blot and anti‐AR N20 antibody incubation. Both Figure 8B and C, indicate the association between STAT3 and AR NTD. 154 could downregulate the IL6‐activated association, whereas the STAT3 inhibitor S3I‐201 did not (Figure 8B). Of note, inhibitor treatment for these experiments was for shorter time than required to reduce the level of AR protein. Therefore, this experiment confirmed that 154 firstly disrupted the association between STAT3 and AR NTD within 4 hours, followed thereafter by reduced expression of AR protein, including AR NTD.
To sum up (Figure 8D), compound 154 reduced phosphorylation of STAT3 at Tyr705 within 2 hours in cells, which results in downregulation of the STAT3 pathway. It is reported that phosphorylated STAT3 at Ser727 and STAT3 could directly interact with the NTD of the AR,17 which contains the major transactivation function of AR. Therefore, downregulation of phosphorylated STAT3 by 154 caused AR pathway downregulation through STAT3‐AR‐NTD dissociation within 4 hours.
3.6. 154 treatment decreases the ability to form colonies
To determine the antitumor activities of 154, we analyzed its effect in in vitro clonogenic assays. In vitro clonogenic assays correlate well with in vivo assays of tumorigenicity in nude mice.31 Figure 9A shows that clonogenicity of three cancer lines, DU145, EPT3M1‐STAT3, and LNCaP cells, was reduced in a concentration‐dependent manner after exposure to 154. Cell cytotoxic effect of 154 was further analyzed by fluorescence signal intensity in EPT2‐D5 miR146a cells with CMV promoter‐driven miR146. 154 did not alter the fluorescence signal activity detected by the multi‐plate reader (Figure 9B) and the mCherry signal by indirect immunofluorescence microscopy analysis (Figure 9C). Furthermore, LNCaP‐207‐1 cells that contain ARE‐driven mCherry reporter and SV40 promoter driven constitutive GFP reporter were utilized to test whether the above effects of 154 in both IL6/STAT3 and AR pathways were due to its cytotoxicity. At 90% confluency, LNCaP‐207‐1 cells were stimulated with 1 nmol/L R1881, simultaneously different doses of 154 compound, ENZ or Abi were added for 24 hours. ARE‐reporter responses were analyzed by fluorescence microscopy (Figure S2A), fluorescence multi‐plate reader (Figure S2B) and flowcytometry (Figure S2C). As shown in Figure S2, 154 decreased the signals of ARE driven mCherry, but not of the SV40‐driven GFP fluorescence. This supports the specificity of 154 targeting.
Figure 9.
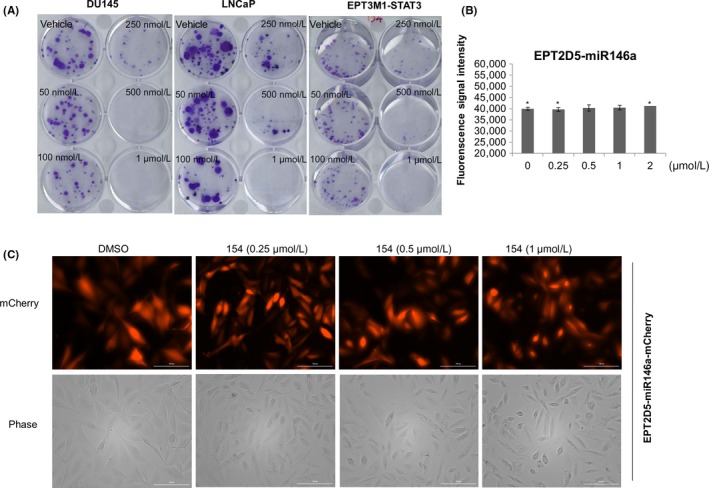
154 reduces cell clonogenicity. A, On the 2nd day after seeding, compound 154 was added and DU145, LNCaP, and EPT3M1‐STAT3 cells were allowed to grow for 2 weeks to form colonies and next stained with 0.4% crystal violet (w/v). EPT2‐D5‐miR146a cells were treated with different doses of 154 for 24 h. Fluorescence signals were analyzed by the multi‐plate reader (B) and indirect immunofluorescence microscopy analysis (C). All data are represented as the average ± SEM (n = 3). Significance was verified by using unpaired two‐tailed Student's t test. *P ≤ 0.05
4. DISCUSSION AND CONCLUSIONS
IL6 is a cytokine that activates STAT3 and contributes to growth regulation of several tumors.32 Overexpression of IL6 was found to correlate with increased metastasis33 and decreased survival34 in PCa patients.17 Constitutively active STAT3 also regulates the progression of PCa and plays a key role in drug resistance, tumor immunoescape, and chemoresistance.35 Recently, it was summarized that STAT3 is crucial to reactivation of AR, regulates interactions between tumor cells and the microenvironment in CRPC progression.17 Don‐Doncow et al36 analyzed 223 metastatic samples from 71 CRPC patients and found that 95% of metastases were positive for pSTAT3 and IL6R and with high STAT3 mRNA levels. Therefore, STAT3 is an attractive target for cancer therapy in advanced PCa. One STAT3 inhibitor, Galiellalactone, was reported to remarkably reduce tumor growth and downregulate metastatic spread of PCa cells to lymph nodes.35
The progression of CRPC relies on AR pathway signaling.37 Strategies for direct targeting the AR pathway have been thoroughly investigated, including inhibition of AR protein levels or reducing AR nuclear translocation and AR target genes by binding of small molecules.38 These strategies have produced compounds with in vivo activity, such as the AR LBD inhibitor ENZ, the CYP17A1 inhibitor Abi, and the AR NTD inhibitor EPI‐001. However, AR gene amplification, mutations in the AR LBD or posttranslational modifications of AR can contribute to increasing AR responses to low levels of androgen, thereby enhancing AR reactivation in CRPC and drug resistance to AR LBD inhibitors or CYP17A1 inhibitors.39 Thus, combination therapies with agents that block other pathways, like dual targeting of both IL6/STAT3 and AR pathways, may be beneficial for CRPC patients by increasing efficacy of the therapy and reducing drug resistance. Meanwhile, as AR splice variants AR‐V7 or ARv567es are very weakly expressed in PCa cell lines, constructs of overexpression with AR‐V7 or ARv567es fused to fluorescent tag might be very useful for the further study.
This study identified, for the first time, a biologically active small molecule with dual targeting of both IL6/STAT3 and AR pathways and investigated its value for drug development against CRPC. We found that 154 was an antagonist of the AR NTD, could bind to AR directly, inhibited both AR and STAT3 transcriptional activity by disrupting the association between STAT3 and AR NTD. Furthermore, 154 downregulated the mRNA level of AR target genes, inhibited AR protein expression in a dose‐ and time‐dependent manner without altering AR mRNA level. Moreover, compound 154 did not reduce AR nuclear translocation.
Thus, 154 presents itself as a leading compound for dual inhibition of IL6/STAT3 and AR pathways, thereby providing a promising new PCa treatment modality. The cytotoxicity of 154 is quite high in several different cell lines and the side effect might also be higher in vivo. Thus, modification of 154 will be needed to reduce cytotoxicity. This study also suggests that STAT3 and AR might be active at the same promoters in an association which may serve as a therapeutic target in CRPC.
DISCLOSURES
None declared.
AUTHOR CONTRIBUTIONS
The experiments were designed and performed by Y.H., who proposed the hypothesis, analyzed the data and wrote the manuscript as well. W.A. generated LNCaP‐207‐1 cells and performed the experiment of Figure S2. W.Z. and Y.S. extracted, analyzed and provided the compound 154. S.Z. was responsible for the computational virtual screening of target validation. J.J. analyzed the data. A.Ø., X.K., K.K. conceived the hypothesis and analyzed the data.
Supporting information
ACKNOWLEDGEMENTS
The authors thank Stephen Plymate for providing ARv567es construct; Dr. Saïd M. Sebti for providing FLAG‐STAT3 plasmids; Marianne Sadar for providing FLAG‐AR‐NTD plasmid, AR‐1‐558‐Gal4DBD and 5xGal4UAS‐TATA‐luciferase vectors and Hua My Hoang for DNA microarray profiling.
Hua Y, Azeem W, Shen Y, et al. Dual androgen receptor (AR) and STAT3 inhibition by a compound targeting the AR amino‐terminal domain. Pharmacol Res Perspect. 2018;e00437 10.1002/prp2.437
Funding information
The work was supported by Bergen Research Foundation, Helse Vest (grants 911778, 911626, 911747, 912062, 990094) and Helse Vest Strategic Programs 2015‐2019, Bergen Stem Cell Consortium (grant 502027) and Personalized Medicine (grant 303484 “Liquid Biopsies”), the Research Council of Norway through its Centres of Excellence funding scheme, project number 223250, Professor of Chang Jiang Scholars Program, Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200). We greatly acknowledge support of this study by Einar Galtung Døsvig, Espen Galtung Døsvig, Bjarne Rieber, Trond Mohn, Herman Friele, Jan Einar Greve, Thorstein Selvik, Bergen Research Foundation (BFS) and Kåre Rommetveit.
Contributor Information
Yaping Hua, Email: yaping.hua@uib.no, Email: kalland@uib.no.
Weidong Zhang, Email: wdzhangy@hotmail.com.
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359‐E386. 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2. Miyamoto DT, Zheng Y, Wittner BS, et al. RNA‐Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amin KS, Jagadeesh S, Baishya G, et al. A naturally derived small molecule disrupts ligand‐dependent and ligand‐independent androgen receptor signaling in human prostate cancer cells. Mol Cancer Ther. 2014;13(2):341‐352. 10.1158/1535-7163.MCT-13-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine‐specific demethylase 1. Cancer Cell. 2011;20(4):457‐471. 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dalal K, Roshan‐Moniri M, Sharma A, et al. Selectively targeting the DNA‐binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289(38):26417‐26429. 10.1074/jbc.M114.553818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bambury RM, Rathkopf DE. Novel and next‐generation androgen receptor‐directed therapies for prostate cancer: beyond abiraterone and enzalutamide. Urol Oncol. 2016;34(8):348‐355. 10.1016/j.urolonc.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 7. Andersen RJ, Mawji NR, Wang J, et al. Regression of castrate‐recurrent prostate cancer by a small‐molecule inhibitor of the amino‐terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535‐546. 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 8. Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287(23):19736‐19749. 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qu Y, Dai B, Ye D, et al. Constitutively active AR‐V7 plays an essential role in the development and progression of castration‐resistant prostate cancer. Sci Rep. 2015;5:7654 10.1038/srep07654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y, Hwang TH, Oseth LA, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31(45):4759‐4767. 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain‐mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007;67(19):9001‐9005. 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- 12. Kelvin J, Lu D, Louw J, et al. Predictive biomarkers of sensitivity to androgen receptor signaling (ARS) and taxane‐based chemotherapy in circulating tumor cells (CTCs) of patients (pts) with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol. 2015;33(suppl 7; abstr 147):1‐1.25332246 [Google Scholar]
- 13. Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand‐dependent full‐length androgen receptor and its splice variants in castration‐resistant prostate cancer. Cancer Res. 2012;72(14):3457‐3462. 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715‐2730. 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antonarakis ES, Lu C, Wang H, et al. AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028‐1038. 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Onstenk W, Sieuwerts AM, Kraan J, et al. Efficacy of cabazitaxel in castration‐resistant prostate cancer is independent of the presence of AR‐V7 in circulating tumor cells. Eur Urol. 2015;68(6):939‐945. 10.1016/j.eururo.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 17. Bishop JL, Thaper D, Zoubeidi A. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers (Basel). 2014;6(2):829‐859. 10.3390/cancers6020829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu C, Zhu Y, Lou W, Cui Y, Evans CP, Gao AC. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate. 2014;74(2):201‐209. 10.1002/pros.22741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qu Y, Øyan AM, Liu R, et al. Generation of prostate tumor‐initiating cells is associated with elevation of reactive oxygen species and IL‐6/STAT3 signaling. Cancer Res. 2013;73(23):7090‐7100. 10.1158/0008-5472.CAN-13-1560. [DOI] [PubMed] [Google Scholar]
- 20. Zhang X, Sun Y, Pireddu R, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922‐1933. 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Azeem W, Hellem MR, Olsen JR, et al. An androgen response element driven reporter assay for the detection of androgen receptor activity in prostate cells. PLoS One. 2017;12(6):e0177861 10.1371/journal.pone.0177861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011;6(1):34‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci USA. 2009;106(51):21984‐21989. 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dysvik B, Jonassen I. J‐Express: exploring gene expression data using Java. Bioinformatics. 2001;17(4):369‐370. [DOI] [PubMed] [Google Scholar]
- 25. Liu L, Hua Y, Wang D, et al. A sesquiterpene lactone from a medicinal herb inhibits proinflammatory activity of TNF‐alpha by inhibiting ubiquitin‐conjugating enzyme UbcH5. Chem Biol. 2014;21(10):1341‐1350. 10.1016/j.chembiol.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 26. Dart DA, Waxman J, Aboagye EO, Bevan CL. Visualising androgen receptor activity in male and female mice. PLoS One. 2013;8(8):e71694 10.1371/journal.pone.0071694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clegg NJ, Wongvipat J, Joseph JD, et al. ARN‐509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72(6):1494‐1503. 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Myung JK, Banuelos CA, Fernandez JG, et al. An androgen receptor N‐terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123(7):2948‐2960. 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tran C, Ouk S, Clegg NJ, et al. Development of a second‐generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787‐790. 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N‐terminal domain by interleukin‐6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277(9):7076‐7085. 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- 31. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi‐solid medium. Cell. 1975;3(4):355‐359. [DOI] [PubMed] [Google Scholar]
- 32. Qiu Y, Ravi L, Kung HJ. Requirement of ErbB2 for signalling by interleukin‐6 in prostate carcinoma cells. Nature. 1998;393:83‐85. [DOI] [PubMed] [Google Scholar]
- 33. Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin‐6 and transforming growth factor‐beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161(1):182‐187. [PubMed] [Google Scholar]
- 34. Jun N, Masaaki T, Yutaka H, et al. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6(7):2702‐2706. [PubMed] [Google Scholar]
- 35. Canesin G, Evans‐Axelsson S, Hellsten R, et al. The STAT3 inhibitor galiellalactone effectively reduces tumor growth and metastatic spread in an orthotopic xenograft mouse model of prostate cancer. Eur Urol. 2016;69(3):400‐404. 10.1016/j.eururo.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 36. Don‐Doncow N, Marginean F, Coleman I, et al. Expression of STAT3 in prostate cancer metastases. Eur Urol. 2017;71(3):313‐316. 10.1016/j.eururo.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8(4):440‐448. 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu C, Nadiminty N, Tummala R, et al. Andrographolide targets androgen receptor pathway in castration‐resistant prostate cancer. Genes Cancer. 2011;2(2):151‐159. 10.1177/1947601911409744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration‐resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33(22):2815‐2825. 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials