

Abstract
Identifying physiological roles of specific signaling pathways that regulate hematopoietic stem cell (HSC) functions may lead to new treatment strategies and therapeutic interventions for hematologic disorders. Here, we provide genetic evidence that constitutive activation of NF-κB in HSCs results in reduced pool size, repopulation capacities, and quiescence of HSCs. Global transcriptional profiling and bioinformatics studies identified loss of ‘stemness’ and ‘quiescence’ signatures in HSCs with deregulated NF-κB activation. In particular, gene set enrichment analysis identified upregulation of cyclin dependent kinase- Ccnd1 and down regulation of cyclin dependent kinase inhibitor p57kip2. Interestingly, constitutive activation of NF-κB is sufficient to alter the regulatory circuits of transcription factors (TFs) that are critical to HSC self-renewal and functions. Molecular studies identified Junb, as one of the direct targets of NF-κB in hematopoietic cells. In essence, these studies demonstrate that aberrant activation of NF-κB signals impairs HSC quiescence and functions and alters the ‘TF networks’ in HSCs.
Keywords: hematopoietic stem cells, NF-κB, quiescence, transcription factors, signal transduction
Introduction
Hematopoiesis is a process through which all the blood cells are generated through sequential cell divisions and differentiation of progeny that originate from hematopoietic stem cells (HSCs) (Kondo et al., 2003). Under steady state conditions HSCs remain largely quiescent (Morrison and Weissman, 1994; Passegue et al., 2005), and divide < 20 times in their entire life span (Wilson et al., 2008; Trumpp et al., 2010). A tight control on quiescence is vital for maintaining the “stemness” of HSCs (Wilson and Trumpp, 2006; Wilson et al., 2008). Indeed, decision of HSCs to remain at a quiescent state or enter into an actively proliferating state is controlled by number of factors through both cell intrinsic and extrinsic mechanisms. In response to extrinsic soluble factors; inflammatory cytokines such as interferon (IFN)-α and IFN-γ; growth factors such as granulocyte colony stimulating factor (GCSF), stem cell factor (SCF), and thrombopoietin (TPO);cytokines such as transforming growth factor (TGF) -β and tumor necrosis factor (TNF)-α; and chemokines such as the stromal cell derived factor (SDF)-1, HSCs can either enter dormancy or cell cycle (Wilson and Trumpp, 2006; Kiel and Morrison, 2008; Trumpp et al., 2010). Intrinsic factors that regulate HSC quiescence includes; cell cycle inhibitors such as p21 and p57; transcription factors (TFs) such as Gfi1, Egr1, FOXOs, and PBX1; and ubiquitin ligases such as c-Cbl, Itch, Fbxw7, and A20 (Orford and Scadden, 2008; Rathinam et al., 2008, 2011; Thompson et al., 2008; Trumpp et al., 2010; Zou et al., 2011; Nakagawa et al., 2015). A harmony between intrinsic and extrinsic factors is essential for proper maintenance of HSCs in the bone marrow niche.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) proteins act as TFs and are regarded as the master regulators of innate and adaptive immunity (Vallabhapurapu and Karin, 2009). They are five key members of this family in mammals: Rel A (p65), Rel B, c-Rel, p50/p105 (also known as NF-κB1) and p100/52 (also known as NF-κB2) (Ghosh and Hayden, 2008). NF-κB signals are activated in response to various upstream stimuli. In the absence of any activating signals, inhibitor of NF-κB (IκB) proteins form complexes with inactive NF-κB proteins and remain in the cytoplasm. Nevertheless, following an activating signal, the IκB kinase (IKK) complex, which is composed of two kinases; IKK1 (IKKα), IKK2 (IKKβ) and a regulatory subunit NEMO (IKKγ), phosphorylates IκB, which leads to ubiquitylation and subsequent degradation of IκB (Rothwarf et al., 1998). This results in the release of NF-κB complexes from the cytoplasm to the nucleus, where they activate expression of target genes (Ghosh and Hayden, 2008). Therefore, in the entire cascade of NF-κB signals IKK complex plays a major role, as decontrolled activation of the IKK complex can lead to detrimental downstream consequences. IKK complex phosphorylates IκB proteins at two amino (N)-terminal regulatory serine residues (Brown et al., 1995). In majority of the canonical NF-κB signaling pathways, IKK2 is necessary and sufficient to phosphorylate IκB and activate NF-κB (Ghosh and Hayden, 2008).
Inflammatory control of HSCs has emerged as a novel mode of HSC regulation in the recent years. HSCs are believed to sense immune insults by both cell intrinsic, and extrinsic mechanisms (Welner et al., 2008; Baldridge et al., 2011; King and Goodell, 2011). Similar to differentiated immune cells, HSCs recognize pathogens through their expression of toll-like receptors (TLRs). Ligation of TLR signals in HSCs leads to proliferation and differentiation (Nagai et al., 2006; Takizawa et al., 2017). On the other hand, cell extrinsic mode of recognition of an immune insult by HSCs involves signaling through receptors for pro-inflammatory cytokines. A spectrum of pro-inflammatory cytokines and chemokines, that includes IL1, IL6, IL8, TNF, CC-Chemokine ligand 2 (CCL2), IFN-α and IFN-γ (Baldridge et al., 2011; King and Goodell, 2011; Mirantes et al., 2014) has been shown to influence HSCs. Of note, prolonged exposure of HSCs to pro-inflammatory cytokines causes diminished self-renewal and quiescence (Essers et al., 2009; Sato et al., 2009; Baldridge et al., 2010). In this regard, NF-κ B can be viewed as a gatekeeper on the inflammatory control of HSCs. Indeed, NF-κ B is the major downstream effector of signals transduced by both TLRs and pro-inflammatory cytokines (Ghosh and Hayden, 2008; Vallabhapurapu and Karin, 2009). In addition, uncontrolled NF-κ B activity results in increased expression of pro-inflammatory cytokines, including TNF, IL1, IL6, and IFN-γ (Boone et al., 2004; Coornaert et al., 2009; Dong et al., 2010; Harhaj and Dixit, 2012). A recent study from our group demonstrated that defects in the negative regulatory circuits of NF-κ B signaling cause loss of quiescence and pre-mature depletion of HSCs (Nakagawa et al., 2015). While these studies highlight the importance of NF-κ B pathways in HSC quiescence, the downstream consequences of NF-κ B signals and the precise mechanisms through which NF-κ B controls HSCs remain largely unknown. More importantly, constitutive activation of NF-κB has been documented in different types of human diseases, including myeloid neoplasms (Brown et al., 1995; Guzman et al., 2001; Panagopoulos et al., 2013). In particular, NF-κB has been shown to be constitutively active in leukemic stem cells (LSCs) (Kagoya et al., 2014). These links to human disease provide a compelling rationale for more detailed investigations into the mechanisms through which NF-κB induces leukemogenesis and transformation of normal HSCs to LSCs. Moreover, these studies were conducted with LSCs and the consequences of constitutive NF-κB signaling in normal HSCs remains unexplored.
Thus, in the present study we attempted to define the role of NF-κB in HSC biology and to gain molecular insights into mechanisms through which NF-κB impacts HSCs. To this end, we followed a ‘gain of function’ approach to delineate NF-κB functions in HSCs. Using mice that express the constitutively active form of IKK2 in HSCs, we show that the HSC pool was reduced and HSC functions were compromised. HSCs from these mice exhibited a hyper-proliferative phenotype associated with loss of quiescence. Genome-wide transcriptional profiling studies combined with ‘in silico’ analysis identified deregulated molecular and genetic signatures of HSCs and ‘TF network’ in HSCs with constitutive activation of NF-κB. Furthermore, molecular studies identified deregulated expression of the quiescence factor-p57 and that Junb as one of the key targets of NF-κB in hematopoietic cells. Taken together, these data indicate that NF-κB signaling plays a key role in the determination of ‘quiescence’ vs. ‘active’ state of HSCs and that fine-tuning of NF-κB signaling preserves the molecular and genetic identities of HSCs.
Materials and Methods
Mice
R26STOPFLIKK2ca (B6-Gt(ROSA)26Sortm1(Ikbkb)Rsky/J Stock #: 008242) transgenic mice (Sasaki et al., 2006) and Vav-Cre(B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J, stock #: 008610) (de Boer et al., 2003) mice were purchased from the Jackson laboratory. B6.CD45.1 congenic (stock #: 002014) congenic animals were purchased from the National Cancer Institute. All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University and University of Maryland School of Medicine.
Bone Marrow Transplantation
1 × 106 of bone marrow cells were injected into lethally irradiated (10 Gy) congenic (CD45.1+) recipient mice. For competitive-repopulation experiments, 5 × 105 of bone marrow cells were mixed with equal numbers of CD45.1+ competitor cells and injected into congenic recipients.
Cell Proliferation and Quiescence
For bromodeoxyuridine (BrdU) assay, 3.33 mg of BrdU (BD Pharmingen) was injected intraperitoneally and mice were maintained on 0.8 mg/ml BrdU in the drinking water. After 16 h of injection, mice were sacrificed and bone marrow cells were stained for BrdU, following the BrdU flow kit manufacturer’s instructions (BD Pharmingen).
Cell Cycle
For pyronin Y staining, cells were first incubated with 5 μg/ml hoechst 33342 (Life technologies) at 37°C for 45 min and then with pyronin Y (Sigma-Aldrich), at 1 μg/ml, for an additional 45 min at 37°C (Cheng et al., 2000). For side population assays, cells were incubated with 5 μg/ml Hoechst 33342 (Life Technologies) at 37°C for 90 min.
Flow Cytometry
Cells were analyzed by flow cytometry with FACS Fortessa or LSR II (BD) and FACSDiva software (BD Biosciences) or FlowJo software (Tree Star). The following monoclonal antibodies were used: anti- CD34 (RAM34), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD48 (HM48-1), anti-CD117 (2B8), anti-Flt3 (A2F10.1), and anti-Sca-1 (D7) from BD Biosciences; anti-CD150 (TC15- 12F12.2) from Biolegend; anti-CD16/32 (93) and anti-CD127 (A7R34) from eBioscience. In all the FACS plots, indicated are the percentages (%) of the gated fraction.
Apoptosis Assay
Apoptotic cells were detected by annexin V PE apoptosis detection kit according to the manufacturer’s instructions (BD Bioscience).
Western Blot Analysis
Cells were lysed with cell lysis buffer (cell signaling) in the presence of protease inhibitor cocktail (complete, Roche) and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Santa Cruz Biotechnologies). Cell lysates were subjected to 10% SDS-PAGE, transferred to PVDF membranes (Bio-Rad) and were treated with primary and secondary antibodies, respectively. The blots were visualized using the protoglow ECL (National Diagnostics) and image station 440 (Kodak). Antibodies used were as follows: anti- IκBα (44D4; Cell Signaling), anti-phospho- IκBα (5A5; Cell Signaling), anti-actin (I-19; Santa Cruz Biotechnologies), HRP-conjugated anti-mouse IgG (Cell Signaling), HRP-conjugated anti-rabbit IgG (Cell Signaling), and HRP-conjugated anti-goat IgG (Santa Cruz Biotechnologies).
RNA Extraction and Real-Time PCR
Total RNA was isolated with RNeasy mini kit (Qiagen), then cDNA was synthesized with oligo (dT) primer and maxima reverse transcriptase (thermo scientific). Real-time PCR was performed in duplicates with a CFX-connect real-time PCR system (Biorad) and SsoAdvanced SYBR green supermix according to the manufacturer’s instructions (BioRad). Relative expression was normalized to the expression levels of the internal control-HPRT.
ChIP Assay
Chromatin immunoprecipitation (ChIP) assay was performed with pierce agarose ChIP kit (Pierce) according to the manufacturer’s instructions. In brief, 1 × 107 of bone marrow cells were fixed and immunoprecipitated with anti-p65 antibody (D14E12; Cell Signaling) or rabbit IgG (Pierce). Immunoprecipitated DNA fragment were quantified by real-time PCR with the use of the following primers, which amplify the Junb enhancer region containing NF-κB binding sites; forward 5′-ATAAGGTTCAGTACAAACGCCC-3′, reverse 5′-GCGTCACTGAGCTGAATAGG-3′. Fold enrichment was normalized to rabbit IgG-precipitated samples.
Microarray
Total RNA of CD150+CD48-LSK cells from either control or IKK2CA mice were isolated using the Qiagen RNAeasy micro kit according to the manufacturer’s instruction (Qiagen). Expression profiling was performed using Illumina’s MouseWG-6 v2.0 Expression BeadChip at Yale center for genome analysis. Normalized expression data were collapsed to gene symbols with max probes by collapsedataset module in Genepattern (Reich et al., 2006). These genes were pre ranked for log2 fold change and analyzed with gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005).
The list of TFs was selected with GO0003700 (sequence-specific DNA binding TF activity) from Quickgo with its default behavior (is_a, part_of, and occurs_in relationships only) (Binns et al., 2009).
GO Analysis
Upregulated genes (more than 1.5 fold) or downregulated genes (less than 0.67 fold) in IKK2CA mice were analyzed with DAVID 6.7 tools (Huang da et al., 2009).
Statistical Analysis
Data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001). For survival curve analysis, log rank test was used to assess statistical significance (∗P < 0.05 and ∗∗P < 0.01).
Results
Generation and Characterization of Mice With Constitutively Active IKK2 in HSCs
To investigate the gain of function role of NF-κB signals in hematopoiesis, we made use of the already available R26StopFLikk2ca transgenic mice that were previously engineered (Sasaki et al., 2006) to express two serine to glutamate substitutions in the activation loop of the kinase domain of IKK2, preceded by a loxP-flanked STOP cassette, into the ubiquitously expressed ROSA26 locus and followed by a frt-flanked IRES-EGFP cassette (Sasaki et al., 2006). IKK2 containing these serine to glutamate mutations (IKK2CA) is constitutively active (Mercurio et al., 1997). We crossed the R26StopFLikk2ca mice with the transgenic mice that express Cre under Vav1 promoter, to obtain heterozygous IKK2CAVavcre (henceforth referred as IKK2CA) mice. Vav1 promoter has been proven to be largely specific to the hematopoietic lineage, in particular, it is expressed at all stages of hematopoiesis, i.e., from HSCs to terminally differentiated myeloid, erythroid, and lymphoid lineage cells (de Boer et al., 2003).
To directly asses if NF-κB is constitutively activated in IKK2CA mice, we measured both total and phosphorylation levels of IκBα protein in hematopoietic cells. Western blot data indicated reduced levels of total IκBα protein and increased levels of phosphorylation IκBα protein in hematopoietic cells of IKK2CA mice, even in the absence of any specific stimulation (Figure 1A). As expected, data of real-time PCR assays indicated increased expression levels of key NF-κB targets, such as Nf-κb2 (∼4.5 fold), IκBα (∼ 6 fold), and IκB𝜀 (∼ 3.5 fold) in IKK2CA CD150 +CD48-LSK cells (Figure 1B). These data suggest that NF-κB signaling is constitutively active in the HSCs of IKK2CA mice.
FIGURE 1.
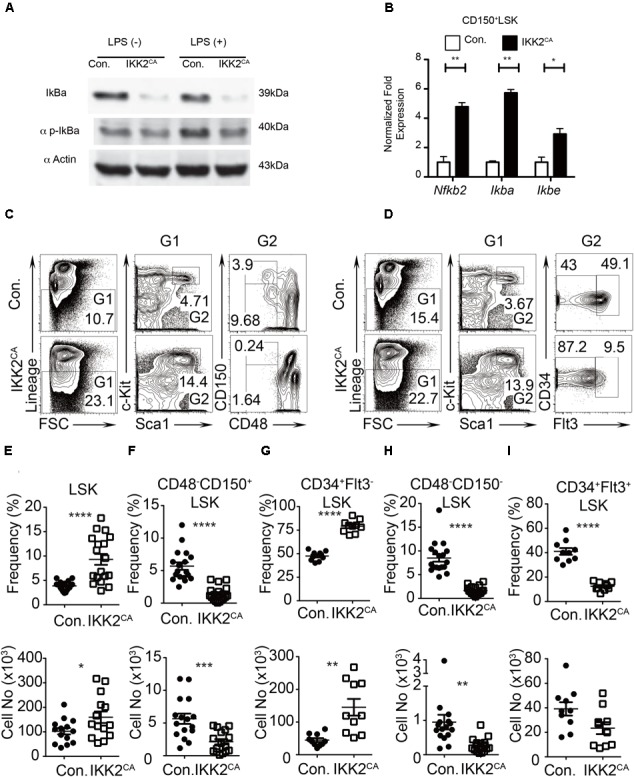
Reduced LT-HSC pool in IKK2CA mice. (A) Western blot analysis of total IκBα protein and phospho-IκBα protein in splenocytes, from 6 weeks old IKK2CA and control mice, either in the presence or absence of LPS stimulation for 1 h. Data are representative of two independent experiments. (B) Real time PCR analysis of NF-κB target genes in HSCs in the BM of 6 weeks old IKK2CA and control mice. Data are representative of two independent experiments. (C) FACS plots of LSK cells, LT-HSCs (CD48-CD150+LSK), and MPPs (CD48-CD150-LSK) from the BM (two femurs and two tibias) of 6 weeks old IKK2CA and control mice. Data are representative of five independent experiments. (D) FACS plots of LSK cells, ST-HSCs (CD34+Flt3-LSK), and MPPs (CD34+Flt3+LSK) from the BM (two femurs and two tibias) of 6 weeks old IKK2CA and control mice. Data are representative of five independent experiments. Relative (top) and absolute (bottom) numbers of LSK (n = 14) (E), LT-HSCs (CD48-CD150+LSK) (n = 16) (F), ST-HSCs (CD34+Flt3-LSK) (n = 10) (G), SLAM-MPPs (CD48-CD150-LSK) (n = 16) (H) and MPPs (CD34+Flt3+LSK) (n = 10) (I) in the BM (two femurs and two tibias) of 6 weeks old IKK2CA and control mice. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001).
Diminished LT-HSC Pool in the BM of IKK2CA Mice
IKK2CA mice were born in mendelian ratios and did not show any obvious gross morphological abnormalities. To investigate whether hematopoietic stem and progenitor cell (HSPC) pool was altered in IKK2CA mice, we measured the frequencies of HPSCs in the BM. Adult murine HSPCs are identified based on their Lineage-Sca1+c-Kit+ (LSK) immunophenotype and contain both HSC and multipotent progenitor (MPP) fractions. Recent studies identified that HSC pool in the BM of mice contains at least two physiologically distinct cell types; (1) long-term (LT) HSCs that are largely quiescent and with long-term capacities, and (2) short-term (ST) HSCs that are actively diving and with reduced long-term capacities (Kiel and Morrison, 2008; Orford and Scadden, 2008; Trumpp et al., 2010; Pietras et al., 2011). Analysis of HSPC pool in the BM of IKK2CA mice indicated an increase in the percentage (4.7% in control vs. 14.4% in IKK2ca mice) of LSK cells (Figure 1C). Distinguishing LSK cells into LT-HSCs (CD150+CD48-LSK) and MPPs (CD150-CD48-LSK) based on SLAM markers identified a decrease of both LT-HSCs (3.9% in control vs. 0.24% in IKK2CA mice) and SLAM-MPPs (9.7% in control vs. 1.6% in IKK2ca mice) (Figure 1C). Similarly, dividing LSK cells into ST-HSCs (CD34+Flt3-LSK) and MPPs (CD34+Flt3+LSK) based on Flt3 and CD34 expression identified a relative increase (43% in control vs. 87% in IKK2CA mice) of ST-HSCs and a decrease of conventional MPPs (49% in control vs. 9.5% in IKK2CA mice) (Figure 1D). Determination of relative and absolute numbers indicated an increase of LSK cells (Figure 1E), decrease of LT-HSCs (Figure 1F), increase of ST-HSCs (Figure 1G) and decrease of MPPs (Figures 1H,I). These data indicated that constitutive activation of NF-κB was sufficient to cause reduction in the LT-HSC pool and to alter the distribution of HSPC numbers.
Normal Numbers of Lineage Committed Progenitors in IKK2CA Mice
To test whether constitutive activation of NF-κB signals impair differentiation of lineage committed progenitors of the myeloid and lymphoid lineage, we performed immunophenotyping studies. Analysis of progenitor compartments revealed comparable relative and absolute frequencies of common myeloid progenitors (CMPs; Lin-c-Kit+CD34+CD16/32-), granulocyte monocyte progenitors (GMPs; Lin-c-Kit+CD34+CD16/32+) and megakaryocyte erythrocyte progenitors (MEPs; Lin-c-Kit+CD34-CD16/32-) between IKK2CA and control littermates (Supplementary Figures 1A,B). Similarly, lymphoid progenitor analysis indicated comparable relative and absolute numbers of common lymphoid progenitors (CLPs; Lin-c-KitmedSca1medIL7Rα+) between control and IKK2CA mice (Supplementary Figures 1C–E). To confirm these results, we performed an alternative surface staining that identifies CLPs based on Lin-c-KitmedSca1medIL7Rα+Flt3+ immunophenotype, and found comparable numbers of CLPs between control and IKK2CA mice (Supplementary Figure 1F). Similarly, lymphoid primed multi-potent progenitor (LMPPs; Lin-c-Kit+Sca1+Flt3+CD34+IL7Rα+) analysis did not reveal any difference between control and IKK2CA mice (Supplementary Figure 1G). Consistent with normal distribution of lineage restricted progenitors, differentiation of myeloid and lymphoid lineage cells was relatively normal in IKK2CA mice, although a modest decrease of B cells was noticed in the BM of these mice (data not shown). These data suggest that, even though the frequencies of MPPs were reduced due to constitutive NF-κB activation, differentiation of MPPs into CMPs, GMPs, MEPs, and CLPs was not impaired, at least, under steady state conditions.
Defective Repopulation Capacities of HSCs From IKK2CA Mice
To evaluate whether the functions of HSCs were intact in IKK2CA mice, we first performed bone marrow transplantation studies. Total BM cells (1 × 106) from either control or IKK2CA mice were injected i.v. into lethally (10 Gy) irradiated congenic (CD45.1+) wild type (WT) recipients. Strikingly, survival curve analysis indicated that ∼30% of the recipients, that received IKK2CA BM, died within 3 weeks of transplantation and further analysis at longer time points suggested that only ∼50% of IKK2CA BM recipients survived after 12 weeks of transplantation (Figure 2A). Whereas recipients that received control BM showed an average donor (CD45.2+) derived chimera of >70–80% in the peripheral blood at 4 weeks of transplantation, recipients that received IKK2CA BM exhibited reduced donor derived chimera, with an average chimera of ∼50% (Figure 2B). Donor derived multi-lineage analysis indicated that both myeloid and lymphoid differentiation was compromised in IKK2CA recipients, even though donor derived B lineage differentiation was more affected than myeloid differentiation (Figure 2C). Consistent with decreased donor derived hematopoiesis at 4 weeks, analysis of chimera at 11 weeks revealed a more severe reduction of IKK2CA derived hematopoiesis (Figure 2D) and multi-lineage analysis suggested that majority of these donor cells were of myeloid lineage (Figure 2E). Interestingly, our analysis indicated that a few IKK2CA recipient mice had a donor chimera of >75%, even after 11 weeks of transplantation (Figure 2D). However, transplantation of these BM cells from primary recipients into lethally irradiated secondary recipients failed to provide radioprotection (Figure 2F), suggesting that the BM of IKK2CA recipients with high donor chimera was devoid of functionally competent HSCs.
FIGURE 2.

Loss of HSC quiescence in IKK2CA mice. (A) Kaplan–Meier survival curve analysis of WT recipients (CD45.1+) that received BM of either IKK2CA (n = 19) or control (n = 8) mice (CD45.2+) following lethal (10 Gy) irradiation. Significance (P = 0.0398) was assessed using the log-rank test. (B) Frequencies of donor (CD45.2+) derived hematopoiesis, at 4 weeks of transplantation, in the PB of WT (CD45.1+) recipients. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 9 mice in control group and 10 mice in IKK2CA group. (C) Frequencies of donor (CD45.2+) derived CD11b+ myeloid cells and CD19+ B cells in the PB of WT (CD45.1+) recipients at 4 weeks of transplantation. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 9 mice in control group and 10 mice in IKK2CA group. (D) Frequencies of donor (CD45.2+) derived hematopoiesis, at 11 weeks of transplantation, in the PB of WT (CD45.1+) recipients. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 9 mice in control group and 8 mice in IKK2CA group. (E) Frequencies of donor (CD45.2+) derived CD11b+ myeloid cells, CD19+ B cells and CD3e+ T cells in the PB of WT (CD45.1+) recipients at 11 weeks of transplantation. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 9 mice in control group and 8 mice in IKK2CA group. (F) Serial transplantation experiments: Kaplan–Meier survival curve analysis of secondary WT recipients (CD45.1+) that received BM of the primary recipients, following lethal (10 Gy) irradiation. Each group represents data from a pool of four animals. Significance (P = 0.0067) was assessed using the log-rank test. (G) Competitive repopulation assays: frequencies of donor (CD45.2+) derived hematopoiesis, at 4 weeks of transplantation, in the PB of WT (CD45.1+) recipients. Recipients received mixed chimera containing donor (CD45.2+) BM cells (from either IKK2CA or control mice) and WT competitor (CD45.1+) BM cells at a ratio of 1:1. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 7 mice in control group and 6 mice in IKK2CA group. (H) Competitive repopulation assays: frequencies of donor (CD45.2+) derived hematopoiesis, at 4 weeks of transplantation, in the CD11b+ myeloid cells and CD19+ B cells in PB of WT (CD45.1+) recipients. Shown are representative FACS plots (left) and average frequencies (right). Data are pool of 7 mice in control group and 6 mice in IKK2CA group. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001). For survival curve analysis, log rank test was used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01).
Next, we tested the repopulation capacities of IKK2CA BM cells under competitive settings. BM cells from either control or IKK2CA mice (CD45.2+) were mixed with the BM cells of WT congenic (CD45.1+) mice at equal proportions (1:1) and were injected into lethally (10 Gy) irradiated WT congenic (CD45.1+) animals. While recipients of the control group showed an average of ∼40% of donor (CD45.2+) derived hematopoiesis, recipients that received IKK2CA BM showed an average chimera of only ∼8%, at 4 weeks of transplantation (Figure 2G). Multi-lineage analysis indicated that both myeloid and lymphoid differentiation was affected under competitive conditions, however, donor derived B cell differentiation was absent in IKK2CA recipients (Figure 2H). Together, these results indicated that constitutive activation of NF-κB results in impaired HSC functions.
Augmented Proliferation and Diminished Quiescence of IKK2CA HSCs
To determine whether the loss of LT-HSC pool in the BM of IKK2CA mice was due to increased apoptosis, we measured the frequencies of Annexin V+ cells in the LSK fraction. Surprisingly, the percentages of Annexin V+ cells were lesser in IKK2CA mice, when compared to the control animals (Figure 3A). Next, we tested whether the proliferation kinetics of HSCs was altered in IKK2CA mice. Our analysis indicated that the frequencies of BrdU+ cells were increased in both LSK and LT-HSC fractions (Figure 3B).
FIGURE 3.

Altered cell cycle properties of HSCs from IKK2CA mice. (A) Representative FACS plots (left) and average frequencies (right) of AnnexinV+ cells in LSK fractions in the BM of 6 weeks old IKK2CA and control mice. Data are pool of 5 mice per group. (B) Representative FACS plots (left) and average frequencies (right) of BrdU+ cells in LSK cells and LT-HSCs in the BM of 6 weeks old IKK2CA and control mice. Data are representative of two independent experiments with 3 mice per group. (C) Representative FACS plots (left) indicating frequencies of HSPCs in G0 (Pyronin Y- and Hoechst-), G1 (Pyronin Y+ and Hoechst-) and G2/S (Pyronin Y+ and Hoechst+) phase of cell cycle in the BM of 6 weeks old IKK2CA and control mice. Shown are average frequencies of HSPCs in G0 phase of cell cycle in the BM of 6 weeks old IKK2CA and control mice (right). Data are representative of two independent experiments with 3 mice per group. Representative FACS plots (left) and average frequencies (right) of side population (SP) fraction in Sca1+c-Kit+ (SK) cells (D) and CD48-CD150+Sca1+c-Kit+ cells (E) in the BM of 6 weeks old IKK2CA and control mice. Data are representative of two independent experiments with 3 mice per group. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01).
Based on these results we hypothesized that constitutive activation of NF-κB impairs HSC quiescence. To validate this, we first measured the frequencies of HSPCs in G0 phase under steady state conditions by using a combination of an RNA dye -pyronin and a DNA dye-Hoechst. Consistent with our hypothesis, we observed 46% of control LSK cells in the G0 phase, whereas only 14% of LSK cells from IKK2CA mice was in G0 phase (Figure 3C). To further investigate HSC quiescence in IKK2CA mice, we performed ‘side population’ (SP) studies, SP cells are a quiescent HSC population of immature LSK cells in the bone marrow (Arai et al., 2004). Strikingly, the data of SP studies identified 5% of quiescent cells in the Sca1+c-Kit+ BM fraction of control mice, while only 0.29% quiescent cells could be found in the same fraction of IKK2CA mice (Figure 3D). Further studies identified 21% of the cells to be quiescent in the LT-HSC enriched CD48-CD150+Sca1+c-Kit+ BM fraction of control mice, whereas only 4% of the same was found to be quiescent in IKK2CA mice (Figure 3E). These data strongly indicated that constitutive activation of NF-κB signals causes hyper-proliferation of HSCs and loss of quiescence in HSCs.
Loss of ‘Stemness’ Signature in IKK2CA HSCs
To uncover the molecular mechanisms through which constitutive NF-κB signals impair HSC quiescence and functions, we performed whole-genome transcriptome analysis. To this end, total RNA from CD150+ CD48-LSK cells from the BM of IKK2CA and control mice was hybridized on to the Illumina MouseWG-6 v2.0 Expression BeadChips. A direct comparison of gene expression profiles between the control and IKK2CA groups identified an upregulation (>1.5 fold) of 475 genes and a downregulation (<0.67 fold) of 672 genes in LT-HSCs of IKK2CA mice (Supplementary Table 1). Next, we performed gene ontology (GO) analysis, to get an overview on the major functional alterations that are caused by constitutive NF-κB signaling in HSCs. Data revealed that the major molecular functions of the upregulated genes (Figure 4A) in IKK2CA LT-HSCs include; ion binding, DNA binding, nucleotide binding, and TF activity. Similarly, the major molecular functions of the downregulated genes (Figure 4B) in IKK2CA LT-HSCs include; ion binding, nucleotide binding, DNA binding, and TF activity. Interestingly, the functions of 115 upregulated genes and of 166 downregulated genes in IKK2CA LT-HSCs remain unknown.
FIGURE 4.

Deregulated molecular signatures in IKK2CA HSCs. Gene ontology (GO) analysis of microarray data: top 10 major GO terms of molecular function among upregulated genes (>1.5 fold) (A) and downregulated genes (<0.67 fold) (B) in IKK2CA LT-HSCs. Gene set enrichment analysis (GSEA) of microarray data from IKK2CA versus control LT-HSCs with the following gene sets: genes enriched in normal, steady-state BM HSCs from WT mice compared to multi potent progenitors (MPPs), leukemic stem cells (LSCs), and mobilized HSCs (C). Genes enriched in HSCs, but not in MPPs (D). Genes enriched in human HSCs of cord blood (E). Quiescent stem cell genes, which are commonly upregulated in HSCs, muscle stem cells and hair follicle stem cells (F). Genes enriched in proliferative HSCs, which are commonly upregulated in fetal liver HSCs (vs. adult HSCs) and 2, 3, and 6 days after 5FU injection (vs. 0, 1, 10, 30 days after 5FU injection) (G).
To gain further insights into the molecular identities of the gene expression programs that are differentially expressed in IKK2CA LT-HSCs, we performed GSEA (Subramanian et al., 2005), as this can provide information about gene clusters shared by the IKK2CA LT-HSCs signature and publicly available data sets. Accordingly, we first compared our gene profiling data with the published HSC ‘stemness’ signature data sets. These data sets were identified as genes enriched in normal, steady-state BM HSCs, when compared to MPP, LSC, and mobilized HSC (Forsberg et al., 2010). Our analysis revealed that the HSC ‘stemness’ signature was almost lost in IKK2CA LT-HSCs (Figure 4C). To corroborate these results, independently, we compared our gene array data with the published (Park et al., 2002) HSC enriched-data sets that were identified as genes highly enriched in HSCs, but not in MPPs. As expected, these results confirmed that the HSC signature was abolished in IKK2CA LT-HSCs (Figure 4D). Furthermore, a direct comparison of our expression profiling data with the data sets generated from human HSCs enriched gene sets, revealed a down regulation of human HSC signature in IKK2CA LT-HSCs (Figure 4E). These data suggest that constant NF-κB signals are disadvantageous to the molecular regulatory circuits of HSCs.
Impaired ‘Quiescence’ Signature in IKK2CA HSCs
One of the major cellular mechanisms by which HSC ‘stemness’ and functions are regulated is through induction of quiescence (Morrison and Weissman, 1994; Passegue et al., 2005; Wilson et al., 2008; Trumpp et al., 2010). Our analayis revealed that HSC quiescence is impaired in IKK2CA mice. Thus, we compared our gene expression data with the quiescence-related data sets (Cheung and Rando, 2013), that were identified as ‘quiescence’ signature that is common in HSCs, muscle stem cells and hair follicle stem cells. In line with our experimental data (Figure 3), this GSEA indicated that the HSC ‘quiescence’ signature was lost in IKK2CA LT-HSCs (Figure 4F). Consistent with lack of “quiescence” signature, comparison of our data with published proliferative HSC-related data sets (Venezia et al., 2004) identified an enrichment of “proliferation” signature in IKK2CA LT-HSCs (Figure 4G).
To explore the molecular mechanisms through which NF-κB signals suppress HSC quiescence, we directly focused on the expression of key genes that may influence cell cycle. Pioneering studies have unequivocally proven that combined actions of both cyclin dependent kinases (CDKs) and cyclin dependent kinase inhibitors (CDKIs) regulate HSC quiescence (Orford and Scadden, 2008; Pietras et al., 2011). To obtain insights into the deregulated molecular mechanisms that might affect HSC quiescence in IKK2CA mice, we analyzed the expression levels of positive regulators of cell cycle, such as Ccnd1, Ccnd2, Ccnd3, Cdk4 and Cdk6, and negative regulators of cell cycle, such as p15ink4b, p16ink4a, p18ink4c, p19ink4d, p19arf, p27kip, and p57kip2 in IKK2CA LT-HSCs. To validate the microarray data through an independent technique, we performed real-time PCR analysis. As expected, expression levels of Ccnd1 were increased and of p57kip2 were reduced in IKK2CA LT-HSCs (Figure 5), even though the fold differences between control and IKK2 mutant LT-HSCs were more striking from the data of real-time PCR assays. Of note, loss of D-cyclins in HSCs results in increased quiescence (Pietras et al., 2011) and a deficiency of p57 causes loss of HSC quiescence and impaired reconstitution of the hematopoietic system (Matsumoto et al., 2011), a phenotype that is very similar to IKK2CA HSCs. These data from GSEA and real-time PCR analysis provide a molecular explanation for impaired HSC quiescence in IKK2CA mice and indicate that the HSC ‘quiescence’ signature is sensitive to constitutive NF-κB signals.
FIGURE 5.

Deregulated gene expression levels of Cell cycle regulators in IKK2CA HSCs. Real time PCR data for Ccnd1, p21, p27, and p57 in IKK2CA and Control LT-HSCs. Expression levels of target genes were normalized to HPRT levels. Data are representative of two independent experiments. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05).
Deregulated ‘Transcription Factor Networks’ in IKK2 Mutant HSCs
Combinatorial actions, that include antagonistic or cooperative effects, of TFs play indispensable roles in the control of self-renewal, differentiation, and functions of HSCs (Orkin, 2000). Based on our in vivo and in silico findings, we hypothesized that a tight control on NF-κB signaling is critical to maintain the ‘stemness’ associated ‘TF networks’ in HSCs. Indeed, a more refined GO analysis on the list of upregulated genes identified changes in transcription regulator activity (GO:0030528) as the major and most significant (P = 0.0006) functional consequence caused by increased NF-κB activity in LT-HSCs (Figure 6A and Supplementary Table 2). Overall, our analysis identified a differential expression of 56 (30 upregulated and 26 downregulated) TFs in IKK2CA LT-HSCs (Figure 6B). To further understand the TF networks that were deregulated by constitutive NF-κB signals in HSCs, we focused on the specific functions of these TFs. Our analysis on upregulated TFs indicated that positive regulators of HSC differentiation; Relb, Gata1, Egr3, Klf2, Klf13, Id3, Hes1, Egr1, Taf10, Batf2 and E2f4, and negative regulators of HSC self-renewal and functions; Junb, Meis2, and Atf4 were upregulated in the IKK2 mutant LT-HSCs (Figure 6B). On the other hand, TFs that positively regulate HSC self-renewal, quiescence and functions; Gfi1, Cited2, Meis1, Satb1, Hoxa7, Tcf7l2, and Sin3a were downregulated in the LT-HSCs of IKK2CA mice (Figure 6B). To validate the expression levels of selected TFs through an independent technique, we performed real-time PCR analysis. In agreement with the microarray data, results of real-time PCR experiment suggested an upregulation of Junb and Relb, and a downregulation of Gfi1 (Figure 6C). In keeping with our results, increased expression of Junb (Passegue et al., 2004) and reduced expression of Gfi1 (Hock et al., 2004) are associated with loss of self-renewal, quiescence, and functions of HSCs. Overall, these data indicated that aberrant activation of NF-κB pathways in HSCs affects its physiology by; (1) inducing expression of multiple TFs associated with lineage differentiation and (2) repressing expression of key TFs that contributes to HSC self-renewal and functions. These results are of unique importance, as it suggests that constitutive activation of NF-κB is sufficient to alter the regulatory circuits of TFs that are critical to HSC self-renewal and functions.
FIGURE 6.

Altered transcription factor networks in IKK2CA HSCs. (A) Gene ontology analysis of microarray data: the 10 most significantly enriched GO terms (function) in upregulated genes (>1.5 fold) in IKK2CA LT-HSCs. P-values were calculated by DAVID 6.7 software. (B) Differentially expressed transcription factors (TFs) in IKK2CA LT-HSCs. Highlighted TFs have been shown to regulate HSC self-renewal, quiescence, and differentiation. (C) Real time PCR data for Relb, Junb, and Gfi1 in IKK2CA or Control LT-HSCs. Expression levels of target genes were normalized to HPRT levels. Data are representative of two independent experiments. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05 and ∗∗P < 0.005).
Augmented NF-κB Binding to Junb in IKK2CA Hematopoietic Cells
NF-κB family of proteins functions as TFs by binding to the RelA binding sites found in the regulatory regions of the target genes (Ghosh and Hayden, 2008; Vallabhapurapu and Karin, 2009). While NF-κB proteins have been documented to bind and activate over 500 target genes, their key transcriptional targets in HSCs remain largely unknown. In an attempt to identify the direct targets of NF-κB in HSCs, we compared the list of differentially genes in IKK2CA HSCs with the public database that contains the list of validated NF-κB target genes. This analysis indicated an upregulation of 33 potential NF-κB target genes in IKK2 mutant LT-HSCs (Figure 7A). In general, NF-κB proteins are believed to act as transcriptional activators, rather than transcriptional repressors, upon binding to the regulatory regions of their target genes (Ghosh and Hayden, 2008; Vallabhapurapu and Karin, 2009). Hence, we focused exclusively on the upregulated NF-κB targets in IKK2CA LT-HSCs, as these genes may represent the key targets of NF-κB in HSCs. Among the 33 candidate genes, we found upregulation of five TFs; Junb, Relb, Egr1, Irf1, and Bcl3. Of these, Junb caught our immediate attention for at least two following reasons; (1) Junb is the most abundantly expressed TF in IKK2CA LT-HSCs and (2) Overexpression of Junb led to the loss of quiescence and functions of HSCs (Passegue et al., 2004). Next, we explored if increased transcription of Junb was a directly associated with increased NF-κB binding in IKK2CA hematopoietic cells. To this end, we followed the ‘in silico’ approach to identify the presence of NF-κB binding site(s) in the regulatory regions of Junb. Our analysis identified three NF-κB binding sites in the enhancer region (+2028 to +2249) of Junb (Figure 7B) and all these three NF-κB binding sites are well-conserved between mouse and humans (Figure 7C). Of note, previous studies highlighted the importance of this enhancer region in NF-κB mediated regulation of Junb expression (Salem et al., 2013). To check if constitutive activation of NF-κB results in augmented NF-κB binding to its target sites in the enhancer region of Junb, we performed ChIP assays. In line with our hypothesis, data of ChIP experiments documented an increase in binding of NF-κB to the enhancer region of Junb (Figure 7D). It is noteworthy that the HSC phenotype of IKK2CA mice is very similar to the phenotype of Junb overexpressing HSCs (Passegue et al., 2004). Even though our data suggest that Junb is overexpressed in IKK2CA HSCs due to increased NF-κB binding to its enhancer region, we believe that deregulated expression of Junb may not be wholly responsible for the defective HSC functions in IKK2CA mice. Based on our gene expression and bioinformatics studies we hypothesize that uncontrolled NF-κB activity introduces several alterations at a genetic and molecular level, which in turn may be responsible for the impaired HSC quiescence and functions.
FIGURE 7.

Increased NF-κB binding to Junb enhancer region in hematopoietic cells from IKK2CA mice. (A) NF-κB target genes that are upregulated in IKK2CA LT-HSCs. (B) Diagrammatic representation of the Junb gene indicating the presence of three NF-κB binding sites in its enhancer region. (C) NF-κB binding sites in enhancer region of the Junb gene of indicated species. (D) Chromatin immunoprecipitation (ChIP) analysis of NF-κB (p65) binding to Junb promoter in the hematopoietic cells of 6 weeks old IKK2CA and control mice. Shown are the Real Time PCR data of p65 immunoprecipitates, that were normalized to IgG control immunoprecipitates. Data are representative of two independent experiments. All data represent mean ± SEM. Two-tailed Student’s t-tests were used to assess statistical significance (∗P < 0.05, ∗∗P < 0.01).
Discussion
In the present study, we investigated the impact of persistent IKK2/NF-κB activation in HSC maintenance and functions. Data reveal that constitutive IKK2/NF-κB activation causes many alterations in the HSPC compartments, such as depletion of the LT-HSC pool, increased numbers of ST-HSCs and reduction in MPP numbers. The decrease of LT-HSCs was not due to increased apoptosis, instead, LT-HSCs of IKK2CA mice showed augmented proliferation and reduced quiescence. A potential explanation for this could be that these LT-HSCs were proliferating more and subsequently differentiating into ST-HSCs, without undergoing self-renewal divisions. However, it was interesting that even though the numbers of ST-HSCs were augmented, the frequencies of MPPs were reduced in IKK2CA mice. This might be due to either a block in the differentiation of ST-HSCs into MPPs or increased differentiation of MPPs into lineage committed progenitors. However, committed progenitors of both myeloid and lymphoid lineages did not show any significant differences between control and constitutively active IKK2 mice. Together, these data suggest that the HSPC fraction, but not lineage committed progenitor faction, is more sensitive to constitutive activation of IKK2.
At a molecular level, the loss of HSC quiescence and defective HSC functions observed in constitutively active IKK2 mice could be explained, at least partially, by the loss of CDKI- p57 expression. Earlier studies identified p57 as a key factor of HSC quiescence and maintenance (Matsumoto et al., 2011). According to this report, p57 deficient HSCs show reduced self-renewal, decreased proportion HSCs in the G0 phase, decreased colony forming unit (CFUs) and defective hematopoietic reconstitution abilities (Matsumoto et al., 2011). Thus, the HSC phenotype of IKK2CA mice is very similar to that of p57 deficient mice. Even though our studies established an interesting link between constitutive activation of IKK2/NF-κB pathway and deregulated p57 expression, understanding the precise molecular mechanisms through which IKK2/NF-κB signaling suppress p57 expression would be an exciting area of future investigation.
In the current study, we document that increased IKK2 activation leads to deregulated expression of TF networks in HSCs. Based on earlier investigations, on the role of TFs in hematopoiesis (Shivdasani and Orkin, 1996; Orkin, 2000; Zhu and Emerson, 2002; Iwasaki et al., 2006; Chen et al., 2014), and our own studies documenting increased binding of NF-κB to the promoters of its target genes; IFN-γ (Nakagawa et al., 2015) and Junb (Figure 7), we speculate that the HSC phenotype of IKK2CA mice is caused, primarily, due to increased binding of NF-κB to its targets in HSCs. However, we cannot rule out the involvement of additional mechanisms that might indirectly influence the physiology of HSCs. Of note, in the current studies we utilized Vavcre transgenic mice to activate NF-κB in HSCs. Even though Vavcre has been used extensively in studies involving HSCs and considered as a faithful cre line to induce recombination of floxed alleles in HSCs and in their progeny, Vavcre mouse strain has also been reported to generate recombination in vascular endothelial cells (Georgiades et al., 2002), albeit at lower efficiencies. Our analysis of IKK2CA Vavcre mice indicated an absence of recombination in endothelial lineage cells of the BM (data not shown). However, we cannot rule out other possibilities through which endothelial cells (Poulos et al., 2016) might affect HSCs of IKK2CA mice, especially during the fetal hematopoiesis. Furthermore, in IKK2CAVavcre mice NF-κB is constitutively active in differentiated hematopoietic lineage cells, including neutrophils, macrophages, dendritic cells, and NK cells. In view of the fact that NF-κB controls expression of inflammatory cytokines in these immune cells and that inflammatory cytokines can directly influence the physiology of HSCs, including quiescence (Essers et al., 2009; Sato et al., 2009; Baldridge et al., 2010), it remains possible that the HSC phenotype of IKK2CAVavcre mice might be influenced, at least in part, by these non-cell intrinsic mechanisms. A future study with a focus on delineating the involvement of cell intrinsic vs. extrinsic pathways of NF-κB mediated regulation of HSC physiology would be essential in understanding the precise role of NF-κB in hematopoiesis.
In the recent years, the importance of non-canonical NF-κB signals in early hematopoietic events has begun to unfold to a greater extent (Vallabhapurapu and Karin, 2009; Gerondakis et al., 2012; Zhao et al., 2012; González-Murillo et al., 2015; Xiu et al., 2017). However, to date, there are only a few studies that described the role of canonical NF-κB signals in HSC biology. Grossmann et al. (1999) reported that the combined loss of c-Rel and RelA did not affect pluripotent stem cells. However, this study did not analyze the ‘bona fide’ HSC compartment and their functions (Grossmann et al., 1999). Stein and Baldwin described that a deficiency of RelA resulted in impaired HSC functions, increased proliferation and reduced apoptosis of HSPCs (Stein and Baldwin, 2013). Taken together, these studies have highlighted that loss of NF-κB signals results in increased HSC cycling and impaired HSC functions. Based on these previous findings, it may be expected that increased NF-κB signaling would promote quiescence and functions of HSCs. However, surprisingly, our data indicate that constitutive activation of NF-κB signals in HSCs causes diminished HSC quiescence and functions. It is interesting to observe that the HSC phenotype of IKK2CA mice is similar to the HSC phenotype of RelA-/- mice. Even though right now it is unclear, as why and how both loss of NF-κB signals and gain of NF-κB signals would cause a similar phenotype, these data suggest that a ‘fine tuning’ of NF-κB signals is critical to HSC biology. More mechanistic studies would be essential to unravel the functional complexities executed by NF-κB in HSCs.
In essence, the study presented here explored the functional consequences of constitutive activation of NF-κB in HSCs and attempted to unravel the molecular mechanisms and through which NF-κB controls HSCs. Our data identify that deregulated NF-κB signals lead to loss of HSC quiescence and functions. Furthermore, these results indicate that increased NF-κB signaling perturbs the TFs networks in HSCs. A clearer understanding on the precise role of NF-κB in normal and pathologic hematopoiesis may provide new therapeutic opportunities.
Author Contributions
MN performed the research, collected and interpreted the data, and prepared the figures. HC performed the research and collected the data. CR designed the research, interpreted data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grant from the NHLBI (Grant No. HL132194) (CR).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2018.00143/full#supplementary-material
Differentiation of lineage committed progenitors remains intact in IKK2CA mice.
Differentially expressed genes in IKK2CA/+ HSCs.
Gene ontology (GO) analysis on upregulated genes in IKK2CA/+ HSCs.
References
- Arai F., Hirao A., Ohmura M., Sato H., Matsuoka S., Takubo K., et al. (2004). Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118 149–161. 10.1016/j.cell.2004.07.004 [DOI] [PubMed] [Google Scholar]
- Baldridge M. T., King K. Y., Boles N. C., Weksberg D. C., Goodell M. A. (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465 793–797. 10.1038/nature09135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge M. T., King K. Y., Goodell M. A. (2011). Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 32 57–65. 10.1016/j.it.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R. (2009). QuickGO: a web-based tool for gene ontology searching. Bioinformatics 25 3045–3046. 10.1093/bioinformatics/btp536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone D. L., Turer E. E., Lee E. G., Ahmad R. C., Wheeler M. T., Tsui C., et al. (2004). The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5 1052–1060. 10.1038/ni1110 [DOI] [PubMed] [Google Scholar]
- Brown K., Gerstberger S., Carlson L., Franzoso G., Siebenlist U. (1995). Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267 1485–1488. 10.1126/science.7878466 [DOI] [PubMed] [Google Scholar]
- Chen L., Kostadima M., Martens J. H. A., Canu G., Garcia S. P., Turro E., et al. (2014). Transcriptional diversity during lineage commitment of human blood progenitors. Science 345:1251033. 10.1126/science.1251033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., et al. (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287 1804–1808. 10.1126/science.287.5459.1804 [DOI] [PubMed] [Google Scholar]
- Cheung T. H., Rando T. A. (2013). Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14 329–340. 10.1038/nrm3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coornaert B., Carpentier I., Beyaert R. (2009). A20: central gatekeeper in inflammation and immunity. J. Biol. Chem. 284 8217–8221. 10.1074/jbc.R800032200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J., Williams A., Skavdis G., Harker N., Coles M., Tolaini M., et al. (2003). Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33 314–325. 10.1002/immu.200310005 [DOI] [PubMed] [Google Scholar]
- Dong J., Jimi E., Zeiss C., Hayden M. S., Ghosh S. (2010). Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 241709–1717. 10.1101/gad.1958410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers M. A., Offner S., Blanco-Bose W. E., Waibler Z., Kalinke U., Duchosal M. A., et al. (2009). IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458 904–908. 10.1038/nature07815 [DOI] [PubMed] [Google Scholar]
- Forsberg E. C., Passegue E., Prohaska S. S., Wagers A. J., Koeva M., Stuart J. M., et al. (2010). Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS One 5:e8785. 10.1371/journal.pone.0008785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades P., Ogilvy S., Duval H., Licence D. R., Charnock-Jones D. S., Smith S. K., et al. (2002). VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 34 251–256. 10.1002/gene.10161 [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Banerjee A., Grigoriadis G., Vasanthakumar A., Gugasyan R., Sidwell T., et al. (2012). NF-kappaB subunit specificity in hemopoiesis. Immunol. Rev. 246 272–285. 10.1111/j.1600-065X.2011.01090.x [DOI] [PubMed] [Google Scholar]
- Ghosh S., Hayden M. S. (2008). New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 8 837–848. 10.1038/nri2423 [DOI] [PubMed] [Google Scholar]
- González-Murillo A., Fernandez L., Baena S., Melen G. J., Sanchez R., Sánchez-Valdepenas C., et al. (2015). The NFKB inducing kinase modulates hematopoiesis during stress. Stem Cells 33 2825–2837. 10.1002/stem.2066 [DOI] [PubMed] [Google Scholar]
- Grossmann M., Metcalf D., Merryfull J., Beg A., Baltimore D., Gerondakis S. (1999). The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc. Natl. Acad. Sci. U.S.A. 96 11848–11853. 10.1073/pnas.96.21.11848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M. L., Neering S. J., Upchurch D., Grimes B., Howard D. S., Rizzieri D. A., et al. (2001). Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98 2301–2307. 10.1182/blood.V98.8.2301 [DOI] [PubMed] [Google Scholar]
- Harhaj E. W., Dixit V. M. (2012). Regulation of NF-kappaB by deubiquitinases. Immunol. Rev. 246 107–124. 10.1111/j.1600-065X.2012.01100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H., Hamblen M. J., Rooke H. M., Schindler J. W., Saleque S., Fujiwara Y., et al. (2004). Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431 1002–1007. 10.1038/nature02994 [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B. T., Lempicki R. A. (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H., Mizuno S., Arinobu Y., Ozawa H., Mori Y., Shigematsu H., et al. (2006). The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 20 3010–3021. 10.1101/gad.1493506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagoya Y., Yoshimi A., Kataoka K., Nakagawa M., Kumano K., Arai S., et al. (2014). Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J. Clin. Invest. 124 528–542. 10.1172/JCI68101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel M. J., Morrison S. J. (2008). Uncertainty in the niches that maintain haematopoietic stem cells. Nat. Rev. Immunol. 8 290–301. 10.1038/nri2279 [DOI] [PubMed] [Google Scholar]
- King K. Y., Goodell M. A. (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 11 685–692. 10.1038/nri3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M., Wagers A. J., Manz M. G., Prohaska S. S., Scherer D. C., Beilhack G. F., et al. (2003). Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu. Rev. Immunol. 21 759–806. 10.1146/annurev.immunol.21.120601.141007 [DOI] [PubMed] [Google Scholar]
- Matsumoto A., Takeishi S., Kanie T., Susaki E., Onoyama I., Tateishi Y., et al. (2011). p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 9 262–271. 10.1016/j.stem.2011.06.014 [DOI] [PubMed] [Google Scholar]
- Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., et al. (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278 860–866. 10.1126/science.278.5339.860 [DOI] [PubMed] [Google Scholar]
- Mirantes C., Passegue E., Pietras E. M. (2014). Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell Res. 329 248–254. 10.1016/j.yexcr.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V. K., Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34 267–273. 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Morrison S. J., Weissman I. L. (1994). The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1 661–673. 10.1016/1074-7613(94)90037-X [DOI] [PubMed] [Google Scholar]
- Nagai Y., Garrett K. P., Ohta S., Bahrun U., Kouro T., Akira S., et al. (2006). Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24 801–812. 10.1016/j.immuni.2006.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M. M., Thummar K., Mandelbaum J., Pasqualucci L., Rathinam C. V. (2015). Lack of the ubiquitin-editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J. Exp. Med. 212 203–216. 10.1084/jem.20132544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford K. W., Scadden D. T. (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 9 115–128. 10.1038/nrg2269 [DOI] [PubMed] [Google Scholar]
- Orkin S. H. (2000). Diversification of haematopoietic stem cells to specific lineages. Nat. Rev. Genet. 1 57–64. 10.1038/35049577 [DOI] [PubMed] [Google Scholar]
- Panagopoulos I., Micci F., Thorsen J., Haugom L., Buechner J., Kerndrup G., et al. (2013). Fusion of ZMYND8 and RELA genes in acute erythroid leukemia. PLoS One 8:e63663. 10.1371/journal.pone.0063663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I. K., He Y., Lin F., Laerum O. D., Tian Q., Bumgarner R., et al. (2002). Differential gene expression profiling of adult murine hematopoietic stem cells. Blood 99 488–498. 10.1182/blood.V99.2.488 [DOI] [PubMed] [Google Scholar]
- Passegue E., Wagers A. J., Giuriato S., Anderson W. C., Weissman I. L. (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 202 1599–1611. 10.1084/jem.20050967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E., Wagner E. F., Weissman I. L. (2004). JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119 431–443. 10.1016/j.cell.2004.10.010 [DOI] [PubMed] [Google Scholar]
- Pietras E. M., Warr M. R., Passegue E. (2011). Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 195 709–720. 10.1083/jcb.201102131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos M. G., Ramalingam P., Gutkin M. C., Kleppe M., Ginsberg M., Crowley M. J., et al. (2016). Endothelial-specific inhibition of NF-kappaB enhances functional haematopoiesis. Nat. Commun. 7:13829. 10.1038/ncomms13829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C., Matesic L. E., Flavell R. A. (2011). The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat. Immunol. 12 399–407. 10.1038/ni.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C., Thien C. B., Langdon W. Y., Gu H., Flavell R. A. (2008). The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 22 992–997. 10.1101/gad.1651408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J. P. (2006). GenePattern 2.0. Nat. Genet. 38 500–501. 10.1038/ng0506-500 [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M., Zandi E., Natoli G., Karin M. (1998). IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395 297–300. 10.1038/26261 [DOI] [PubMed] [Google Scholar]
- Salem T., Gomard T., Court F., Moquet-Torcy G., Brockly F., Forne T., et al. (2013). Chromatin loop organization of the junb locus in mouse dendritic cells. Nucleic Acids Res. 41 8908–8925. 10.1093/nar/gkt669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y., Derudder E., Hobeika E., Pelanda R., Reth M., Rajewsky K., et al. (2006). Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 24 729–739. 10.1016/j.immuni.2006.04.005 [DOI] [PubMed] [Google Scholar]
- Sato T., Onai N., Yoshihara H., Arai F., Suda T., Ohteki T. (2009). Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 15 696–700. 10.1038/nm.1973 [DOI] [PubMed] [Google Scholar]
- Shivdasani R. A., Orkin S. H. (1996). The transcriptional control of hematopoiesis. Blood 87 4025–4039. [PubMed] [Google Scholar]
- Stein S. J., Baldwin A. S. (2013). Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood 121 5015–5024. 10.1182/blood-2013-02-486142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H., Fritsch K., Kovtonyuk L. V., Saito Y., Yakkala C., Jacobs K., et al. (2017). Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21 225.e5–240.e5. 10.1016/j.stem.2017.06.013 [DOI] [PubMed] [Google Scholar]
- Thompson B. J., Jankovic V., Gao J., Buonamici S., Vest A., Lee J. M., et al. (2008). Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205 1395–1408. 10.1084/jem.20080277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A., Essers M., Wilson A. (2010). Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10 201–209. 10.1038/nri2726 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Karin M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27 693–733. 10.1146/annurev.immunol.021908.132641 [DOI] [PubMed] [Google Scholar]
- Venezia T. A., Merchant A. A., Ramos C. A., Whitehouse N. L., Young A. S., Shaw C. A., et al. (2004). Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2:e301. 10.1371/journal.pbio.0020301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welner R. S., Pelayo R., Nagai Y., Garrett K. P., Wuest T. R., Carr D. J., et al. (2008). Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood 112 3753–3761. 10.1182/blood-2008-04-151506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Laurenti E., Oser G., van der Wath R. C., Blanco-Bose W., Jaworski M., et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135 1118–1129. 10.1016/j.cell.2008.10.048 [DOI] [PubMed] [Google Scholar]
- Wilson A., Trumpp A. (2006). Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol. 6 93–106. 10.1038/nri1779 [DOI] [PubMed] [Google Scholar]
- Xiu Y., Xue W. Y., Lambertz A., Leidinger M., Gibson-Corley K., Zhao C. (2017). Constitutive Activation of NIK impairs the self-renewal of hematopoietic stem/progenitor cells and induces bone marrow failure. Stem Cells 35 777–786. 10.1002/stem.2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Xiu Y., Ashton J., Xing L., Morita Y., Jordan C. T., et al. (2012). Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells 30 709–718. 10.1002/stem.1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Emerson S. G. (2002). Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 21 3295–3313. 10.1038/sj.onc.1205318 [DOI] [PubMed] [Google Scholar]
- Zou P., Yoshihara H., Hosokawa K., Tai I., Shinmyozu K., Tsukahara F., et al. (2011). p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell 9 247–261. 10.1016/j.stem.2011.07.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentiation of lineage committed progenitors remains intact in IKK2CA mice.
Differentially expressed genes in IKK2CA/+ HSCs.
Gene ontology (GO) analysis on upregulated genes in IKK2CA/+ HSCs.