

Abstract
Monoclonal antibodies and derived fragments are used extensively both experimentally and therapeutically. Thorough characterization of such antibodies is necessary and includes assessment of their thermal and storage stabilities. Thus, assessment of the underlying conformational stabilities of the antibodies is also important. We recently documented that non-reducing SDS-PAGE can be used to assess both monoclonal and polyclonal IgG domain thermal unfolding in SDS. Utilizing this same h2E2 anti-cocaine mAb, in this study we generated and analyzed various mAb antibody fragments to delineate the structural domains of the antibody responsible for the observed discrete bands following various heating protocols and analysis by non-reducing SDS-PAGE. Previously, these domain unfolding transitions and gel bands were hypothesized to stem from known mAb structural domains based on the relative thermal stability of those CH2, CH3, and Fab domains in the absence of SDS, as measured by differential scanning calorimetry. In this study, we generated and analyzed F(ab’)2, Fab, and Fc fragments, as well as a mAb consisting of only heavy chains, and examined the thermally induced domain unfolding in each of these fragments by non-reducing SDS-PAGE. The results were interpreted and integrated to generate an improved model of thermal unfolding for the mAb IgG in SDS. These results and the model presented should be generally applicable to many monoclonal and polyclonal antibodies and allow novel comparisons of conformational stabilities between chemically or genetically modified versions of a given antibody. Such modified antibodies and antibody drug conjugates are commonly utilized and important for experimental and therapeutic applications.
Abbreviations: mAb, monoclonal antibody; h2E2, humanized anti-cocaine monoclonal antibody; IPA, isopropanol; CHO, Chinese Hamster Ovary; MALDI-Tof MS, matrix-assisted laser desorption ionization/time-of-flight mass spectrometry
Keywords: Monoclonal antibody, Antibody fragments, Non-reducing SDS-PAGE, Antibody analysis, Electrophoretic migration, Antibody domain unfolding
Graphical abstract
Highlights
-
•
mAb F(ab’)2 fragments exhibit multiple unfolded states in non-reducing SDS-PAGE.
-
•
Fab and Fc mAb fragments do not exhibit similar multiple unfolded state bands.
-
•
Previous mAb domain unfolding pathway in SDS is revised based on fragment analyses.
-
•
A heavy chain only mAb variant is detected and exhibits multiple unfolded states.
-
•
These results are likely relevant to analyses of many monoclonal and polyclonal Abs.
1. Introduction
Monoclonal antibodies (mAbs) are heavily utilized in basic and translational research, as well as in therapeutics. In recent years, a substantial percentage of FDA newly approved drugs is based on monoclonal antibodies, and such antibodies are widely used in the treatment of autoimmune diseases and cancer. Thorough characterization of such therapeutic mAbs is necessary, and mandated by the FDA.
Despite decades of research that have identified dopaminergic neurotransmission as a primary mediator of the actions of cocaine, there is no effective pharmacological treatment available for cocaine abuse [1]. An alternative approach is to use anti-cocaine antibodies that bind cocaine and sequester it in the peripheral circulation [2]. A cocaine vaccine that generates anti-cocaine antibodies decreased cocaine use in some cocaine abusers [3], [4], [5]. However, the response to the vaccine is unpredictable with regards to the levels of antibodies, their affinities and specificities and, therefore, efficacy differs widely between individuals [3], [4]. An alternative approach is to use a human or humanized anti-cocaine monoclonal antibody (mAb) [6]. With passive immunization immediate immunity is conferred with a well-characterized mAb with a known high affinity and specificity for cocaine. Similarly, the dosage and pharmacokinetics of the mAb will be well established and treatment responses predictable. To this end, our laboratory has characterized a humanized anti-cocaine mAb (h2E2) [7], which is under development for treatment of cocaine abuse and addiction. For this purpose, this anti-cocaine mAb has been produced in gram quantities in Chinese hamster ovary (CHO) cells [8].
Many protocols and methodologies have been applied to characterize monoclonal antibodies, as well as fragments and derivatives of these antibodies. SDS-PAGE is one of the simplest, least expensive, and most commonly used techniques to analyze antibodies for purity. When analyzed without reduction of disulfide bonds, IgG antibodies should give rise to a single band on SDS-PAGE, i.e., the intact antibody consisting of 2 heavy and 2 light chains, with a combined size of approximately 150 kDa. Recently, we noted and published that there are apparent overestimated molecular weights and size heterogeneities, as evidenced by multiple bands on non-reducing gels [9]. The results were very similar when analyzing a commercially available polyclonal IgG antibody in an identical manner, indicating that this behavior is common to many polyclonal and monoclonal antibodies currently in use.
In the current study, we investigated the domains which were previously hypothesized to be involved in these unfolding transitions in SDS, i.e., the CH2, CH3, and Fab antibody domains, by generating and analyzing F(ab’)2, Fab, and Fc fragments. In addition, we also identified and characterized an antibody species consisting of only two heavy chains, present at a very low level in some preparations of the CHO produced anti-cocaine h2E2 mAb. As a result of these experiments, we have revised, updated, and improved our model for the unfolding of the domains responsible for the generation of the discrete molecular weight bands observed upon heating for different amounts of time in non-reducing SDS-PAGE sample buffer, prior to non-reducing SDS-PAGE analyses. Application of the unfolding of antibodies and antibody fragments in SDS prior to non-reducing SDS-PAGE should contribute an additional novel antibody characterization technique, and may represent a unique technique to analyze the effects of modifications, fragmentations, and mutations on the stability of IgG antibodies that is rapid, inexpensive, and widely available.
2. Materials and methods
2.1. Materials
The generation and production of the h2E2 anti-cocaine monoclonal antibody was previously described [10]. The recombinant h2E2 mAb was purified by protein A affinity chromatography and used as supplied by the manufacturer, Catalent. The purity, structure, and function of the resultant mAb protein has been well characterized in our laboratory [8], [11], [12], [13]. Generation and purification of the Fab fragment of this h2E2 mAb by Endo-Lys-C cleavage was described previously [8]. Generation and purification of the F(ab’)2 and Fc h2E2 mAb fragments was accomplished by IdeS cleavage, as previously described [12], using the commercial Frag-It kit from Genovis (catalog # A2-FR2–025). Acrylamide, bisacrylamide, and all reagents used to pour, run, and stain SDS-PAGE gels were from BioRad. HPLC grade 2-propanol (isopropanol, IPA) was from Fisher. Pre-stained SDS-PAGE molecular weight standards (10–240 kDa) were purchased from SMOBIO Technology, Inc., catalog # PM1700.
2.2. Methods
The 7% SDS-PAGE gels were poured, run, and stained with Coomassie Blue, essentially as described by Laemmli [14]. Antibody samples were typically diluted to a final protein concentration of 0.2 mg/mL in non-reducing sample buffer (containing 5% SDS, and 12.5% glycerol). Some samples also contained a low concentration (5%) of 2-propanol (isopropanol, IPA). After incubation at 50 °C for varying times (boiled samples were incubated in boiling water for 3 min) prior to electrophoresis, typically 10 μL (2 μg) of each sample was loaded into individual wells of 15 well, 1.5 mm thick 7% acrylamide gels. Experiments were designed so that comparisons of conditions and effects on electrophoretic mobility were done on the same gel. All gels were used at least 24 h after they were poured (polymerized), and all gel sample wells were washed 3 times with Laemmli running buffer, prior to loading samples, to eliminate possible artefacts due to reaction of the protein samples with any residual unpolymerized acrylamide or residual free radicals. Electrophoresis was performed until the bromophenol blue tracking dye reached the bottom of the gel. Pre-stained molecular weight standards (10 μL) were also run on each gel. Gels were stained at room temperature for 30–45 min with Coomassie blue, and destained for 4–20 h prior to photography. Each panel in each figure in this study depicts a single SDS-PAGE gel, to enable direct comparisons of bands. The vertical line in Fig. 1B was overlaid on the gel image as a visual aid to facilitate reader understanding and interpretation of the gel and does not represent lanes from different gels that were aligned following cutting and pasting from different photographic images.
Fig. 1.

The effect of time of pre-electrophoretic heating at 50 °C on non-reducing SDS-PAGE analyses of h2E2 anti-cocaine mAb fragments. Panel A (top) – results using F(ab’)2, Panel B (bottom, left) – results using Fab, Panel B (bottom, right) – results using Fc. A 1.5 mm thick, 7% acrylamide gel was loaded with 2 μg (10 μL) of antibody fragment per well, electrophoresed until the tracking dye reached the bottom of the gel, and stained with Coomassie blue. The entire gels are shown. Pre-electrophoretic incubation in SDS-PAGE non-reducing sample buffer was carried out for the indicated times at 50 °C. The samples in the lanes labeled “boil” were boiled for 3 min prior to electrophoresis. In Panel A, discrete bands/folded states noted for the F(ab’)2 fragment are labeled “A”, “B”, and “C”. See Fig. 4A for the hypothesized identification of these 3 bands/differentially folded states.
MALDI-ToF MS was used to evaluate and identify the protein composition of selected bands from the Coomassie blue stained gels. This was accomplished by in-gel digestion of the protein bands, recovery of the peptides and analysis of the peptides by MALDI-ToF MS as described previously [15] Briefly, proteins bands were excised from the gel, washed and dehydrated followed by reduction of the disulfide bonds with DTT and blocking of the reduced cysteines with iodoacetamide to produce carbamidomethyl-cysteine (CAM-cysteine). After trypsin digestion, the extracted peptides were desalted on c18 micro ZipTips using the vendor's instructions (Millipore). MALDI-ToF MS of the desalted peptides was done by mixing 20% sample (1 μL of the ZipTip elution) into 4 μL of α-cyano-4-hydroxycinnamic acid (4-HCCA) MALDI matrix in 60% acetonitrile containing 0.1% formic acid and 10 mM ammonium phosphate (monobasic). Mass spectra were collected in reflector positive ion mode on a Sciex 4800 MALDI-ToF/ToF system. Peptide spectra were collected by the accumulation of 1000 laser shots over a mass to charge range (m/z) of 800–4000. Spectra were collected with a default instrument calibration and then recalibrated using 4 internal tryptic peptides from the heavy chain of the mAb. Peptide masses detected are reported in table format (see Table 1) with the theoretical peptide masses and the actual peptide masses detected for each protein band (100 and 120 kDa proteins). The percentage of the mAb heavy chain amino acid sequence accounted for by these peptides is reported as the total sequence coverage percentage.
Table 1.
MALDI-TOF mass spectral identification and listing of tryptic peptides derived from the 100 and 120 kDa h2E2 mAb gel bands.
| Expected MH+ (monoisotopic) | Detected MH+ (100 kDa) | Detected MH+ (120 kDa) | Mod. | Start AA | End AA | Missed cleavages | Peptide sequence (heavy chain) |
|---|---|---|---|---|---|---|---|
| 836.4737 | 836.4769 | 836.4813 | 66 | 72 | 1 | (K)GRFTISR(D) | |
| 838.5033 | 838.5037 | 838.5075 | 323 | 330 | 0 | (K)ALPAPIEK(T) | |
| 851.4291 | 851.4292 | 851.4340 | Met-ox | 245 | 251 | 0 | (K)DTLMISR(T) |
| 873.4353 | 873.4333 | 873.4421 | 59 | 65 | 0 | (K)YYVDSVK(G) | |
| 970.5680 | 970.5698 | 970.5778 | 207 | 214 | 2 | (K)VDKRVEPK(S) | |
| 1086.5578 | 1086.5518 | 1086.5753 | 59 | 67 | 1 | (K)YYVDSVKGR(F) | |
| 1118.5623 | 1118.5149 | 1118.5284 | 210 | 218 | 2 | (K)RVEPKSC(CAM)DK(T) | |
| 1161.6296 | 1161.6302 | 1161.6364 | 357 | 366 | 0 | (K)NQVSLTC(CAM)LVK(G) | |
| 1186.6467 | 1186.6551 | 1186.6514 | 118 | 129 | 0 | (K)GPSVFPLAPSSK(S) | |
| 1286.6739 | 1286.6737 | 1286.6802 | 341 | 351 | 0 | (R)EPQVYTLPPSR(E) | |
| 1290.5671 | 1290.5667 | 1290.5739 | 88 | 98 | 0 | (R)AEDTAVYYC(CAM)AK(E) | |
| 1321.6780 | 1321.6754 | 1321.6814 | 130 | 143 | 0 | (K)STSGGTAALGC(CAM)LVK(D) | |
| 1659.7973 | 1659.7960 | 1659.8055 | 44 | 58 | 0 | (K)GLEWVANINQDGSEK(Y) | |
| 1677.8020 | 1677.8081 | 1677.8088 | 271 | 284 | 0 | (K)FNWYVDGVEVHNAK(T) | |
| 1766.8490 | 1766.8372 | 1766.8396 | 73 | 87 | 0 | (R)DNAQNSLYLQMNSLR(A) | |
| 1782.8439 | 1782.8441 | 1782.8538 | Met-ox | 73 | 87 | 0 | (R)DNAQNSLYLQMNSLR(A) |
| 1808.0065 | 1808.0018 | 1808.0070 | 298 | 313 | 0 | (R)VVSVLTVLHQDWLNGK(E) | |
| 1873.9218 | 1873.9220 | 1873.9286 | 389 | 405 | 0 | (K)TTPPVLDSDGSFFLYSK(L) | |
| 1882.0029 | 1882.0023 | 1882.0123 | 1 | 19 | 0 | (-)EVQLVESGGGLVQPGGSLR(L) | |
| 1917.9916 | 1917.9883 | 1917.9969 | 99 | 117 | 0 | (K)ELGPWGQGTLVTVSSASTK(G) | |
| 1920.9372 | 1920.9379 | 1920.9449 | Met-ox | 341 | 356 | 1 | (R)EPQVYTLPPSREEMTK(N) |
| 2250.0107 | 2250.0112 | 2255.0242 | Met-ox | 20 | 38 | 0 | (R)LSC(CAM)AASGFIFSSDWMNWVR(Q) |
| 2359.1711 | 2359.1731 | 2359.1843 | Met-ox | 337 | 356 | 2 | (K)GQPREPQVYTLPPSREEMTK(N) |
| 2514.2147 | 2514.2188 | 2514.2312 | 44 | 65 | 1 | (K)GLEWVANINQDGSEKYYVDSVK(G) | |
| 2544.1314 | 2544.1382 | 2544.1389 | 367 | 388 | 0 | (K)GFYPSDIAVEWESNGQPENNYK(T) | |
| 2817.2620 | 2817.2678 | 2817.2937 | Met-ox | 413 | 435 | 0 | (R)WQQGNVFSC(CAM)SVMHEAL |
| HNHYTQK(S) | |||||||
| 2844.4575 | 2844.4500 | 2844.4575 | 219 | 244 | 0 | (K)THTC(CAM)PPC(CAM)PAPELLGG | |
| PSVFLFPPKPK(D) | |||||||
| 3054.3932 | 3054.4094 | 3054.3097 | Met-ox | 73 | 98 | 1 | (R)DNAQNSLYLQMNSLRAEDTA |
| VYYC(CAM)AK(E) | |||||||
| 3334.6421 | 3334.6440 | 3334.6501 | 215 | 244 | 1 | (K)SC(CAM)DKTHTC(CAM)PPC(CAM) | |
| PAPELLGGPSVFLFPPKPK(D) | |||||||
| 3797.8116 | 3797.8013 | 3797.8337 | 252 | 284 | 1 | (R)TPEVTC(CAM)VVVDVSHEDPEVKF | |
| NWYVDGVEVHNAK(T) |
All identified peptides listed in the Table are derived from the mAb heavy chain, and no light chain peptide peaks were identified in the tryptic digest. The % sequence coverage of the 443 amino acids comprising the heavy chain for the listed peptides is 76% (i.e., 336 of 443 amino acids were identified in the peptides), and includes the N-terminal peptide and a peptide ending just 8 amino acids from the C-terminus of the intact heavy chain. AA = amino acid residue number, CAM = carbamidomethyl (iodoacetamide modified cysteine), Met-ox = oxidized methionine.
3. Results
We previously generated a hypothetical model of the sequential thermal unfolding of an intact anti-cocaine mAb and a commercially available polyclonal antibody [9] by assigning discrete bands observed on non-reducing SDS-PAGE after heating for different times in SDS based on the known relative thermal stabilities of antibody domains, as measured by differential scanning calorimetry (DSC) in the absence of SDS [16], [17], [18]. To investigate the nature of these SDS unfolding domains and bands experimentally, we generated, purified, and analyzed F(ab’)2, Fab, and Fc fragments of this anti-cocaine mAb. Fig. 1 shows the kinetics of unfolding in SDS at 50 °C for these antibody fragments. Three discrete, different apparent molecular weight species are evident for the F(ab’)2 fragment (Panel A). For the Fab and Fc fragments (Panel B), the results are less striking, with smearing of the Fab band from higher molecular weights to about 40 kDa noted without heating in SDS, and gradual loss of Fab band smearing observed upon heating. For the Fc fragment, there is a faint band observed with no heating at about 45 kDa, just below the major band. This minor species also disappears with increasing time of heating, leading to a single band at about 47 kDa after about 10 min at 50 °C – identical to that seen upon boiling the Fc fragment in SDS for 3 min.
To assess the reproducibility and independence of these fragment unfolding events, as well as to compare them kinetically to the intact mAb on the same gel, the purified intact anti-cocaine mAb, purified F(ab’)2, and purified Fc, all at 0.2 mg/mL in non-reducing sample buffer, were combined in one sample and heated for various times at 50 °C (or boiled for 3 min for comparison), as shown in Fig. 2. The 4 discrete bands observed previously for the intact antibody are evident, and the same bands for the fragments which were observed when heating these individual components separately as shown in Fig. 1 were also observed for the F(ab’)2 and Fc fragments in Fig. 2.
Fig. 2.

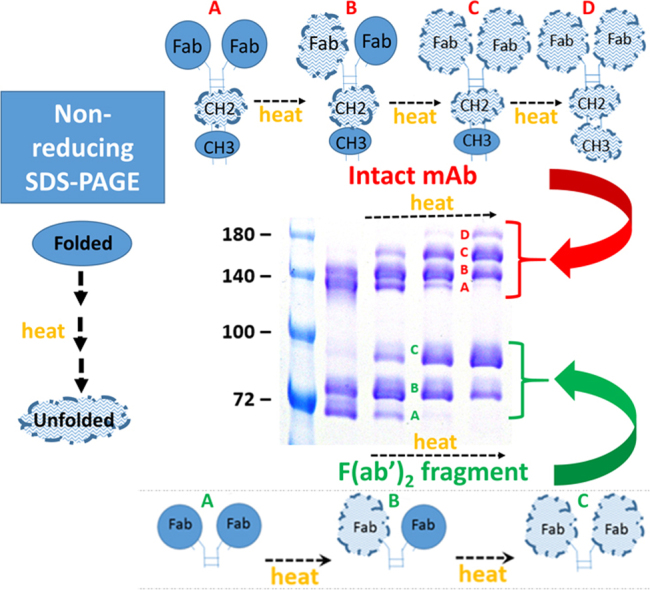
The effect of time of pre-electrophoretic heating at 50 °C on non-reducing SDS-PAGE analyses of a mixture of the intact h2E2 anti-cocaine mAb and its F(ab’)2, and Fc fragments. A 1.5 mm thick, 7% acrylamide gel was loaded with samples containing 2 μg each of intact h2E2 anti-cocaine mAb, F(ab’)2, and Fc fragments (10 μL total volume) per well, electrophoresed until the tracking dye reached the bottom of the gel, and stained with Coomassie blue. The entire gel is shown. Pre-electrophoretic incubation in SDS-PAGE non-reducing sample buffer was carried out for the indicated times at 50 °C. The sample in the lane labeled “boil” was boiled for 3 min prior to electrophoresis. The electrophoretic migration positions of various unfolded stages of each species are indicated to the right of the gel, and discrete bands identifiable in each sample are labeled by letters between gel lanes (see 0–3 min sample lanes). In the Figure, discrete bands/folded states noted for the F(ab’)2 fragment are labeled with green lettering; “A”, “B”, and “C”, while the discrete bands/folded states noted for the intact mAb are labeled with red lettering; “A”, “B”, “C”, and “D”. See Fig. 4A for the hypothesized identification of the 3 bands/differentially folded states for the F(ab’)2 fragment, and Fig. 4B for the hypothesized identification of the 4 bands/differentially folded states for the intact mAb.
In separate experiments, we sometimes noted a small amount of an approximately 100 kDa band after non-reducing SDS-PAGE of some CHO expressed h2E2 mAb samples. This band was also noted to shift its apparent molecular weight to approximately 120 kDa upon heating in non-reducing SDS-PAGE buffer. In addition, this apparent SDS unfolding transition (100–120 kDa shift) was largely blocked by the inclusion of 5% isopropanol (IPA) in the sample buffer (Fig. 3). We previously observed similar inhibitory effects of isopropanol and other short chain alcohols on the thermal SDS unfolding of the intact mAb [9]. To elucidate the nature of this 100 kDa band seen on non-reducing SDS-PAGE, this same sample of mAb was run on another gel, with or without heating at 50 °C, and the 100 and 120 kDa bands were excised from that gel after staining and de-staining. These proteins were reduced and alkylated in the gel, and then digested with trypsin in the gel. The resulting tryptic peptides were extracted and analyzed by mass spectral methods. The results were essentially identical for both the 100 and 120 kDa bands, indicating the interconvertible nature of these bands. As is evident from Table 1, both of these bands consisted of intact mAb heavy chains (76% coverage of the heavy chain sequence was observed in the identified tryptic peptides, including the N-terminus and a peptide ending just 8 amino acids from the C-terminus of the heavy chain). Importantly, there were no light chain peptides identified in either sample, indicating that the 100/120 kDa band consists of 2 heavy chains, but lacks the two light chains contained in the intact mAb. Consistent with these mass spectral results, no light chain bands were observed in several (6) separate samples when analyzed in the lower molecular weight range on 10% non-reducing SDS-PAGE gels, even when overloading the gels with sample (see Supplementary data, Fig. 1).
Fig. 3.

The effect of isopropanol (IPA) and time of pre-electrophoretic heating at 50 °C on non-reducing SDS-PAGE analyses of a preparation of the intact h2E2 anti-cocaine mAb containing the 100 kDa mAb fragment. Each lane of a 1.5 mm thick, 7% acrylamide gel was loaded with 2 μg h2E2 anti-cocaine mAb preparation (10 μL total volume) per well, electrophoresed until the tracking dye reached the bottom of the gel, and stained with Coomassie blue. The entire gel is shown. Pre-electrophoretic incubation in SDS-PAGE non-reducing sample buffer was carried out for various times at 50 °C, with or without 5% isopropanol (IPA), as indicated in the figure. The electrophoretic migration positions of the 100 and 120 kDa species are indicated to the right of the gel, as are the positions of the various unfolded species for the intact mAb.
A model explaining the multiple discrete bands seen for the F(ab’)2 fragment is presented in Fig. 4A, with stochastic unfolding of each of the two Fab fragments in the F(ab’)2 fragment giving rise to the 3 discrete bands seen in Figs. 1A and 2. Fig. 4B is an updated and improved model for the unfolding domain transitions observed for the intact mAb, with the unfolding of the two Fab fragments of the F(ab’)2 portion and the CH3 domain of the Fc fragment all contributing to the discrete bands observed for the intact mAb in Fig. 2, and with the more unstable CH2 domain being essentially completely unfolded in the absence of heating in the presence of SDS.
Fig. 4.

Cartoon models of the conformational identities of the non-reducing SDS-PAGE gel bands observed for the F(ab’)2 fragment (A) and the intact mAb (B). Stochastic unfolding in SDS of both of the Fab domains observed in both F(ab’)2 fragment and the intact mAb is followed by unfolding of the very stable CH3 domain in the Fc portion of the intact mAb to explain the 3 and 4 discrete molecular weight bands observed for the F(ab’)2 fragment and the intact h2E2 anti-cocaine mAb, respectively. More compact “Folded” domains are shown as filled circles or ellipses, while more extended “Unfolded” domains are depicted as larger, uneven cloud-like structures. Letter labeling of the gel bands is the same as used in Figs. 1A and 2.
4. Discussion
Variable results upon analysis by non-reducing SDS-PAGE have been observed in our laboratory when analyzing not only the intact h2E2 anti-cocaine mAb, but also F(ab’)2 and Fab fragments derived from this mAb. Specifically, for the Fab fragment a smeared band was sometimes observed, and for the F(ab’)2 fragment multiple discrete bands were sometimes evident on non-reducing SDS-PAGE. These observations are not consistent with mass spectral data demonstrating a high degree of purity and a lack of size heterogeneity that would be measurable on SDS-PAGE gels for both the intact mAb and the F(ab’)2 and Fab fragments.
Therefore, we conducted studies to determine the nature and identities of these gel bands observed by non-reducing SDS-PAGE. For this, we extended and improved upon the non-reducing SDS-PAGE analyses of the intact mAb which was previously published [9]. Thus, Fab fragments were generated by Endo-Lyc-C digestion and purification [8], and Fc and F(ab’)2 fragments were generated by IdeS digestion and purified as previously described [12]. Fig. 1A clearly shows that the F(ab’)2 fragment undergoes a two-step unfolding process in SDS, as evidenced by 2 discrete higher apparent molecular weight bands appearing as a function of time of heating at 50 °C. The only reasonable explanation for this observation is the independent denaturation of the 2 Fab fragments which comprise the F(ab’)2. Interestingly, the whole of the F(ab’)2 fragment seems to be greater than the sum of its parts, since the untethered Fab fragment itself does not appear to undergo a similar, simple unfolding transition in SDS, but rather changes its migration on non-reducing SDS-PAGE from a smeared band to a compact band of the expected molecular weight upon heating (Fig. 1B). Also seen in Fig. 1B is that the Fc fragment seems to be largely denatured by SDS without heating, with only the conversion of a small amount of Fc to a higher molecular weight band upon heating.
Close inspection of non-reducing SDS-PAGE gels of some samples of the expressed and purified intact h2E2 mAb revealed a very small, and varying amount of a 100 kDa band. Like the intact mAb, the electrophoretic migration of this band decreased with heating, and this thermally induced change in electrophoretic migration could be attenuated by inclusion of isopropanol (and other short chain alcohols, data not shown) in the sample buffer (Fig. 3). To determine if the 100 and 120 kDa bands were identical in composition and to elucidate what modification of the mAb generated these bands, mass spectral analyses of these bands were performed after reduction, alkylation and trypsin digestion (results shown in Table 1). The results indicated that these bands were indeed identical in composition and consisted of the heavy chain only, and the mass spectrometry data supports that these bands contain identical protein sequences, only varying by their migration on the non-reducing gel. This is consistent with two distinct forms of the same heavy chain dimer resulting from a differentially folded/denatured domain, presumably due to thermal unfolding of the CH3 domain. The approximately 100 kDa molecular weight observed is consistent with 2 heavy chains without the presence of the 2 light chains found in the intact antibody. It is possible that such a species could be generated by selective partial reduction involving the single disulfide bond connecting each heavy chain to its light chain, but there is no evidence for partial reduction of these antibody preparations, and intentionally overloaded, non-reducing gels run to examine the possibility of light chains separated from the heavy chains by selective reduction in these samples did not reveal any traces of light chain bands at about 25 kDa (as determined on 10% acrylamide gels, see Supplementary Figure 1). Thus, it seems likely that the very small amount of this dimeric heavy chain only 100/120 kDa species is likely due to CHO expression rather than partial selective reduction occurring after expression, upon purification or storage of the mAb.
The straight-forward interpretation of the unfolding of the F(ab’)2 fragment shown in Figs. 1A and 2 is that the two Fab arms of the fragment unfold stochastically, leading to the observation of three possible bands, depending upon the extent of heating. The explanation for the 4th discrete band present in the intact mAb (Fig. 2) is less clear. The purified Fc fragment does exhibit a lower apparent molecular weight band with little or no heating in SDS (Fig. 1B), but there is very little of this faster migrating Fc band evident. The heavy chain only 100 kDa species does present an obvious single thermally-induced unfolding in SDS (Fig. 3). This may suggest that the part of the heavy chain normally associated with the light chain to form the Fab portion of the antibody is what is unfolding, or, more likely, it may indicate that the heavy chain-heavy chain interaction in the Fc part of the molecule is stabilized by the presence of the remainder of the heavy chains. Given the observed high stability towards thermal denaturation of the CH3 region of the Fc fragment in the absence of SDS [16], [17], [18], the model we developed to describe the thermal unfolding of the intact mAb in SDS suggests that the stochastic thermal unfolding of the Fab regions precedes the unfolding of the Fc CH3 domain. The lack of much unfolding observed for the Fc fragment (Fig. 1B) suggests that the less stable domain of the Fc region, i.e., the CH2 domain, may not require any heating in SDS to unfold, and thus is already unfolded in all samples in SDS prior to any heating. The CH3 domain is depicted as unfolding last in the intact antibody. This seems likely, since the kinetics of the appearance of two of the bands in the intact antibody are very similar to the appearance of the two unfolding transitions/bands in the F(ab’)2 fragment, which must be attributable to the Fab domains in common between the intact antibody and the F(ab’)2 fragment. Thus, from Fig. 2 it is evident that both Fab unfolding transitions are essentially complete by 7 min of heating at 50 °C, as are the first two of the three transitions observed for the intact antibody. The third domain unfolding transition for the intact antibody is still not complete by 20 min at 50 °C, suggesting that it is due to the very thermally stable CH3 domain of the intact mAb, which is absent in the F(ab’)2 fragment.
The current study confirms, extends, and improves the simple, inexpensive, and ubiquitous method of non-reducing SDS-PAGE for assessing the relative stability of the independently unfolding domains in IgGs, i.e., the CH2, CH3 and Fab domains in both intact antibodies and their fragments. A possible use of this approach is to examine the same antibody that has been produced using different conditions, cell types, or expression systems. In addition, comparisons of different batches of the same monoclonal antibodies, which may also vary in the extent of glycosylation and therefore could differ in their stabilities, is another possible use. However, we think non-reducing SDS-PAGE thermal unfolding analyses may be more useful to detect changes in stability after performing possibly destabilizing procedures such as chemical modification or conjugation of the mAb with small molecules or drugs, such as the construction of antibody drug conjugates (ADCs). This non-reducing SDS-PAGE thermal stability technique should also be useful to compare the relative stabilities of various domains of different mAbs. Such experiments can be easily quantified by gel densitometry to compare the kinetics of unfolding of the domains of mutants, conjugates, or other derivatives of antibodies and antibody fragments that are generated for therapeutic or experimental purposes. Thus, these simple, inexpensive, and widely available analyses should prove generally useful for the characterization of antibodies and their fragments and derivatives.
Acknowledgments
This work was supported in part by the United States National Institutes of Health National Institute on Drug Abuse Grant U01DA039550. We are grateful to Catalent PharmaSolutions, Inc. (Madison, WI) for providing the recombinant humanized h2E2 anti-cocaine mAb protein expressed using their GPex® technology. Support for the mass spectrometry analyses in the UC Proteomics Laboratory is provided in part by the UC College of Medicine, Cincinnati Children's Hospital Research Foundation and the shared instrumentation grant (SIG) program from the United States National Institutes of Health (NIH) Grant (S10RR027015).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.bbrep.2018.10.004.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.bbrep.2018.10.004.
Appendix A. Transparency document
Supplementary material
Appendix A. Supplementary material
Fig. S1.

The heavy chain only dimer species is present in multiple preparations of the h2E2 anti-cocaine mAb. Six samples (5 ug each per well) of the h2E2 mAb were analyzed on a non-reducing 10% SDS-PAGE gel either without heating ("-") or after boiling for 5 min ("+") in non-reducing sample buffer. Visible in the stained gel are varying amounts of the 100/120 kDa heavy chain dimer bands in all samples. No trace of light chain was detected at the expected 20–25 kDa range in any of the preparations.
References
- 1.Vocci F., Ling W. Medications development: successes and challenges. Pharmacol. Ther. 2005;108:94–108. doi: 10.1016/j.pharmthera.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Kosten T., Owens S.M. Immunotherapy for the treatment of drug abuse. Pharmacol. Ther. 2005;108:76–85. doi: 10.1016/j.pharmthera.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Kosten T.R., Domingo C.B., Shorter D., Orson F., Green C., Somoza E., Sekerka R., Levin F.R., Mariani J.J., Stitzer M., Tompkins D.A., Rotrosen J., Thakkar V., Smoak B., Kampman K. Vaccine for cocaine dependence: a randomized double-blind placebo-controlled efficacy trial. Drug Alcohol Depend. 2014;140:42–47. doi: 10.1016/j.drugalcdep.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martell B.A., Orson F.M., Poling J., Mitchell E., Rossen R.D., Gardner T., Kosten T.R. Cocaine vaccine for the treatment of cocaine dependence in methadone-maintained patients: a randomized, double-blind, placebo-controlled efficacy trial. Arch. Gen. Psychiatry. 2009;66:1116–1123. doi: 10.1001/archgenpsychiatry.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martell B.A., Mitchell E., Poling J., Gonsai K., Kosten T.R. Vaccine pharmacotherapy for the treatment of cocaine dependence. Biol. Psychiatry. 2005;58:158–164. doi: 10.1016/j.biopsych.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 6.Norman A.B., Ball W.J., Jr. Predicting the clinical efficacy and potential adverse effects of a humanized anticocaine monoclonal antibody. Immunotherapy. 2012;4:335–343. doi: 10.2217/imt.12.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paula S., Tabet M.R., Farr C.D., Norman A.B., Ball W.J., Jr. Three-dimensional quantitative structure-activity relationship modeling of cocaine binding by a novel human monoclonal antibody. J. Med. Chem. 2004;47:133–142. doi: 10.1021/jm030351z. [DOI] [PubMed] [Google Scholar]
- 8.Kirley T.L., Norman A.B. Characterization of a recombinant humanized anti-cocaine monoclonal antibody and its Fab fragment. Hum. Vaccines Immunother. 2015;11:458–467. doi: 10.4161/21645515.2014.990856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirley T.L., Norman A.B. Unfolding of IgG domains detected by non-reducing SDS-PAGE. Biochem. Biophys. Res. Commun. 2018;503:944–949. doi: 10.1016/j.bbrc.2018.06.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norman A.B., Gooden F.C., Tabet M.R., Ball W.J. A recombinant humanized anti-cocaine monoclonal antibody inhibits the distribution of cocaine to the brain in rats. Drug Metab. Dispos. 2014;42:1125–1131. doi: 10.1124/dmd.114.057034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wetzel H.N., Webster R.P., Saeed F.O., Kirley T.L., Ball W.J., Norman A.B. Characterization of a recombinant humanized anti-cocaine monoclonal antibody produced from multiple clones for the selection of a master cell bank candidate. Biochem. Biophys. Res. Commun. 2017;487:690–694. doi: 10.1016/j.bbrc.2017.04.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirley T.L., Greis K.D., Norman A.B. Structural characterization of expressed monoclonal antibodies by single sample mass spectral analysis after IdeS proteolysis. Biochem. Biophys. Res. Commun. 2016;477:363–368. doi: 10.1016/j.bbrc.2016.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirley T.L., Greis K.D., Norman A.B. Selective disulfide reduction for labeling and enhancement of Fab antibody fragments. Biochem. Biophys. Res. Commun. 2016;480:752–757. doi: 10.1016/j.bbrc.2016.10.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 15.Eismann T., Huber N., Shin T., Kuboki S., Galloway E., Wyder M., Edwards M.J., Greis K.D., Shertzer H.G., Fisher A.B., Lentsch A.B. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G266–G274. doi: 10.1152/ajpgi.90583.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demarest S.J., Rogers J., Hansen G. Optimization of the antibody C(H)3 domain by residue frequency analysis of IgG sequences. J. Mol. Biol. 2004;335:41–48. doi: 10.1016/j.jmb.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 17.Garber E., Demarest S.J. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem. Biophys. Res. Commun. 2007;355:751–757. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 18.Johnson C.M. Differential scanning calorimetry as a tool for protein folding and stability. Arch. Biochem. Biophys. 2013;531:100–109. doi: 10.1016/j.abb.2012.09.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material