

The monohydrated chloride, bromide and iodide salt forms of the amino acid l-asparagine form an isostructural series.
Keywords: crystal structure, amino-acid, salt selections, isostructural series
Abstract
The structures of three monohydrated halide salt forms of l-asparagine are presented, viz. l-asparaginium chloride monohydrate, C4H9N2O3 +·Cl−·H2O, (I), l-asparaginium bromide monohydrate, C4H9N2O3 +·Br−·H2O, (II), and l-asparaginium iodide monohydrate, C4H9N2O3 +·I−·H2O, (III). These form an isomorphous and isostructural series. The C—C—C—C backbone of the amino acid adopts a gauche conformation in each case [torsion angles for (I), (II) and (III) = −55.4 (2), −55.6 (5) and −58.3 (7)°, respectively]. Each cation features an intramolecular N—H⋯O hydrogen bond, which closes an S(6) ring. The extended structures feature chains of cations that propagate parallel to the b-axis direction. These are formed by carboxylic acid/amide complimentary O—H⋯O + N—H⋯O hydrogen bonds, which generate R 2 2(8) loops. These chains are linked by further hydrogen bonds mediated by the halide ions and water molecules to give a layered structure with cation and anion layers parallel to the ab plane. Compound (III) was refined as an inversion twin.
Chemical context
Changing the salt form of an organic material is a well known way of altering the material’s physical properties whilst retaining many of the chemical properties inherent to the organic fragment. Selection of the salt form with the most suitable properties is thus an important consideration in the development of pharmaceutical materials and indeed of other fine chemicals (Stahl & Wermuth, 2008 ▸; Bastin et al., 2000 ▸; Kennedy et al., 2012 ▸). Often, the main property of interest is solubility, but salt selection may also be used to alter properties such as crystal morphology, hygroscopicity or stability, as well as mechanical properties such as hardness and strength (Stahl & Wermuth, 2008 ▸; Sun & Grant, 2001 ▸; Hao & Iqbal, 1997 ▸; de Moraes et al., 2017 ▸). In short, any bulk property that depends in some way on the packing or on the intermolecular forces within the crystalline array structure may be altered by changing the salt-forming counter-ion. Despite the common usage of salt selection strategies, our understanding of what effect on properties any particular change of counter-ion will have is extremely limited. This means, for example, that it is not currently possible to predict which salt form of an active pharmaceutical ingredient (API) will be the most soluble or have the best compaction properties. In this area, isostructural series of structures are especially interesting as they allow changes in properties to be related to changes in intermolecular interaction strength or type without the complication of changes to the overall gross structure (Galcera & Molins, 2009 ▸; Allan et al., 2018 ▸). Here we present the structures of three isostructural halide salts of l-asparagine, namely the monohydrates [HAsp][Cl]·H2O, (I), [HAsp][Br]·H2O, (II) and [HAsp][I]·H2O, (III), (HAsp = C4H9N2O3 cation). l-asparagine is a non-essential amino acid, the bioavailability of which is associated with altered rates of breast cancer progression (Knott et al., 2018 ▸).
Structural commentary
The crystals isolated from all three reactions of l-asparagine with HX (X = Cl, Br, I) solutions were found to be hydrated compounds with the formula [HAsp][X]·H2O with protonation occurring at N1 as well as at the carboxylic acid. The starting material used was labelled l-asparagine and in all cases the refined Flack parameter confirmed that, as expected, this is S-asparagine.
Crystals (I), (II) and (III) were found to adopt the same space group and to have similar unit-cell dimensions. They thus represent an isostructural series, with the unit-cell dimensions increasing as expected in line with increasing halide ion size. The HAsp cations are found to have near identical geometries. All equivalent bond lengths are statistically similar and all cations adopt the same general conformation with both C=O units syn with respect to the NH3 group, see Figs. 1 ▸–3 ▸ ▸. There are some small differences within this general conformation. The largest of these differences occurs between the iodide salt and the others, as indicated by the torsion angles involving the NH3 group [N1—C2—C1—O1 (acid C=O) = 24.6 (2), 20.2 (5) and 12.5 (8) and N1—C2—C4—O3 (amide) 27.1 (2), 27.73 (5) and 33.38 (8)°, for Cl, Br and I respectively].
Figure 1.

View of the contents of the asymmetric unit of (I). Non-H atoms are drawn as 50% probability ellipsoids and H atoms as spheres of arbitrary size.
Figure 2.

View of the contents of the asymmetric unit of (II). Non-H atoms are drawn as 50% probability ellipsoids and H atoms as spheres of arbitrary size.
Figure 3.

View of the contents of the asymmetric unit of (III). Non-H atoms are drawn as 50% probability ellipsoids and H atoms as spheres of arbitrary size.
Supramolecular features
Isostructurality is also indicated by examination of the hydrogen bonding, Tables 1 ▸–3 ▸ ▸ and Fig. 4 ▸. The three compounds all make the same number and type of hydrogen bonds, with the main difference being the increasing D⋯A distances caused by the different anion sizes. Where A = X there is a 7.4 to 11.5% increase in D⋯A distance from Cl to I, whereas where A = O there is a smaller 0.6 to 4.0% increase. The only exception is the sole intramolecular interaction. The D⋯A distance of this NH3 to amide contact decreases by about 1.5% from Cl to I.
Table 1. Hydrogen-bond geometry (Å, °) for (I) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H1H⋯O3i | 0.88 (1) | 1.66 (2) | 2.533 (2) | 172 (4) |
| N1—H1N⋯Cl1 | 0.91 (1) | 2.27 (1) | 3.1663 (17) | 166 (2) |
| N1—H2N⋯Cl1ii | 0.89 (1) | 2.56 (2) | 3.2909 (17) | 140 (2) |
| N1—H2N⋯O3 | 0.89 (1) | 2.19 (2) | 2.809 (2) | 126 (2) |
| N1—H3N⋯O1W | 0.90 (1) | 1.97 (1) | 2.867 (2) | 172 (2) |
| N2—H4N⋯Cl1iii | 0.90 (1) | 2.89 (2) | 3.4056 (17) | 118 (2) |
| N2—H4N⋯O1iv | 0.90 (1) | 2.21 (2) | 3.051 (2) | 156 (2) |
| N2—H5N⋯O1W v | 0.88 (1) | 2.08 (1) | 2.949 (2) | 167 (2) |
| O1W—H1W⋯Cl1vi | 0.87 (1) | 2.41 (1) | 3.2650 (18) | 169 (2) |
| O1W—H2W⋯Cl1vii | 0.87 (1) | 2.40 (2) | 3.2184 (17) | 157 (2) |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  ; (vi)
; (vi)  ; (vii)
; (vii) 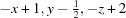 .
.
Table 2. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H1H⋯O3i | 0.88 (1) | 1.68 (2) | 2.543 (4) | 167 (6) |
| N1—H1N⋯Br1 | 0.90 (1) | 2.46 (2) | 3.314 (5) | 159 (4) |
| N1—H2N⋯Br1ii | 0.89 (1) | 2.59 (3) | 3.408 (5) | 153 (4) |
| N1—H2N⋯O3 | 0.89 (1) | 2.30 (5) | 2.787 (6) | 114 (4) |
| N1—H3N⋯O1W | 0.90 (1) | 1.99 (2) | 2.886 (4) | 173 (5) |
| N2—H4N⋯Br1iii | 0.90 (1) | 2.95 (4) | 3.479 (4) | 119 (4) |
| N2—H4N⋯O1iv | 0.90 (1) | 2.26 (3) | 3.081 (5) | 152 (4) |
| N2—H5N⋯O1W v | 0.90 (1) | 2.07 (2) | 2.959 (6) | 170 (5) |
| O1W—H1W⋯Br1vi | 0.88 (1) | 2.51 (2) | 3.362 (4) | 167 (5) |
| O1W—H2W⋯Br1vii | 0.88 (1) | 2.61 (4) | 3.323 (4) | 138 (5) |
Symmetry codes: (i) ; (ii) ; (iii) ; (iv) ; (v) ; (vi) ; (vii) .
Table 3. Hydrogen-bond geometry (Å, °) for (III) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H1H⋯O3i | 0.88 (1) | 1.71 (3) | 2.549 (6) | 160 (9) |
| N1—H1N⋯I1 | 0.91 | 2.65 | 3.528 (7) | 164 |
| N1—H2N⋯I1ii | 0.91 | 2.89 | 3.591 (8) | 135 |
| N1—H2N⋯O3 | 0.91 | 2.11 | 2.766 (8) | 129 |
| N1—H3N⋯O1W | 0.91 | 2.03 | 2.905 (6) | 160 |
| N2—H4N⋯I1iii | 0.90 (1) | 3.07 (6) | 3.659 (5) | 125 (5) |
| N2—H4N⋯O1iv | 0.90 (1) | 2.37 (4) | 3.171 (7) | 149 (6) |
| N2—H5N⋯O1W v | 0.90 (1) | 2.12 (3) | 2.983 (9) | 160 (7) |
| O1W—H1W⋯I1vi | 0.88 (1) | 2.68 (2) | 3.526 (8) | 164 (5) |
| O1W—H2W⋯I1vii | 0.88 (1) | 2.76 (4) | 3.504 (7) | 143 (6) |
Symmetry codes: (i) ; (ii) ; (iii) ; (iv) ; (v) ; (vi) ; (vii) .
Figure 4.

View of all the unique hydrogen-bonding contacts made by the contents of the asymmetric unit of (I).
The only HAsp to HAsp hydrogen bonds form the classic carboxylic acid to amide O—H⋯O + N—H⋯O heterodimer motif [R(8)2 2]. With two such contacts per cation, this motif generates a one-dimensional hydrogen-bonded chain running parallel to the b-axis direction, see Fig. 5 ▸. Additionally, each halide ion accepts five unique hydrogen bonds, two bonds from water molecules, two from NH3 groups and one from NH2. The water molecules donate two hydrogen bonds to the halide ions and accept two from the NH3 and NH2 groups. The water molecules thus form fourfold nodes, as is typical for organic hydrates (Gillon et al., 2003 ▸; Briggs et al., 2012 ▸). These interactions combine to give the structure shown in Fig. 6 ▸ with alternating layers of organic cations and halide anions lying parallel to the ab plane.
Figure 5.

Chain of cations in (II) propagating parallel to the b-axis direction via O—H⋯O and O—H⋯N carboxylic acid to amide hydrogen bonds.
Figure 6.

Packing diagram of (III) as viewed down the a-axis direction.
Database survey
The only other known structure of a simple salt of S-asparagine is that of the nitrate (Aarthy et al., 2005 ▸). Here both the cations in a Z′ = 2 structure adopt different conformations from that found for the halides: compare N—C—C—O(acid C=O) of −176.9 (6) and 173.2 (5)° and N—C—C—O(amide) of −123.2 (7) and 77.0 (4)° with the equivalent values given above. The structures of two simple salts of racemic asparagine have also been reported. These are the nitrate and the perchlorate forms (Moussa Slimane et al., 2009 ▸; Guenifa et al., 2009 ▸). All these literature forms are anhydrous, but despite this difference and further differences in anion type and cation geometry, all form the same R(8)2 2-based, one-dimensional hydrogen-bonded chain motif seen in the halide salts (I), (II) and (III).
Synthesis and crystallization
Salt forms of l-asparagine were prepared by dissolving 29 mmol of the amino acid in 90 ml of distilled water. The solution was stirred and heated slightly until complete dissolution had occurred. The solution was then equally divided between three vials. To each vial was added 1 ml of concentrated acid, either hydrochloric acid, hydrobromic acid or hydroiodic acid. The first crystals appeared after 24 h of sitting at room temperature. Crystals suitable for analyses [colourless prisms for (I), colourless tablets for (II) and colourless rods for (III)] were obtained directly from the mother liquors and were removed from these solutions just prior to data collection.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 4 ▸. Structure solution for (III) was by substitution from the Br equivalent. All H atoms bound to C were placed in calculated positions and refined in riding modes. C—H distances were 0.99 and 1.00 Å for CH2 and CH groups respectively, with U(H)iso = 1.2U eq(C). With the exception noted below, all other H atoms were observed and positioned as found. For (I) these were refined isotropically, but for (II) restraints were required for the NH3 and OH2 atoms. For (III) all H atoms required restraints to be applied. N—H distances were restrained to 0.90 (1) Å and O—H distances to 0.88 (1) Å. U(H)iso = 1.2U eq of the parent atom. The exception was the NH3 group of (III). The best model involved treating this as a rigid tetrahedral group and allowing only rotation around the C—N bond. For this group, U iso(H) = 1.5U eq(N). Compound (III) was refined as an inversion twin.
Table 4. Experimental details.
| (I) | (II) | (III) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C4H9N2O3 +·Cl−·H2O | C4H9N2O3 −·Br+·H2O | C4H9N2O3 +·I−·H2O |
| M r | 186.60 | 231.06 | 278.05 |
| Crystal system, space group | Monoclinic, P21 | Monoclinic, P21 | Monoclinic, P21 |
| Temperature (K) | 123 | 123 | 123 |
| a, b, c (Å) | 5.0922 (1), 10.1450 (2), 8.1950 (2) | 5.2167 (2), 10.2784 (5), 8.3063 (4) | 5.3668 (5), 10.6744 (8), 8.4532 (6) |
| β (°) | 103.834 (2) | 103.606 (5) | 102.772 (8) |
| V (Å3) | 411.08 (2) | 432.88 (4) | 472.28 (7) |
| Z | 2 | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| μ (mm−1) | 0.44 | 4.72 | 3.37 |
| Crystal size (mm) | 0.45 × 0.30 × 0.25 | 0.5 × 0.3 × 0.12 | 0.6 × 0.35 × 0.15 |
| Data collection | |||
| Diffractometer | Oxford Diffraction Xcalibur E | Oxford Diffraction Xcalibur E | Oxford Diffraction Xcalibur E |
| Absorption correction | Multi-scan [CrysAlis PRO (Agilent, 2014 ▸), based on expressions derived by Clark & Reid (1995 ▸)] | Analytical [CrysAlis PRO (Agilent, 2014 ▸), based on expressions derived by Clark & Reid (1995 ▸)] | Analytical [CrysAlis PRO (Agilent, 2014 ▸), based on expressions derived by Clark & Reid (1995 ▸)] |
| T min, T max | 0.900, 1.000 | 0.205, 0.487 | 0.286, 0.612 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 4053, 2079, 2032 | 4320, 2232, 2118 | 5854, 2458, 2288 |
| R int | 0.013 | 0.032 | 0.039 |
| (sin θ/λ)max (Å−1) | 0.698 | 0.700 | 0.702 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.022, 0.058, 1.07 | 0.028, 0.062, 1.03 | 0.029, 0.058, 1.02 |
| No. of reflections | 2079 | 2232 | 2458 |
| No. of parameters | 128 | 128 | 117 |
| No. of restraints | 9 | 9 | 7 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.20 | 0.60, −0.38 | 0.87, −0.65 |
| Absolute structure | Flack x determined using 897 quotients [(I +)−(I −)]/[(I +)+(I −)] (Parsons et al., 2013 ▸) | Flack x determined using 908 quotients [(I +)−(I −)]/[(I +)+(I −)] (Parsons et al., 2013 ▸) | Refined as an inversion twin |
| Absolute structure parameter | −0.02 (2) | −0.022 (11) | −0.07 (4) |
Supplementary Material
Crystal structure: contains datablock(s) I, II, III, general. DOI: 10.1107/S2056989018014603/hb7779sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018014603/hb7779Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989018014603/hb7779IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2056989018014603/hb7779IIIsup4.hkl
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779Isup5.cml
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779IIsup6.cml
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779IIIsup7.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
L-Asparaginium chloride monohydrate (I) . Crystal data
| C4H9N2O3+·Cl−·H2O | F(000) = 196 |
| Mr = 186.60 | Dx = 1.508 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.0922 (1) Å | Cell parameters from 3444 reflections |
| b = 10.1450 (2) Å | θ = 3.3–29.7° |
| c = 8.1950 (2) Å | µ = 0.44 mm−1 |
| β = 103.834 (2)° | T = 123 K |
| V = 411.08 (2) Å3 | Prism, colourless |
| Z = 2 | 0.45 × 0.30 × 0.25 mm |
L-Asparaginium chloride monohydrate (I) . Data collection
| Oxford Diffraction Xcalibur E diffractometer | 2032 reflections with I > 2σ(I) |
| Radiation source: sealed tube | Rint = 0.013 |
| ω scans | θmax = 29.8°, θmin = 3.3° |
| Absorption correction: multi-scan [CrysAlis PRO (Agilent, 2014), based on expressions derived by Clark & Reid (1995)] | h = −7→7 |
| Tmin = 0.900, Tmax = 1.000 | k = −14→13 |
| 4053 measured reflections | l = −10→10 |
| 2079 independent reflections |
L-Asparaginium chloride monohydrate (I) . Refinement
| Refinement on F2 | H atoms treated by a mixture of independent and constrained refinement |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0331P)2 + 0.0289P] where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.022 | (Δ/σ)max < 0.001 |
| wR(F2) = 0.058 | Δρmax = 0.26 e Å−3 |
| S = 1.07 | Δρmin = −0.20 e Å−3 |
| 2079 reflections | Extinction correction: SHELXL-2014/7 (Sheldrick 2015), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 128 parameters | Extinction coefficient: 0.029 (8) |
| 9 restraints | Absolute structure: Flack x determined using 897 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: −0.02 (2) |
| Hydrogen site location: mixed |
L-Asparaginium chloride monohydrate (I) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
L-Asparaginium chloride monohydrate (I) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.36571 (8) | 0.90075 (4) | 0.92401 (5) | 0.02062 (13) | |
| O1 | 1.1145 (3) | 0.51257 (16) | 0.77258 (18) | 0.0258 (3) | |
| O2 | 0.8130 (3) | 0.48931 (15) | 0.52673 (18) | 0.0237 (3) | |
| O3 | 0.8617 (3) | 0.83494 (16) | 0.56812 (17) | 0.0269 (3) | |
| O1W | 1.1611 (3) | 0.58702 (15) | 1.1607 (2) | 0.0242 (3) | |
| H1W | 1.275 (4) | 0.528 (2) | 1.142 (3) | 0.029* | |
| H2W | 1.034 (4) | 0.532 (2) | 1.170 (3) | 0.029* | |
| N1 | 0.8288 (3) | 0.71004 (16) | 0.8685 (2) | 0.0162 (3) | |
| H1N | 0.699 (4) | 0.758 (2) | 0.902 (3) | 0.019* | |
| H2N | 0.936 (4) | 0.764 (2) | 0.827 (3) | 0.019* | |
| H3N | 0.927 (4) | 0.664 (2) | 0.957 (2) | 0.019* | |
| N2 | 0.5625 (4) | 0.78473 (17) | 0.3247 (2) | 0.0218 (4) | |
| C1 | 0.8969 (4) | 0.53507 (18) | 0.6792 (2) | 0.0153 (3) | |
| C2 | 0.6864 (4) | 0.61977 (17) | 0.7318 (2) | 0.0140 (3) | |
| H1 | 0.5703 | 0.5597 | 0.7813 | 0.017* | |
| C3 | 0.5009 (4) | 0.69279 (18) | 0.5859 (2) | 0.0162 (3) | |
| H2 | 0.3900 | 0.6278 | 0.5091 | 0.019* | |
| H3 | 0.3765 | 0.7499 | 0.6304 | 0.019* | |
| C4 | 0.6548 (4) | 0.77587 (19) | 0.4888 (2) | 0.0170 (4) | |
| H1H | 0.936 (6) | 0.437 (3) | 0.503 (5) | 0.067 (11)* | |
| H4N | 0.643 (4) | 0.8399 (19) | 0.266 (3) | 0.019 (6)* | |
| H5N | 0.423 (4) | 0.736 (2) | 0.273 (3) | 0.023 (6)* |
L-Asparaginium chloride monohydrate (I) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.01624 (19) | 0.0241 (2) | 0.0209 (2) | 0.00077 (17) | 0.00325 (14) | −0.00539 (18) |
| O1 | 0.0182 (7) | 0.0346 (8) | 0.0224 (7) | 0.0096 (6) | 0.0008 (5) | −0.0059 (6) |
| O2 | 0.0211 (7) | 0.0281 (8) | 0.0204 (7) | 0.0067 (6) | 0.0020 (5) | −0.0079 (6) |
| O3 | 0.0222 (7) | 0.0359 (8) | 0.0199 (7) | −0.0128 (6) | 0.0000 (6) | 0.0078 (6) |
| O1W | 0.0252 (8) | 0.0200 (7) | 0.0270 (8) | −0.0019 (6) | 0.0051 (6) | −0.0003 (6) |
| N1 | 0.0150 (8) | 0.0182 (8) | 0.0156 (8) | 0.0008 (6) | 0.0040 (6) | −0.0019 (6) |
| N2 | 0.0265 (9) | 0.0217 (8) | 0.0163 (7) | −0.0029 (7) | 0.0034 (7) | 0.0011 (6) |
| C1 | 0.0149 (8) | 0.0149 (8) | 0.0168 (8) | −0.0014 (6) | 0.0052 (6) | 0.0014 (6) |
| C2 | 0.0123 (8) | 0.0149 (8) | 0.0152 (8) | −0.0006 (6) | 0.0039 (6) | −0.0007 (6) |
| C3 | 0.0129 (8) | 0.0186 (8) | 0.0166 (8) | 0.0004 (7) | 0.0026 (6) | 0.0023 (7) |
| C4 | 0.0168 (8) | 0.0163 (8) | 0.0183 (8) | 0.0023 (6) | 0.0046 (7) | 0.0023 (6) |
L-Asparaginium chloride monohydrate (I) . Geometric parameters (Å, º)
| O1—C1 | 1.209 (2) | N2—C4 | 1.317 (2) |
| O2—C1 | 1.305 (2) | N2—H4N | 0.897 (12) |
| O2—H1H | 0.876 (13) | N2—H5N | 0.882 (13) |
| O3—C4 | 1.251 (2) | C1—C2 | 1.515 (2) |
| O1W—H1W | 0.869 (13) | C2—C3 | 1.527 (2) |
| O1W—H2W | 0.868 (13) | C2—H1 | 1.0000 |
| N1—C2 | 1.493 (2) | C3—C4 | 1.502 (3) |
| N1—H1N | 0.913 (13) | C3—H2 | 0.9900 |
| N1—H2N | 0.891 (13) | C3—H3 | 0.9900 |
| N1—H3N | 0.901 (12) | ||
| C1—O2—H1H | 110 (3) | N1—C2—C3 | 112.85 (15) |
| H1W—O1W—H2W | 97 (3) | C1—C2—C3 | 113.57 (15) |
| C2—N1—H1N | 107.3 (15) | N1—C2—H1 | 107.3 |
| C2—N1—H2N | 108.8 (17) | C1—C2—H1 | 107.3 |
| H1N—N1—H2N | 110 (2) | C3—C2—H1 | 107.3 |
| C2—N1—H3N | 111.2 (16) | C4—C3—C2 | 112.56 (15) |
| H1N—N1—H3N | 109 (2) | C4—C3—H2 | 109.1 |
| H2N—N1—H3N | 110 (2) | C2—C3—H2 | 109.1 |
| C4—N2—H4N | 119.5 (15) | C4—C3—H3 | 109.1 |
| C4—N2—H5N | 119.9 (17) | C2—C3—H3 | 109.1 |
| H4N—N2—H5N | 121 (2) | H2—C3—H3 | 107.8 |
| O1—C1—O2 | 125.44 (17) | O3—C4—N2 | 123.25 (18) |
| O1—C1—C2 | 122.06 (16) | O3—C4—C3 | 118.34 (16) |
| O2—C1—C2 | 112.48 (15) | N2—C4—C3 | 118.40 (17) |
| N1—C2—C1 | 108.17 (14) | ||
| O1—C1—C2—N1 | 24.6 (2) | N1—C2—C3—C4 | 68.1 (2) |
| O2—C1—C2—N1 | −156.61 (16) | C1—C2—C3—C4 | −55.4 (2) |
| O1—C1—C2—C3 | 150.72 (17) | C2—C3—C4—O3 | −37.7 (2) |
| O2—C1—C2—C3 | −30.5 (2) | C2—C3—C4—N2 | 143.49 (18) |
L-Asparaginium chloride monohydrate (I) . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1H···O3i | 0.88 (1) | 1.66 (2) | 2.533 (2) | 172 (4) |
| N1—H1N···Cl1 | 0.91 (1) | 2.27 (1) | 3.1663 (17) | 166 (2) |
| N1—H2N···Cl1ii | 0.89 (1) | 2.56 (2) | 3.2909 (17) | 140 (2) |
| N1—H2N···O3 | 0.89 (1) | 2.19 (2) | 2.809 (2) | 126 (2) |
| N1—H3N···O1W | 0.90 (1) | 1.97 (1) | 2.867 (2) | 172 (2) |
| N2—H4N···Cl1iii | 0.90 (1) | 2.89 (2) | 3.4056 (17) | 118 (2) |
| N2—H4N···O1iv | 0.90 (1) | 2.21 (2) | 3.051 (2) | 156 (2) |
| N2—H5N···O1Wv | 0.88 (1) | 2.08 (1) | 2.949 (2) | 167 (2) |
| O1W—H1W···Cl1vi | 0.87 (1) | 2.41 (1) | 3.2650 (18) | 169 (2) |
| O1W—H2W···Cl1vii | 0.87 (1) | 2.40 (2) | 3.2184 (17) | 157 (2) |
Symmetry codes: (i) −x+2, y−1/2, −z+1; (ii) x+1, y, z; (iii) x, y, z−1; (iv) −x+2, y+1/2, −z+1; (v) x−1, y, z−1; (vi) −x+2, y−1/2, −z+2; (vii) −x+1, y−1/2, −z+2.
L-Asparaginium bromide monohydrate (II) . Crystal data
| C4H9N2O3−·Br+·H2O | F(000) = 232 |
| Mr = 231.06 | Dx = 1.773 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.2167 (2) Å | Cell parameters from 3145 reflections |
| b = 10.2784 (5) Å | θ = 3.2–29.8° |
| c = 8.3063 (4) Å | µ = 4.72 mm−1 |
| β = 103.606 (5)° | T = 123 K |
| V = 432.88 (4) Å3 | Tablet, colourless |
| Z = 2 | 0.5 × 0.3 × 0.12 mm |
L-Asparaginium bromide monohydrate (II) . Data collection
| Oxford Diffraction Xcalibur E diffractometer | 2232 independent reflections |
| Radiation source: fine-focus sealed tube | 2118 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.032 |
| ω scans | θmax = 29.8°, θmin = 3.2° |
| Absorption correction: analytical [CrysAlis PRO (Agilent, 2014), based on expressions derived by Clark & Reid (1995)] | h = −7→6 |
| Tmin = 0.205, Tmax = 0.487 | k = −14→14 |
| 4320 measured reflections | l = −11→11 |
L-Asparaginium bromide monohydrate (II) . Refinement
| Refinement on F2 | H atoms treated by a mixture of independent and constrained refinement |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0274P)2] where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.028 | (Δ/σ)max = 0.001 |
| wR(F2) = 0.062 | Δρmax = 0.60 e Å−3 |
| S = 1.03 | Δρmin = −0.38 e Å−3 |
| 2232 reflections | Extinction correction: SHELXL2014 (Sheldrick, 2015), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 128 parameters | Extinction coefficient: 0.019 (3) |
| 9 restraints | Absolute structure: Flack x determined using 908 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: −0.022 (11) |
| Hydrogen site location: mixed |
L-Asparaginium bromide monohydrate (II) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
L-Asparaginium bromide monohydrate (II) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.36490 (6) | 0.90306 (8) | 0.92345 (4) | 0.01479 (13) | |
| O1 | 1.1176 (6) | 0.5163 (3) | 0.7609 (4) | 0.0196 (7) | |
| O2 | 0.8106 (6) | 0.4779 (3) | 0.5270 (4) | 0.0187 (7) | |
| O3 | 0.8665 (6) | 0.8262 (3) | 0.5653 (4) | 0.0226 (7) | |
| O1W | 1.1602 (8) | 0.5964 (4) | 1.1565 (6) | 0.0196 (9) | |
| H1W | 1.274 (8) | 0.536 (4) | 1.147 (7) | 0.024* | |
| H2W | 1.059 (10) | 0.541 (5) | 1.193 (7) | 0.024* | |
| N1 | 0.8287 (9) | 0.7034 (5) | 0.8580 (6) | 0.0124 (9) | |
| H1N | 0.716 (8) | 0.749 (4) | 0.903 (6) | 0.015* | |
| H2N | 0.951 (8) | 0.757 (4) | 0.836 (6) | 0.015* | |
| H3N | 0.932 (8) | 0.663 (5) | 0.947 (4) | 0.015* | |
| N2 | 0.5699 (7) | 0.7822 (4) | 0.3244 (4) | 0.0169 (8) | |
| C1 | 0.8998 (8) | 0.5314 (4) | 0.6731 (5) | 0.0124 (8) | |
| C2 | 0.6900 (8) | 0.6143 (4) | 0.7242 (5) | 0.0109 (8) | |
| H1 | 0.5765 | 0.5549 | 0.7729 | 0.013* | |
| C3 | 0.5124 (8) | 0.6858 (4) | 0.5811 (5) | 0.0130 (8) | |
| H2 | 0.4071 | 0.6215 | 0.5044 | 0.016* | |
| H3 | 0.3884 | 0.7411 | 0.6243 | 0.016* | |
| C4 | 0.6627 (8) | 0.7697 (4) | 0.4866 (5) | 0.0132 (8) | |
| H1H | 0.938 (9) | 0.428 (5) | 0.509 (8) | 0.046 (18)* | |
| H4N | 0.645 (9) | 0.837 (4) | 0.265 (5) | 0.016 (12)* | |
| H5N | 0.431 (7) | 0.733 (4) | 0.276 (6) | 0.016 (14)* |
L-Asparaginium bromide monohydrate (II) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.01205 (18) | 0.01658 (19) | 0.01513 (18) | 0.0006 (2) | 0.00197 (12) | −0.0034 (2) |
| O1 | 0.0120 (15) | 0.0263 (18) | 0.0185 (16) | 0.0072 (13) | −0.0002 (13) | −0.0061 (13) |
| O2 | 0.0148 (14) | 0.0238 (17) | 0.0159 (15) | 0.0046 (13) | 0.0003 (12) | −0.0062 (13) |
| O3 | 0.0176 (16) | 0.0309 (19) | 0.0159 (15) | −0.0127 (13) | −0.0031 (13) | 0.0076 (14) |
| O1W | 0.015 (2) | 0.018 (2) | 0.025 (2) | −0.0018 (17) | 0.0022 (16) | 0.0001 (17) |
| N1 | 0.013 (2) | 0.012 (2) | 0.012 (2) | 0.0026 (18) | 0.0035 (17) | −0.0018 (17) |
| N2 | 0.0205 (19) | 0.0169 (18) | 0.0123 (17) | −0.0031 (16) | 0.0013 (15) | 0.0014 (14) |
| C1 | 0.014 (2) | 0.0102 (18) | 0.0142 (19) | −0.0021 (15) | 0.0056 (16) | 0.0019 (15) |
| C2 | 0.0072 (18) | 0.0129 (19) | 0.0124 (18) | −0.0021 (15) | 0.0020 (15) | −0.0005 (15) |
| C3 | 0.0092 (18) | 0.014 (2) | 0.016 (2) | −0.0008 (16) | 0.0018 (16) | 0.0003 (16) |
| C4 | 0.0135 (19) | 0.0099 (19) | 0.0158 (19) | 0.0025 (16) | 0.0024 (16) | 0.0017 (15) |
L-Asparaginium bromide monohydrate (II) . Geometric parameters (Å, º)
| O1—C1 | 1.207 (5) | N2—C4 | 1.326 (5) |
| O2—C1 | 1.314 (5) | N2—H4N | 0.898 (14) |
| O2—H1H | 0.879 (14) | N2—H5N | 0.897 (14) |
| O3—C4 | 1.252 (5) | C1—C2 | 1.524 (6) |
| O1W—H1W | 0.875 (14) | C2—C3 | 1.515 (6) |
| O1W—H2W | 0.879 (14) | C2—H1 | 1.0000 |
| N1—C2 | 1.490 (6) | C3—C4 | 1.504 (6) |
| N1—H1N | 0.900 (14) | C3—H2 | 0.9900 |
| N1—H2N | 0.894 (14) | C3—H3 | 0.9900 |
| N1—H3N | 0.904 (14) | ||
| C1—O2—H1H | 106 (4) | N1—C2—C1 | 107.3 (3) |
| H1W—O1W—H2W | 93 (5) | C3—C2—C1 | 113.5 (3) |
| C2—N1—H1N | 112 (3) | N1—C2—H1 | 107.7 |
| C2—N1—H2N | 118 (3) | C3—C2—H1 | 107.7 |
| H1N—N1—H2N | 109 (5) | C1—C2—H1 | 107.7 |
| C2—N1—H3N | 115 (4) | C4—C3—C2 | 113.0 (3) |
| H1N—N1—H3N | 102 (4) | C4—C3—H2 | 109.0 |
| H2N—N1—H3N | 98 (5) | C2—C3—H2 | 109.0 |
| C4—N2—H4N | 121 (3) | C4—C3—H3 | 109.0 |
| C4—N2—H5N | 118 (3) | C2—C3—H3 | 109.0 |
| H4N—N2—H5N | 121 (5) | H2—C3—H3 | 107.8 |
| O1—C1—O2 | 125.7 (4) | O3—C4—N2 | 123.2 (4) |
| O1—C1—C2 | 122.6 (4) | O3—C4—C3 | 118.4 (4) |
| O2—C1—C2 | 111.7 (4) | N2—C4—C3 | 118.4 (4) |
| N1—C2—C3 | 112.8 (3) | ||
| O1—C1—C2—N1 | 20.2 (5) | N1—C2—C3—C4 | 66.7 (5) |
| O2—C1—C2—N1 | −161.4 (4) | C1—C2—C3—C4 | −55.6 (5) |
| O1—C1—C2—C3 | 145.4 (4) | C2—C3—C4—O3 | −35.7 (5) |
| O2—C1—C2—C3 | −36.1 (5) | C2—C3—C4—N2 | 145.6 (4) |
L-Asparaginium bromide monohydrate (II) . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1H···O3i | 0.88 (1) | 1.68 (2) | 2.543 (4) | 167 (6) |
| N1—H1N···Br1 | 0.90 (1) | 2.46 (2) | 3.314 (5) | 159 (4) |
| N1—H2N···Br1ii | 0.89 (1) | 2.59 (3) | 3.408 (5) | 153 (4) |
| N1—H2N···O3 | 0.89 (1) | 2.30 (5) | 2.787 (6) | 114 (4) |
| N1—H3N···O1W | 0.90 (1) | 1.99 (2) | 2.886 (4) | 173 (5) |
| N2—H4N···Br1iii | 0.90 (1) | 2.95 (4) | 3.479 (4) | 119 (4) |
| N2—H4N···O1iv | 0.90 (1) | 2.26 (3) | 3.081 (5) | 152 (4) |
| N2—H5N···O1Wv | 0.90 (1) | 2.07 (2) | 2.959 (6) | 170 (5) |
| O1W—H1W···Br1vi | 0.88 (1) | 2.51 (2) | 3.362 (4) | 167 (5) |
| O1W—H2W···Br1vii | 0.88 (1) | 2.61 (4) | 3.323 (4) | 138 (5) |
Symmetry codes: (i) −x+2, y−1/2, −z+1; (ii) x+1, y, z; (iii) x, y, z−1; (iv) −x+2, y+1/2, −z+1; (v) x−1, y, z−1; (vi) −x+2, y−1/2, −z+2; (vii) −x+1, y−1/2, −z+2.
L-Asparaginium iodide monohydrate (III) . Crystal data
| C4H9N2O3+·I−·H2O | F(000) = 268 |
| Mr = 278.05 | Dx = 1.955 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.3668 (5) Å | Cell parameters from 4015 reflections |
| b = 10.6744 (8) Å | θ = 3.8–29.9° |
| c = 8.4532 (6) Å | µ = 3.37 mm−1 |
| β = 102.772 (8)° | T = 123 K |
| V = 472.28 (7) Å3 | Fragment cut from long rod, colourless |
| Z = 2 | 0.6 × 0.35 × 0.15 mm |
L-Asparaginium iodide monohydrate (III) . Data collection
| Oxford Diffraction Xcalibur E diffractometer | 2288 reflections with I > 2σ(I) |
| Radiation source: sealed tube | Rint = 0.039 |
| ω scans | θmax = 29.9°, θmin = 3.8° |
| Absorption correction: analytical [CrysAlis PRO (Agilent, 2014), based on expressions derived by Clark & Reid (1995)] | h = −7→7 |
| Tmin = 0.286, Tmax = 0.612 | k = −14→14 |
| 5854 measured reflections | l = −11→11 |
| 2458 independent reflections |
L-Asparaginium iodide monohydrate (III) . Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.029 | w = 1/[σ2(Fo2) + (0.0215P)2] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.058 | (Δ/σ)max = 0.001 |
| S = 1.02 | Δρmax = 0.87 e Å−3 |
| 2458 reflections | Δρmin = −0.65 e Å−3 |
| 117 parameters | Absolute structure: Refined as an inversion twin |
| 7 restraints | Absolute structure parameter: −0.07 (4) |
| Primary atom site location: structure-invariant direct methods |
L-Asparaginium iodide monohydrate (III) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refined as a two-component inversion twin. |
L-Asparaginium iodide monohydrate (III) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| I1 | 0.36921 (6) | 0.90844 (10) | 0.91430 (3) | 0.01907 (10) | |
| O1 | 1.1192 (8) | 0.5186 (4) | 0.7440 (5) | 0.0243 (10) | |
| O2 | 0.8070 (8) | 0.4561 (4) | 0.5384 (5) | 0.0258 (11) | |
| H1H | 0.936 (10) | 0.415 (8) | 0.516 (7) | 0.031* | |
| O3 | 0.8904 (9) | 0.7986 (4) | 0.5464 (5) | 0.0282 (11) | |
| O1W | 1.1491 (15) | 0.6160 (6) | 1.1460 (9) | 0.0274 (16) | |
| H1W | 1.253 (10) | 0.553 (4) | 1.143 (9) | 0.033* | |
| H2W | 1.029 (9) | 0.575 (5) | 1.181 (8) | 0.033* | |
| N1 | 0.8348 (14) | 0.6975 (7) | 0.8380 (8) | 0.0165 (16) | |
| H3N | 0.9372 | 0.6546 | 0.9205 | 0.025* | |
| H1N | 0.7172 | 0.7420 | 0.8773 | 0.025* | |
| H2N | 0.9315 | 0.7508 | 0.7926 | 0.025* | |
| N2 | 0.5817 (11) | 0.7743 (5) | 0.3201 (6) | 0.0224 (12) | |
| H5N | 0.428 (7) | 0.740 (6) | 0.277 (8) | 0.027* | |
| H4N | 0.636 (13) | 0.835 (5) | 0.262 (7) | 0.027* | |
| C1 | 0.9010 (11) | 0.5232 (5) | 0.6669 (7) | 0.0161 (12) | |
| C2 | 0.7013 (12) | 0.6073 (5) | 0.7127 (7) | 0.0155 (12) | |
| H2 | 0.5882 | 0.5537 | 0.7640 | 0.019* | |
| C3 | 0.5321 (12) | 0.6737 (6) | 0.5676 (7) | 0.0177 (12) | |
| H3A | 0.4349 | 0.6100 | 0.4937 | 0.021* | |
| H3B | 0.4074 | 0.7271 | 0.6067 | 0.021* | |
| C4 | 0.6816 (11) | 0.7537 (6) | 0.4743 (7) | 0.0181 (12) |
L-Asparaginium iodide monohydrate (III) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| I1 | 0.01668 (17) | 0.01986 (16) | 0.01982 (16) | 0.0013 (3) | 0.00223 (11) | −0.0036 (2) |
| O1 | 0.018 (2) | 0.029 (2) | 0.024 (2) | 0.009 (2) | −0.0010 (17) | −0.006 (2) |
| O2 | 0.016 (2) | 0.031 (2) | 0.029 (2) | 0.0061 (18) | 0.0031 (18) | −0.0140 (18) |
| O3 | 0.023 (3) | 0.034 (3) | 0.024 (2) | −0.011 (2) | −0.0023 (18) | 0.011 (2) |
| O1W | 0.025 (4) | 0.022 (4) | 0.034 (3) | −0.006 (3) | 0.003 (3) | 0.001 (3) |
| N1 | 0.013 (4) | 0.019 (4) | 0.018 (3) | 0.007 (3) | 0.005 (2) | 0.001 (3) |
| N2 | 0.025 (3) | 0.022 (3) | 0.019 (3) | −0.003 (2) | 0.002 (2) | 0.002 (2) |
| C1 | 0.017 (3) | 0.013 (3) | 0.018 (3) | 0.001 (2) | 0.004 (2) | 0.003 (2) |
| C2 | 0.015 (3) | 0.015 (3) | 0.016 (2) | −0.002 (2) | 0.003 (2) | −0.001 (2) |
| C3 | 0.014 (3) | 0.018 (3) | 0.020 (3) | 0.002 (3) | 0.002 (2) | 0.001 (2) |
| C4 | 0.016 (3) | 0.016 (3) | 0.022 (3) | 0.002 (2) | 0.004 (2) | 0.001 (2) |
L-Asparaginium iodide monohydrate (III) . Geometric parameters (Å, º)
| O1—C1 | 1.209 (7) | N2—C4 | 1.314 (7) |
| O2—C1 | 1.305 (7) | N2—H5N | 0.900 (14) |
| O2—H1H | 0.876 (14) | N2—H4N | 0.896 (14) |
| O3—C4 | 1.247 (7) | C1—C2 | 1.513 (8) |
| O1W—H2W | 0.880 (14) | C2—C3 | 1.529 (8) |
| O1W—H1W | 0.877 (14) | C2—H2 | 1.0000 |
| N1—C2 | 1.492 (9) | C3—C4 | 1.509 (9) |
| N1—H3N | 0.9100 | C3—H3A | 0.9900 |
| N1—H1N | 0.9100 | C3—H3B | 0.9900 |
| N1—H2N | 0.9100 | ||
| C1—O2—H1H | 106 (5) | N1—C2—C3 | 112.1 (5) |
| H1W—O1W—H2W | 98 (3) | C1—C2—C3 | 113.5 (5) |
| C2—N1—H3N | 109.5 | N1—C2—H2 | 107.7 |
| C2—N1—H1N | 109.5 | C1—C2—H2 | 107.7 |
| H3N—N1—H1N | 109.5 | C3—C2—H2 | 107.7 |
| C2—N1—H2N | 109.5 | C4—C3—C2 | 113.1 (5) |
| H3N—N1—H2N | 109.5 | C4—C3—H3A | 109.0 |
| H1N—N1—H2N | 109.5 | C2—C3—H3A | 109.0 |
| C4—N2—H5N | 118 (5) | C4—C3—H3B | 109.0 |
| C4—N2—H4N | 124 (5) | C2—C3—H3B | 109.0 |
| H4N—N2—H5N | 117 (7) | H3A—C3—H3B | 107.8 |
| O1—C1—O2 | 125.2 (6) | O3—C4—N2 | 123.1 (6) |
| O1—C1—C2 | 122.9 (5) | O3—C4—C3 | 119.1 (5) |
| O2—C1—C2 | 111.9 (5) | N2—C4—C3 | 117.8 (5) |
| N1—C2—C1 | 107.9 (5) | ||
| O1—C1—C2—N1 | 12.5 (8) | N1—C2—C3—C4 | 64.3 (7) |
| O2—C1—C2—N1 | −168.8 (5) | C1—C2—C3—C4 | −58.3 (7) |
| O1—C1—C2—C3 | 137.5 (6) | C2—C3—C4—O3 | −27.3 (8) |
| O2—C1—C2—C3 | −43.9 (7) | C2—C3—C4—N2 | 153.8 (6) |
L-Asparaginium iodide monohydrate (III) . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1H···O3i | 0.88 (1) | 1.71 (3) | 2.549 (6) | 160 (9) |
| N1—H1N···I1 | 0.91 | 2.65 | 3.528 (7) | 164 |
| N1—H2N···I1ii | 0.91 | 2.89 | 3.591 (8) | 135 |
| N1—H2N···O3 | 0.91 | 2.11 | 2.766 (8) | 129 |
| N1—H3N···O1W | 0.91 | 2.03 | 2.905 (6) | 160 |
| N2—H4N···I1iii | 0.90 (1) | 3.07 (6) | 3.659 (5) | 125 (5) |
| N2—H4N···O1iv | 0.90 (1) | 2.37 (4) | 3.171 (7) | 149 (6) |
| N2—H5N···O1Wv | 0.90 (1) | 2.12 (3) | 2.983 (9) | 160 (7) |
| O1W—H1W···I1vi | 0.88 (1) | 2.68 (2) | 3.526 (8) | 164 (5) |
| O1W—H2W···I1vii | 0.88 (1) | 2.76 (4) | 3.504 (7) | 143 (6) |
Symmetry codes: (i) −x+2, y−1/2, −z+1; (ii) x+1, y, z; (iii) x, y, z−1; (iv) −x+2, y+1/2, −z+1; (v) x−1, y, z−1; (vi) −x+2, y−1/2, −z+2; (vii) −x+1, y−1/2, −z+2.
References
- Aarthy, A., Anitha, K., Athimoolam, S., Bahadur, S. A. & Rajaram, R. K. (2005). Acta Cryst. E61, o2042–o2044.
- Agilent (2014). CrysAlis PRO. Agilent Technologies Ltd, Yarnton, Oxfordshire, England.
- Allan, P., Arlin, J.-B., Kennedy, A. R. & Walls, A. (2018). Acta Cryst. C74, 131–138. [DOI] [PubMed]
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst. 27, 435.
- Bastin, R. J., Bowker, M. J. & Slater, B. J. (2000). Org. Process Res. Dev. 4, 427–435.
- Briggs, N. E. B., Kennedy, A. R. & Morrison, C. A. (2012). Acta Cryst. B68, 453–464. [DOI] [PubMed]
- Clark, R. C. & Reid, J. S. (1995). Acta Cryst. A51, 887–897.
- Galcera, J. & Molins, E. (2009). Cryst. Growth Des. 9, 327–334.
- Gillon, A. L., Feeder, N., Davey, R. J. & Storey, R. (2003). Cryst. Growth Des. 3, 663–673.
- Guenifa, F., Bendjeddou, L., Cherouana, A., Dahaoui, S. & Lecomte, C. (2009). Acta Cryst. E65, o2264–o2265. [DOI] [PMC free article] [PubMed]
- Hao, Z. & Iqbal, A. (1997). Chem. Soc. Rev. 26, 203–213.
- Kennedy, A. R., Stewart, H., Eremin, K. & Stenger, J. (2012). Chem. Eur. J. 18, 3064–3069. [DOI] [PubMed]
- Knott, S. R. V., Wagenblast, E., Khan, S., Kim, S. Y., Soto, M., Wagner, M., Turgeon, M. O., Fish, L., Erard, N., Gable, A. L., Maceli, A. R., Dickopf, S., Papachristou, E. K., D’Santos, C. S., Carey, L. A., Wilkinson, J. E., Harrell, J. C., Perou, C. M., Goodarzi, H., Poulogiannis, G. & Hannon, G. J. (2018). Nature, 554, 378–381. [DOI] [PMC free article] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Moraes, L. S. de, Edwards, D., Florence, A. J., Johnston, A., Johnston, B. F., Morrison, C. A. & Kennedy, A. R. (2017). Cryst. Growth Des. 17, 3277–3286.
- Moussa Slimane, N., Cherouana, A., Bendjeddou, L., Dahaoui, S. & Lecomte, C. (2009). Acta Cryst. E65, o2180–o2181. [DOI] [PMC free article] [PubMed]
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Stahl, P. H. & Wermuth, C. G. (2008). Handbook of Pharmaceutical Salts: Properties, Selection and Use. VHCA: Zurich.
- Sun, C. C. & Grant, D. J. W. (2001). Pharm. Res. 18, 281–286. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, II, III, general. DOI: 10.1107/S2056989018014603/hb7779sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018014603/hb7779Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989018014603/hb7779IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2056989018014603/hb7779IIIsup4.hkl
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779Isup5.cml
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779IIsup6.cml
Supporting information file. DOI: 10.1107/S2056989018014603/hb7779IIIsup7.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report