

SUMMARY
The DNA helicase FANCJ is mutated in hereditary breast and ovarian cancer and Fanconi anemia (FA). Nevertheless, how loss of FANCJ translates to disease pathogenesis remains unclear. We addressed this question by analyzing proteins associated with replication forks in cells with or without FANCJ. We demonstrate that FANCJ-knockout (FANCJ-KO) cells have alterations in the replisome that are consistent with enhanced replication stress, including an aberrant accumulation of the fork remodeling factor helicase-like transcription factor (HLTF). Correspondingly, HLTF contributes to fork degradation in FANCJ-KO cells. Unexpectedly, the unrestrained DNA synthesis that characterizes HLTF-deficient cells is FANCJ dependent and correlates with S1 nuclease sensitivity and fork degradation. These results suggest that FANCJ and HLTF promote replication fork integrity, in part by counteracting each other to keep fork remodeling and elongation in check. Indicating one protein compensates for loss of the other, loss of both HLTF and FANCJ causes a more severe replication stress response.
In Brief
Peng et al. find that loss of FANCJ enhances the replisome association of helicase-like transcription factor (HLTF). HLTF depletion suppresses fork degradation in FANCJ-deficient cells, and FANCJ depletion suppresses aberrant fork elongation in HLTF-deficient cells. However, the combined loss of HLTF and FANCJ causes severe replication stress.
Graphical Abstract
INTRODUCTION
Preserving genomeintegrity is absolutely essential for cell survival and to prevent disease. BRCA1 and BRCA2 are tumor suppressors with central functions in the DNA damage response that preserve genome integrity. In double-strand break repair, they mediate distinct steps of homology-directed repair (HDR). Genome preservation functions for BRCA1 and BRCA2 also involve roles in the replication stress response, which enables cells to cope with perturbations to replication. When forks stall, BRCA1 and BRCA2 protect nascent DNA from degradation. In BRCA1- and BRCA2-deficient cells, MRE11-dependent nucleolytic processing of reversed forks leads to fork degradation (Schlacher et al., 2011; Schlacher et al., 2012). Preventing fork reversal through depletion of fork remodelers such as SMARCAL1, ZRANB3, or helicase-like transcription factor (HLTF) restores fork protection to BRCA1 and BRCA2-deficient cells and in some cases improves resistance to stress-inducing agents (Kolinjivadi et al., 2017; Taglialatela et al., 2017; Cantor and Calvo, 2017; Mijic et al., 2017).
Given this understanding, it is proposed that perturbations in the replication stress response along with defects in DNA repair underlie BRCA-Fanconi anemia (FA) pathway maladies. Indeed, hereditary breast and ovarian cancer cells as well as cells from FA patients have proliferation defects. In conjunction with sources of endogenous replication stress, especially in rapidly dividing cells, FA cells may ultimately lose proliferation capacity and develop anemia or bone marrow failure as found in FA (Cheung and Taniguchi, 2017). Loss of the BRCA-FA pathway could also elevate replication stress. However, the underlying cause of exacerbated replication stress aside from elevated DNA damage responses in FA cells remains unclear, because little is known about how the BRCA-FA pathway contributes to the replisome function.
The BRCA-associated FANCJ DNA helicase is mutated in hereditary breast and ovarian cancer as well as in FA (Cantor et al., 2004; Litman et al., 2005; Minion et al., 2015). Although experimental analyses have focused largely on FANCJ function in response to genotoxic agents, it is clear that FANCJ is needed for endogenous replication problems as well. For example, knockdown of FANCJ causes increased DNA damage in otherwise unperturbed S-phase cells (Kumaraswamy and Shiekhattar, 2007). The endogenous source of replication stress is unknown but could be unusual DNA structures that have a propensity to form at stalled forks. In support of this point, along with induction of γ-H2AX and slower growth, FANCJ-deficient cells display microsatellite instability (Matsuzaki et al., 2015). FANCJ could counteract replication perturbations as it travels with the elongating replication fork (Alabert et al., 2014; Sirbu et al., 2011).
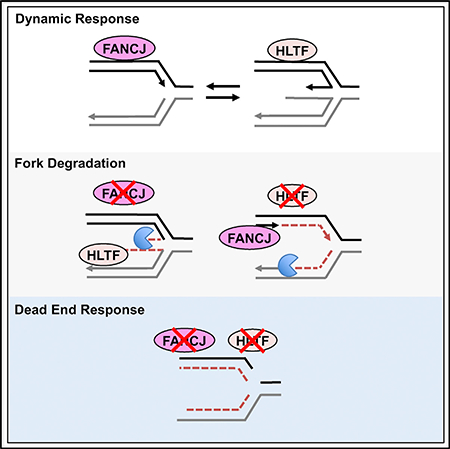
Here, we used DNA fiber analysis to uncover a function for FANCJ in fork protection. Through an unbiased proteomics approach, we also identify proteins that associate with replication forks in an FANCJ-dependent manner. We present evidence that FANCJ limits fork degradation by suppressing HLTF, which normally slows and remodels DNA replication forks (Kile et al., 2015). In addition, we find that HLTF fork remodeling limits permissive replication mediated by FANCJ. We propose that FANCJ and HLTF participate in a general surveillance mechanism by counteracting each other to maintain unperturbed DNA replication. In response to stress, these opposing activities are critical for replication forks to have dynamic response.
RESULTS
FANCJ Is Required for Fork Protection
To determine if FANCJ functions in fork protection, we measured replication-fork elongation using DNA fiber spreading analysis. First, we generated human FANCJ-knockout (FANCJ-KO) 293T cells using clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated protein 9 (Cas9) technology (Figure 1A). FANCJ loss generated the expected sensitivity to mitomycin C (MMC) (Peng et al., 2007), and complementation with FANCJWT elevated MMC resistance (Figure S1A). Next, fork degradation was assessed by measuring the ratio of 5-chloro-2ʹ-deoxyuridine (CldU) to 5-iodo-2ʹ-deoxyuridine (IdU) tract lengths following sequential pulses with IdU and CldU prior to hydroxyurea (HU) treatment. FANCJ-KO cells had significantly greater fork degradation than control cells with FANCJ (Figure 1B). In these assay conditions, fork protection was restored by inhibition of the nuclease MRE11 with mirin (Dupré et al., 2008) (Figure 1B). As with FANCJ-KO 293T cells, we found that FANCJ-null Fanconi anemia complementation group J (FA-J) patient fibroblasts had significant fork degradation. Both fork degradation and MMC sensitivity were suppressed by complementation with FANCJWT, but not the cat-alytic FANCJ K52R mutant FANCJK52R (Figures 1C, 1D, and S1B). Based on a complementary assay in which IdU tract lengths were measured only when also labeled with CldU, which was incorporated concurrently with the HU treatment assay (Lemaçon et al., 2017), we could conclude that the enhanced degradation in 293T or FA-J cells without FANCJ or its catalytic activity was not due to premature termination of replication and/or unintentional breaks (Figures S1C and S1D). Furthermore, using this assay, we detected fork degradation in CRISPR-Cas9-generated FANCJ-KO U2OS cells. The MMC sensitivity of FANCJ-KO U2OS cells was suppressed by reexpression of FANCJWT, but not vector (Figure S1E), validating functional loss of FANCJ. A similar level of fork degradation was also observed upon depletion of BRCA2 (Lemaçon et al., 2017; Schlacher et al., 2011) (Figure S1F). In summary, these findings are consistent with the idea that FANCJ and its helicase activity are required to protect forks from MRE11-dependent degradation.
Figure 1. FANCJ and Its Helicase Activity Protect Nascent DNA at Replication Forks from MRE11-Dependent Degradation.

(A) Western blot analysis with the indicated antibodies (Abs) of lysates from control and FANCJKO 293T cells.
(B) Schematic, representative images, and quantification of the CldU/IdU ratio after HU treatment with or without mirin.
(C) Western blot analysis with indicated Abs of lysates from FANCJ-null FA-J cells complemented with vector (V), wild-type (WT), or a catalytically inactive FANCJ (K52R) mutant.
(D) Schematic, representative images, and quantification of the CldU/IdU ratio after HU treatment. Each dot represents one fiber.
For each analysis, at least 200 fibers are quantified from two independent experiments. Red bars represent the median. Statistical analysis according to two-tailed Mann-Whitney test; ****p < 0.0001. Scale bars, 10 μm.
Identification of Proteins at Replication Forks in Unchallenged FANCJ-KO Cells
To mechanistically explain the role of FANCJ in replication, we sought to analyze how FANCJ loss altered the composition of the replisome. Isolation of proteins on nascent DNA (iPOND) has revealed the accumulation of FA proteins at active replication forks including FANCJ (Alabert et al., 2014; Sirbu et al., 2011). We detected mild, but reproducible, FANCJ association with replication forks (Figure 2A). Upon thymidine chase, FANCJ association was lost, along with other replisome components including proliferating cell nuclear antigen (PCNA), confirming the presence of FANCJ only at the active replication fork (Figure 2A). Despite a growth defect and higher levels of g-H2AX (Figures S2A and S2B) consistent with endogenous DNA damage, unchallenged FANCJ-KO 293T cells had similar median length of CldU (Figure S2C) and precipitated similar amounts of PCNA and histone H2B by iPOND compared to control 293T cells (Figure 2B). Thus, iPOND in FANCJ-KO and control 293T cells is a tractable system to address how FANCJ contributes to the composition of the replisome.
Figure 2. Isolation of Proteins on Nascent DNA (iPOND) from FANCJ-KO and Control 293T Cells.

(A) Western blot analysis with the indicated Abs of input or iPOND samples following non-click, click, or thymidine chase from 293T cells.
(B) Western blot analysis with indicated Abs of input or iPOND samples from control and FANCJ-KO 293T cells.
(C) Schematic representation of the SILAC iPOND technique.
(D) Volcano plots of the p values versus the log2 protein abundance differences between purified replisomes of 293T control (heavy) and FANCJ-KO (light) cells. Significantly enriched and reduced proteins are highlighted in red and blue, respectively. p values are calculated from the data of three biological repeats.
(E) Table shows the light/heavy ratio in chromatin fractions for indicated proteins.
(F) Western blot analysis with the indicated Abs of WCE and chromatin preparations from 293T cells.
(G) Western blot analysis with the indicated Abs of cells transfected with vector (V) or wild-type FANCJ (WT) 293T cells. Asterisk indicates nonspecific band.
To quantitatively compare the composition of replication-fork-associated factors between FANCJ-proficient and deficient cells, we combined iPOND and stable isotope labeling by amino acids in cell culture (SILAC). Following successful incorporation of isotopes, cells “light” (FANCJ KO) or “heavy” (control) were labeled for 10 min with 5-ethynyl-2′-deoxyuridine (EdU). EdU was similarly incorporated within a 10-min pulse (Figure S2D). Equal cell numbers were combined and processed together for coupling to biotin azide and precipitation of DNA-bound proteins for mass spectrometry (MS) analyses (Figure 2C). For each protein identified, we plotted the light versus heavy ratio and p value. Proteins found after an EdU pulse labeling and thymidine chase (Table S1) were excluded from the analysis to ensure that only proteins at replicating forks were compared. We successfully isolated active replication forks, as illustrated by the majority of high-confidence proteins from iPOND-MS with low p values and multiple peptide identifications that are also known replisome components. According to the iPOND-SILAC ratios, the majority of proteins quantitated from the two cell populations did not substantially change between FANCJ-KO and control cells (Figure 2D).
While the majority of proteins that are directly involved in DNA synthesis accumulate similarly on nascent DNA in FANCJ-KO cells (Table S2), we did observe changes of several factors known to function in genomic stability, DNA damage repair, and the replication stress response. In particular, we found reductions in the splicing-associated factor THOC2 (Chi et al., 2013) and the chromatin architecture factors KDM1A and SMARCA1 (Mohrmann and Verrijzer, 2005; Yuan et al., 2012). Other proteins that were reduced included phosphoglycerate kinase (PGK1), of which deficiency causes anemia (Beutler, 2007); KIF4A, which is a protein that promotes homologous recombination (HR) and binds BRCA2 (Lee and Kim, 2003; Wu et al., 2008); and KDM1A/LSD1 (lysine-specific histone demethylase 1A) (Mosammaparast et al., 2013; Peng et al., 2015), which functions in the 53BP1 DNA damage response (Thorslund et al., 2015; Torres et al., 2016). The factors most enriched included HLTF, which functions in replication fork remodeling, and GNL3 (nucleostemin) (Lin et al., 2014) and PSPC1 (Knott et al., 2016), which function in the DNA damage response. In addition, FANCJ-KO cells were enriched for PSIP1 (Baude et al., 2016) and CDCA7L, both of which form a complex and function in DNA end resection and HR (Chan et al., 2016). Thus, even under nonchallenged conditions, iPOND revealed that FANCJ contributes to the composition of the replisome.
HLTF Is Enriched in the Chromatin of FANCJ-KO Cells
We focused on HLTF because of its role in regulating fork dynamics. HLTF is a fork remodeler that restrainsDNAreplication in response to stress and promotes fork reversal (Kile et al., 2015). Moreover, similar to the iPOND results, SILAC-chromatin-MS revealed that HLTF was enriched in the chromatin fraction of FANCJ-KO 293T cells (Figure 2E). Furthermore, immunoblots validated enriched HLTF in chromatin of FANCJ-KO cells and showed slight enrichment in whole-cell extracts (WCEs) as compared to SMARCA1, which was reduced in both WCE and chromatin (Figure 2F). HLTF levels did not change and remained enriched in FANCJ-KO 293T cells upon treatment with HU (Figure S2E), suggesting that HLTF enrichment was due to FANCJ deficiency as opposed to DNA damage in FANCJ-KO cells. The chromatin of FA-J patient immortalized fibroblast cells complemented with vector also trended toward enriched HLTF as compared to FA-J cells complemented with FANCJWT (Figure S2F), suggesting that these findings were independent of cell type. HLTF chromatin enrichment was also suppressed by transfection of FANCJ-KO cells with FANCJWT as compared to vector (Figure 2G). Collec-tively, these findings are consistent with FANCJ counteracting HLTF chromatin enrichment in a cell-type-independent manner.
HLTF Contributes to Fork Degradation in FANCJ-KO Cells
Depletion of fork remodelers such as HLTF restores fork protection to BRCA1- and BRCA2-deficient cells (Kolinjivadi et al., 2017; Taglialatela et al., 2017; Mijic et al., 2017). To determine the contribution of HLTF to fork degradation in FANCJ-KO cells, we measured the ratio of CldU to IdU tract lengths following HU treatment in FANCJ-KO cells with or without HLTF depletion. HLTF depletion in either control or FANCJ-KO 293T cells did not alter the tract lengths in unchallenged conditions (Figures 3A, S3A, and S3B). After HU treatment, while HLTF depletion did not alter fork degradation in control cells, HLTF depletion did significantly reduce fork degradation in FANCJ-KO 293T cells (Figure 3B), consistent with HLTF contributing to fork degradation in this background.
Figure 3. HLTF Contributes to Fork Degradation in FANCJ-KO Cells, and FANCJ Contributes to Fork Degradation in HLTF-Depleted Cells following Prolonged Stress.

(A) Western blot analysis with the indicated Abs of lysates from control and FANCJ-KO 293T cells expressing shRNA against HLTF or NSC.
(B) Schematic, representative images, and quantification of the CldU/IdU ratio after HU treatment. (C) Schematic, representative images, and quantification of the CIdU tract length.
Each dot represents one fiber; at least 200 fibers are quantified from two independent experiments. Red bars represent the median. Statistical analysis according to two-tailed Mann-Whitney test; ****p < 0.0001.
Given that HLTF depletion leads to unrestrained replication during stress (Kile et al., 2015), we considered that this phenotype could generate longer tract lengths and therefore mask fork degradation. To mitigate this possible issue, we extended the duration of stress before tract lengths were measured. FANCD2-depleted cells also fail to slow in response to stress, but with the extension of low-dose HU, forks degrade (Lossaint et al., 2013; Schlacher et al., 2011). As with short-term, highdose HU, FANCJ-KO 293T cells underwent enhanced degradation as compared to control cells upon the extension of low-dose HU (Figures 3C and S3C). Again, nascent-strand degradation in FANCJ-KO 293T cells was suppressed by HLTF depletion with either of two distinct small hairpin RNAs (shRNAs), suggesting that enriched HLTF contributes to fork degradation in FANCJKO 293T cells (Figures 3C and S3C). Notably, however, during extended replication stress, HLTF depletion, similar to FANCJKO cells, had a significant degradation of nascent DNA (Figures 3C and S3C). In agreement, similar findings were found in U2OS cells (Figures S3D and S3E). Collectively, these findings indicate that FANCJ and HLTF are critical to protect nascent DNA during prolonged replication stress and that loss of either factor leads to fork degradation, which depends on the other factor.
HTLF Supports the Recovery of FANCJ-KO Cells from HU-Induced Stress
We sought to address the relationship between FANCJ and HLTF with respect to cell growth and survival following replication stress. Both control or FANCJ-KO U2OS cells exhibited similar reductions in growth kinetics with HLTF depletion (Figures 4A and S4A). There was a significant gain in MMC resistance with HLTF depletion in FANCJ-KO cells (Figure 4B). By comparison, there was a significant loss in HU resistance in HLTF-depleted FANCJ-KO cells. Indeed, HLTF depletion highly sensitized FANCJ-KO cells to HU, whereas HLTF depletion in control cells enhanced resistance to HU (Figure 4B). Significant sensitivity to HU was also found in FANCJ-null FA-J cells upon HLTF depletion (Figure S4B), further suggesting that loss of FANCJ and HLTF was detrimental for recovery from nucleotide depletion. To further evaluate replication stress, we queried the level of γ-H2AX found in EdU-positive cells as a marker of replication-associated breaks (Sirbu et al., 2013). As compared to control cells, FANCJ-KO cells exhibit increased levels of replication stress. Moreover, the γ-H2AX/EdU ratio in FANCJ-KO cells was significantly enhanced upon HLTF depletion (Figure 4C). Increased replication stress was also detected in HLTF-depleted FANCJ-KO cells in a modified alkaline comet assay. Newly synthesized BrdU-positive DNA appears as a halo-like structure of “loose DNA” when replication is discontinuous due to stress (McGlynn et al., 1999; Mórocz et al., 2013) (Figure 4D). Collectively, these data reveal that the combined loss of FANCJ and HLTF elevates replication stress following HU treatment.
Figure 4. HLTF Counteracts Replication Stress in FANCJ-KO Cells.

(A) Western blot analysis with the indicated Abs of lysates from control and FANCJ-KO U2OS cells and when expressing shRNA against HLTF or NSC.
(B) Cell survival assays with FANCJ-KO and control U2OS cells expressing shRNA against HLTF or NSC under increasing concentrations of MMC or HU. Data represent the mean percent ± SD of survival from three independent experiments.
(C) γ-H2AX/EdU ratio in FANCJ-KO and control U2OS cells expressing shRNA against HLTF or NSC following treatment with HU. At least 200 cells were measured for each. The assay was completed in triplicate for each shRNA.
(D) Quantification of the percentage of comet tail DNA in FANCJ-KO and control 293T cells expressing shRNA against HLTF or NSC following treatment with HU, and images of comet tail formation. 100 cells were measured for each. Statistical analysis according to two-tailed Mann-Whitney test; ****p < 0.0001; *p < 0.05.
To assess whether fork degradation in FANCJ-KO cells is suppressed by loss of a distinct remodeler, we depleted SMARCAL1 using shRNA reagents (Figure S4C). FANCJ-KO U2OS cells were more sensitive to SMARCAL1 depletion than control cells in unchallenged conditions. Indeed, with one of the shRNA reagents that had the greatest SMARCAL1 depletion (as observed in FANCJ-proficient cells), the survival of FANCJ-KO U2OS cells was dramatically reduced (Figure S4C). However, SMARCAL1 depletion with either of three shRNA reagents in FANCJ-KO 293T cells did not dramatically reduce viability. Therefore, we tested whether fork protection was enhanced (Figure S4D). SMARCAL1 depletion enhanced fork protection in FANCJ-KO 293T cells (Figure S4E). These findings further suggest that the reversed fork substrate is important for degradation in FANCJ-deficient cells.
FANCJ Is Required for Unrestrained Replication that Generates S1-Nuclease-Sensitive Tracts in HLTF-Deficient Cells
The replication stress induction and heightened HU sensitivity in HLTF depleted FANCJ-KO cells predicted that the unrestrained replication during stress that characterizes HLTF depletion (Kile et al., 2015) was prohibited in FANCJ-KO cells. To test this idea, we first confirmed that HLTF depletion, as previously achieved in HCT116 cells (Achar et al., 2015), maintained fork speed despite the presence of HU (Figures 5A and 5B). We also confirmed that HCT116 cells with or without FANCJ and HLTF depletion did not have significant differences in tract lengths in unchallenged conditions (Figure S5A). We observed a reduction in fork progression in cells deficient in FANCJ, indicating that FANCJ is not required for restraining replication elongation in HU (Figures 5A, 5B, S5B, and S5C). Notably, HLTF depletion in these FANCJ-deficient cells (FANCJ-depleted HCT116 or FANCJ-deleted 293T and U2OS) did not generate the previously noted unrestrained replication found in control cells (Figures 5A, 5B, S5B, and S5C). These findings indicate that HLTF deficiency causes permissive replication that is dependent on FANCJ.
Figure 5. FANCJ Contributes to Unrestrained Replication and S1 Nuclease Sensitivity in HLTF-Depleted Cells.

(A) Western blot analysis with the indicated Abs of lysates from HCT116 cells expressing shRNA against FANCJ, HLTF, or NSC.
(B) Schematic, representative images, and quantification of CldU tract length during HU treatment.
(C) Schematic and quantification of the CldU/IdU ratio in the indicated shRNA-expressing HCT116 cells with or without S1 nuclease incubation. Each dot represents one fiber; at least 200 fibers are quantified from two independent experiments. Red bars represent the median. Statistical analysis according to two-tailed Mann-Whitney test; ****p < 0.0001.
(D) Model of opposing roles of FANCJ and HLTF at forks.
Unrestrained replication is associated with single-stranded DNA (ssDNA) gap formation (Lossaint et al., 2013). To detect gaps not directly observable in DNA fiber assays, we treated nuclei with the S1 nuclease after the second pulse (CIdU) and before spreading the DNA onto the glass slide (Quinet et al., 2016, 2017) (Figure 5C). When gaps are present, ssDNA regions were nicked by the nuclease, generating shorter CIdU tracts. In the absence of the S1 nuclease, HLTF-depleted HCT116 cells had longer CldU fibers than control cells and therefore a higher CldU/IdU ratio, as expected. However, after the addition of S1 nuclease, only HLTF-deficient cells presented a decrease in the CldU fiber lengths, which significant decreased the CldU/ IdU ratio (Figures 5C and S5D). The effect of the S1 nuclease was similar in HLTF-depleted 293T cells (Figure S5D). These data indicate that in HLTF-deficient cells, ssDNA regions accumulate in DNA tracts exposed to HU. In FANCJ-deficient cells, the addition of S1 nuclease with control or HLTF depletion had no effect on the CldU/IdU ratio (Figure 5C). Therefore, ssDNA gaps were generated upon exposure to HU in HLTF-deficient cells in a manner dependent on FANCJ.
DISCUSSION
In this study, we report that FANCJ loss significantly alters the replisome. We utilize iPOND and quantitative proteomics to show that among other changes, the remodeling factor HLTF is enriched in FANCJ-KO cells. We find that following replication stress induced by HU, FANCJ-KO cells have shorter replication tracts, and this loss of nascent DNA is dependent on HLTF. These findings are consistent with FANCJ promoting fork protection by countering HLTF fork reversal. However, depletion of HLTF and restoration of fork protection while improving MMC resistance dramatically sensitizes FANCJ-KO cells to HU. These findings indicate that HLTF-dependent fork degradation is beneficial to the restoration of replication in FANCJ-KO cells. We also report that failure to slow replication in response to stress that characterizes HLTF-depleted cells (Kile et al., 2015) corresponds with nascent DNA being not only S1 nuclease sensitive but also vulnerable to degradation, outcomes that are dependent on FANCJ. Collectively, these findings indicate that a dynamic response to replication stress requires both FANCJ and HLTF. Loss of one or the other skews the response to enhance fork degradation, whereas loss of both proteins generates a toxic “dead end” replication stress response (Figure 5D).
The finding that fork degradation is observed in HLTF-depleted cells during prolonged stress, but not during shortterm stress, could reflect that unrestrained replication initially conceals degradation. This is reminiscent of the finding that FANCD2-depleted cells fail to slow in response to stress, but forks degrade under long-term stress (Lossaint et al., 2013). A defect in fork reversal has been proposed to account for the effect of HLTF depletion on fork progression (Kile et al., 2015). Rather than slowing and reversing upon stress, the unrestrained DNA synthesis could skip over difficult-to-replicate regions, generating “bad-quality” replication with gaps that serve as entry points for nucleases that degrade nascent DNA. We show that unrestrained replication in HLTF-deficient cells correlates with S1 nuclease sensitivity suggesting formation of ssDNA gaps. FANCJ-dependent ssDNA induction could drive senescence and chromosomal aberrations in HLTF-deficient cells. This idea, however, is at odds with the role of FANCJ combatting senescence by localizing BRCA1 to chromatin (Tu et al., 2011). Moreover, we did not detect chromosomal aberrations in HLTF-deficient cells in response to HU (Figure S5E), suggesting that conditions that are sufficient to induce gap formation and fork degradation may not be sufficient to induce gross genomic instability detectable in metaphase spreads. Nevertheless, the finding that unrestrained replication, gap formation, and fork degradation in HLTF-deficient cells are dependent on FANCJ suggests these phenotypes are functionally linked. To limit replication during stress and prevent genomic instability, HLTF may regress the replication fork by annealing the stalled nascent strand to the undamaged newly synthesized strand (Achar et al., 2015; Blastyák et al., 2010; Kile et al., 2015). In addition, with its remodeling or translocase activity, HLTF may displace FANCJ at stalled replication forks to limit replication.
Our findings raise interesting questions about the role of FANCJ in regulating HLTF chromatin localization and function in the cell and how FANCJ may function together with other components of post-replication repair to restrict fork reversal proteins. Fork remodelers have distinct substrates. Replication of G4s, repeat regions, and other natural barriers present in heterochromatin that tend to form secondary structures can slow or stall replication and reverse replication forks. The propensity for these natural triggers of replication fork reversal to form in FANCJ-KO cells could lead to greater replication stress, induction of HLTF, and dependence on it for recovery. Therefore, despite extensive resection, FANCJ-KO cells are able to restart stalled forks, which may reflect HLTF-dependent fork restart pathways contributing to cell survival. Likewise, BRCA2 mutant cells are dependent on MUS81 for survival and restart following HU treatment (Rondinelli et al., 2017). It is also possible that the survival of FANCJ-KO cells following HU could be linked to HLTF because of nonreplication functions in cell cycle regulation, sister chromatid cohesion, or chromosome condensation (Dhont et al., 2016).
Although enriched HLTF provides fitness benefits during HU stress, the failure to regulate HLTF fork reversal activity under conditions when FANCJ is absent or mutated could compromise the genome. HLTF fork reversal activity could protect the replication fork by limiting ssDNA accumulation. However, in the absence of FANCJ, overactive HLTF-dependent fork remodeling and resection may be detrimental to recovery from interstrand cross-link (ICL)-induced lesions that require a series of distinct processing events for replication to restart. This provides an explanation for why HLTF depletion enhances MMC resistance in FANCJ-KO cells. An overabundance of HLTF in FANCJ-KO mice could contribute to the subfertility, germ cell attrition, and hypersensitivity to replication inhibitors (Matsuzaki et al., 2015; Sun et al., 2016). Problems restoring replication due to enriched HLTF could contribute to the defects in proliferation and self-renewal that lead to a G2/M arrest and the exhaustion of bone marrow cells in FA. Problematic replication caused by an overabundance of replication-forkslowing factors could also be a driving force in transformation. FA patients that do not succumb to bone marrow failure often develop leukemia and other cancers. The finding that HLTF is both disrupted and amplified in cancer suggests that the consequence of loss of HLTF and unrestrained replication are comparable to enriched HLTF and unrestrained replication fork reversal.
We propose that FANCJ serves to limit HLTF-dependent reversed forks that are extensively degraded by MRE11. If true, FANCJ loss should magnify the degradation in cells lacking BRCA1 or BRCA2 in which MRE11 is unrestricted. The depletion of BRCA2 in control or FANCJ-KO U2OS cells did not provide much evidence for distinct roles in fork protection. However, to understand the relationship between FANCJ, BRCA2, and other fork-protection proteins, additional experiments, including electron microscopy, will be critical. It is possible that despite enriched HLTF, fork remodeling is compromised in FANCJ-null cells because other fork remodelers are deficient. For example, FANCJ promotes RPA chromatin loading (Gong et al., 2010), and RPA recruits SMARCAL1 to stalled forks (Bhat et al., 2015; Gong et al., 2010); therefore, SMARCAL1 may not be maintained at forks as robustly in FANCJ-KO cells. The reduced viability upon loss of SMARCAL1 in FANCJ-KO U2OS cells suggests that residual SMARCAL1 at forks may be critical for viability, perhaps for telomere maintenance. In contrast, SMARCAL1 depletion in FANCJ-KO 293T cells did not dramatically reduce viability and enhanced fork protection. While these findings suggest that the reversed fork structure is degraded in FANCJ-deficient cells, fork degradation could be furthered by aberrantly activated MRE11 activity given that FANCJ interacts with MRE11 and inhibits its exonuclease activity (Suhasini et al., 2013).
In summary, we show that FANCJ associates specifically with nascent DNA in cells, consistent with other reports (Alabert et al., 2014; Sirbu et al., 2011). These findings suggest that FANCJ travels with the replication fork, which would enable it to respond rapidly to DNA damage or replication stress. Characterization of the replisome in FANCJ-KO cells revealed that the fork reversal factor HLTF is elevated and causative in replication fork degradation associated with FANCJ deficiency. FANCJ joins the growing list of FA proteins that function beyond ICL repair in the replication stress response. Similar to BRCA1, BRCA2, FANCD2, FANCA, and FANCM (Blackford et al., 2012; Schlacher et al., 2011, 2012), FANCJ also protects nascent DNA at stalled forks under stressful conditions. In addition, we report that FANCJ is causative in unrestrained replication, gap formation, and fork degradation associated with HLTF deficiency. Whether this insight provides tools for targeting FANCJ- or HLTF-associated cancer remains to be determined.
EXPERIMENTAL PROCEDURES
Cell Lines
293T and U2OS cell lines were grown in DMEM supplemented with 10% fetal bovine serum and penicillin and streptomycin (100 U/mL each). FA-J (EUFA30-F) was immortalized with human telomerase reverse transcriptase (hTERT) (Peng et al., 2007), and HCT116 cells were cultured as previously described (Achar et al., 2015; Litman et al., 2005).
shRNA
FANCJ-KO and control 293T, U2OS, HCT116 cells were infected with pLK 0.1 vectors containing shRNAs against non-silencing control (NSC) or one of three shRNAs against HLTF: (A) mature antisense sequence 5′-TTTGTGATGA TAACTTCTTGC-3′, (B) 5′-TAAGAAGGTAAGTATGGCAAC-3′, and as described (Achar et al., 2015) with changes shown. The shRNAs against SMARCAL1 include (a) mature antisense sequence 5′-AAACACTGCAATGA GTTCCGC-3′, (b) 5′-TTTGGTCAGCATTAGATGAGC-3′, or (c) 5′- ATGAG TTGGGTTAGCAAAGGG-3′. shRNAs were obtained from the University of Massachusetts Medical School (UMMS) shRNA core facility. HCT116 cells were infected with FSIPPW vectors containing shRNAs against NSC or FANCJ (Litman et al., 2005). Stable shRNA cell lines were selected with puromycin (0.5–1 μg/mL) or G418 (300–600 mg/mL).
CRISPR-Cas9 KO Generation
The CRISPR-sgRNA (single guide RNA) construct was generated ligating the sgRNA sequence targeting two different sites on exon 2 of FANCJ (5′-GG TCTGAATATACAATTGGTGGG-3′ [guide 1] or 3′-TCATCATAGCAAGCTGT-5′ [guide 2]) onto the pX330 plasmid containing the Cas9 gene (Addgene catalog number 42230). The target sequence was bioinformatically designed to minimize off-target effects. To assess the specificity of the CRISPR endonuclease activity, a GFP reporter construct was generated ligating the M427 vector (Wilson et al., 2013) with the same sgRNA sequence used previously. Briefly, 293T or U2OS cells were transfected with both the reporter and the CRISPR constructs. 48 hr after transfection, GFP+ cells were sorted using the FACSAria II (BD Biosciences) and subsequently seeded at one cell per well in 96-well plates. Clones were analyzed for FANCJ protein levels, and those without expression of the protein were genotyped following PCR using forward primer (5′-CATTACCACAATCCTATGGG-3′) and reverse primer (5′-CTGGAAAGCT GGTTTACTC-3′). The exon 2 ATG start site and remaining sequence is similar to the native sequence, but in KO clone 1, there is a 1-nt insertion (bold), and in KO clone 2, there is a deletion of 2 nt before the protospacer adjacent motif (PAM) site (underlined) of guide 1 (italics). WT: ATGTCTTCAATGTGGTCTGAA TATACATTGGTGGGGTGAAGATTT; KO clone 1: ATGTCTTCATTTGTGGTCT GAATATACATTGGTGGGGTGAAGATTT; KO clone 2: ATGTCTTCA-GTGGTC TGAATATACATTGGTGGGGTGAAGATTT
Immunoblotting and Abs
Cells were harvested, lysed, and processed for western blot analysis as described previously using 150mM NETN lysis buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, 10 mg/mL leupeptin, and 10 mg/mL aprotinin). For cell fractionation, we isolated cytoplasmic and soluble nuclear fractions with the NE-PER Kit (Thermo Scientific) according to the manufacturer’s protocol; to isolate the chromatin fraction, the insoluble pellet was resuspended in RIPA buffer and sonicated in a BioRuptor according to the manufacturer’s protocol (high power, 15 min, 30 s on and 30 s off at 4°C). Proteins were separated using SDSPAGE and electrotransferred to nitrocellulose membranes. Membranes were blocked in 5% milk PBS-Tween and incubated with primary antibody for 1 hr. Abs for western blot analysis included anti-PCNA (Abcam), anti-H2B (Cell Signaling Technology), anti-β-actin (Sigma), anti-FANCJ (E67), anti-HLTF (Abcam), anti-SMARCA1 (Abcam), and anti-HLTF (Santa Cruz Biotechnology). Membranes were washed, incubated with horseradish-peroxidase-linked secondary antibodies (Amersham), and detected by chemiluminescence (Amersham).
Viability Assays
Cells were seeded onto 96-well plates (500 cells per well, performed in triplicate for each experiment) and incubated overnight. The next day, cells were treated with increasing doses of MMC for 1 hr in serum-free media or HU and maintained in complete media for 5 days. Percentage survival was measured photometrically using a CellTiter-Glo viability assay (Promega) in a microplate reader (Beckman Coulter DTX 880 Multimode Detector). For the growth assay, cells were seeded onto 12-well plates and counted at the indicated times using a hemocytometer.
Immunofluorescence
Immunofluorescence was performed as described previously (Cantor et al., 2001). Cells were grown on coverslips. The next day, cells were fixed and permeabilized. After incubation with primary antibodies against γ-H2AX (Millipore), cells were washed and then incubated with secondary antibody. After washing, coverslips were mounted onto glass slides using Vectashield mounting medium containing DAPI (Vector Laboratories). For EdU labeling, cells were left untreated or treated with 0.5 mM HU for 3 hr and released at 1 hr. EdU labeling was carried out with a Click-iT EdU imaging kit (Invitrogen) according to the manufacturer’s instructions.
DNA Fiber Assays
To directly visualize replication fork dynamics, we established single-molecular-DNA fiber analysis. In this assay, progressing replication forks in cells were labeled by sequential incorporation of two different nucleotide analogs, IdU (50 μM) and CldU (50 μM), into nascent DNA strands for the indicated time and conditions. After nucleotide analogs were incorporated in vivo, the cells were collected, washed, spotted (2.5 μL of 105 cells/mL PBS cell suspension), and lysed on positively charged microscope slides (Globe Scientific, 1358W) by 7.5 μL spreading buffer (0.5% SDS, 200 mM Tris-HCl [pH 7.4], and 50 mM EDTA) for 8 min at room temperature. For experiments with the ssDNA-specific endonuclease S1, after the CldU pulse, cells were treated with CSK100 buffer (100 mM NaCl, 10 mM MOPS, 3 mM MgCl2 [pH 7.2], 300 mM sucrose, and 0.5% Triton X-100) for 10 min at room temperature, then incubated with S1 nuclease buffer (30 mM sodium acetate [pH 4.6], 10 mM zinc acetate, 5% glycerol, and 50 mM NaCl) with or without 20 U/mL S1 nuclease (Invitrogen, 18001–016) for 30 min at 37°C. The cells were then scraped in PBS + 0.1% BSA and centrifuged at 7,000 rpm for 5 min at 4°C (Quinet et al., 2016, 2017). Cell pellets were resuspended at ~1,500 cells/mL and lysed with lysis solution on slides. Individual DNA fibers were released and spread by tilting the slides at 45 degrees. After air-drying, fibers were fixed by 3:1 methanol/acetic acid at room temperature for 3 min. After air-drying again, fibers were rehydrated in PBS, denatured with 2.5 M HCl for 30 min, washed with PBS, and blocked with blocking buffer (3% BSA and 0.1% Trition in PBS) for 1 hr. Next, slides were incubated for 2.5 hr with primary antibodies for (IdU:1:100, mouse monoclonal anti-BrdU, Becton Dickinson 347580; CldU: 1:100, rat monoclonal anti-BrdU, Abcam 6326) diluted in blocking buffer, washed several times in PBS, and then incubated with secondary antibodies (IdU:1:200, goat anti-mouse, Alexa 488; CldU: 1:200, goat anti-rat, Alexa Fluor 594) in blocking buffer for 1 hr. After washing and air-drying, slides were mounted with Prolong (Invitrogen, P36930). Finally, visualization of green and/or red signals by fluorescence microscopy (Axioplan 2 imaging, Zeiss) provided information about the active replication directionality at the singlemolecule level. Representative cropped images are shown on a black backdrop.
iPOND
iPOND was performed as described previously (Dungrawala and Cortez, 2015; Sirbu et al., 2011). The click reaction was completed in 2 hr. Capture of DNAprotein complexes utilized streptavidin-coupled C1 magnabeads for 1 hr. Beads were washed with lysis buffer (1% SDS in 50 mM Tris [pH 8.0]), lowsalt buffer (1% Triton X-100, 20 mM Tris [pH 8.0], 2 mM EDTA, and 150 mM NaCl), high-salt buffer (1% Triton X-100, 20 mM Tris [pH 8.0], 2 mM EDTA, and 500 mM NaCl), and lithium chloride wash buffer (100 mM Tris [pH 8.0], 500 mM LiCl, and 1% Igepal) and then twice in lysis buffer. For most experimental samples, 4 × 108 HEK293T cells were used; light and heavy labeled cells were mixed 1:1 prior to the click reaction.
iPOND samples were separated by SDS-PAGE. Gel regions above and below the streptavidin band were excised and treated with 45 mM DTT for 30 min, and available cysteine residues were carbamidomethylated with 100 mM iodoacetamide for 45 min. After destaining the gel pieces with 50% acetonitrile (MeCN) in 25 mM ammonium bicarbonate, proteins were digested with trypsin (Promega) in 25 mM ammonium bicarbonate at 37°C. Peptides were extracted by gel dehydration (60% MeCN, 0.1% trifluoroacetic acid [TFA]), vacuum dried, and reconstituted in 0.1% formic acid.
SILAC and Liquid Chromatography Tandem MS
2 mL tryptic digests was analyzed on the Thermo Q-Exactive mass spectrometer coupled to an EASY-nLC system (Thermo Fisher). Peptides were separated on a fused silica capillary (12 cm × 100 μm ID) packed with Halo C18 (2.7 mm particle size, 90 nm pore size; Michrom Bioresources) at a flow rate of 300 nL/min. Peptides were introduced into the mass spectrometer via a nanospray ionization source at a spray voltage of 2.2 kV. Mass spectrometry data were acquired in a data-dependent top-10 mode, and the lock mass function was activated (m/z, 371.1012; use lock masses, best; lock mass injection, full MS). Full scans were acquired from m/z 350 to 1,600 at 70,000 resolution (automatic gain control [AGC] target, 1e6; maximum ion time [max IT], 100 ms; profile mode). Resolution for dd-MS2 spectra was set to 17,500 (AGC target: 1e5) with a maximum ion injection time of 50 ms. The normalized collision energy was 27 eV. A gradient of 0 to 40% acetonitrile (0.1% FA) over 55 min was applied.
MS Data Analysis
SILAC experiments were performed in triplicate and analyzed in MaxQuant using the requantify option with two unique peptide required to identify a protein. The peptide search was performed with the UniProt Human Proteome AUP000005640 that includes both canonical protein sequences and isoforms. The search parameters permitted a 10 ppm precursor MS tolerance and a 0.02 Da MS/MS tolerance. Carboxymethylation of cysteines was set up as fixed modifications, and oxidation of methionine (M), SILAC labeling (13C615N2) at lysine, and (13C615N4) at arginine were allowed as variable modifications. Up to two missed tryptic cleavages of peptides were considered with the false-discovery rate set to 1% at the peptide level. The SILAC ratio for each identified peptide was calculated by the quantitation node. Known contaminants and reverse hits were removed prior to statistical analysis. Low-scoring proteins were automatically removed by MaxQuant and did not receive a ratio value. As a quality control, we confirmed that the isotopic labels were fully incorporated and observed that heavy lysine +8 and heavy arginine +10 isotopic labels were incorporated at greater than 95% in 95% of all peptides. For statistical analysis, we recorded the light-to-heavy (L/H) ratio for each identified protein from MaxQuant, which compares the area of the heavy and light isotopic peaks for each identified peptide. These ratios were log2 transformed, sorted by rank, and plotted in a rank order plot versus log2 ratio. In addition, for proteins identified in at least two experiments, we determined p values by the limma statistical analysis package for R Bioconductor and plotted the data in a volcano plot of log2 L/H ratio versus −log10 p value to identify statistically significant changes in the proteome with a substantial log2 fold
Alkaline BrdU Comet Assay
The alkaline BrdU comet assay was performed as described previously (Mórocz et al., 2013) and following treatment with 4 mM HU for 2 hr.
Chromsome Spreads
Chromosome spreads were prepared as previously described (Guillemette et al., 2015). Briefly, cells were treated with HU as indicated. Cells were washed and recovered in fresh media for 8 hr before Colcemid arrest (1:200, KaryoMAX, 4 hr). Cells were harvested by trypsinization, incubated in 75 mM KCl for 18 m at 37°C and fixed in a 3:1 methanol/acetic acid solution. The fixed suspension was dropped onto slides to obtain chromosome spreads and mounted with VectaShield mounting medium with DAPI (Vector Laboratories) and visualized using fluorescence microscopy (Axioplan 2 imaging, Zeiss).
Statistical Methods
Statistical differences in DNA fiber assays, immunofluorescence, and the alkaline BrdU comet assay were determined using a two-tailed Mann-Whitney test. Statistical analysis was performed using GraphPad Prism (Version 7.0). MS data analysis is described above. In all cases, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Highlights.
FANCJ suppresses the accumulation of HLTF in replisomes
HLTF promotes fork degradation in FANCJ-deficient cells
FANCJ promotes fork elongation, ssDNA gaps, and degradation in HLTF-deficient cells
Combined loss of FANCJ and HLTF enhances replication stress
ACKNOWLEDGMENTS
We thank the members of the Cantor laboratory for helpful discussions and Jianhong Ou for help with data analysis. This work was supported by NIH grant R01 CA176166–01A1, as well as charitable contributions from Mr. and Mrs. Edward T. Vitone, Jr. and the Lipp Family Foundation. The Vermont Genetics Network Proteomics Facility is supported through NIH grant P20GM103449 from the INBRE Program of the National Institute of General Medical Science. The Lajos laboratory is supported by the National Research, Development and Innovation Office (grants GINOP-2.3.2–15-2016–00026 and GINOP-2.3.2–15-2016–00024).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Raw images and data have been deposited in Mendeley Data and are available at https://data.mendeley.com/datasets/zjtbfxhkg3/1.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.065.
REFERENCES
- Achar YJ, Balogh D, Neculai D, Juhasz S, Morocz M, Gali H, Dhe-Paganon S, Venclovas Č, and Haracska L (2015). Human HLTF mediatespostreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 43, 10277–10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Aaes TL, Zhai B, Al-Nakouzi N, Oo HZ, Daugaard M, Rohde M, and Jäättelä M (2016). Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 44, 2214–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler E (2007). PGK deficiency. Br. J. Haematol 136, 3–11. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Bétous R, and Cortez D (2015). High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J. Biol. Chem 290, 4110–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Schwab RA, Nieminuszczy J, Deans AJ, West SC, and Niedzwiedz W (2012). The DNA translocase activity of FANCM protects stalled replication forks. Hum. Mol. Genet 21, 2005–2016. [DOI] [PubMed] [Google Scholar]
- Blastyák A, Hajdú I, Unk I, and Haracska L (2010). Role of doublestranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol 30, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, and Calvo JA (2017). Fork protection and therapy resistance in hereditary breast cancer. Cold Spring Harb. Symp. Quant. Biol 82, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, and Livingston DM (2001). BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160. [DOI] [PubMed] [Google Scholar]
- Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, and Livingston DM (2004). The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 101, 2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TS, Hawkins C, Krieger JR, McGlade CJ, and Huang A (2016). JPO2/CDCA7L and LEDGF/p75 are novel mediators of PI3K/AKT signaling and aggressive phenotypes in medulloblastoma. Cancer Res. 76, 2802–2812. [DOI] [PubMed] [Google Scholar]
- Cheung RS, and Taniguchi T (2017). Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int. J. Hematol 106, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, and Cheng H (2013). Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res. 41, 1294–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhont L, Mascaux C, and Belayew A (2016). The helicase-like transcription factor (HLTF) in cancer: loss of function or oncomorphic conversion of a tumor suppressor? Cell. Mol. Life Sci 73, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, and Cortez D (2015). Purification of proteins on newly synthesized DNA using iPOND. Methods Mol. Biol 1228, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, and Gautier J (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Kim JE, Leung CC, Glover JN, and Chen J (2010). BACH1/ FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol. Cell 37, 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, and Cantor SB (2015). Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 29, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, and Cimprich KA (2015). HLTF’s ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol. Cell 58, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Bond CS, and Fox AH (2016). The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 44, 3989–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraswamy E, and Shiekhattar R (2007). Activation of BRCA1/BRCA-2associated helicase BACH1 is required for timely progression through S phase. Mol. Cell. Biol 27, 6733–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, and Kim W (2003). Association of human kinesin superfamily protein member 4 with BRCA2-associated factor 35. Biochem. J 374, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, and Vindigni A (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Meng L, Lin TC, Wu LJ, Pederson T, and Tsai RY (2014). Nucleostemin and GNL3L exercise distinct functions in genome protection and ribosome synthesis, respectively. J. Cell Sci 127, 2302–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, and Cantor SB (2005). BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8, 255–265. [DOI] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, and Constantinou A (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K, Borel V, Adelman CA, Schindler D, and Boulton SJ (2015). FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev 29, 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn AP, Wasson G, O’Connor J, McKerr G, McKelvey-Martin VJ, and Downes CS (1999). The bromodeoxyuridine comet assay: detection of maturation of recently replicated DNA in individual cells. Cancer Res. 59, 5912–5916. [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, and Lopes M (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minion LE, Dolinsky JS, Chase DM, Dunlop CL, Chao EC, and Monk BJ (2015). Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol 137, 86–92. [DOI] [PubMed] [Google Scholar]
- Mohrmann L, and Verrijzer CP (2005). Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim. Biophys. Acta 1681, 59–73. [DOI] [PubMed] [Google Scholar]
- Mórocz M, Gali H, Raskó I, Downes CS, and Haracska L (2013). Single cell analysis of human RAD18-dependent DNA post-replication repair by alkaline bromodeoxyuridine comet assay. PLoS ONE 8, e70391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, et al. (2013). The histone demethylase LSD1/KDM1A promotes the DNA damage response. J. Cell Biol 203, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr., and Cantor SB (2007). The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 26, 3238–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Wang J, Hu Y, Zhao H, Hou W, Zhao H, Wang H, Liao J, and Xu X (2015). Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res. 43, 5936–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res. 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA fiber analysis: mind the gap!. Methods Enzymol. 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, and D’Andrea AD (2017). EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 19, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/ 2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, and Cortez D (2013). Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem 288, 31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson J-Y, Seidman MM, and Brosh RM Jr. (2013). Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol. Cell. Biol 33, 2212–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Brieno-Enriquez MA, Cornelius A, Modzelewski AJ, Maley TT, Campbell-Peterson KM, Holloway JK, and Cohen PE (2016). FancJ (Brip1) loss-of-function allele results in spermatogonial cell depletion during embryogenesis and altered processing of crossover sites during meiotic prophase I in mice. Chromosoma 125, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, and Mailand N (2015). Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527, 389–393. [DOI] [PubMed] [Google Scholar]
- Torres CM, Biran A, Burney MJ, Patel H, Henser-Brownhill T, Cohen AS, Li Y, Ben-Hamo R, Nye E, Spencer-Dene B, et al. (2016). The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 353, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Aird KM, Bitler BG, Nicodemus JP, Beeharry N, Xia B, Yen TJ, and Zhang R (2011). Oncogenic RAS regulates BRIP1 expression to induce dissociation of BRCA1 from chromatin, inhibit DNA repair, and promote senescence. Dev. Cell 21, 1077–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KA, Chateau ML, and Porteus MH (2013). Design and development of artificial zinc finger transcription factors and zinc finger nucleases to the hTERT locus. Mol. Ther. Nucleic Acids 2, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Zhou L, Khidr L, Guo XE, Kim W, Lee YM, Krasieva T, and Chen PL (2008). A novel role of the chromokinesin Kif4A in DNA damage response. Cell Cycle 7, 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, and Chen J (2012). The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell 47, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.