

Abstract
Background
It was recently demonstrated that the size of cell-free DNA (cfDNA) fragments that originates from tumor cells are shorter than cfDNA fragments that originates from non-malignant cells. We investigated whether cfDNA fragment size and cfDNA levels might have prognostic value in patients with advanced pancreatic cancer.
Methods
Blood samples were obtained from patients with advanced pancreatic cancer, before (n = 61) initiation of chemotherapy and after the first cycle of chemotherapy (n = 39). Samples were separated with density centrifugation and plasma DNA was isolated. Mode cfDNA fragment size and cfDNA levels were then determined using a 2100 Bioanalyzer. A cohort of partially age-matched healthy volunteers (n = 28) constituted the control group.
Results
Both a pre-treatment cfDNA fragment size of ≤ 167 bp (mode) and high pre-treatment cfDNA levels were associated with shorter progression-free survival (PFS) (p = 0.002 and p < 0.001, respectively) and overall survival (OS) (p = 0.001 and p = 0.001, respectively). Furthermore, multivariable Cox regression analyses demonstrated that pre-treatment cfDNA levels could independently predict prognosis for both PFS (HR = 3.049, p = 0.005) and OS (HR = 2.236, p = 0.028).
Conclusion
This study demonstrates that cfDNA fragment size and cfDNA levels can be used to predict disease outcome in patients with advanced pancreatic cancer. The described approach, using a rapid, economic and simple test to reveal prognostic information, has potential for future treatment stratification and monitoring.
Electronic supplementary material
The online version of this article (10.1186/s12967-018-1677-2) contains supplementary material, which is available to authorized users.
Keywords: Cell-free DNA, cfDNA, cfDNA fragment size, Pancreatic cancer
Background
Previous studies have demonstrated that cell-free DNA (cfDNA) levels are elevated in blood from patients with cancer compared to the levels in blood from healthy individuals [1]. Moreover, high levels of cfDNA have been associated with poor survival in several cancers, including pancreatic cancer [2–4]. Using surrogate markers, such as point mutations [5–10], copy-number aberrations [11, 12], microsatellite alterations [13] and methylations [12], it has been possible to detect the fraction of cfDNA that originates from tumor cells. This fraction, better known as circulating tumor DNA (ctDNA), can be detected in most patients with cancer, and both the fraction of patients with detectable ctDNA and the levels of ctDNA in blood have been shown to increase with increasing tumor stages [5]. In recent years, ctDNA has also been demonstrated to provide prognostic value and monitoring potential for patients with different cancers, including patients with pancreatic cancer [6–10].
Studies of cfDNA in maternal plasma and transplant recipients have indicated that cfDNA is predominantly of hematopoietic origin, and that cfDNA from non-hematopoietic cells are shorter in size than hematopoietically derived cfDNA [14–16]. In accordance with these observations, studies on cfDNA from patients with cancer have demonstrated that the fragments of cfDNA that originates from tumor cells (i.e. ctDNA) are, in fact, shorter in size than fragments that originate from non-malignant cells [17–20]. Another study recently demonstrated that size-selection of smaller cfDNA fragments could be used to increase the amount of tumor-derived cfDNA fragments in cfDNA samples [20]. Given the reported shorter size of tumor-derived cfDNA fragments, we reasoned that increasing numbers of short cfDNA fragments might indicate higher fractions of tumor-derived cfDNA and thus, might reflect an increased tumor load. Accordingly, we hypothesized that a cfDNA fragment size analysis might hold prognostic value in cancer. The present study aimed to investigate the prognostic value of cfDNA fragment size in cancer. We employed simple laboratory techniques to determine the fragment size and total levels of cfDNA isolated from patients with locally advanced or metastatic pancreatic cancer.
Materials and methods
Patients and samples
Between September 2012 and July 2017, we included 61 patients with locally advanced (n = 6) or metastatic (n = 55) pancreatic cancer that were admitted to Stavanger University Hospital (SUH) and Haukeland University Hospital (HUH). Patients received first-line treatment with gemcitabine (SUH only), nab-paclitaxel plus gemcitabine (SUH only), or FOLFIRINOX (both centers). Peripheral venous blood samples (9 ml EDTA tubes) were drawn before initiation of chemotherapy (n = 61; denoted B1) and after the first cycle of chemotherapy (n = 39; denoted B2). Treatment response was defined with a standard disease evaluation of radiological images, based on the RECIST 1.1 criteria [21]. We also included a control group (median age = 54), which comprised 28 healthy individuals with no prior or current cancer diagnosis. Blood samples collected at SUH were processed within 2 h; samples collected at HUH were processed at SUH within 24 h, after overnight transport. All patients and healthy controls provided written informed consent to participate in the study. The project was approved by the Regional Committee for Medical and Health Research Ethics (REK-Vest 2011/475, REK-Vest 2013/1743).
Isolation of cfDNA from plasma
Blood samples were separated with density centrifugation using Lymphoprep™ (Axis Shield) density gradient media, according to the manufacturer’s instructions. We used 4 mL (1–2 mL for the first 8 patients) of plasma (diluted 1:1 with 0.9% NaCl for the density gradient separation protocol) for isolating total cfDNA with the QIAamp Circulating Nucleic Acid kit (Qiagen), as described by the manufacturer. cfDNA was eluted in 50 µL of Buffer AVE (Qiagen) and stored at − 80 °C until further analysis.
Determination of cfDNA fragment size and cfDNA level
The cfDNA fragment size and cfDNA level were determined for each sample with an Agilent 2100 Bioanalyzer and the Agilent High Sensitivity DNA chip, according to the manufacturer’s instructions. The fragment size of cfDNA was determined, with the Agilent 2100 Bioanalyzer software, and defined as the mode of the main peak (corresponding to one nucleosome plus linker, derived from apoptotic cells) in the electropherogram (Additional file 1: Figure S1). cfdna level were also determined with the software, calculated as the area under the main peak in the electropherogram (Additional file 1: Figure S1). Determination of cfDNA fragment size and cfDNA levels were performed by individuals blinded to clinical data.
Statistical analysis
All statistical analyses were performed with IBM SPSS version 24.0 (http://www.spss.com/). All tests were two-sided, and p-values less than 0.05 were considered statistically significant. Missing data were automatically excluded from the analyses. Clinicopathological patient data were compared with the χ2 or Fisher’s exact test, for categorical data, and with the independent-samples t-test or Mann–Whitney U test for continuous data. The Mann–Whitney U test was also used to compare cfDNA fragment size and cfDNA levels between control and patient samples, and between centers. The Wilcoxon signed rank test or the sign test were used to compare samples taken before and after initiation of chemotherapy. Correlation tests were performed between cfDNA fragment size and cfDNA levels with the non-parametric Spearman’s rank correlation coefficient for the continuous variables, and Cohen’s Kappa coefficient for the categorical variables. Data from the two study centers were compared statistically (Additional file 1: Table S1 and Figure S2). We found significant differences between centers in first-line treatment and cfDNA fragment size.
Survival analyses were performed with Kaplan–Meier estimates, the log-rank test, and Cox proportional hazards regression. The primary endpoints were progression-free survival (PFS) and overall survival (OS). PFS was defined as the time between inclusion (or sampling date for the B2 sample) and progression or death due to any cause, when the patient died before evidence of progression was obtained. OS was defined as the elapsed time between inclusion (or sampling date for the B2 sample) and death due to any cause.
Univariable Cox regression analyses were used to investigate the effects of single variables on survival. We tested variables measured before initiation of chemotherapy, including the B1 cfDNA fragment size (both continuous and dichotomized), B1 cfDNA levels (both continuous and dichotomized), the combination of B1 cfDNA fragment size and B1cfDNA levels, carbohydrate antigen 19-9 (CA 19-9) status, study center, age, sex, tumor size, tumor location, T-stage, clinical stage, metastatic location, N-stage, Eastern Cooperative Oncology Group (ECOG) performance status, first line treatment, second line treatment, and prior anti-cancer surgery. We also tested selected variables 1–2 months after the start of chemotherapy: the B2 cfDNA fragment size (both continuous and dichotomized) and B2 cfDNA levels (both continuous and dichotomized).
Multivariable Cox regression modeling was also performed for selected variables, including the B1 cfDNA fragment size (dichotomized), B1 cfDNA levels (dichotomized), CA 19-9 status, study center, age, sex, tumor size, tumor location, clinical stage, ECOG performance status, first line treatment, and prior anti-cancer surgery. Several variables were excluded from the multiple regression model, due to associations with other markers, including the B1 cfDNA fragment size (continuous), B1 cfDNA level (continuous), the combination of B1 cfDNA fragment size and B1 cfDNA levels. Other variables were excluded due to missing data (T-stage, metastatic location, N-stage), or because they were not acquired at baseline (B2 cfDNA fragment size, B2 cfDNA levels, and second-line treatment). The multivariable analyses were performed with both forward and backward stepwise selection of covariates.
Results
Patient characteristics
The 61 patients included in this study had either locally advanced (n = 6) or metastatic tumors (n = 55). The cohort included a slight preponderance of men (n = 35, 57.4%), and the mean age was 64 years at diagnosis. Primary tumors were predominately located in the head of the pancreas (55.7%), and the median size was 37 mm. The first six (9.8%) patients included in the study received Gemcitabine monotherapy; the remaining patients received either FOLFIRINOX (n = 30, 49.2%) or nab-paclitaxel plus Gemcitabine (n = 25, 41%). All baseline patient characteristics and clinicopathological data are summarized in Table 1.
Table 1.
Baseline characteristics of patients with pancreatic cancer
| Variable | All patients (n = 61) |
|---|---|
| Median age | 64 (41–81) |
| Sex | |
| Male | 35 (57%) |
| Female | 26 (43%) |
| Median tumor size (mm) | 37 (11–100) |
| Median CA 19-9 levels | 385.5 (5–102041) |
| CA 19-9 | |
| < 37U/mL | 13 (21%) |
| ≥ 37U/mL | 47 (77%) |
| Missing data | 1 (2%) |
| Tumor location | |
| Pancreas head | 34 (56%) |
| Pancreas body | 4 (7%) |
| Pancreas tail | 13 (21%) |
| Multiple | 10 (16%) |
| T-stage | |
| T2 | 11 (18%) |
| T3 | 15 (25%) |
| T4 | 25 (41%) |
| TX | 2 (3%) |
| Missing data | 8 (13%) |
| Clinical stage | |
| Stage III | 6 (10%) |
| Stage IV | 55 (90%) |
| Metastatic location | |
| Liver | 25 (41%) |
| Lung | 4 (7%) |
| Multiple | 16 (26%) |
| Other | 6 (10%) |
| Missing data | 10 (16%) |
| N-stage | |
| N0 | 14 (23%) |
| N1 | 25 (41%) |
| NX | 21 (34%) |
| Missing data | 1 (2%) |
| ECOG status | |
| 0 | 13 (21%) |
| 1 | 34 (56%) |
| 2 | 10 (16%) |
| Missing data | 4 (7%) |
| First-line treatment | |
| Gemcitabine | 6 (10%) |
| FOLFIRINOX | 30 (49%) |
| Nab-Paclitaxel + | 25 (41%) |
| Gemcitabine | |
| Second-line treatment | |
| Yes | 17 (28%) |
| No | 44 (72%) |
| Prior anti-cancer surgery | |
| Yes | 9 (15%) |
| No | 51 (84%) |
| Missing data | 1 (2%) |
Measurements of cfDNA fragment size and cfDNA level
cfDNA fragment size and cfDNA levels were measured in plasma samples from the patients and from partially age-matched healthy controls (n = 28). The cfDNA fragment size in healthy control samples were longer (median 176.5 bp, range 168–185 bp) than the fragment size in patient samples (locally advanced: median 170 bp, range 167–173 bp, p = 0.001; metastatic: median 167 bp, range 148–180 bp, p < 0.001; Figs. 1 and 2). Moreover, the levels of cfDNA in healthy control samples (median 0.33 ng/mL plasma, range 0.03–1.61 ng/mL plasma) were significantly lower than those in patient samples (locally advanced: median 3.26 ng/mL plasma, range 1.16–7.98 ng/mL plasma, p < 0.001; metastatic: median 6.58 ng/mL plasma, range 0.53–1911.63 ng/mL plasma, p < 0.001; Figs. 1 and 2). When samples from patients with locally advanced tumors were compared to samples from patients with metastatic tumors, we found significant differences in cfDNA fragment size (p = 0.044; Fig. 2a), but not in cfDNA levels (p = 0.257; Fig. 2b).
Fig. 1.
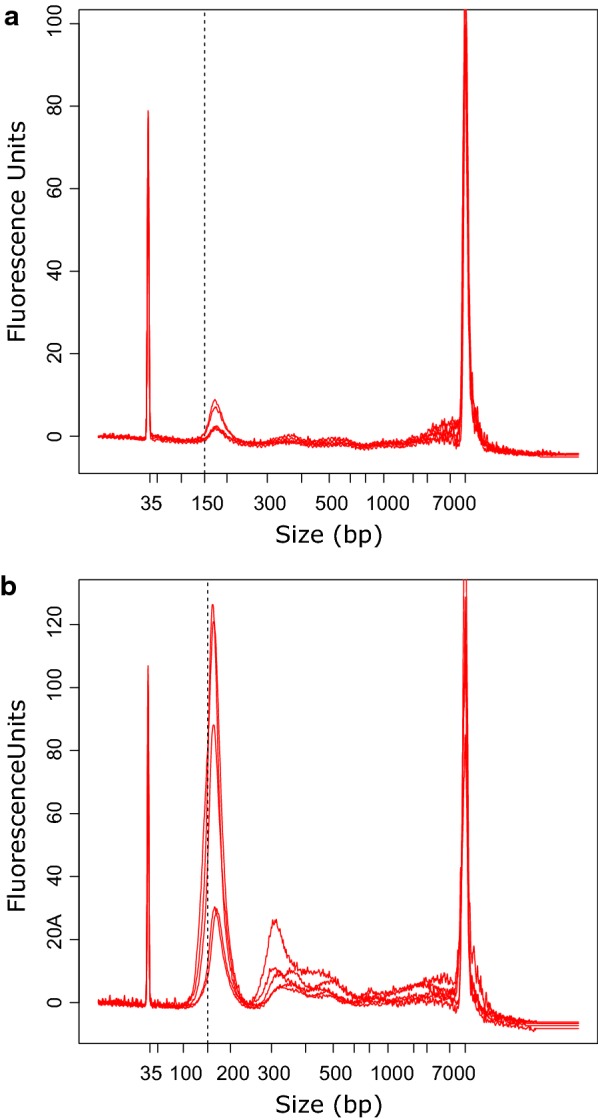
Distribution of cfDNA fragment size and cfDNA level in plasma. a cfDNA from healthy controls (n = 10) displayed larger fragment size (X-axis) and lower cfDNA level (Y-axis) compared to b cfDNA from patients with pancreatic cancer (n = 10). The dotted line placed at 150 bp indicates a shift towards shorter fragment size in patient samples
Fig. 2.

Distributions of cfDNA fragment size and cfDNA levels in patient and control samples. Boxplot shows differences in a cfDNA fragment size and b cfDNA levels between samples from healthy controls (n = 28), patients with localized disease (n = 6), and patients with metastatic disease (n = 55). Abbreviation: n.s, not significant
Changes in cfDNA fragment size and cfDNA levels during therapy
cfDNA fragment size and cfDNA levels were determined in plasma samples obtained before the initiation of chemotherapy (n = 61 patients), and in plasma samples obtained after the first cycle of chemotherapy (n = 39 patients). Neither the median cfDNA fragment (p = 0.956) nor the median cfDNA level (p = 0.110) changed significantly during treatment (Additional file 1: Figure S3).
Survival analyses
Patients were followed for a median of 7.7 months (range 0.3–25.8 months). During follow-up, progression was observed in 54 (88.5%) patients, and 51 (83.6%) patients died. We dichotomized cfDNA fragment size and cfDNA levels in patient samples based on the median values of all patient samples; thus a short fragment size was defined as ≤ 167 bp, and a high cfDNA level was defined as > 4.66 ng/mL plasma. We found that a short pre-treatment cfDNA fragment size (≤ 167 bp) was highly associated with both shorter PFS (4.0 vs 7.7 months; log-rank p = 0.003; Fig. 3a) and shorter OS (4.6 vs 10.5 months; log-rank p = 0.001; Fig. 3b). Analyses of in-treatment cfDNA fragment size demonstrated that short fragment size were again associated with shorter PFS (3.1 vs 6.8 months; log-rank p = 0.049; Additional file 1: Figure S4A), but not with shorter OS (7.0 vs 7.0 months; log-rank p = 0.634; Additional file 1: Figure S4B). Similarly, patients with high pre-treatment cfDNA levels had both shorter PFS (3.3 vs 7.7 months; log-rank p < 0.001; Fig. 3c) and shorter OS (5.4 vs 8.8 months; log-rank p = 0.002; Fig. 3d) compared to patients with low cfDNA levels. In samples obtained after initiation of chemotherapy, high cfDNA levels were neither associated with shorter PFS (4.5 vs 6.9 months; log-rank p = 0.185; Additional file 1: Figure S4C), nor shorter OS (7.0 vs 7.4 months; log-rank p = 0.368; Additional file 1: Figure S4D).
Fig. 3.

Kaplan-Meier analyses of pre-treatment samples. a, c, e Progression-free survival and b, d, f overall survival are shown for a, b patients with short (≤ 167 bp) vs those with long (> 167 bp) cfDNA fragment size; c, d patients with high vs those with low cfDNA levels, and e, f patients positive for two markers, vs those with one or no markers
We found a moderately negative correlation between pre-treatment cfDNA fragment size and cfDNA levels (spearman correlation − 0.608, p < 0.001). Short fragment size (≤ 167 bp) and high cfDNA levels (> 4.66 ng/mL plasma) were also concordant in 45/61 (74%; Kappa = 0.475; 95% CI 0.253–0.696) patients. To see if it provided additional prognostic value, we performed survival analysis on the combination of the pre-treatment cfDNA fragment size and cfDNA levels. The patients were grouped according to whether they had neither short cfDNA fragment size nor high cfDNA levels (negative for both markers), either short cfDNA fragment size or high cfDNA levels (positive for 1 marker), or both (positive for two markers). The survival analyses demonstrated that both PFS (8.1 vs 4.6 vs 2.6 months; log-rank p < 0.001; Fig. 3e) and OS (10.6 vs 7.9 vs 3.8 months; log-rank p = 0.001; Fig. 3f) were significantly prolonged in patients negative for both markers, compared to patients that were positive for one or two markers.
Both univariable and multivariable Cox proportional hazards regression analyses were performed to estimate the prognostic impact of cfDNA fragment size and cfDNA levels on survival, relative to other clinicopatological parameters. The univariable regression analyses (Table 2) confirmed the prognostic impact of cfDNA fragment size and cfDNA levels, as demonstrated by the Log-Rank test. In addition, the univariable analysis showed that radiologically determined tumor size, ECOG performance status, first-line treatment, second-line treatment and study center also had prognostic value. The multivariable Cox regression analyses (Table 3) demonstrated that the dichotomized cfDNA level was an independent prognostic factor for both PFS (HR = 3.049, p = 0.005) and OS (HR = 2.236, p = 0.028). In addition, CA 19-9 was an independent prognostic factor for OS. Finally, the ECOG performance status and first-line treatment were independent prognostic factors for both PFS and OS. No other covariates were identified as significant predictors of survival in the multivariable model.
Table 2.
Univariable Cox regression
| Parameter | Progression-free survival | Overall survival | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p-value | Hazard ratio (95% CI) | p-value | |
| cfDNA fragment size B1 (continuous) | 0.913 (0.865–0.962) | 0.001 | 0.897 (0.848–0.949) | < 0.001 |
| cfDNA fragment size B1 (> 167 bp vs ≤ 167 bp) | 0.409 (0.225–0.744) | 0.003 | 0.394 (0.220–706) | 0.002 |
| cfDNA level B1 (continuous) | 1.002 (1.001–1.003) | 0.001 | 1.002 (1.001–1.003) | 0.001 |
| cfDNA level B1 (> median vs ≤ median) | 3.773 (2.009–7.084) | < 0.001 | 2.418 (1.350–4.329) | 0.003 |
| Combination cfDNA fragment size and cfDNA levels | < 0.001 | 0.002 | ||
| 0 markersa | Reference | Reference | ||
| 1 marker | 2.825 (1.275–6.258) | 2.032 (0.943–4.379) | ||
| 2 markers | 4.476 (2.139–9.364) | 3.326 (1.695–6.526) | ||
| CA 19-9 (< 37U/mL vs ≥ 37U/mL) | 0.665 (0.319–1.385) | 0.276 | 0.854 (0.412–1.771) | 0.672 |
| cfDNA fragment size B2 (continuous) | 0.935 (0.860–1.017) | 0.116 | 0.981 (0.899–1.070) | 0.662 |
| cfDNA fragment size B2 (> 167 bp vs ≤ 167 bp) | 0.498 (0.245–1.011) | 0.054 | 0.841 (0.413–1.715) | 0.635 |
| cfDNA levels B2 (continuous) | 1.061 (1.028–1.095) | < 0.001 | 1.062 (1.026–1.099) | 0.001 |
| cfDNA levels B2 (> median vs ≤ median) | 1.626 (0.786–3.365) | 0.190 | 1.412 (0.664–3.002) | 0.371 |
| Age at diagnosis | 1.003 (0.973–1.035) | 0.833 | 0.990 (0.957–1.023) | 0.539 |
| Sex (male vs female) | 1.194 (0.688–2.070) | 0.529 | 1.194 (0.671–2.124) | 0.546 |
| Center (SUH vs HUH) | 2.176 (1.153–4.105) | 0.016 | 2.213 (1.130–4.337) | 0.021 |
| Tumor size (mm) | 1.030 (1.012–1.049) | 0.001 | 1.021 (1.001–1.041) | 0.042 |
| Tumor location | 0.075 | 0.075 | ||
| Pancreas head | Reference | Reference | ||
| Pancreas body | 0.616 (0.182–2.088) | 0.657 (0.197–2.195) | ||
| Pancreas tail | 2.314 (1.165–4.596) | 2.239 (1.125–4.458) | ||
| Multiple | 1.135 (0.531–2.425) | 1.581 (0.732–3.434) | ||
| T-stage | 0.342 | 0.350 | ||
| T2 | Reference | Reference | ||
| T3 | 1.116 (0.471–2.645) | 0.922 (0.367–2.318) | ||
| T4 | 0.664 (0.314–1.403) | 0.603 (0.281–1.294) | ||
| Clinical stage (Stage IV vs Stage III) | 2.147 (0.836–5.515) | 0.112 | 1.829 (0.715–4.678) | 0.207 |
| Metastatic location | 0.457 | 0.735 | ||
| Liver | Reference | Reference | ||
| Lung | 0.382 (0.113–1.288) | 0.590 (0.195–1.781) | ||
| Multiple | 0.726 (0.271–1.944) | 0.767 (0.262–2.240) | ||
| Other | 0.841 (0.439–1.610) | 0.740 (0.366–1.496) | ||
| N-stage (N1 vs N0) | 1.745 (0.760–4.006) | 0.189 | 1.566 (0.703–3.488) | 0.273 |
| ECOG performance status | 0.003 | < 0.001 | ||
| 0 | Reference | Reference | ||
| 1 | 1.656 (0.809–3.389) | 1.634 (0.782–3.415) | ||
| 2 | 5.057 (1.932–13.235) | 6.810 (2.537–18.277) | ||
| First-line treatment | < 0.001 | < 0.001 | ||
| FOLFIRINOX | Reference | Reference | ||
| Gemcitabine | 14.760 (4.874–44.697) | 15.099 (4.951–46.047) | ||
| Nab-Pac + gemcitabine | 2.796 (1.481–5.280) | 3.201 (1.665–6.155) | ||
| Second-line treatment (yes vs no) | 0.822 (0.453–1.490) | 0.519 | 0.494 (0.255–0.955) | 0.036 |
| Prior anti-cancer surgery (yes vs no) | 0.601 (0.287–1.260) | 0.178 | 0.722 (0.324–1.612) | 0.427 |
a A positive marker indicates either short cfDNA fragment size or high cfDNA levels
Italic indicates a significant p-value
Table 3.
Multivariable Cox regression
| Parameter | Progression-free survival | Overall survival | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p-value | Hazard ratio (95% CI) | p-value | |
| cfDNA level B1 (> median vs ≤ median) | 3.049 (1.398–6.649) | 0.005 | 2.236 (1.089–4.590) | 0.028 |
| CA 19-9 (< 37U/mL vs ≥ 37U/mL) | n.s. | 0.345 (0.148–0.804) | 0.014 | |
| ECOG performance status | 0.043 | 0.011 | ||
| 0 | Reference | Reference | ||
| 1 | 1.122 (0.486–2.589) | 1.149 (0.540–2.446) | ||
| 2 | 3.108 (1.066–9.056) | 4.066 (1.443–11.461) | ||
| First line treatment | 0.003 | < 0.001 | ||
| FOLFIRINOX | Reference | Reference | ||
| Gemcitabine | 7.765 (2.313–26.068) | 12.064 (3.385–42.996) | ||
| Nab-Pac + gemcitabine | 2.021 (1.002–4.076) | 2.901 (1.411–5.965) | ||
n.s. not significant
Discussion
In this study, we demonstrate that cfDNA fragment size and cfDNA level have prognostic value for patients with advanced pancreatic cancer. Previous studies have shown that increased levels of cfDNA measured before the initiation of chemotherapy are associated with worse survival in patients with pancreatic cancer [3] and with increased tumor load in patients with colorectal cancer [2]. Similarly to our approach, it was also recently demonstrated that shorter cfDNA fragment size are associated with shorter PFS in patients with renal cell carcinoma [22]. The fragment size of cfDNA that originates from non-hematopoietic cells [14–16], including tumor cells [19, 20], have been shown to be shorter than cfDNA that originates from cells of hematopoietic origin. That finding suggests that increased numbers of short cfDNA fragments primarily reflects increased relative levels of tumor DNA in the blood of patients with cancer. The mechanisms underlying the shorter fragment size of non-hematopoietically derived cfDNA are not well understood. However, it has been speculated that different cell types might have differences in intracellular DNA degradation and different mechanisms of DNA release, and that DNA methylation could be a contributing factor [16, 23].
Interestingly, our multivariable Cox regression model indicated that the cfDNA level was an independent prognostic factor, but not the cfDNA fragment size. Although cfDNA fragment size and cfDNA levels were moderately correlated in our cohort, we found that cfDNA fragment size was not an independent prognostic factor even when we tested cfDNA fragment size in the absence of cfDNA levels in the regression model (Additional file 1: Table S2). Nevertheless, it would be interesting to compare the levels of the tumor-derived cfDNA with cfDNA fragment size in future studies, to clarify the association between cfDNA fragment size and prognosis.
Both cfDNA fragment size and cfDNA levels were also measured 1 month after the initiation of chemotherapy. Previous studies have shown that patients that receive chemotherapy display significant increases in cfDNA levels at 24 h and 8 days after initiation of chemotherapy [24], and after several cycles of chemotherapy [25]. In this study, however, we did not observe any change in either cfDNA fragment size or cfDNA levels after treatment initiation. Further, we did not observe any association between cfDNA levels and survival after initiation of chemotherapy. In contrast, we did observe a weak association between cfDNA fragment size and PFS.
In this study, we found significant differences in the PFS and OS between the two study centers. The first six patients recruited at SUH were all treated with Gemcitabine monotherapy and died within 4 months after initiation of treatment. These patients were the only patients treated with Gemcitabine monotherapy, and their inclusion explained most of the difference in survival between the two centers. However, FOLFIRINOX treatment was given to a higher proportion of patients at HUH (100%) than at SUH (26%). In this study, FOLFIRINOX treatment was more beneficial for both PFS and OS than nab-paclitaxel treatment. The increase in survival among patients that received FOLFIRINOX compared to patients that received nab-paclitaxel could not be directly explained by differences in either ECOG performance status (p = 0.768) or clinical stage (p = 0.271). However, it should be mentioned that most patients that received FOLFIRINOX were recruited at HUH, and that we only recruited patients that were expected to tolerate FOLFIRINOX treatment from that center. Consequently, the patient selection method at HUH might have been biased. After we adjusted for covariates in the multivariable model, the study center was no longer an independent factor (Table 3).
This study had some limitations. First, we used the Agilent 2100 Bioanalyzer and Agilent High Sensitivity DNA kit to determine cfDNA fragment size and cfDNA levels. This kit was reported (from the manufacturer) to have a sizing accuracy of ± 10% coefficient of variation (CV) and a sizing reproducibility of 5% CV or better; moreover, its quantification accuracy was 20% CV and its quantification reproducibility was 15% CV or better. Thus, measurements close to the values of dichotomization might have represented false positives or false negatives. Alternatively, we might have obtained more precise cfDNA fragment size and cfDNA level estimates by utilizing next-generation sequencing (NGS) for size estimations [19, 20], and qPCR-based kits for quantification [19]. Higher precision might have increased the prognostic value of our markers. However, the Agilent 2100 Bioanalyzer is much simpler and faster than NGS and qPCR, and it provided reasonable accuracy and reproducibility. From a clinical perspective, a rapid, economic, simple test offers important advantages. However, it would be interesting, in future, to assess cfDNA size and cfDNA levels with more precise techniques to validate the current findings.
Another study limitation was the excess of patients with metastatic disease compared to patients with locally advanced disease, and the total lack of patients with localized disease. Consequently, our results are not broadly generalizable. Future studies should investigate whether cfDNA fragment size and cfDNA level also provide prognostic information for patients with resectable pancreatic cancer. It would also be of interest to investigate the prognostic potential of these markers in other cancer types. Most previous investigations of cfDNA in pancreatic cancer have focused on detecting mutated cfDNA, which show prognostic significance in patients with both resectable and unresectable pancreatic cancer [9, 26, 27]. Thus, a side-by-side comparison of the cfDNA fragment size and cfDNA level approach and the mutated cfDNA detection approach should be performed to investigate further the clinical potential of our findings.
Conclusion
We demonstrated that the determination of cfDNA fragment size and cfDNA levels is a non-invasive, simple method for predicting clinical outcome in patients with advanced pancreatic cancer. Shorter cfDNA fragment size was associated with worse PFS and OS, although it was not an independent prognostic marker. In contrast the cfDNA level was a strong independent prognostic factor for both PFS and OS in our patient cohort. Future investigations are needed to elaborate these findings with regard to clinical relevance, and to establish whether the determination of cfDNA fragment size and cfDNA level might also have prognostic value for patients with localized pancreatic cancer.
Additional file
Additional file 1: Figure S1. Patient sample electropherogram. Main peak corresponds to nucleosomes released from apoptotic cells (corresponding to one nucleosome plus linker). Table S1. Baseline characteristics of patients with pancreatic cancer. Figure S2. Comparison of cfDNA fragment size and cfDNA levels in plasma between centers. Boxplot showing differences in (A) cfDNA fragment size and (B) cfDNA levels between samples obtained at SUH versus samples obtained at HUH. Figure S3. Distributions of cfDNA fragment size and cfDNA levels in plasma at baseline and during treatment. Boxplot showing differences in (A) cfDNA fragment size and (B) cfDNA levels between samples obtained before initiation of chemotherapy versus samples obtained during treatment. Figure S4. Kaplan-Meier analyses of samples obtained after the first cycle of chemotherapy. (A, C) Progression-free survival and (B, D) overall survival are shown for (A, B) patients with short (≤167 bp) vs those with long (>167 bp) cfDNA fragment size, and (C, D) patients with high vs those with low cfDNA levels. Table S2. Multivariable Cox regression without cfDNA level.
Authors’ contributions
ML and ON designed the study; ML analyzed the cfDNA samples, performed the statistical analysis, and drafted the manuscript; SO and KT isolated the cfDNA from patient samples; MJ provided scientific advice and contributed to results interpretations; TB, RS, HG, NIG, BKA, and BG recruited patients and provided patient samples and data; ON supervised the study and contributed to results interpretations and the statistical analysis. All authors reviewed and commented on the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank participating patients and their families.
Competing interests
Celgene Corporation provided research support in the form of drugs free of charge for some of the patients. They played no role in the design of the study, in the collection, analysis, or interpretation of data, or in writing the manuscript.
Availability of data and materials
The data supporting the conclusions of this study is included in the article and its Additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All patients and healthy controls provided written informed consent to participate in the study. The project was approved by the Regional Committee for Medical and Health Research Ethics (REK-Vest 2011/475, REK-Vest 2013/1743).
Funding
This project was supported by the Folke Hermansen Foundation, the Norwegian Cancer Society, and the Western Norway Health Authorities. This project was part of a strategic program called “Personalized medicine-biomarkers and clinical studies”, which was supported by the Western Norway Regional Health Authority. Additional support was provided by Celgene Corporation. The funding bodies played no role in the design of the study, in the collection, analysis, or interpretation of data, or in writing the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- cfDNA
cell-free DNA
- PFS
progression-free survival
- OS
overall survival
- HR
hazard ratio
- ctDNA
circulating tumor DNA
- FOLFIRINOX
folinic acid, fluorouracil, irinotecan, oxaliplatin
- CA 19-9
carbohydrate antigen 19-9
- T-stage
tumor stage
- N-stage
nodal stage
- ECOG
Eastern Cooperative Oncology Group
Contributor Information
Morten Lapin, Phone: +47 47233151, Email: morten.lapin@sus.no.
Satu Oltedal, Email: satu.oltedal@sus.no.
Kjersti Tjensvoll, Email: kjersti.tjensvoll@sus.no.
Tove Buhl, Email: tove.buhl@sus.no.
Rune Smaaland, Email: runesmaaland@me.com.
Herish Garresori, Email: herish.garresori@sus.no.
Milind Javle, Email: mjavle@mdanderson.org.
Nils Idar Glenjen, Email: nils.idar.glenjen@helse-bergen.no.
Bente Kristin Abelseth, Email: bente.kristin.abelseth@helse-bergen.no.
Bjørnar Gilje, Email: bjornar.gilje@sus.no.
Oddmund Nordgård, Email: oddmund.nordgard@sus.no.
References
- 1.Wu TL, Zhang D, Chia JH, Tsao K, Sun CF, Wu JT. Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin Chim Acta. 2002;321(1–2):77–87. doi: 10.1016/S0009-8981(02)00091-8. [DOI] [PubMed] [Google Scholar]
- 2.Spindler KL, Appelt AL, Pallisgaard N, Andersen RF, Brandslund I, Jakobsen A. Cell-free DNA in healthy individuals, noncancerous disease and strong prognostic value in colorectal cancer. Int J Cancer. 2014;135(12):2984–2991. doi: 10.1002/ijc.28946. [DOI] [PubMed] [Google Scholar]
- 3.Singh N, Gupta S, Pandey RM, Chauhan SS, Saraya A. High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest. 2015;33(3):78–85. doi: 10.3109/07357907.2014.1001894. [DOI] [PubMed] [Google Scholar]
- 4.Shaw JA, Guttery DS, Hills A, Fernandez-Garcia D, Page K, Rosales BM, et al. Mutation analysis of cell-free DNA and single circulating tumor cells in metastatic breast cancer patients with high circulating tumor cell counts. Clin Cancer Res. 2017;23(1):88–96. doi: 10.1158/1078-0432.CCR-16-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 8.Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 2015;5(10):1040–1048. doi: 10.1158/2159-8290.CD-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016;10(4):635–643. doi: 10.1016/j.molonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5(4):30. doi: 10.1186/gm434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A. 2013;110(47):18761–18768. doi: 10.1073/pnas.1313995110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw JA, Smith BM, Walsh T, Johnson S, Primrose L, Slade MJ, et al. Microsatellite alterations in plasma DNA of primary breast cancer patients. Clin Cancer Res. 2000;6(3):1119–1124. [PubMed] [Google Scholar]
- 14.Lui YY, Chik KW, Chiu RW, Ho CY, Lam CW, Lo YM. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48(3):421–427. [PubMed] [Google Scholar]
- 15.Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem. 2004;50(1):88–92. doi: 10.1373/clinchem.2003.024893. [DOI] [PubMed] [Google Scholar]
- 16.Zheng YW, Chan KC, Sun H, Jiang P, Su X, Chen EZ, et al. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem. 2012;58(3):549–558. doi: 10.1373/clinchem.2011.169318. [DOI] [PubMed] [Google Scholar]
- 17.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102(45):16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE. 2011;6(9):e23418. doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A. 2015;112(11):E1317–E1325. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12(7):e1006162. doi: 10.1371/journal.pgen.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto Y, Uemura M, Nakano K, Hayashi Y, Wang C, Ishizuya Y, et al. Increased level and fragmentation of plasma circulating cell-free DNA are diagnostic and prognostic markers for renal cell carcinoma. Oncotarget. 2018;9(29):20467–20475. doi: 10.18632/oncotarget.24943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun K, Jiang P, Wong AIC, Cheng YKY, Cheng SH, Zhang H, et al. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc Natl Acad Sci U S A. 2018;115(22):E5106-e14. doi: 10.1073/pnas.1804134115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost. 2011;9(11):2313–2321. doi: 10.1111/j.1538-7836.2011.04465.x. [DOI] [PubMed] [Google Scholar]
- 25.Kwee S, Song MA, Cheng I, Loo L, Tiirikainen M. Measurement of circulating cell-free DNA in relation to 18F-fluorocholine PET/CT imaging in chemotherapy-treated advanced prostate cancer. Clin Transl Sci. 2012;5(1):65–70. doi: 10.1111/j.1752-8062.2011.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietrasz D, Pecuchet N, Garlan F, Didelot A, Dubreuil O, Doat S, et al. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res. 2017;23(1):116–123. doi: 10.1158/1078-0432.CCR-16-0806. [DOI] [PubMed] [Google Scholar]
- 27.Kim MK, Woo SM, Park B, Yoon KA, Kim YH, Joo J, et al. Prognostic implications of multiplex detection of KRAS mutations in cell-free DNA from patients with pancreatic ductal adenocarcinoma. Clin Chem. 2018;64(4):726–734. doi: 10.1373/clinchem.2017.283721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Patient sample electropherogram. Main peak corresponds to nucleosomes released from apoptotic cells (corresponding to one nucleosome plus linker). Table S1. Baseline characteristics of patients with pancreatic cancer. Figure S2. Comparison of cfDNA fragment size and cfDNA levels in plasma between centers. Boxplot showing differences in (A) cfDNA fragment size and (B) cfDNA levels between samples obtained at SUH versus samples obtained at HUH. Figure S3. Distributions of cfDNA fragment size and cfDNA levels in plasma at baseline and during treatment. Boxplot showing differences in (A) cfDNA fragment size and (B) cfDNA levels between samples obtained before initiation of chemotherapy versus samples obtained during treatment. Figure S4. Kaplan-Meier analyses of samples obtained after the first cycle of chemotherapy. (A, C) Progression-free survival and (B, D) overall survival are shown for (A, B) patients with short (≤167 bp) vs those with long (>167 bp) cfDNA fragment size, and (C, D) patients with high vs those with low cfDNA levels. Table S2. Multivariable Cox regression without cfDNA level.
Data Availability Statement
The data supporting the conclusions of this study is included in the article and its Additional files.