

Abstract
Deposition of amyloid plaques in the brain is one of the two main pathological hallmarks of Alzheimer’s disease (AD). Amyloid positron emission tomography (PET) is a neuroimaging tool that selectively detects in vivo amyloid deposition in the brain and is a reliable endophenotype for AD that complements cerebrospinal fluid biomarkers with regional information. We measured in vivo amyloid deposition in the brains of ~1000 subjects from three collaborative AD centers and ADNI using 11C-labeled Pittsburgh Compound-B (PiB)-PET imaging followed by meta-analysis of genome-wide association studies, first to our knowledge for PiB-PET, to identify novel genetic loci for this endophenotype. The APOE region showed the most significant association where several SNPs surpassed the genome-wide significant threshold, with APOE*4 being most significant (P-meta = 9.09E-30; β = 0.18). Interestingly, after conditioning on APOE*4, 14 SNPs remained significant at P < 0.05 in the APOE region that were not in linkage disequilibrium with APOE*4. Outside the APOE region, the meta-analysis revealed 15 non-APOE loci with P < 1E-05 on nine chromosomes, with two most significant SNPs on chromosomes 8 (P-meta = 4.87E-07) and 3 (P-meta = 9.69E-07). Functional analyses of these SNPs indicate their potential relevance with AD pathogenesis. Top 15 non-APOE SNPs along with APOE*4 explained 25–35% of the amyloid variance in different datasets, of which 14–17% was explained by APOE*4 alone. In conclusion, we have identified novel signals in APOE and non-APOE regions that affect amyloid deposition in the brain. Our data also highlights the presence of yet to be discovered variants that may be responsible for the unexplained genetic variance of amyloid deposition.
Subject terms: Genetics, Psychology
Introduction
Genomic efforts mainly through large-scale genome-wide association studies (GWAS), as part of the Alzheimer’s Disease Genetics Consortium (ADGC) [1] and the International Genomics of Alzheimer’s Project (IGAP) [2], have identified over 20 genes/loci for late-onset Alzheimer’s disease (AD). However, known common AD variants account for only ~30% of the AD genetic variance [3], and they also do not provide definitive information about the underlying disease mechanisms. Genetic studies focusing on AD-related quantitative phenotypes/endophenotypes may help to identify additional AD-related genes. One such AD-related phenotype is deposition of amyloid-beta (Aβ) in the brain, which is one of the two main pathologic hallmarks of AD; the other being the formation of tau deposits in the form of neurofibrillary tangles, neuropil threads, and dystrophic neurites (tau pathology) in the brain [4]. According to the current model for sporadic AD, Aβ pathology occurs independently of tau pathology, is detectable earlier, and is believed to accelerate neocortical tau pathology and neurodegeneration [5]. Recent longitudinal studies on cognitively normal subjects also confirm that amyloidosis is an early process in AD [6, 7]. The in vivo detection of Aβ deposition in the brain, as measured by positron emission tomography (PET) scanning with 11C-labeled Pittsburgh Compound-B (PiB) and the increased retention of PiB observed in the brains of AD patients compared with cognitively normal controls, was first reported by Klunk and colleagues [8, 9] and since has been confirmed in many studies [10]. There is a high correlation between amyloid PET imaging and neuritic plaque frequency as confirmed by autopsy studies [11–13]. Multiple studies have shown that amyloid PET has a high value for the clinical diagnosis of AD and in clinical trials aiming to reduce brain Aβ burden [14].
There is a well-established association of APOE variants with risk [1, 2] and age-at-onset [15, 16] of AD. Likewise, APOE genetic variation is also strongly associated with Aβ deposition in the brain as measured by PiB retention [17–19], indicating a genetic basis of Aβ deposition in the brain. Here, we used PiB-PET as an endophenotype to identify novel genetic loci for AD pathology using meta-analysis of three GWAS, the first to our knowledge, using the largest sample with the PiB-PET imaging from three different centers and the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Materials and methods
Sample description
All subjects with PiB-PET data were European-Americans and derived from three sites: University of Pittsburgh (PITT), Washington University (WU), and Indiana University (IU) combined with the initial phase of the multicenter ADNI PiB-PET add-on study (here they are referred to as ADNI/IU). All subjects provided informed consent, and all studies were approved by their local Institutional Review Boards. The summary statistics of these samples are included in Supplementary Table S1 and their description is given Supplementary Text.
Amyloid-PET data
Detailed methods for acquisition and processing of PiB-PET scans are described in previous reports for the PITT [17, 18], WU [19], ADNI [20–22], and IU [23] studies. PiB retention was measured in four cortical regions of the brain, including medial frontal cortex (MFC; anterior cingulate/gyrus rectus), lateral frontal cortex (LFC), precuneus cortex (PRC), and parietal cortex (PAR) and expressed as a ratio to the cerebellum. In the GWAS meta-analysis, the PiB retention values from these four cortical regions were averaged in each subject to calculate a mean global score (GBL4) as the quantitative endophenotype. PiB retention was expressed as standardized uptake volume ratio (SUVR) in the PITT and ADNI/IU data [23, 24] and as binding potential (BP) in the WU data [25]. BP is approximately equal to SUVR-1. Because of this inconsistency in the PiB measurement methods, the GWAS data were analyzed via P-value-based meta-analysis as described below.
Genotyping, imputation, and quality control
The genotyping platforms used for each study sample are listed in Supplementary Table S1. Imputation of non-genotyped single-nucleotide polymorphisms (SNPs) was performed with IMPUTE2 [26] using the 1000 Genomes Project [27] Phase III (May 2013 release) data as the reference panel for PITT and Phase I (November 2010 release) data for WU and ADNI/IU datasets. Full description of these procedures is given in Supplementary Text.
Meta-analysis
METAL [28] software was used to perform meta-analysis on three GWAS, using the mean PiB-PET GBL4 value. METAL performs a P-value-based meta-analysis, which is appropriate when the effects being estimated are different in different cohorts. It does, however, account for differences in sample size between cohorts and for the direction of effects. The summary effect size was calculated by averaging the study-specific effect sizes, with weights reflecting the standard errors from the study-specific effect sizes.
Functional analyses
To evaluate the biological significance of PiB-associated signals, we conducted five different analyses: differential gene expression in AD versus non-AD in relevant tissues, brain gene expression, expression quantitative trait loci (eQTL) analyses, and summary-data-based Mendelian randomization (SMR) analyses to test for pleiotropic association between gene expression and PiB, and pathway analyses. Detailed description of these analyses is given in Supplementary Text.
Results
Amyloid PET data characteristics
The characteristics of participants in each of the three datasets included in the meta-analysis are shown in Supplementary Table S1. The WU sample was younger with less male participants. The distribution of mean global PiB retention is shown in Fig. 1.
Fig. 1.
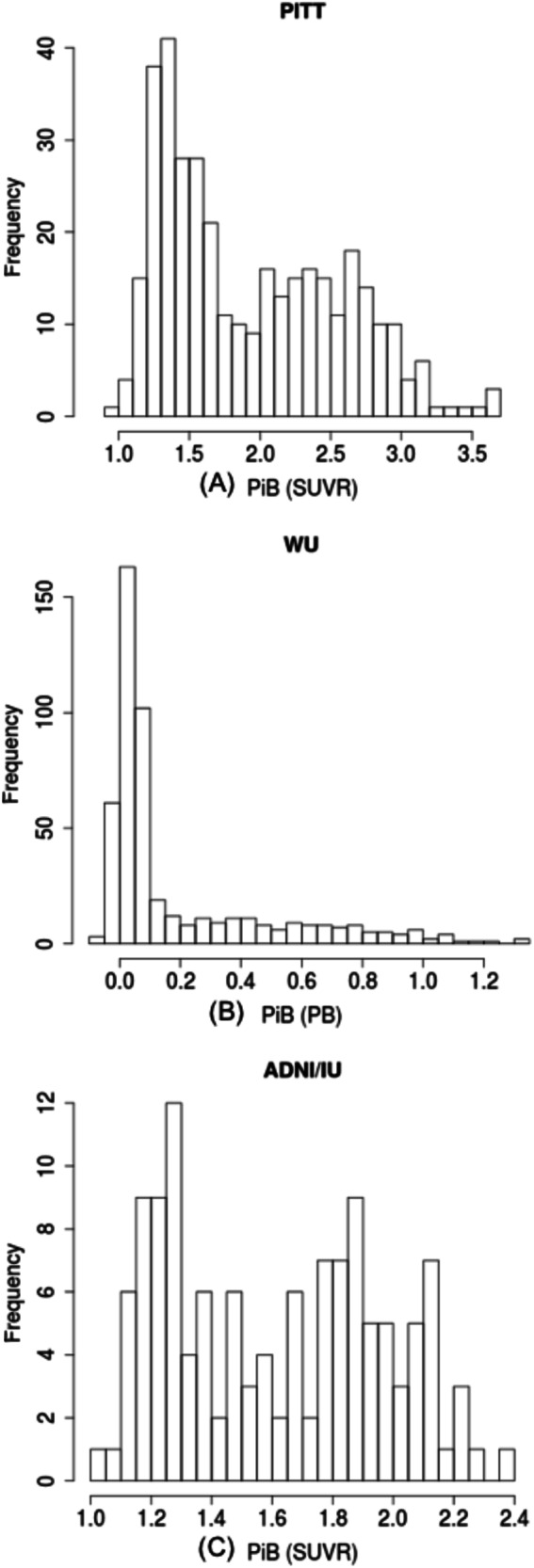
Distribution of PiB retention in the University of Pittsburgh (PITT) (a), Washington University (WU) (b), and the Alzheimer’s disease Neuroimaging Initiative (ADNI) and the Indiana Memory and Aging Study (ADNI/IU) (c) samples. SUVR standardized uptake volume ratio, BP binding potential
GWAS analysis
Quantile–quantile (QQ) plots and lambda values for the meta-analysis showed that neither the results from each of the three component studies nor the combined results from meta-analysis were inflated in their test statistics (Fig. 2a). Meta-analysis revealed 27 genome-wide significant SNPs (P < 5E-08) in a four-gene region on chromosome 19: PVRL2-TOMM40-APOE-APOC1(Fig. 2b, and Supplementary Table S2). As expected, APOE*4/rs429358 showed the most significant association with the average global PiB retention (P-meta = 9.09E-30; β = 0.18; Fig. 3, Supplementary Figure S1).
Fig. 2.

a Quantile–quantile plot for the individual GWAS results in the University of Pittsburgh (PITT), Washington University (WU), and the Alzheimer’s disease Neuroimaging Initiative (ADNI) and the Indiana Memory and Aging Study (ADNI/IU) datasets and in the meta-analysis. λ is the genomic control value. b Manhattan plot showing the P-values in the meta-analysis. The blue line represents the suggestive significance line (P < E-05). The red line represents the significance threshold (P < 5E-08)
Fig. 3.

Regional plot of the APOE region on chromosome 19 in the meta-analysis. The relative location of genes and the direction of transcription are shown in the lower portion of the figure, and the chromosomal position is shown on the x -axis. The light blue line shows the recombination rate across the region (right y -axis) and the left y-axis shows the significance of the associations. The purple diamond shows the P-value for rs429358 that is the most significant SNP in the meta-analysis. The circles show the P-values for all other SNPs and are color coded according to the level of LD with rs429358 in the 1000 Genome Project EUR population
Outside of the APOE region, no genome-wide significant signal was observed. However, the meta-analysis revealed 15 non-APOE loci with P < 1E-05 on chromosomes 8, 3, 15, 4, 21, 13, 2, 12, and 1 (Table 1). Most of these loci show quite consistent results across all datasets. The regional plots of these 15 non-APOE loci are shown in Supplementary Figures S2.1–S2.15. The most significant SNP outside the APOE region is intergenically located between ADCY8 and EFR3A on chromosome 8 (rs13260032; P = 4.87E-07, Supplementary Figure S2.1). The next most significant SNP is also intergenically located between RAP2B and C3orf79 on chromosome 3 (rs4680057; P = 9.69E-07, Supplementary Figure S2.2). Chromosome 3 also harbors two additional signals: one in ncRNA (LINC00971/rs9831119; P = 2.98E-06, Supplementary Figure S2.6) and another near MAGEF1/ rs11923588 (P = 5.66E-06, Figure S2.9). The third most significant SNP is located in the DAPK2 gene on chromosome 15 (rs12908891; P = 1.39E-06, Supplementary Figure S2.3). We also analyzed the data after adjusting for the effect of APOE*4/rs429358 in these non-APOE regions, which showed a slight attenuation of the association strengths (Table 1).
Table 1.
Genetic loci associated with PiB-PET with P < 1E-05 in the meta-analysis
| SNP | Chr | Position | A1 | A2 | Gene | Region | PITT | WU | ADNI/IU | Meta | Meta adjusted for APOE*4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | Beta | P-value | MAF | Beta | P-value | MAF | Beta | P-value | Beta | P-value | Beta | P-value | |||||||
| rs429358 | 19 | 45411941 | C | T | APOE | Exonic | 0.20 | 0.35 | 5.20E-12 | 0.21 | 0.16 | 5.36E-16 | 0.28 | 0.19 | 7.88E-05 | 0.18 | 9.09E-30 | NA | NA |
| rs13260032 | 8 | 132451455 | C | A | ADCY8,EFR3A | Intergenic | 0.43 | −0.17 | 4.90E-05 | 0.44 | −0.06 | 1.34E-02 | 0.43 | −0.07 | 6.76E-02 | −0.08 | 4.87E-07 | −0.07 | 1.15E-05 |
| rs4680057 | 3 | 153096985 | A | G | RAP2B,C3orf79 | Intergenic | 0.45 | 0.16 | 2.60E-04 | 0.42 | 0.04 | 1.93E-02 | 0.46 | 0.11 | 8.07E-03 | 0.06 | 9.69E-07 | 0.05 | 2.61E-05 |
| rs12908891 | 15 | 64236441 | G | A | DAPK2 | Intronic | 0.46 | 0.12 | 6.38E-03 | 0.49 | 0.06 | 5.77E-06 | 0.50 | 0.01 | 9.01E-01 | 0.06 | 1.39E-06 | 0.04 | 5.30E-04 |
| rs7377304 | 4 | 187129780 | G | T | CYP4V2 | Intronic | 0.42 | 0.08 | 8.84E-02 | 0.46 | 0.06 | 5.98E-05 | 0.47 | 0.09 | 2.83E-02 | 0.06 | 2.46E-06 | 0.06 | 1.59E-05 |
| rs55708341 | 21 | 45627581 | T | A | C21orf33,ICOSLG | Intergenic | 0.23 | 0.18 | 1.88E-04 | 0.18 | 0.05 | 4.65E-03 | 0.19 | 0.07 | 2.26E-01 | 0.07 | 2.51E-06 | 0.06 | 6.74E-05 |
| rs9831119 | 3 | 84712077 | C | T | LINC00971 | ncRNA_intronic | 0.12 | −0.13 | 3.66E-02 | 0.14 | −0.07 | 5.15E-04 | 0.12 | −0.16 | 8.89E-03 | −0.08 | 2.98E-06 | −0.07 | 1.80E-05 |
| rs9531483 | 13 | 84244873 | A | C | SLITRK1 | Intergenic | 0.30 | −0.12 | 7.81E-03 | 0.31 | −0.06 | 5.62E-04 | 0.33 | −0.07 | 1.12E-01 | −0.06 | 3.65E-06 | −0.05 | 8.45E-05 |
| rs6722000 | 2 | 209075957 | G | A | C2orf80,IDH1 | Intergenic | 0.21 | 0.14 | 7.27E-03 | 0.20 | 0.07 | 3.92E-04 | 0.22 | 0.06 | 2.37E-01 | 0.07 | 4.96E-06 | 0.07 | 1.71E-05 |
| rs11923588 | 3 | 184459667 | T | C | MAGEF1,LOC101928992 | Intergenic | 0.07 | −0.20 | 2.19E-02 | 0.06 | −0.19 | 6.50E-05 | 0.06 | −0.14 | 1.53E-01 | −0.18 | 5.66E-06 | −0.13 | 1.56E-03 |
| rs66837203 | 4 | 36897136 | T | C | DTHD1,MIR4801 | Intergenic | 0.06 | 0.24 | 9.85E-03 | 0.08 | 0.10 | 3.53E-04 | 0.04 | 0.13 | 2.36E-01 | 0.11 | 6.03E-06 | 0.09 | 4.66E-04 |
| rs200028958 | 4 | 70923661 | A | G | HTN1 | Intronic | 0.10 | 0.29 | 2.99E-05 | 0.11 | 0.09 | 1.29E-03 | 0.09 | −0.05 | 5.54E-01 | 0.10 | 6.25E-06 | 0.09 | 8.02E-05 |
| rs4526799 | 12 | 57280586 | T | C | HSD17B6,SDR9C7 | Intergenic | 0.34 | −0.19 | 3.58E-05 | 0.34 | −0.04 | 3.09E-02 | 0.36 | −0.04 | 4.18E-01 | −0.05 | 7.26E-06 | −0.06 | 1.16E-06 |
| rs17105538 | 1 | 81315043 | G | A | ELTD1,LPHN2 | Intergenic | 0.13 | 0.13 | 3.65E-02 | 0.15 | 0.08 | 2.08E-04 | 0.17 | 0.09 | 1.10E-01 | 0.08 | 7.66E-06 | 0.07 | 1.58E-05 |
| rs62121100 | 2 | 3093952 | G | T | LINC01250 | ncRNA_intronic | 0.19 | −0.20 | 1.09E-04 | 0.16 | −0.05 | 1.03E-02 | 0.21 | −0.04 | 4.79E-01 | −0.07 | 8.44E-06 | −0.06 | 2.90E-05 |
| rs1809136 | 2 | 11152180 | G | C | KCNF1,FLJ33534 | Intergenic | 0.07 | 0.28 | 5.91E-04 | 0.07 | 0.17 | 5.45E-04 | 0.08 | 0.00 | 9.95E-01 | 0.16 | 9.99E-06 | 0.15 | 6.36E-05 |
A1 minor allele, A2 major allele
Conditional analysis in the APOE region
In order to check if there were independent SNPs associated with the PiB retention in the APOE region, we performed conditional analysis by adjusting for the top SNP (APOE*4/rs429358). A total of 14 SNPs remained significant at P < 0.05 (Table 2), including three SNPs that showed genome-wide significance before adjusting for APOE*4 (rs75627662, rs483082, and rs438811; Supplementary Table S2). Supplementary Figure S3 shows LD structure of these 14 SNPs along with APOE*4/rs429358 and APOE*2/rs7412 SNPs. APOE*4 and APOE*2 have essentially no LD with nine of the 14 SNPs that are located in the PVRL2 gene (SNPs 1–9 in Supplementary Figure S3). One SNP located in the APOE/APOC1 intergenic region (rs59325138) has only very weak correlation with APOE*4 (R2 = 0.15) and APOE*2 (R2 = 0.03), while three SNPs located downstream of APOE and APOE/APOC1 intergenic region have weak to moderate LD with APOE*4 (R2 = 0.42, 0.64, 0.65 for rs75627662, rs483082, and rs438811, respectively).
Table 2.
Conditional analysis on SNPs reaching P < 0.05 in the APOE region with additional adjustment for APOE*4 (rs429358) in the meta-analysis
| SNP | Chr | Position | A1 | A2 | Gene | Region | PITT | WU | ADNI/IU | Meta | LD (R2) with | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | Beta | P-value | MAF | Beta | P-value | MAF | Beta | P-value | Beta | P-value | E*4 | E*2 | |||||||
| rs7412 | 19 | 45412079 | T | C | APOE | Exonic | 0.07 | −0.21 | 7.63E-03 | 0.07 | −0.04 | 1.45E-01 | 0.04 | −0.06 | 5.49E-01 | −0.06 | 3.69E-03 | 0.01 | NA |
| rs3852859 | 19 | 45379309 | C | T | PVRL2 | Intronic | 0.20 | 0.08 | 1.14E-01 | 0.20 | 0.05 | 1.10E-01 | 0.24 | 0.07 | 1.52E-01 | 0.06 | 8.81E-03 | 0.004 | 0.01 |
| rs2075642 | 19 | 45377467 | A | G | PVRL2 | Intronic | 0.20 | 0.07 | 1.78E-01 | 0.20 | 0.06 | 8.44E-02 | 0.22 | 0.07 | 1.33E-01 | 0.06 | 1.11E-02 | 0.004 | 0.01 |
| rs3852856 | 19 | 45361574 | A | G | PVRL2 | Intronic | 0.21 | 0.06 | 2.43E-01 | 0.20 | 0.07 | 3.16E-02 | 0.22 | 0.06 | 2.03E-01 | 0.06 | 1.15E-02 | 0.006 | 0.01 |
| rs75627662 | 19 | 45413576 | T | C | APOE | Downstream | 0.23 | −0.15 | 1.57E-02 | 0.23 | −0.02 | 2.92E-01 | 0.26 | −0.03 | 6.19E-01 | −0.03 | 1.50E-02 | 0.42 | 0.21 |
| rs4803767 | 19 | 45372959 | T | C | PVRL2 | Intronic | 0.26 | 0.05 | 2.63E-01 | 0.28 | 0.04 | 1.56E-01 | 0.31 | 0.07 | 8.34E-02 | 0.05 | 2.06E-02 | 0.002 | 0.01 |
| rs60389450 | 19 | 45372184 | C | A | PVRL2 | Intronic | 0.26 | 0.04 | 3.20E-01 | 0.26 | 0.02 | 1.72E-01 | 0.31 | 0.07 | 8.34E-02 | 0.03 | 2.72E-02 | 0.003 | 0.01 |
| rs59325138 | 19 | 45416291 | T | C | APOE,APOC1 | Intergenic | 0.38 | 0.06 | 2.16E-01 | 0.36 | 0.02 | 2.50E-01 | 0.36 | 0.07 | 1.17E-01 | 0.03 | 3.10E-02 | 0.15 | 0.03 |
| rs483082 | 19 | 45416178 | T | G | APOE,APOC1 | Intergenic | 0.26 | −0.10 | 1.60E-01 | 0.29 | −0.04 | 1.59E-01 | 0.32 | −0.06 | 4.51E-01 | −0.05 | 3.35E-02 | 0.64 | 0.16 |
| rs8104483 | 19 | 45372354 | G | T | PVRL2 | Intronic | 0.27 | 0.03 | 4.65E-01 | 0.27 | 0.02 | 2.04E-01 | 0.32 | 0.09 | 4.20E-02 | 0.03 | 3.67E-02 | 0.003 | 0.01 |
| rs3729640 | 19 | 45381917 | T | C | PVRL2 | UTR3 | 0.20 | 0.08 | 9.33E-02 | 0.19 | 0.03 | 3.35E-01 | 0.23 | 0.04 | 4.33E-01 | 0.05 | 3.79E-02 | 0.004 | 0.01 |
| rs8104292 | 19 | 45372707 | A | G | PVRL2 | Intronic | 0.27 | 0.04 | 4.14E-01 | 0.28 | 0.03 | 2.62E-01 | 0.32 | 0.09 | 4.20E-02 | 0.04 | 3.80E-02 | 0.003 | 0.01 |
| rs438811 | 19 | 45416741 | T | C | APOE,APOC1 | Intergenic | 0.26 | −0.10 | 1.60E-01 | 0.30 | −0.04 | 1.84E-01 | 0.32 | −0.06 | 4.51E-01 | −0.04 | 3.84E-02 | 0.65 | 0.16 |
| rs58521715 | 19 | 45372129 | T | A | PVRL2 | Intronic | 0.27 | 0.03 | 4.65E-01 | 0.27 | 0.02 | 2.53E-01 | 0.32 | 0.09 | 4.20E-02 | 0.03 | 4.50E-02 | 0.004 | 0.01 |
A1 minor allele, A2 major allele
The most significant SNP in meta-conditional analysis was APOE*2/rs7412 (P-meta = 3.69E-03; β = −0.06; Table 2), though it was not genome-wide significant before adjusting for APOE*4 (P-meta = 6.57E-05; β = −0.09). A similar strength of association was seen with an intronic PVRL2/rs3852859 SNP after adjusting for APOE*4 (P-meta = 8.8E-03; β = 0.06; Table 2) that was in LD with three additional SNPs (SNPs 1, 7, and 9 in Supplementary Figure S3). Three additional apparently independent associations were seen with rs4803767 (P-meta = 2.06E-02; β = 0.05 Table 2) that was in LD with four additional SNPs (SNPs 2–5 in Supplementary Figure S3), rs75627662 (P-meta = 1.50E-02; β = −0.03; Table 2) that was in LD with two additional SNPs (SNPs 13, 15 in Supplementary Figure S3), and rs59325138 (P-meta = 3.10E-02; β = 0.03; Table 2) that has very weak correlation with all other SNPs (R2 = 0.01–0.24).
Association of known AD risk loci with amyloid burden and association of amyloid loci with AD risk
We examined the top IGAP genome-wide significant SNPs (Supplementary Table S3.1) and the associated gene regions (Supplementary Table S3.2) in relation to amyloid burden and found only some nominally significant SNPs. Likewise, we examined the suggestive non-APOE amyloid loci in our PITT-ADRC case–control sample of >2200 subjects [29] and found association of two top amyloid-associated SNPs with AD risk (Supplementary Table S4.1). When we examined additional Aβ-associated SNPs in each region with AD risk, we found multiple associations with P < 0.05 (Supplementary Table S4.2), indicating that our suggestive Aβ-associated loci are also associated with AD risk (see Supplementary Text for more details).
Estimation of amyloid-PET variance by APOE and non-APOE loci
The genetic variance was estimated based on the R-square calculated from a linear regression model regressing global PiB retention on six independent APOE SNPs (rs429358, rs7412, rs3852859, rs4803767, rs75627662, and rs59325138), as described above, and 15 non-APOE SNPs given in Table 1. The contribution of six APOE SNPs to the variance of global PiB retention was 28.0, 17.3, and 17.12% in the PITT, WU, and ADNI/IU datasets, respectively; APOE*4/rs429358 alone explained 17.5, 16.5, and 13.9%, respectively. The top 15 non-APOE SNPs explained 22.6, 21.6, and 21.7% of the amyloid variance in the PITT, WU, and ADNI/IU datasets, respectively. The consistency of these estimates across the different datasets gives confidence that the difference in measurement of PiB across the datasets does not affect the bottom-line results.
Functional analyses
We performed five analyses (Methods section) to evaluate the biological significance of PiB-implicated signals/genes. We considered all genes within ±500 kb of the top variant in each locus from Table 1 plus any eQTL-controlled genes outside the ±500 kb boundary as target genes (Fig. 3, Supplementary Figures S2.1-S2.15) and selected a total of 257 genes.
Of 257 target genes, we found 20 upregulated and 25 downregulated genes that were differentially expressed in the same direction in two or more AD studies and no opposite directions were reported (Fig. 4 and Supplementary Table S6 marked in green color). Brain RNA-seq data reveals that many of these differentially expressed candidate genes are expressed in AD-relevant cell types (Fig. 4 and Supplementary Table S6 marked in yellow color).
Fig. 4.


The functional analysis results for target genes. Out of 257 target genes, only genes meeting at least three functional criteria are listed. The criteria include: (1) differential expression in at least two Alzheimer disease studies that up- or downregulated consistently in different studies; (2) expression in the brain cells (Barres website); (3) having cis-eQTL effects in any brain tissues using GTEx database (P < 0.05); 4) mediating genetic effects on PiB (SMR analysis with P < 0.05) in any brain tissues; (5) having cis-eQTL effects in whole blood (P < 0.05); (6) mediating genetic effects on PiB (SMR analysis with P < 0.05) in whole blood; and (7) included in nominally significant pathways. The detailed results are summarized in Supplementary Table S6
For eQTL analyses, we identified SNPs in LD (R2 ≥ 0.5) with the top SNP for each locus in Table 1. For these SNPs, there were cis-acting eQTLs (eQTL P < 0.05) for 151 of the 257 target genes in various brain tissues and 36 genes in whole blood available in GTEx. Supplementary Table S5 gives the eQTL results for each top SNP in 15 non-APOE loci, and the detailed results of LD SNPs (R2 ≥ 0.80) with top SNPs are given in Supplementary Table S7. With the exception of SLITRK1/rs9831119, the other 14 top SNPs were eQTLs in different brain regions; 11 of them were eQTL in anterior cingulate cortex/frontal cortex/cortex where PiB intake is highest [30], indicating their role in affecting amyloid deposition in the brain.
For SMR analyses, only the gene/variant pairs identified in the cis-eQTL analyses were considered. For these gene/variant pairs, 99 genes in any brain tissue and 19 in whole blood were shown to mediate genetic effects on PiB by cis-regulating gene expression (SMR P < 0.05; Fig. 4, Supplementary Table S6).
We conducted pathway analyses (MAGMA [31]) using four gene set resources, including and excluding target genes in the APOE region, and detected nine genome-wide significant pathways: NDK dynamin pathway, FDR = 4.6E-04; synaptic vesicle recycling, FDR = 3.5E-07; synaptic vesicle endocytosis, FDR = 3.1E-04; protein depolymerization, FDR = 3.1E-04; inositol tetrakisphosphate phosphatase activity, FDR = 5.7E-03; positive regulation of vacuole organization, FD = 5.7E-03; inositol trisphosphate phosphatase activity, FDR = 0.033; regulation of clathrin-mediated endocytosis, FDR = 0.038; and clathrin-mediated endocytosis, FDR = 0.043. Although none of the 257 target genes, including APOE, are included in these nine genome-wide significant pathways, 71 target genes are included in the nominally significant pathways, and 46 target genes are included in the non-APOE region-related nominally significant pathways (P < 0.05. Fig. 4 and Supplementary Table S6 marked in pink color).
Discussion
In this investigation, we have used the largest PiB-PET imaging data (n = ~1000), available from multiple collaborative centers, as an endophenotype to identify novel genetic loci for AD pathology using the GWAS meta-analysis approach, the first to our knowledge for PiB-PET.
The APOE region showed the most significant association where several SNPs surpassed the genome-wide significant threshold (P < 5E-08), with APOE*4 as the top hit that was associated with higher PiB retention in the brain (P-meta = 9.09E-30; β = 0.18). APOE*2, a protective genetic factor against AD, was associated with lower PiB retention, albeit, not genome-wide significant (P-meta = 6.57E-05; β = −0.09). This observation is consistent with earlier reports of the association of the APOE* 2/3/4 polymorphism with Aβ deposition in the brain as measured by PiB-PET [17–19] or florbetapir-PET [32]. Likewise, a GWAS of cerebrospinal fluid (CSF) Aβ has identified a genome-wide significant SNP that was a proxy for APOE*4 [33]. Numerous prior studies have investigated the role of the APOE* 2/3/4 polymorphism on Aβ production, aggregation, and clearance in the brain [34], but recent studies provide solid mechanistic clues into the role of APOE genetic variation in affecting APP transcription and Aβ production [35], and seeding of amyloid pathology [36]. In addition to the APOE*2/3/4 association, conditional analysis on APOE*4 identified 14 independent signals in the APOE region that also affect brain amyloidosis. Nine of 14 SNPs had essentially no LD with APOE*4 and APOE*2, and the remaining five showed moderate to weak LD with APOE*4. Thus, our meta-analysis indicates the presence of additional signals in the APOE region, beyond the APOE*4/rs429358 and APOE*2/rs7412 SNPs, that affect Aβ deposition in the brain.
Outside the APOE region, the meta-analysis revealed 15 suggestive non-APOE loci with P < 1E-05 on nine chromosomes. Although they do not meet the established genome-wide significance criteria, their consistent and directional associations in three independent datasets (Table 1) suggest that at least some of them are likely candidate loci for brain amyloidosis process and/or AD risk and variants in these loci may have achieved the genome-wide significance threshold in larger datasets. Credence to this idea was provided by our observation that most of these suggestive loci were also associated with AD risk when we examined the Aβ-associated SNPs in a published AD GWAS [29] (Supplementary Tables S4.1-S4.2). The most significant non-APOE SNP (rs13260032; P = 4.87E-07) on chromosomes 8 is intergenic, and this was an eQTL for a nearby ADCY8 gene in frontal cortex, which is one of the highest PiB uptake cortical regions [30]. ADCY8 is essential to long-term potentiation and synaptic plasticity and is implicated in memory and learning [37]. Genetic variation in or around ADCY8 has shown to be associated with dissociation symptoms in subjects with post-traumatic stress disorder [37], abdominal visceral [38] and alcohol-dependent depression [39]. The second top SNP (rs4680057; P = 9.69E-07) resides near C3orf79 and was an eQTL for a nearby long noncoding RNA (lncRNA) gene in anterior cingulate cortex and hippocampus in the brain and for ARHGEF26 in blood. lncRNAs play a critical role in gene regulatory networks and may affect diverse biological processes and diseases [40], including AD where several IncRNAs have been shown to regulate Aβ production/generation [41, 42]. A recent GWAS has identified ARHGEF26 as a new genetic factor for coronary artery disease risk that influences the transendothelial migration of leukocytes [43]. The third top SNP (rs12908891; P = 1.39E-06) is located DAPK2 on chromosome 15 that belongs to a family of related serine/threonine kinases shown to be involved in multiple functions, including apoptosis, autophagy, tumor suppression, and inflammation [44]. Although the role of DAPK2 in amyloidosis in unknown, another family member, DAPK1, promotes the phosphorylation and amyloidogenic processing of APP [45]. The DAPK2 region contains other candidate genes, such as GSNK1G1 and TRIP4. While TRIP4 is a known gene for AD [46], GSNK1G1 has been implicated in the formation of Aβ [47]. The top SNP was the most significant eQTL for HERC1 gene expression in anterior cingulate cortex (P_eQTL= 7.02E-05; P_SMR = 1.94E-03). HERC1 belongs to the ubiquitin–proteasome system that plays a key role in the protein degradation pathway essential for neuronal homeostasis, synaptic development and maintenance. Mutations in HERC1 have been associated with intellectual disability [48] and autism spectrum disorders [49].
To identify additional PiB-relevant candidate genes, we combined results from the brain expression, differential brain expression in AD, eQTL/SMR in the brain, and pathway analyses. Four genes meeting all these functional criteria were identified: RPS27L in the DAPK2 region, CYP4V2 and TLR3 in the CYP4V2 region, and IDH1 in the IDH1/C2orf80 region (Fig. 4, Supplementary Table S6). RPS27L is an evolutionarily conserved ribosomal protein and a physiological regulator of transcription factor p53 that is involved in genomic stability and tumor suppression [50]. p53 has also been implicated in AD progression, in part, due to its interaction with Aβ in AD progression [51]. p53 also interact with IDH1 in glioblastoma [52]. It seems that the involvement of RPS27L and IDH1 in the amyloidogenic process is through their effect on or interaction with p53. Although the role of CYP4V2 in amyloidosis is currently unclear, activated TLR3, along with some members of the toll-like receptors family, can induce Aβ uptake or inflammatory response during the AD progression [53]. Further functional characterization of these candidate genes may help to elucidate their roles in brain amyloidosis.
A recent GWAS using CSFAβ42 as an endophenotype has identified two novel loci in addition to the APOE locus [33]. One locus is near GLIS1 on chromosome 1 and the other in SERPINB1 on chromosome 6. The reported GLIS1/185031519 SNP was neither present in our genotyping array nor was it imputed. This SNP was also not in high LD with other SNPs. On the other hand, the reported SERPINB1/rs316341 SNP was present in our data, but it was not significant (P = 0.148). We also examined four additional reported SERPINB1 SNPs with P < 1E-05 (rs316339, rs316337, rs392120, rsrs2293772) [33] and found one of them to be nominally significant in our data (rs392120; P = 0.033).
We estimated the genetic variance of global PiB retention explained by the APOE and top 15 non-APOE SNPs with P < 1E-05 using a linear regression model. The non-APOE SNPs along with APOE*4 explained 25–35% of the amyloid variance; of which 14–17% was explained by APOE*4 alone. A previous study using a different amyloid tracer (florbetapir-PET) [32] found a similar contribution of APOE*4 (11%) to amyloid variance. However, a GWAS on CSF Aβ42 found a smaller contribution of APOE*4 (4%) to amyloid variance [33]. This may be due to the use of different methods to estimate the amyloid variance. While the CSF study used the Genome-wide Complex Trait Analysis (GCTA) that requires >3000 sample size [54], the two amyloid tracer studies with smaller sample sizes used linear regression. Our data, in conjunction with previous studies, highlight the presence of yet to be discovered variants that may be responsible for the unexplained genetic variance of amyloid deposition.
As with any genome-wide study, this study has limitations. Although the present study used the largest combined sample of PiB-PET imaging data reported to-date (from three different centers and ADNI), the sample size was relatively small to achieve genome-wide significance for loci with small effect sizes. We predict that at least some of our suggestive loci with P < 1E-05 might have achieved genome-wide significance with a larger sample size, as the direction of allelic effects for all suggestive loci were consistent in all datasets. Unlike some other phenotypes where data could be obtained readily on large numbers of subjects at a relatively low-cost, this is not the case with amyloid PET. Thus, the lack of a very large PiB-PET imaging database for a genome-wide study was a significant constraint. As more PiB-PET imaging data are obtained by different centers, future collaborative studies, as done here, on larger samples may allow the identification of additional genes for brain amyloidosis.
In conclusion, this is the first GWAS on PiB-PET that has confirmed the established association of the APOE locus with in vivo brain amyloidosis. In addition to the known association, we have identified novel variants in the APOE region that affect amyloidosis. A combination of genetic and functional approaches has also led to the identification of additional putative candidate genes that warrant follow-up genetic and functional studies to confirm their role in brain amyloidosis.
Electronic supplementary material
Acknowledgements
This study was supported by the US National Institutes of Health grants AG030653, AG041718, AG005133, AG025516, AG025204, AG044546, AG003991, AG053303, AG054936, LM012535, AG05681, G026276, and AG024904, and DOD ADNI (Department of Defense award W81XWH-12–2–0012). See full acknowledgments in Supplementary Material.
Compliance with ethical standards
Conflict of interest
GE Healthcare holds a license agreement with the University of Pittsburgh based on the PiB-PET technology described in this manuscript. Drs. Klunk and Mathis are co-inventors of PiB and, as such, have a financial interest in this license agreement. The remaining authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41380-018-0246-7) contains supplementary material, which is available to authorized users.
References
- 1.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:e213–200 e220. doi: 10.1016/j.neurobiolaging.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jack CR, Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR, Jr, Therneau TM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, et al. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol. 2016;15:56–64. doi: 10.1016/S1474-4422(15)00323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnham SC, Bourgeat P, Dore V, Savage G, Brown B, Laws S, et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol. 2016;15:1044–53. doi: 10.1016/S1474-4422(16)30125-9. [DOI] [PubMed] [Google Scholar]
- 8.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 9.Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–47. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 10.Jack CR, Jr, Barrio JR, Kepe V. Cerebral amyloid PET imaging in Alzheimer’s disease. Acta Neuropathol. 2013;126:643–57. doi: 10.1007/s00401-013-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–78. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 12.Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11:964–74. doi: 10.1016/j.jalz.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Curtis C, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72:287–94. doi: 10.1001/jamaneurol.2014.4144. [DOI] [PubMed] [Google Scholar]
- 14.Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36:297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Kamboh MI, Barmada MM, Demirci FY, Minster RL, Carrasquillo MM, Pankratz VS, et al. Genome-wide association analysis of age-at-onset in Alzheimer’s disease. Mol Psychiatry. 2012;17:1340–6. doi: 10.1038/mp.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park YS, et al. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol. 2014;71:1394–404. doi: 10.1001/jamaneurol.2014.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73:751–61. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nebes RD, Snitz BE, Cohen AD, Aizenstein HJ, Saxton JA, Halligan EM, et al. Cognitive aging in persons with minimal amyloid-beta and white matter hyperintensities. Neuropsychologia. 2013;51:2202–9. doi: 10.1016/j.neuropsychologia.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swaminathan S, Shen L, Risacher SL, Yoder KK, West JD, Kim S, et al. Amyloid pathway-based candidate gene analysis of [(11)C]PiB-PET in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. Brain Imaging Behav. 2012;6:1–15. doi: 10.1007/s11682-011-9136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimers Dement. 2015;11:792–814. doi: 10.1016/j.jalz.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, et al. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6:221–9. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deters KD, Risacher SL, Yoder KK, Oblak AL, Unverzagt FW, Murrell JR, et al. [(11)C]PiB PET in Gerstmann-Straussler-Scheinker disease. Am J Nucl Med Mol Imaging. 2016;6:84–93. [PMC free article] [PubMed] [Google Scholar]
- 24.Risacher SL, Kim S, Shen L, Nho K, Foroud T, Green RC, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI) Front Aging Neurosci. 2013;5:11. doi: 10.3389/fnagi.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 26.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamboh MI, Demirci FY, Wang X, Minster RL, Carrasquillo MM, Pankratz VS, et al. Genome-wide association study of Alzheimer’s disease. Transl Psychiatry. 2012;2:e117. doi: 10.1038/tp.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen AD, Mowrey W, Weissfeld LA, Aizenstein HJ, McDade E, Mountz JM, et al. Classification of amyloid-positivity in controls: comparison of visual read and quantitative approaches. Neuroimage. 2013;71:207–15. doi: 10.1016/j.neuroimage.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2014;19:351–7. doi: 10.1038/mp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–56. doi: 10.1007/s00401-017-1685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- 35.Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and abeta secretion. Cell. 2017;168:427–41 e421. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96:1024–32. doi: 10.1016/j.neuron.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf EJ, Rasmusson AM, Mitchell KS, Logue MW, Baldwin CT, Miller MW. A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depress Anxiety. 2014;31:352–60. doi: 10.1002/da.22260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sung YJ, Perusse L, Sarzynski MA, Fornage M, Sidney S, Sternfeld B, et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int J Obes (Lond) 2016;40:662–74. doi: 10.1038/ijo.2015.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Procopio DO, Saba LM, Walter H, Lesch O, Skala K, Schlaff G, et al. Genetic markers of comorbid depression and alcoholism in women. Alcohol Clin Exp Res. 2013;37:896–904. doi: 10.1111/acer.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo Q, Chen Y. Long noncoding RNAs and Alzheimer’s disease. Clin Interv Aging. 2016;11:867–72. doi: 10.2147/CIA.S107037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi C, Zhang L, Qin C. Long non-coding RNAs in brain development, synaptic biology, and Alzheimer’s disease. Brain Res Bull. 2017;132:160–9. doi: 10.1016/j.brainresbull.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–7. doi: 10.1038/ng.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geering B. Death-associated protein kinase 2: regulator of apoptosis, autophagy and inflammation. Int J Biochem Cell Biol. 2015;65:151–4. doi: 10.1016/j.biocel.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Kim BM, You MH, Chen CH, Suh J, Tanzi RE, Ho Lee T. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum Mol Genet. 2016;25:2498–513. doi: 10.1093/hmg/ddw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz A, Heilmann S, Becker T, Hernandez I, Wagner H, Thelen M, et al. Follow-up of loci from the International Genomics of Alzheimer’s Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry. 2014;4:e358. doi: 10.1038/tp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flajolet M, He G, Heiman M, Lin A, Nairn AC, Greengard P. Regulation of Alzheimer’s disease amyloid-beta formation by casein kinase I. Proc Natl Acad Sci USA. 2007;104:4159–64. doi: 10.1073/pnas.0611236104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Utine GE, Taskiran EZ, Kosukcu C, Karaosmanoglu B, Guleray N, Dogan OA, et al. HERC1 mutations in idiopathic intellectual disability. Eur J Med Genet. 2017;60:279–83. doi: 10.1016/j.ejmg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Hashimoto R, Nakazawa T, Tsurusaki Y, Yasuda Y, Nagayasu K, Matsumura K, et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J Hum Genet. 2016;61:199–206. doi: 10.1038/jhg.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong X, Zhao Y, Tang F, Wei D, Thomas D, Wang X, et al. Ribosomal protein S27-like is a physiological regulator of p53 that suppresses genomic instability and tumorigenesis. Elife. 2014;3:e02236. doi: 10.7554/eLife.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jazvinscak Jembrek M, Slade N, Hof PR, Simic G. The interactions of p53 with tau and Ass as potential therapeutic targets for Alzheimer’s disease. Prog Neurobiol. 2018;168:104–127. doi: 10.1016/j.pneurobio.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Chaurasia A, Park SH, Seo JW, Park CK. Immunohistochemical analysis of ATRX, IDH1 and p53 in glioblastoma and their correlations with patient survival. J Korean Med Sci. 2016;31:1208–14. doi: 10.3346/jkms.2016.31.8.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gambuzza ME, Sofo V, Salmeri FM, Soraci L, Marino S, Bramanti P. Toll-like receptors in Alzheimer’s disease: a therapeutic perspective. CNS Neurol Disord Drug Targets. 2014;13:1542–58. doi: 10.2174/1871527313666140806124850. [DOI] [PubMed] [Google Scholar]
- 54.Visscher PM, Hemani G, Vinkhuyzen AA, Chen GB, Lee SH, Wray NR, et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 2014;10:e1004269. doi: 10.1371/journal.pgen.1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.