

Abstract
Refinement of macromolecular X-ray crystal structures involves using complex software with hundreds of different settings. The complexity of underlying concepts and the sheer amount sof instructions may make it difficult for less experienced crystallographers to achieve optimal results in their refinements. This tutorial review offers guidelines for choosing the best settings for the reciprocal-space refinement of macromolecular models and provides practical tips for manual model correction. To help aspiring crystallographers navigate the process, some of the most practically important concepts of protein structure refinement are described. Among the topics covered are the use and purpose of R-free, geometrical restraints, restraints on atomic displacement parameters (ADPs), refinement weights, various parametrizations of ADPs (full anisotropic refinement and TLS), and omit maps. We also give practical tips for manual model correction in Coot, modelling of side-chains with poor or missing density, and ligand identification, fitting, and refinement.
Keywords: structural biology, X-ray crystallography, protein crystal structure, refinement, ligands, reproducibility
Introduction
X-ray crystal structure refinement, which is the process of achieving agreement between the structural model and the experimental data (structure factors and electron density maps), used to take months and, sometimes, years to complete. Today, an experienced crystallographer can complete the process in a matter of days or even hours (for a small to medium-size structure refined at an average resolution of ~2 Å). This impressive progress has been achieved mainly due to the availability and constant improvement of a) highly automated model building tools such as Buccaneer [1], ARP/wARP [2], and SOLVE/RESOLVE [3], b) reciprocal-space refinement programs such as REFMAC [4], phenix.refine [5], SHELX [6], BUSTER [7], and CNS [8], c) streamlined software suites such as CCP4 [9,10], PHENIX [11], and HKL-3000 [12,13], and d) the excellent molecular graphics system Coot [14] and less popular MAIN [15]. These programs have hundreds of different settings, which usually work well with the default parameters, but can (and sometimes should) be tuned for the most optimal refinement for each particular structure. Extensive manuals and FAQs can help in figuring out the best settings, but the sheer amount of instructions and the complexity of underlying concepts may make the process of refinement difficult for less experienced crystallographers.
The purpose of this tutorial is to give guidance for choosing the best settings for the reciprocal-space refinement and practical tips for manual model correction. Although the covered concepts are applicable to all major program suites, REFMAC and Coot are, for the main part, used as practical examples. The other software packages for structure refinement and visualization often have very similar underlying ideas, most of which are easily transferable.
R-factors: global measures of model quality
The crystallographic R-factor serves as a major measure of agreement between the amplitudes of the structure factors calculated from a crystallographic model (Fc) and those derived from the X-ray diffraction data (Fo) [16]:
However, the R-factor is not a completely reliable guide to accuracy: it is not a fully independent parameter because the optimization of the model is carried out to minimize the discrepancies between Fo and Fc and, in effect, is driven by the reduction of the R-factor. Moreover, the R-factor is based on unweighted statistics and thus can be easily manipulated by the use of too many refinement parameters (e.g., adding too many water molecules or using an incorrect atomic displacement parameters (ADP) model), which leads to overfitting [17]. In 1992, to solve these problems, Axel Brunger introduced R-free: the R-factor computed for a ‘free’ set of randomly selected reflections that are omitted from and hence independent of the refinement process [16]. Traditionally, 5% of reflections are selected for this purpose, which is the default in the CCP4 utility FreeRFlag. Usually, 1,000 reflections are sufficient to obtain a better than 1% precision for an overall R-free (see [18] p. 625). The fraction of reflections used for R-free calculations should be decreased for datasets with more than ~30,000 unique reflections because a higher number of free reflections does not confer a statistical advantage but rather diminishes the power of the minimization procedure [19,20]. The fraction, however, may need to be increased for low-resolution datasets to avoid having fewer than 500 reflections that are used to calculate R-free. The R-factor calculated for the ‘working’ set of reflections is usually called R-work.
After its introduction in 1992, R-free became a major parameter used to monitor the progress of the refinement. The nature of crystallographic diffraction data is such that every reflection contains information about the entire structure. Therefore, changes to a model that do not improve the model’s ability to describe the diffraction data will not improve the fit of the model to the test set. In such instances, the R-free value will remain constant or increase regardless of whether the R-work decreases [21]. Moreover, R-free is highly correlated with the phase accuracy of the atomic model [22]. Too large of a difference between R-work and R-free is an indication of overfitting. A common rule of thumb states that the difference should not exceed 5%; however, we do not recommend rigidly adhering to this rule, as statistically reasonable differences can be much larger for low-resolution datasets but lower for those of atomic resolution. Instead, the ratio of the R-free and the R-work can be checked against theoretically expected values presented by Tickle et al. as a function of the ratio between the number of atoms used in refinement and the number of reflections for unrestrained and different types of restrained refinement (e.g., isotropic or anisotropic refinement of ADPs) [17]. Notably, wwPDB [23,24] uses R-free as one of the five measures of overall protein structure quality in the validation report [25,26]. Because R-free is used for cross-validation, it is important to keep the same R-free set throughout the entire structure determination and refinement process.
Despite their importance, the values of R-work and R-free should never be treated as the sole justification of the correctness of the model and/or completeness of the refinement. First, when non-crystallographic symmetry (NCS) is present, the free reflections are not fully independent of the reflections in the working set (unless selected in thin resolution shells) [27]. For further information on complications with translational NCS and pseudosymmetry and the importance of having the correct choice of space group, the reader is referred to specialized articles [28,29]. Second, R-factors for twinned and non-twinned crystals behave very differently and should not be compared; R-values for cases of hemihedral twinning are systematically lower than those for single crystals [30,31]. Third, if the data are noisy, R-factors may be objectively high even if the structure is very well-refined; this fact serves as a reminder that the process of achieving low R-factors starts with optimal data collection and diligent integration and scaling of the diffraction images in a correct space group [30,32–34]. Finally, one should always remember that the ultimate judgment for the correctness of the model should be its agreement with electron density maps, not the numerical values of global measures such as R-work and R-free alone.
Electron density maps
Various versions of electron density maps are commonly calculated and used during structure refinement. The most informative and least biased model-phased maps currently used are the maximum-likelihood σA [35] weighted maps: a map with model bias correction (2mFo-DFc) and a difference map to show errors in model (mFo-DFc), where m is the figure of merit and D is the Luzzati coefficient (see [18] p. 619 for review). These maps are routinely used during manual model building and presentation of the evidence for structural details in a model (e.g., ligands). The maps are displayed as isosurfaces contoured at specified electron density levels usually expressed in units of rmsds from zero. The symbol of standard deviation σ is commonly used instead of rmsd to denote the same quantity, but we will use rmsd for consistency with modern versions of Coot and to avoid assumptions about the normal distribution of the maps. Customarily, values above +1.0 rmsd for 2mFo-DFc are considered to be good indicators of a structural detail’s presence, and values above +3.0 rmsd and below −3 rmsd for mFo-DFc strongly indicate map fragments that are not explained by the model or do not support a structural detail, respectively. These rmsd levels are regularly used by default when inspecting models in Coot. However, the map levels are not always directly comparable between datasets and even different regions [36], and maps below suggested levels may also contain useful information. When inspecting a questionable region, both maps should be scrolled to levels as low as those when noise becomes nearly prevalent. In addition, map levels used for interpretation depend on the significance of the given fragment; for example, more stringent values should be used for validating a ligand than for modelling disordered side-chains.
Omit maps are special types of maps used to present the significance of the evidence supporting a structural detail (especially a ligand) during model building and in publications. This term covers a broad spectrum of maps that a) demonstrate the presence of a structural detail without bias introduced by using that particular detail for calculation of phases and/or b) have the minimal bias of the whole model. The simplest types of omit maps are 2mFo-DFc and mFo-DFc maps calculated from a model refined for several cycles without a side-chain(s) or a ligand. Because parameters (positions, ADPs) of every atom are optimized simultaneously during refinement in the reciprocal space, simple removal of the analysed structural detail and several cycles of refinement only partially remove bias from the maps [37]. Although these types of omit maps are not ideal for the purpose of analysing and presenting the strength of evidence for a particular structural detail, they provide an adequate and practical way for making an informed decision during model building and refinement as outlined in subsequent sections. Unless otherwise noted, we will use the term ‘omit maps’ to refer to mFo-DFc and 2mFo-DFc maps calculated in that way.
To generate omit maps that are relatively unbiased and suitable for demonstrating the evidence that the omitted region of the model is indeed present in the electron density map, it is necessary to fully remove the influence of the removed fragment. If the fragment is relatively small, such as a ligand bound to a protein, it is usually sufficient to reset all ADPs of the model to some constant value and slightly randomize all atoms’ positions before running several cycles of the refinement (e.g., by using keyword noise 0.1 in CCP4 utility PDBSET). Simulated annealing (SA) refinement can be used to generate SA-omit maps, but SA tends to degrade the quality of the final model and is recommended only during the initial stages of refinement [38].
Alternatively, one can calculate the composite omit maps (implemented in both PHENIX and CCP4). These maps are generated by removing random fragments of the model and then combining the resulting omit maps into one map, which is relatively bias-free from the model. This method is the only suitable method for generating omit maps when large regions need to be omitted, as the removal of large portion of the model would significantly degrade the quality of the phases.
It has to be noted that refinement programs use bulk solvent modelling based on a mask of the macromolecule to account for the disordered solvent; every point that is considered to be disordered solvent is modelled as constant density. When omit maps are generated, the removed fragment would not contribute to the protein mask, and consequently, the corresponding density would be modelled as disordered solvent. This, in turn, will decrease the local value of the 2mFo-DFc and mFo-DFc maps, which has to be taken into consideration. The alternative is to use the part in question for the mask calculation but exclude it from the refinement (either by setting the occupancy to zero or by specifying proper keywords, e.g., refinement exclude all from [residue] [chain] to [residue] [chain] for REFMAC). The problem with this alternative approach is that if the density for the modelled part is lacking and indeed represents disordered solvent, then the resulting mFo-DFc density will be relatively strong and adopt the exact shape of the fragment in question even if it is not present. To overcome this problem, a more sophisticated approach can be employed, such as the use of composite omit maps that compensate for the bulk solvent modelling, e.g., POLDER omit maps implemented in PHENIX [39]. In addition to removing the omitted region from the map, POLDER omit maps are calculated by excluding large volumes around the omitted region (5 Å around each atom) from the solvent mask calculation. This approach is preferable for regions with weak density because it will not mask weak density by modelling the bulk solvent and will not bias the difference density map by excluding the exact shape of the region from the solvent mask calculation [39]. In addition to removing the omitted region from the map, POLDER omit maps are calculated by excluding large volumes around the omitted region (5 Å around each atom) from the solvent mask calculation. This approach is preferable for regions with weak density because it will not mask weak density by modelling the bulk solvent and will not bias the difference density map by excluding the exact shape of the region from the solvent mask calculation.
Atomic displacement parameters (ADPs)
ADPs, traditionally referred to as B-factors, temperature factors, or thermal parameters, define the thermal motions and static displacements of atoms in the crystallographic model in the form of a displacement sphere (in the isotropic model) or ellipsoid (in the anisotropic model) [20]. It is crucial to understand that the typical X-ray experiment measures time- and space-averaged structures of billions or even trillions of macromolecules within the crystal; since we do not work with perfectly ordered crystals, this arrangement is imperfect. Thus, the total atomic displacement ellipsoid is the sum of an individual atom’s thermal motions and its thermal and space-averaged movements as a part of an amino acid residue, a secondary structure element, a domain, macromolecule as a whole, and the mosaic blocks that comprise the crystal [20]. It is important to realize that for crystal structures of small molecules, ADPs are dominated by thermal motion, but in the case of macromolecular structures, ADPs are dominated by the imperfections of crystal specimens.
In the full anisotropic model, which is used only at atomic resolution, the ADP ellipsoid is defined by six parameters that form a 3×3 matrix. Thus, combined with the three position coordinates (x, y, z) for every non-hydrogen atom, the full description of each atom requires 9 parameters [20,40]. At a resolution lower than atomic, the ratio of data terms to refinable atomic parameters (data-to-parameters ratio) may be insufficient to determine refinement of the macromolecule in question. Therefore, isotropic ADP refinement, often supplemented with TLS (Translation, Libration, Screw) parametrization, is commonly used in macromolecular refinement, reducing the number of parameters from nine to approximately four per atom [41]. The levels of detail used to describe the ADPs depending on the data-to-parameter ratio can be roughly inferred from the resolution of the dataset (Table 1).
Table 1.
Data resolution categories and suggested ADP parametrization. The resolution ranges are approximate. In borderline cases, the selection of ADP parametrization also depends on the data quality and properties (e.g., solvent content, anisotropy, presence of NCS, space group, etc.) and is best assessed by the Hamilton test.
| Category | Data resolution (dmin), Å | Reflections per non-hydrogen atom | ADP parametrization |
|---|---|---|---|
| Atomic | dmin < 1.2 Å (1.2–1.5 Å if the reflections per atom condition is fulfilled) |
> 18 (> 25–30 if the dataset has many weak reflections) |
Full anisotropic |
| High | 1.2 Å ≤ dmin < 1.7 Å | Does not satisfy the condition for atomic resolution | Isotropic BRESIDUAL with TLS; number of TLS groups per protein chain depends on chain length, presence of separate domains, data resolution, and stability of the refinement |
| Medium | 1.7 Å ≤ dmin < 2.7 Å | – | |
| Low | 2.7 Å ≤ dmin < 3.5 Å | – | |
| Extra-low | 3.5 Å ≤ dmin | < 1 | BOVERALL / BOVERALL with TLS |
Refinement at atomic resolution
Atomic resolution is often defined as a resolution at which there are sufficient accurately measured observables (reflections) to justify the refinement of the ordered part of the structure with full anisotropic ADPs [20]. Because R-free is not a weighted statistic, a drop in R-free is a necessary but not always sufficient condition to justify anisotropic ADP refinement. The Hamilton R-factor ratio test is currently the best tool that can be used to find whether the use of the more complex model is justified [41]. Simply put, the Hamilton test checks whether the drop of the R-free is statistically significant in relation to the increase in the number of parameters. In practice, the Hamilton test for full anisotropic ADP refinement is certainly satisfied at a resolution higher than 1.2 Å (Table 1). Structures ranging in resolution from 1.2–1.5 Å may also fall into this category depending on the solvent content, data completeness, and space group [41]. The Hamilton test is currently implemented in HKL-3000 [12] and PDB_REDO [42]. We highly recommend introducing and testing full anisotropic ADP refinement only at the latest stages of refinement when most of the protein and solvent atoms have been built. The Hamilton test depends on the number of atoms in the model (which defines the number of refined parameters and used restraints); therefore, the conclusion may be different if the model is not complete. The introduction of the anisotropic refinement at the latest stages of refinement also allows avoiding potential problems with extreme ADP anisotropy (see below).
If using the Hamilton test is not feasible, we recommend using two independent criteria based on our experience. First, the ratio of unique reflections to non-hydrogen atoms should be higher than 18; this ratio stands for a ‘data-to-parameters ratio’ higher than two. If the dataset has many weak reflections, the ratio may need to be higher than 25 to provide enough data points for justified and stable refinement. A ratio above 30 will most certainly guarantee justification for full anisotropic refinement. Second, the change in the R-factors that results from introducing the anisotropic refinement has to be meaningful. While a drop in the R-free of at least 1% can be considered significant in most cases, the anisotropic refinement can easily lead to overfitting, in which case such a drop in R-free would not justify the introduction of anisotropic ADPs. The warning signs for overfitting are a much higher drop in R-work as compared to R-free and, consequently, too large of a difference between R-work and R-free (see section ‘R-factors: global measures of model quality’).
Refinement of anisotropic ADPs of all atoms can be unstable, especially if the data-to-parameters ratio is relatively low. Often, unstable refinement manifests as the presence of a large number of atoms with an extreme ADP anisotropy – virtually flat or elongated (cigar-like) thermal ellipsoids (in Coot, use Draw>Anisotropic Atoms to display the ellipsoids). To monitor the distribution of ADP anisotropy, we recommend using the PARVATI server (http://skuld.bmsc.washington.edu/parvati/) [43]. The server presents distributions of ADP anisotropy for the protein, ligand(s), and water molecules in the submitted model, as well as a list of atoms with extreme ADP anisotropy. A comparison with the typical ADP distribution for protein structures, which is also presented on the PARVATI server, helps to assess the adequacy of the model. If the distribution is skewed towards extreme anisotropy or if too many atoms have extreme anisotropy, the anisotropy should be further restrained during the refinement. REFMAC has the keyword sphericity with the default value of 5, which may be too lax for structures refined against data at resolution lower than 1.2 Å; a lower value for this parameter results in more spherical atoms.
If many water molecules have extreme anisotropy in spite of adjusting anisotropy restraints, it may be a good idea to exclude all water molecules from the anisotropic ADP parametrization. Ligands can be excluded as well. Sometimes, the anisotropy becomes extreme only after many cycles of REFMAC refinement. In this case, we recommend resetting the anisotropy by running 5–10 cycles of isotropic ADP refinement and then turning the anisotropic mode back on. After setting optimal anisotropy restraints, we recommend visual inspection of all atoms with extreme anisotropy detected by the PARVATI server, as these outliers may be caused by factors other than lax restraints, e.g., incorrect placement or assignment of atoms. Note that persistent problems with extreme anisotropy for many atoms may also mean that full anisotropic refinement is not justified. For more detailed descriptions of refinement at atomic resolution, the reader is referred to an excellent article by Jaskolski [44] (the referred article uses a different and stricter definition of atomic resolution) and other sources [20,40,41].
Refinement with TLS
When working with datasets at lower than atomic resolution, TLS parametrization is often used to account for the anisotropy of ADPs, which results in a significant decrease in R-free [44,45]. The TLS tensor represents the collective displacement of a rigid group of atoms, usually a whole protein chain, domain, or a secondary structure element. Because one TLS tensor contains just 20 refinable parameters, adding several TLS groups to a model does not significantly increase the number of parameters used for the refinement of the protein. When many TLS groups are used, we suggest using the Hamilton test to check if the addition of the extra parameters results in a sufficient decrease in R-free [41].
TLS parametrization splits the ADPs of each atom into two fractions: ADPTLS represents the values derived from the application of the TLS tensor, and ADPRESIDUAL represents the so-called ‘residual’ values that are refined after TLS contribution is established (ADPTOTAL = ADPTLS + ADPRESIDUAL). It is important to remember that REFMAC produces the .pdb file with only residual isotropic B-factors; this file should be used for further refinement. For the structural analysis and structure deposition, a special keyword has to be added (tlsout addu), which results in a .pdb file with anisotropic ADPTOTAL values.
High- and medium-resolution (1.2–2.7 Å) data usually allow for the refinement of individual isotropic ADPs supplemented with multiple TLS groups per protein chain (Table 1). With low- and extra low-resolution (> 2.7 Å) data, the whole content of the asymmetric unit can be given a single residual ADP value (BOVERALL) supplemented with TLS (Table 1). Importantly, TLS should never be combined with full anisotropic ADPs for the same atoms, as this results in over-parameterization [44].
TLS groups can be specified manually or by an automated tool such as the TLSMD server (http://skuld.bmsc.washington.edu/parvati/) [46]. The server will perform an analysis of the effect of introducing different numbers of groups that can be used as a guidance in choosing the best set of TLS groups. In addition, it will produce a starting TLS file for a chosen set of TLS groups and a corresponding .pdb file with initial ADPRESIDUAL values set to a chosen value [46]. The structure submitted for analysis has to contain well-refined isotropic ADPs because the analysis is based on these values. After the initial tensors are set, the refinement is continued with consecutive separate refinements of TLS and the atomic model. In each round, several cycles of TLS tensors refinement (with atoms and ADPRESIDUAL fixed) are followed by several cycles of model refinement (with TLS tensors fixed). In order to reach convergence, two or three rounds of refinement are recommended before working on the model. After further refinement reaches relative convergence (no significant improvement of R-free), we recommend fixing the TLS (0 TLS refinement cycles).
TLS ADP refinement can be very useful in intermediate stages of refinement, when parts of the model have not yet been built. The resultant improvement of the map in regions of weak density often allows multiple residues to be added to the model manually. As the isotropic ADP values for these residues have not been refined, they should not be included into the TLS groups for subsequent rounds of refinement (however, REFMAC will include these residues if they are within a residue range for a previously defined TLS group). Therefore, after several new residues have been added, we recommend re-setting the TLS by refining the model without the TLS (isotropic ADP model) for 20–30 REFMAC cycles and setting new TLS groups as described above.
Sometimes, TLS refinement can be unstable: R-free can increase without an apparent reason, and significant positive peaks of the difference map can appear around main chain atoms. Resetting TLS (as described above) can help in alleviating the problem. In order to avoid recurrence of these problems, consider using a different number of TLS groups. In our experience, decreasing the number of TLS groups to one per chain (or per large domain) for structures with unstable TLS can result in stable refinement and still provide a sufficient improvement in R-free. As with refinement of full anisotropic ADPs, we recommend using the PARVATI server [43] to monitor the distribution of ADP anisotropy when TLS parametrization is used. In the final cycles of refinement, it should be standard practice to test the number and nature of TLS groups to find the optimum ADP model.
TLS parametrization can be extended to water molecules and ligands. By default, REFMAC adds water molecules in the first hydration shell to the nearest TLS groups; this behaviour can be changed by keyword tlsd waters add/exclude. Conversely, HKL-3000 explicitly removes water molecules from the TLS (unless otherwise specified) because the TLS refinement with less ordered water molecules is often less stable and results in extreme ADP anisotropy.
If heavy atoms that are not part of the protein are present (e.g., transition metals such as Zn, Ni, Co, or Pt and even lighter elements P, S, Cl, K, and Ca), it is usually better to refine these heavy scatterers with individual anisotropic ADPs [40] than to include them in TLS groups or refine isotropically. For this, use the REFMAC keyword brefine mixed anisou atoms PT CO (PT and CO indicate atom types to be refined with anisotropic ADPs). The significant anisotropy of the heavy atoms may still be observed even at medium and low resolution (up to ~2.7 Å). In such cases, if the ADPs are refined as isotropic, the difference between the observed ellipsoidal and modelled spherical density would be noticeable as a characteristic ripple-like pattern of the difference density (Figure 1).
Figure 1.
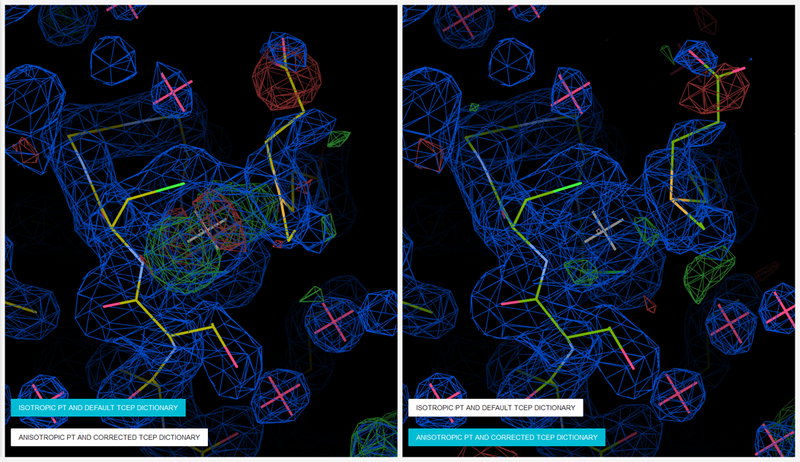
Characteristic ripples of difference density map (positive and negative peaks) are observed around the platinum atom with significant anisotropy (left, 3iwl). Refinement with anisotropic ADPs for this atom results in much cleaner density (right, 4ydx). 2mFo – DFc maps are displayed in blue contoured at a level of 1.0 rmsd. mFo - DFc difference maps are contoured at the 3.0 rmsd level in green (positive) and red (negative). The TCEP molecule in the original model was refined with a default dictionary that incorrectly defined the phosphorus atom (left, 3iwl). After a corrected dictionary was used, the density fit of TCEP significantly improved (right, 4ydx). Figure generated by Molstack from project http://molstack.bioreproducibility.org/project/view/9XPRJFZR9S9vBTNNHBpY/.
Geometrical restraints in macromolecular refinement
The resolution (which is related to the information content) of diffraction data does not usually allow for the refinement of a chemically correct model of a macromolecule without prior knowledge about the geometry of building blocks (amino acid residues and nucleotides), ligands, and non-covalent interatomic distances (van der Waals interactions and hydrogen bonds). Thus, the majority of macromolecular structures are modelled and refined using ‘restrained refinement’ [47,48]. The positions of the atoms during restrained refinement are optimized using not only the experimental data but also the allowed geometry (derived from small molecules, macromolecules determined at atomic resolution, and theoretical calculations [49]) as well as non-bonding interactions.
Several classes of geometrical restraints are commonly used; their particular uses depend mostly on the resolution of the experimental data. The principal restraints that are used in refinement of macromolecular structures are stereochemical restraints that define the monomer residues and ligands. These restraints are primarily derived from databases of small molecules determined at very high resolution: the Cambridge Structural Database (CSD) [51] and Crystallography Open Database (COD) [52]. A typical dictionary of restraints consists of expected bond lengths, bond angles, and torsion angles with corresponding standard deviations, as well as chiral centres and coplanar groups of atoms, if applicable. Hydrogen atoms are usually present in these dictionaries; however, the refinement programs do not normally refine their positions. Instead, they use the ‘riding-hydrogen’ description: positions and ADPs of hydrogen atoms are derived from the atoms they are bonded to (if possible) and used to calculate the structure factors. These dictionaries are a primary source of correct geometries for the refined residues and ligands, and as a result, errors in the dictionaries propagate to the refined structure [4].
The majority of compounds that have been deposited in the PDB [23] have restraints already automatically generated and distributed with crystallographic suites. While the majority of protein amino acid residues and nucleotides are well-validated, it is prudent to check the correctness of dictionaries for other compounds because these restraints may sometimes have errors (Figure 1), as not all of them are fully validated [53,54]. The most common problems arise with the chirality and planarity of chemical groups (see section ‘Ligand in protein crystal structures’ for tips about how to easily check the restraints). In case of errors or a lack of pre-generated restraints (e.g., a completely new compound), either the new set of restraints should be generated or existing ones should be modified.
Significant effort was dedicated toward the development of several programs and web servers that automatically generate new sets of restraints. The CCP4 suite offers LIBCHECK [49], PRODRG [55], and AceDRG [56], PHENIX uses eLBOW [57], and Coot can use either LIBCHECK, AceDRG, PRODRG, or an internal tool, Pyrogen, depending on the setup. Web servers include GRADE [58], PURY [59], and PRODRG [55]. Similarly, several editors for defining new molecules and editing restraints manually were developed as components of different software suites: Sketcher [10] and JLigand [60] (CCP4), Lidia [61] (Coot), and REEL [62] (PHENIX). While these tools enable easy and automatic generation of restraints, it must be stressed that any automatically generated ligand restraints should be checked manually for (stereo)chemical correctness. If a new restraints dictionary is generated, the location of the file should then be specified in both REFMAC and Coot. See section ‘Ligands in protein crystal structures’ as well as an excellent recent review [63] for further details regarding restraints for ligands.
The relative weights between the experimental data and geometrical restraints used usually need to be adjusted in every structure to achieve an optimal balance between molecular geometry and R-free [50]. Today, the weight adjustment is mostly done automatically but may require fine-tuning in the final stages of refinement. The weight optimization of the geometric restraints should be coupled with the optimization of the restraints for ADPs [50]. These weights can be simultaneously optimized by PHENIX; in REFMAC, we have been adjusting the weights of restraints for ADPs manually based on practical considerations (see section ‘Modelling of side-chains’) and then adjusting the weight for geometric restraints [50]. The optimal geometry of the residues can be measured both locally (per residue) and globally (per structure) as a deviation from the expected values; for a global geometry, it is usually expressed as the rmsd of the bond lengths and bond angles. We recommend manually adjusting geometry weights to meet target global bond length rmsd between 0.010–0.015 Å (in REFMAC, use keyword weight matrix value; a lower value results in stricter geometry).
Stereochemical and non-bonding restraints (the latter are usually set by the refinement programs in correlation with stereochemical restraints and do not need special adjustments) are usually sufficient to restrain geometry for the refinement of structures at high resolution. However, it is often beneficial to include additional sources of information, especially as the information content in the experimental data decreases with its resolution. One of the possible sources is the macromolecule itself: if NCS is present, it is possible to restrain the local geometry of the macromolecule to the macromolecule’s other copies in the asymmetric unit, thus reducing the number of independent parameters to be refined, stabilising the refinement, and improving the signal-to-noise ratio of electron density maps [64].
While the groups and weights for the NCS used to be adjusted manually in REFMAC (e.g., by specifying tight, medium, or loose restraints), they are now quite well adjusted automatically by using so-called local NCS restraints; in our practice, we rarely perform manual adjustments of NCS weights. In REFMAC, due to the way in which the local NCS weight is calculated, it is possible (and recommended) to use the local NCS restraints across all resolution ranges (keyword: nscr local). We have observed, though, that after extensive reciprocal refinement against data at high resolution, it may be necessary to inspect the model and perform real-space refinement for problematic residues (those with poor density fit or corrupt geometry) in each chain to reintroduce the differences between the chains. For data resolution higher than medium, we recommend testing removal/relaxation of NCS restraints by the Hamilton test in the final cycles of refinement to see if the restraints are still warranted.
It is also possible to restrain the refinement to the initial structure or external reference structure by using methods such as DEN [65] (in CNS and PHENIX), ‘jelly body’ refinement [4] (in REFMAC), refinement with reference structure (in PHENIX [48] and BUSTER [7]), or external restraints in REFMAC generated by ProSMART [66]. At resolutions lower than 3.0 Å or after initial model placement after molecular replacement, these methods can greatly improve the convergence of the refinement, as they will keep the original (or derived from a reference structure) local relationship between the atoms largely intact. Similarly, during the final cycles of refinement, these external restraints should be relaxed and/or removed to test if they are still valid to use.
Oligomeric assemblies and standardized placement within the unit cell
Often, the asymmetric unit of a crystal structure contains more than one monomer of a macromolecule. As mentioned above, the use of local NCS restraints is highly recommended in these cases. For the purpose of making the structure analysis more convenient, it is recommended to arrange the content of the asymmetric unit according to either a known or predicted oligomeric state of the protein. For example, if the protein is a dimer and the asymmetric unit contains two monomers, they should be placed as a dimer but not as two separated monomers from different dimers. The PISA server (http://www.ebi.ac.uk/pdbe/pisa/) is an excellent tool for determining the oligomeric assemblies based on contacts between monomers [67,68]. The interface to PISA in Coot enables automatic placement of the monomers according to predicted assemblies. In addition, we recommend using the ACHESYM server (http://achesym.ibch.poznan.pl/) for selecting a standardized location of a unique molecule in the unit cell [69].
Manual model correction
When starting work on a large model, the amount of corrections to be done can appear daunting. Fortunately, major improvements in crystallographic software within the last decade have enabled automatic/semi-automatic fixing of many model issues. Automated model building software tools, such as Buccaneer [1], ARP/wARP [2], and SOLVE/RESOLVE [3], can produce almost complete models. After the model building or molecular replacement procedure, a better side-chain placement can be automatically achieved with Fitmunk [70], as incorporated into HKL-3000 [12] or as an online service (http://fitmunk.bioreproducibility.org/fitmunk). However, models built by software tools may or may not be correct, especially in regions of poor density. In any case, the crystallographer needs to manually inspect the electron density maps to verify the sequence assignment and side-chain placement.
Traditionally, it was common to start working on a structure by manually ‘walking’ through the protein chains and inspecting the model and the maps. We recommend leaving this step for later and starting the manual corrections by addressing the most significant issues first. Coot offers several excellent and highly efficient tools under the Validate menu that enable fast detection of the largest inconsistencies between the model and the data. We suggest the following protocol for addressing the most significant issues:
-
(1)
Review the unmodelled electron density blobs, which can represent ligands and/or residues missing from the model (see section ‘Ligands in protein crystal structures’).
-
(2)
Inspect the difference map peaks above 5.0 or even 4.0 rmsd, depending on how many peaks are discovered.
-
(3)
Inspect rotamer outliers, which may indicate incorrect placement of side-chains, and residues with missing atoms to see if some of them can be easily rebuilt, e.g. using the ‘k’ key (Table 2).
-
(4)
Review density fit graphs and inspect poorly fitting residues; on the same graphs, navigate to terminal residues and verify their correct placement; inspect any gaps in the sequence.
-
(5)
Inspect Ramachandran outliers; keep in mind that some outliers can be justifiable if they are placed well in the electron density.
-
(6)
Inspect cis-peptides using Extensions > Modelling > Residues with CIS Peptide Bonds.
-
(7)
Once major issues with the protein backbone and the sidechains are addressed, add water molecules using Calculate > Other Modelling Tools > Find Waters. We recommend choosing the 2mFo-DFc map and search for peaks above 1.1 rmsd, with distance to protein atoms ranging from 2.4–4.0 Å.
Table 2.
Most useful key bindings in Coot. If not present by default, files with various key bindings can be downloaded from https://strucbio.biologie.uni-konstanz.de/ccp4wiki/index.php/Coot and added as an editable file in .coot-preferences directory in the user main directory. Alternatively, using Extensions > Settings > Install Template keybindings from Coot will result in similar file to be used. The key bindings available in different Coot versions, websites and this table may differ; the exact settings can be verified by clicking Extensions > Settings > Key Bindings.
| Key | Function | Comment |
|---|---|---|
| w | add a water molecule | By default, water molecules are added as a new molecule, which is saved as a separate file. Add a first water molecule in each Coot session with a ‘place atom at pointer’ function and choose to affix it to the .pdb file under refinement. Then, all subsequent water molecules will be added to the same file. |
| x | refine and accept | Refines a currently selected monomer: a residue, a water molecule, or a ligand. |
| t | triple refine | Refines three residues at once: currently selected residue, upstream, and downstream. |
| h | triple refine and accept | Same as ‘t’ but with automatic acceptance of the results. |
| k | rebuild side-chain | Truncates the side-chain of a selected residue and then rebuilds a best conformer that fits the electron density map |
| K | remove side-chain | Truncates the side-chain of a selected residue |
| N | copy current residue in all the NCS related chains | Copies the residue in exactly same conformation to all chains. Best used when NCS ghosts are displayed |
| o | toggle through NCS ghosts | Best used when NCS ghosts are displayed |
| p | park on a residue | Use when toggling through NCS ghosts does not behave well |
| V | park on a symmetrical residue | Use when inspecting residues at an interface with a symmetry-related protein chain |
To ease the manual work, make use of the multiple key bindings when correcting the model (Table 2). If NCS is present, display NCS ghosts (Draw>NCS Ghost Control) on the chain under review and inspect any apparent differences in other chains; to toggle between chains, use the ‘o’ key. In order to take advantage of the improvement of phases, we suggest saving the model and running 4–10 cycles of REFMAC refinement after significant changes are applied (e.g., 10–30 corrected residues). It is a good idea to write down the residues to which the changes were applied and inspect them in Coot once again after REFMAC refinement; sometimes, they may require further adjustments. Because the phases improve with the model, all the steps described in this protocol can be repeated a few times until no significant issues are detected. Only then do we recommend starting to ‘walk’ through the protein chains.
Modelling of poorly-resolved regions
The crystallographic data represents an average state of billions or even trillions of macromolecules. Due to their dynamic nature, some regions of these macromolecules (e.g., flexible loops or termini) may have multiple conformations, resulting in less defined electron density than the rigid “core” of the macromolecule. These regions are particularly challenging to model. The disordered regions can be roughly divided into three groups of decreasing interpretability: (1) regions with two or three distinct alternative conformations that can be clearly traced, (2) regions that are moderately mobile and adopt multiple conformations that result in an average density that is above the noise level but still allow the average or major conformation to be traced, and (3) highly mobile regions that result in density indistinguishable from the disordered solvent. In the first and second cases, it may not be possible to unambiguously place the conformations of the side-chains. These regions may refine with high ADPs that indicate the high mobility and uncertainty of their positions. In the third case, it is impossible to trace the main-chain, unless the region is very short. Although it may be possible to model an ensemble of models that would show possible conformations, the usual practice is to skip speculation and not model these regions at all. Most common examples of the third case are long disordered loops and N- and C-termini, including small purification tags such as His-tags [71].
Modelling of side-chains
Side-chain placement can be challenging when the electron density is ambiguous or there is no visible density for some atoms of the side-chain. The density can be partially (or even completely) missing due to radiation damage or intrinsic disorder, but it is usually not possible to distinguish between these two. The most radiation damage-prone residues are Asp, Glu, Cys, Met, and SeMet. Intrinsic disorder is the most typical reason for the absence of electron density for long side-chains (Lys, Arg, Glu, Gln) located on the surface of the protein and not involved in intermolecular or crystal contacts.
There is no single set of rules for modelling residues with poor or missing density; different research groups use different guidelines. Two approaches are common: (1) keeping the disordered side-chain in the most likely conformation and allowing the ADPs to be refined to high values that will reflect the disorder and (2) removing atoms without density from the side-chain and allowing the residual density to be modelled as the disordered solvent (see section ‘Electron density maps’). The first approach has the advantage of being more chemically true to the nature of the protein that was crystallized (e.g., a charge surface would be calculated more accurately because all residues are included in the model without additional processing from the user side), but some users of crystal structures may not realize that some side-chains are not defined for certain or may have higher position uncertainty than what is modelled by ADPs. The second approach has the advantage of avoiding “guessing” side-chain conformations but results in an incomplete model.
Regardless of the approach, omit maps should be used to decide on the best-fitting conformation, the presence of an alternative conformation, and, if the second approach is used, whether to truncate the side-chain. When using REFMAC, delete the questionable side-chains, run the reciprocal-space refinement, and review the resulting maps (see section ‘Electron density maps’ for limitations and other options). We suggest that if 2mFo-DFc and/or mFo-DFc omit maps show some density with a shape similar to a possible conformer of the residue at any rmsd level that is not dominated by noise, then this conformer should be modelled. If additional conformers are clearly seen on the omit maps, these conformers should be modelled too; however, there is usually no need to model an alternative conformation if the density is ambiguous and the peaks are low.
If the first approach is used, the best conformer should be modelled based on any clues from the omit maps and the environment (e.g., a Lys residue can form a salt bridge with an Asp or Glu). Often, placement of side-chains with poor or missing density results in negative peaks of the difference map and/or in a large number of RSRZ outliers in the PDB validation report, which might discourage some crystallographers from modelling side-chains with weak density. The most common reason for these issues is over-restrained ADP values, which are normally restrained to resemble those of nearby atoms. In this case, loosening ADP restraints may help; in REFMAC, this is achieved by using the keyword bfactor 0.3 or lower (default is 1). Apart from eliminating negative peaks on correctly placed side-chains and decreasing the number of RSRZ outliers, this tweak often results in a lower R-free value. Importantly, negative peaks may also indicate the presence of alternative conformations, too low of a limit set on the highest possible ADP value (default value is 200 Å in REFMAC), or significant radiation damage. Thus, reasonable relaxation of ADPs will not necessarily eliminate all such positive peaks, and lowering the restraint too much to eliminate these peaks should be discouraged.
If the second approach (removing atoms without density) is used, we suggest using the following two rules for truncating side-chains that do not have conclusive density on the omit maps (Figure 2). First, do not delete atoms if their positions can be deducted from the positions of other atoms based on the stereochemistry of the residue. For example, do not remove only the OD1 atom of an aspartate residue if the OD2 atom can be located: the location of OD1 is defined by the OD2 and CG atoms because the carboxyl group is planar. The choice to be made should be between truncating/keeping the whole carboxyl group. If questionable atoms belong to a rigid chemical group – carboxyl, isopropyl, phenyl, indole, guanidine, or imidazole – place the whole group based on the most probable conformer that most closely matches the observed density. Second, avoid truncating single atoms, especially for long side-chains; usually, there are some features of the omit maps that suggest placement of the last atom in the side-chain of residues such as Met and Lys if other atoms are located. Similarly, Ala, Pro, and Cβ atoms of any residue should never be truncated because the positions of these atoms are defined by the main-chain atoms. Long and flexible residues, such as Arg, Lys, Gln, and Glu, sometimes have density for the end of the side-chain, especially if it is bound by hydrogen bonds or a salt-bridge, but not for the intermediate carbon atoms. In such cases, we suggest choosing the best-fitting allowed conformer and modelling the whole side-chain. If the ADPs are restrained properly (see the first approach), this second approach may be rarely needed. A recent study showed that 64% of incomplete residues in a wide subset of structures from the PDB could be reasonably modelled into electron density, suggesting that most of the side-chains were truncated unnecessarily [70]. In particular, side-chains of short amino acids such as Ser, Cys, Asn, and Asp could be rebuilt in 80–89% of cases. However, the second approach can certainly be justified for long residues, such as Arg and Lys, that do not have any clues from the omit maps for their conformations.
Figure 2.
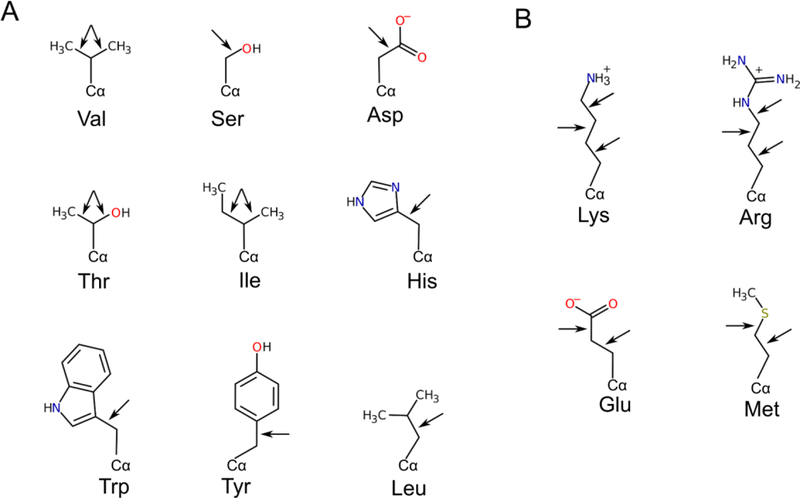
Suggested ways of truncating side-chains of various amino acids are indicated by arrows. Double arrows indicate that if a decision is made to truncate the side-chain, it has to be at both places. A) Examples of residues that have only one option for truncation according to the rules proposed in this article. B) Examples of residues with multiple options for truncation.
It is critical to review side-chains again after their placement and the subsequent refinement in the reciprocal space. Depending on the approach chosen, the atoms that do not have 2mFo-DFc density at a density level that is higher than noise would either be kept at the positions corresponding to the best-fitting allowed conformer (the most probable conformer if there is no density at all) or truncated according to the rules described above. If the side-chains are truncated, do not place water molecules in the remaining peaks; instead, consider rebuilding these side-chains if the densities are continuous. Keep in mind that phases improve over the course of the refinement; thus, reviewing the side-chains with missing atoms at the latest stages of refinement is highly recommended.
Adding alternative conformations to residues should be guided by the same principle: the omit maps should give some evidence for the conformations. In some cases, the presence of an alternative conformation is obvious, and the use of omit maps can be unnecessary. In more difficult cases, however, the use of an omit map is crucial to place the additional conformation based on the experimental evidence.
Ligands in protein crystal structures
The analysis of the small molecules bound to the protein (‘ligands’), especially functionally important molecules such as receptor ligands, cofactors, substrates, or inhibitors, is quite often the culmination of several years of work and forms the basis for further studies. It is, therefore, easy to succumb to human cognitive biases and see the desired or expected molecules (Figure 3) even if the evidence is lacking or inconclusive and then model them in haste without considering various methodological limitations [72,73]. The ligand modelling may, in some circumstances, be the most important part of the structure refinement and interpretation process. As such, it should be done to the highest standards, as improper identification or modelling may lead to false conclusions, which may culminate in the retraction of a paper.
Figure 3.

Even small peaks in the mFo-DFc difference density maps may indicate incorrect identification of the ligand. Here is the example from the work by Wlodawer et al. [72], showing that the multiple conformations of mannobiose (left) can be better reinterpreted as trehalose (right). Figure generated by Molstack from project http://molstack.bioreproducibility.org/project/view/MPYO83KA6I78W8HZSIC0/. 2mFo – DFc maps are displayed in blue contoured at a level of 1.0 rmsd. mFo - DFc difference maps are contoured at the 3.0 rmsd level in green (positive) and red (negative).
The electron density fragments (‘blobs’) that may correspond to the bound ligand can be identified and visually inspected using Coot. Although several automatic methods exist to identify and model the electron density fragments, they have not yet achieved the accuracy needed for unsupervised automated ligand recognition. It is, however, possible to use these methods to differentiate among several possible candidates [74–76].
Some of the blobs in electron density maps with datasets at high and medium resolution may be easily identified based on their shapes, especially those corresponding to tightly bound cofactors. However, the protein environment and the nature of the crystal are heterogeneous, and quite often, the interpretation is not fully unambiguous. Therefore, it is imperative to consider possible molecules that can be bound to the protein. The interpretation of density fragments can be highly subjective; consequently, different options should be considered to maximize objectivity and minimize cognitive bias. The considerations should include both physiological ligands that were added or possibly retained during protein purification and non-physiological ones used for protein preparation and crystallization. Users should prepare to analyse densities that are possibly heterogeneous (e.g., representing multiple states such as bound, unbound, with bound molecule 1, bound molecule 2, etc., or a single ligand bound in several different conformations) by answering questions such as: Can the small molecules hydrolyse or be processed by the enzyme? Which small molecules from the cell lysate can bind to the protein and be retained during purification? Do several compounds that were used for crystallization have similar properties/shapes, and how do they relate to the function of the macromolecule?
Once the candidate molecule for a blob is identified and the restraints and 3D model of the molecule are generated (for an overview of available software, see section ‘Geometrical restraints in macromolecular refinement’ and a review about the accuracy of different programs [63]), fit the proper conformation and orientation of the molecule and proceed to validate whether the identification and modelling are correct.
The first criterion in the model verification is the data-model correspondence. Inspect the electron density maps after placement of the ligand and reciprocal-space refinement with a sufficient number of cycles (no fewer than 10). If there are positive or negative peaks present on the difference map near the modelled ligand (~ 3 Å), there is a chance that it has been sub-optimally modelled or even misidentified. In addition, the ADPs of the ligand should be checked; they should correspond to the ligand environment, especially for the moieties that are tightly bound. If the ADPs differ significantly (>20%), the actual occupancy of the ligand may be lower than modelled or some parts of the ligand have been placed wrongly/misidentified. The data–model correspondence can also be evaluated numerically by calculating different real-space correlation statistics such as RSCC (real-space correlation coefficient) or significance statistics (ZO, ZD) calculated by EDSTATS.
The poor fit to the density may not necessarily mean that the ligand has been wrongly identified. There are several considerations that have to be made before accepting or rejecting the interpretation of a density blob. First, the heterogeneity of the crystal may contribute to the problems with the interpretation and achieved fit. Recently, it was demonstrated that the modelling of some bound ligands can be improved by considering several states simultaneously [77]. Second, the quality of the fit should correlate with the significance of claims that are made based on the structure. The fit to the density of the functionally relevant molecule, such as an enzyme inhibitor, should withstand verification, while the disordered crystallization buffer component, loosely bound to the protein surface, may have average fit indicators.
The second criterion in model verification is adherence to current chemical knowledge. The refined bond lengths and angles, torsion angles, planes, and chirality (if present) should match the expected values derived from the small molecule databases unless there is strong electron density evidence to suggest otherwise. The fit to the expected values can be easily checked using MOGUL [78] distributed together with CSD (using it either as a standalone program or through the interface from Coot) or by running the wwPDB validation pipeline [25]. The interactions between the ligand and the macromolecule should also conform to chemical knowledge. The ligand should make the appropriate chemical interactions with the protein, i.e. hydrophobic moieties of the ligand should make hydrophobic contacts with the protein, and charged moieties and hydrogen bond donors or acceptors should make the appropriate interactions with the protein. The chemically implausible interactions that are indicated, for example, by the presence of steric clashes calculated by MolProbity [79,80] have to be scrutinized.
Sometimes, the electron densities cannot be unambiguously identified. In such cases, we recommend modelling the density as UNL (unknown ligand) if the connectivity between atoms can be established or as multiple UNX (unknown atom) if not. Even if the density is not fully identifiable, it may be possible to provide several reasonable alternatives and the most probable explanations. While the PDB repository [24,25] is not a place to provide such speculative interpretations, we recommend using Molstack [81] to provide multiple interpretations of the ligands bound (Figure 4).
Figure 4.
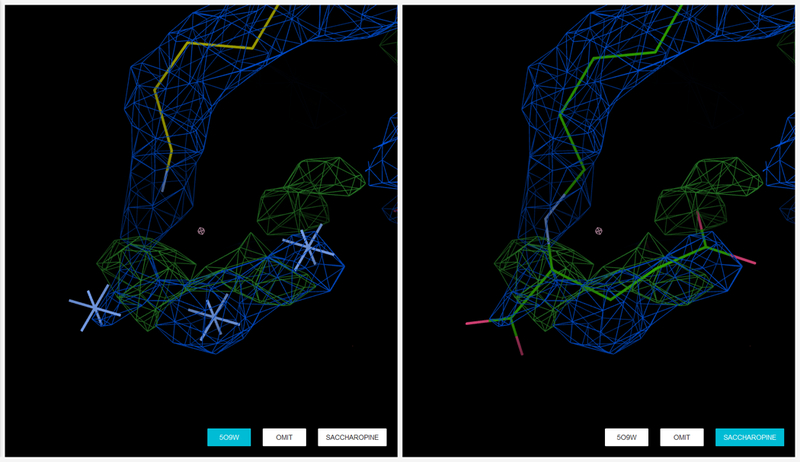
A more speculative interpretation can be provided by submitting the files to Molstack (http://molstack.bioreproducibility.org). Due to disorder and weak density, the possible modification of the lysine was marked as unknown atoms in the deposited structure (left), but using Molstack (http://molstack.bioreproducibility.org/project/view/OJM60NCMQF1VMKW0IBUF/), Kluza et. al [99] provided a probable interpretation that the modified lysine is a saccharopine resulting from the crystallization conditions (right). 2mFo – DFc maps are displayed in blue contoured at a level of 1.0 rmsd. mFo - DFc difference maps are contoured at the 3.0 rmsd level in green (positive) and red (negative).
We suggest the following ligand modelling steps (if using Coot and REFMAC):
-
(1)Identify the electron density fragments that may correspond to small molecules.
-
In Coot use: Validate > Unmodelled blobs…; check for the blobs twice using 2mFo-DFc and mFo-DFc (difference) maps at 1.0 and 3.0 rmsd, respectively.TIP: Try to identify ligands as soon as possible, before automatically adding water molecules, which may ‘flood’ the blobs and make it harder to identify them.
- Near the final stages of refinement, look for peaks in the difference density map. In Coot, you can use Validate > Difference Map Peaks to identify peaks above 4.0 rmsd. These peaks may indicate the presence of moieties larger or heavier than water molecules.
- In the final stages of the refinement, inspect water molecules (especially their clusters placed in continuous density), difference map peaks, and any weak continuous densities near the protein. During the whole model building and refinement process, the phases will improve, and some densities may be easier to interpret.
-
-
(2)
Try to guess the identity of the ligand based on the density shape, possible interactions, crystallization buffer composition, and functional considerations. Check if the molecule is already present in the PDB (e.g., using ligand search at the RCSB PDB website: http://www.rcsb.org/pdb/ligand/chemAdvSearch.do) [23]. If positively identified, import the molecule in Coot using its 3-letter code: File > Get Monomer. If not, then use Lidia (Calculate > Ligand Builder) to generate one. In both cases, verify the restraints by regularizing the ligand (Regularize Zone from refinement toolbar).
-
(3)
Fit the ligand to the density. Run several cycles (no fewer than 10) of refinement using REFMAC. If the ligand was imported using its 3-letter code and the restraints are acceptable, it is not necessary to provide REFMAC with the restraints in a separate file. If the restraints were generated, provide the same restraints for refinement in both Coot (File > Import cif dictionary) and REFMAC (libin my_ligand.cif).
-
(4)
Inspect the resulting density visually for the presence of negative and positive difference peaks larger than 3.0 rmsd. Adjust the model if necessary and refine using REFMAC.
-
(5)
If MOGUL is available, validate the ligand using the tools present in Coot by Ligand > Ligand Metric Slides. If not, run the validation from wwPDB and check the clashes, LLDF, and bond and angle outliers for the ligand. Fix if necessary.
Metal identification and refinement
Metal ions pose a special challenge in macromolecular structure refinement; according to some studies, a significant fraction of metal-containing structures have an incorrect metal assignment or modelling [54,82]. For details of metal identification and modelling, the reader is referred to a recently published protocol for characterizing metal-binding sites in proteins with X-ray crystallography [83] and other sources [82,84–87]. In particular, we would like to stress the use of anomalous maps calculated with data collected above and below the X-ray absorption edge to prove the identity of the metal (if metal absorption edge is within the energy range available at the X-ray source) [83,87,88]. We also recommend using services such as CMM (https://csgid.org/csgid/metal_sites/) [82,86] for metal identification and validation (based on coordination geometry, metal-ligand distances taken into account with the bond-valence method [89,90], and ADP values) and generation of metal-ligand restraints. In addition, we suggest that the Coot function Validate > Highly Coordinated Waters should be routinely used in the latest stages of refinement to assist in identification of metal ions.
Water molecules
As described in the ‘Manual model correction in Coot’ section, most water molecules can be added automatically in Coot. However, this tool is usually not enough to add all ordered water molecules. When inspecting various issues in the model (e.g., as advised by the tools in the Validate menu in Coot) or ‘walking’ through the polypeptide chain, water molecules with weaker density can be conveniently added with the ‘w’ key and immediately placed inside the electron density peak with the ‘x’ key (Table 2). We suggest the following approximate guidelines for placing water molecules:
-
(1)
Place water molecules only in omit difference map (mFo - DFc) peaks present at 3.0–2.5 rmsd or in 2mFo – DFc map peaks at 1.5–2.0 rmsd if they have an approximately spherical shape. Ideally, both maps should display such peaks at least at some rmsd levels.
-
(2)
After placing water molecules and structure refinement in the reciprocal space, inspect those molecules. At least some 2mFo – DFc density at 1.0–0.9 rmsd should be present and ADPs should be below than doubled average ADP for the structure (or doubled Wilson B-factor for the dataset).
-
(3)
At least one (but maximum four) hydrogen bonds with the macromolecule, a ligand, or other confirmed water molecules should be present for every water molecule. Hydrogen bonds should be no shorter than 2.5 Å and no longer than 3.6 Å. Contacts with carbon atoms should not be shorter than 2.8 Å.
Validate > Check/Delete Waters in Coot is a handy tool for checking these requirements for water molecules. Note that in special situations, these guidelines should be relaxed (e.g., when placing all water molecules necessary to complete the coordination sphere of a metal ion).
Refinement and validation should go together
Traditionally, structure validation was a separate step performed after refinement. Currently, these two steps are performed simultaneously. For example, the approach we suggest in ‘Manual model correction in Coot’ starts with validation. In addition, validation should be performed with web services such as PARVATI [43], MolProbity [79], and the wwPDB validation tools [25,26] in an iterative manner. If problems are detected, one should come back to the visual inspection, address the problems, and run validation once again. Some of the validation and correction algorithms are used internally in automated refinement pipelines; for example, MolProbity in PHENIX [91] and in HKL-3000 further facilitate the process of validation during the refinement. We should stress that careful visual inspection of the agreement of the model with the electron density maps is an absolute must if a scientist would like to minimize the possibility of a PDB deposit correction that may, in extreme cases, lead to paper retraction. High quality of protein structures, especially modelling of ligands, is a prerequisite for reproducibility of experiments and findings in many areas of biomedical research [72,92,93].
Concluding remarks
This review has provided multiple guidelines and tips for manual model correction and reciprocal-space refinement to help less experienced crystallographers navigate the model refinement process. To ease understanding, some of the most practically important concepts of macromolecular structure refinement are described. While we hope that this set of the guidelines will help aspiring crystallographers, we also acknowledge that this set is by no means complete or fully applicable to all cases. Moreover, we understand that some members of the crystallographic community, even our close friends, might disagree with some details of the suggestions presented herein, and we are always open to discussion on best practices (most of the guidelines outlined here were presented by the authors at the 2017 meeting of the American Crystallographic Association during the session ‘Apply Macromolecular Crystallography Best Practices to your Challenging Diffraction Data’). We have to note that our own rules, guidelines, and tips have changed over time due to increased experience and the perpetual evolution of crystallographic software. As software improved and new features were added, some aspects of model improvement became much easier to apply and more practical. We expect that this evolution and improvement will continue and that the recommendations will change accordingly with time.
When working on this article, we once again realized the plurality of views and the absence of established guidelines for modelling regions of macromolecular structures with weak or absent density. In our experience, the lack of such guidelines is especially noticeable and impeding when teaching practical refinement. We have tried to outline our recommendations for modelling of poorly resolved regions and side-chains without imposing any particular approach. We hope that the IUCr Commission on Biological Macromolecules will initiate public discussion about possible approaches and adopt a policy that would allow for consistent and verifiable modelling of a crystallographic disorder.
The ongoing improvement of crystallographic software has greatly facilitated corrections of some of the previously deposited structures [53,72,73,76,92,94]. In particular, the most impressive improvements were achieved when the original raw data (diffraction images) were available, thus enabling re-processing of these images with upgraded data-processing software [51,95,96]. In order to enable these kinds of improvements and foster better reproducibility of experiments in structural biology, we highly recommend depositing the diffraction images to one of the available repositories, such as https://proteindiffraction.org/ [96] and https://data.sbgrid.org/ [97]. With the same aspirations, we advocate for manual (by eye) inspection of the reported macromolecular structures by the referees conducting peer review of manuscripts [98]. The inspection can be enabled by submitting the .mtz and .pdb files via the manuscript submission systems, by releasing the reported structures from the PDB prior to manuscript submission, or by depositing the structures to repositories such as Molstack [81] that allow private sharing and multiple interpretations.
Acknowledgements
We thank Mariusz Jaskólski, Barat S. Venkataramany, Garib Murshudov, Zbigniew Dauter, Alexander Wlodawer, Magdalena Woinska, Joanna Loch, Mateusz Czub, and Joanna Lipowska for critical reading of this manuscript and invaluable feedback. We also thank the anonymous reviewers for their excellent comments and suggestions. This work was supported by the National Institute of General Medical Sciences under Grants GM117080 and GM117325, National Institutes of Health BD2K program under grant HG008424, and the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under Contract No. HHSN272201700060C.
Biographies

Dr. Ivan G. Shabalin is a Research Scientist at the University of Virginia. Following his Ph.D. on structural studies of format dehydrogenase from plants and bacteria (Chemistry, A.N. Bach Institute of Biochemistry, Russian Academy of Sciences, 2010), he joined Wladek Minor’s laboratory at the University of Virginia, where he continued to develop his expertise in protein crystallography. This position has enabled him to collaborate with several brilliant scientists, participate in large-scale structure determination projects, and contribute to the development of several software tools for structural biology. His scientific interests include optimization of protein crystallization techniques, determination of difficult structures, quality assessment of crystal structures, verification of identity and proper modelling of metal ions and small molecules, biochemical protein characterization, antimicrobial resistance, and drug discovery. Cumulatively, he has contributed to more than 110 structures deposited in the PDB. He also enjoys teaching structural biology; he has taught a tutorial on Data Reduction and Structure Determination at RapiData2017 and RapiData2018 (Practical Course in Macromolecular X-ray Diffraction Measurement at Stanford University). He has also presented talks discussing the best practices in structural biology research in various meetings.

Dr. Przemyslaw J. Porebski is a Research Associate at the University of Virginia. He completed his Ph.D. research of algorithms to improve the refinement of macromolecular structures at Wladek Minor’s laboratory at the University of Virginia and defended his thesis at Jagiellonian University, Poland. After studying dioxygenases as a postdoc at the Jerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, he rejoined Wladek Minor’s group to develop strategies and pipelines for storing and processing raw diffraction data. He is one of the developers of the HKL-3000 suite. His research interests primarily focus on the development of methods for determination, refinement and validation of macromolecular structures, data mining, knowledge-based algorithms, and structural bioinformatics.

Dr. Wladek Minor is a Harrison Distinguished Professor of Molecular Physiology and Biological Physics at the University of Virginia. Education: M.Sc., University of Warsaw, 1969; Ph.D., University of Warsaw, 1978. Employment: University of Warsaw 1969–1985; Purdue University 1985–1995; University of Virginia 1995-present. Fields of interest - development of methods for structural biology, particularly macromolecular structure determination by protein crystallography, data management in structural biology, reproducibility, data mining as applied to drug discovery, bioinformatics. Member of Center of Structural Genomics of Infectious Diseases. >200 publications, >42,000 total citations, >420 structures in PDB. Trained >65 students, >25 post-docs (trainees) and research faculty. There are five PIs among lab alumni. Recipient of the Edlich-Henderson Inventor of the Year Award.
Footnotes
Disclosure statement
One of the authors (WM) notes that he has also been involved in the development of state-of-the-art software, data management and mining tools; some of them were commercialized by HKL Research and are mentioned in the paper. WM is the co-founder of HKL Research and a member of the board. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- [1].Cowtan K The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:1002–1011. [DOI] [PubMed] [Google Scholar]
- [2].Langer G, Cohen SX, Lamzin VS, et al. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Terwilliger T SOLVE and RESOLVE: automated structure solution, density modification and model building. J. Synchrotron Radiat. 2004;11:49–52. [DOI] [PubMed] [Google Scholar]
- [4].Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Afonine P V, Grosse-Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sheldrick GM. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. [DOI] [PubMed] [Google Scholar]
- [7].Smart OS, Womack TO, Flensburg C, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brünger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. [DOI] [PubMed] [Google Scholar]
- [9].Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Potterton E, Briggs P, Turkenburg M, et al. A graphical user interface to the CCP4 program suite. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003;59:1131–1137. [DOI] [PubMed] [Google Scholar]
- [11].Adams PD, Afonine P V., Bunkóczi G, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Minor W, Cymborowski M, Otwinowski Z, et al. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:859–866. [DOI] [PubMed] [Google Scholar]
- [13].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- [14].Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Turk D MAIN software for density averaging, model building, structure refinement and validation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:1342–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. [DOI] [PubMed] [Google Scholar]
- [17].Tickle IJ, Laskowski RA, Moss DS. Rfree and the Rfree ratio. I. Derivation of expected values of cross-validation residuals used in macromolecular least-squares refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:547–557. [DOI] [PubMed] [Google Scholar]
- [18].Rupp B Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. Garland Science; 2010. [Google Scholar]
- [19].Wlodawer A, Minor W, Dauter Z, et al. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008;275:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dauter Z, Murshudov GN. Refinement at atomic resolution Int. Tables Crystallogr. Chester, England: International Union of Crystallography; 2006. p. 393–402. [Google Scholar]
- [21].Kleywegt GJ, Brünger AT. Checking your imagination: applications of the free R value. Structure. 1996;4:897–904. [DOI] [PubMed] [Google Scholar]
- [22].Brünger AT. Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr. Sect. D Biol. Crystallogr. 1993;49:24–36. [DOI] [PubMed] [Google Scholar]
- [23].Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003;10:980–980. [DOI] [PubMed] [Google Scholar]
- [25].Gore S, Sanz García E, Hendrickx PMS, et al. Validation of Structures in the Protein Data Bank. Structure. 2017;25:1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gore S, Velankar S, Kleywegt GJ. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fabiola F, Korostelev A, Chapman MS. Bias in cross-validated free R factors: mitigation of the effects of non-crystallographic symmetry. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62. [DOI] [PubMed] [Google Scholar]
- [28].Zwart PH, Grosse-Kunstleve RW, Lebedev AA, et al. Surprises and pitfalls arising from (pseudo)symmetry. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008;64:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang J On the validation of crystallographic symmetry and the quality of structures. Protein Sci. 2015;24:621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Murshudov GN. Some properties of crystallographic reliability index - R-factor: effect of twinning. Appl. Comput. Math. 2011;10:250–261. [Google Scholar]
- [32].Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Evans P Resolving some old problems in protein crystallography. Science. 2012;336:986–987. [DOI] [PubMed] [Google Scholar]
- [34].Weiss MS. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001;34:130–135. [Google Scholar]
- [35].Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. Sect. A. 1986;42:140–149. [Google Scholar]
- [36].Urzhumtsev A, Afonine P V., Lunin VY, et al. Metrics for comparison of crystallographic maps. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014;70:2593–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hodel A, Kim SH, Brünger AT, et al. Model bias in macromolecular crystal structures. Acta Crystallogr. Sect. A Found. Crystallogr. 1992;48:851–858. [Google Scholar]
- [38].Adams PD, Pannu NS, Read RJ, et al. Extending the limits of molecular replacement through combined simulated annealing and maximum-likelihood refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:181–190. [DOI] [PubMed] [Google Scholar]
- [39].Liebschner D, Afonine P V., Moriarty NW, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Merritt EA. Expanding the model: anisotropic displacement parameters in protein structure refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:1109–1117. [DOI] [PubMed] [Google Scholar]
- [41].Merritt EA. To B or not to B: a question of resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Joosten RP, Joosten K, Murshudov GN, et al. PDB_REDO: constructive validation, more than just looking for errors. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zucker F, Champ PC, Merritt EA. Validation of crystallographic models containing TLS or other descriptions of anisotropy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Urzhumtsev A, Afonine P V, Adams PD. TLS from fundamentals to practice. Crystallogr. Rev. 2013;19:230–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:439–450. [DOI] [PubMed] [Google Scholar]
- [46].Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006;39:109–111. [Google Scholar]
- [47].Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997;53:240–255. [DOI] [PubMed] [Google Scholar]
- [48].Headd JJ, Echols N, Afonine P V., et al. Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vagin AA, Steiner RA, Lebedev AA, et al. REFMAC 5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2184–2195. [DOI] [PubMed] [Google Scholar]
- [50].Tickle IJ. Experimental determination of optimal root-mean-square deviations of macromolecular bond lengths and angles from their restrained ideal values. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007;63:1274–1281. [DOI] [PubMed] [Google Scholar]
- [51].Groom CR, Bruno IJ, Lightfoot MP, et al. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016;72:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gražulis S, Daškevič A, Merkys A, et al. Crystallography Open Database (COD): An open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res. 2012;40:420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shabalin I, Dauter Z, Jaskolski M, et al. Crystallography and chemistry should always go together: a cautionary tale of protein complexes with cisplatin and carboplatin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015;71:1965–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zheng H, Shabalin IG, Handing KB, et al. Magnesium-binding architectures in RNA crystal structures: validation, binding preferences, classification and motif detection. Nucleic Acids Res. 2015;43:3789–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:1355–1363. [DOI] [PubMed] [Google Scholar]
- [56].Long F, Nicholls RA, Emsley P, et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009;65:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Smart OS, Womack TO, Flensburg C, et al. Better ligand representation in BUSTER protein–complex structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2011;67:C134–C134. [Google Scholar]
- [59].Andrejašič M, Pražnikar J, Turk D. PURY: a database of geometric restraints of hetero compounds for refinement in complexes with macromolecular structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008;64:1093–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lebedev AA, Young P, Isupov MN, et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Emsley P Tools for ligand validation in Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Moriarty NW, Draizen EJ, Adams PD. An editor for the generation and customization of geometry restraints. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Steiner RA, Tucker JA. Keep it together: restraints in crystallographic refinement of macromolecule-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kleywegt GJ, IUCr. Use of Non-crystallographic Symmetry in Protein Structure Refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996;52:842–857. [DOI] [PubMed] [Google Scholar]
- [65].Schröder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nicholls RA, Long F, Murshudov GN. Low-resolution refinement tools in REFMAC5. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Krissinel E Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015;43:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- [69].Kowiel M, Jaskolski M, Dauter Z. ACHESYM: an algorithm and server for standardized placement of macromolecular models in the unit cell. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014;70:3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Porebski PJ, Cymborowski M, Pasenkiewicz-Gierula M, et al. Fitmunk: improving protein structures by accurate, automatic modeling of side-chain conformations. Acta Crystallogr. Sect. D Biol. Crystallogr. 2016;72:266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Djinovic-Carugo K, Carugo O. Missing strings of residues in protein crystal structures. Intrinsically Disord. proteins. 2015;3:e1095697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wlodawer A, Dauter Z, Porebski PJ, et al. Detect, correct, retract: How to manage incorrect structural models. FEBS J. 2018;285:444–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kutner J, Shabalin IG, Matelska D, et al. Structural, Biochemical, and Evolutionary Characterizations of Glyoxylate/Hydroxypyruvate Reductases Show Their Division into Two Distinct Subfamilies. Biochemistry. 2018;57:963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cymborowski M, Klimecka M, Chruszcz M, et al. To automate or not to automate: this is the question. J. Struct. Funct. Genomics. 2010;11:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shumilin IA, Cymborowski M, Chertihin O, et al. Identification of Unknown Protein Function Using Metabolite Cocktail Screening. Structure. 2012;20:1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kowiel M, Brzezinski D, Porebski PJ, et al. Automatic Recognition of Ligands in Electron Density by Machine Learning. Bioinformatics. 2018;[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pearce NM, Krojer T, von Delft F. Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bruno IJ, Cole JC, Kessler M, et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 2004;44:2133–2144. [DOI] [PubMed] [Google Scholar]
- [79].Williams CJ, Headd JJ, Moriarty NW, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Davis IW, Leaver-Fay A, Chen VB, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Porebski PJ, Sroka P, Zheng H, et al. Molstack-Interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps. Protein Sci. 2018;27:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zheng H, Chordia MD, Cooper DR, et al. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2014;9:156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Handing KB, Niedzialkowska E, Shabalin IG, et al. Characterizing metal-binding sites in proteins with X-ray crystallography. Nat. Protoc. 2018;13:1062–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bowman SEJ, Bridwell-Rabb J, Drennan CL. Metalloprotein Crystallography: More than a Structure. Acc. Chem. Res. 2016;49:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Russo Krauss I, Ferraro G, Pica A, et al. Principles and methods used to grow and optimize crystals of protein-metallodrug adducts, to determine metal binding sites and to assign metal ligands. Metallomics. 2017;9:1534–1547. [DOI] [PubMed] [Google Scholar]
- [86].Zheng H, Cooper DR, Porebski PJ, et al. CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017;73:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Volbeda A X-ray crystallographic studies of metalloproteins. Fontecilla-Camps JC, Nicolet Y, editors. Methods Mol. Biol. 2014;1122:189–206. [DOI] [PubMed] [Google Scholar]
- [88].Handing KB, Shabalin IG, Kassaar O, et al. Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chem. Sci. 2016;7:6635–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Brese NE, O’Keeffe M, IUCr. Bond-valence parameters for solids. Acta Crystallogr. Sect. B Struct. Sci. 1991;47:192–197. [Google Scholar]
- [90].Brown ID. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 2009;109:6858–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Headd JJ, Immormino RM, Keedy DA, et al. Autofix for backward-fit sidechains: using MolProbity and real-space refinement to put misfits in their place. J. Struct. Funct. Genomics. 2009;10:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rupp B, Wlodawer A, Minor W, et al. Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J. 2016;283:4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dauter Z, Wlodawer A, Minor W, et al. Avoidable errors in deposited macromolecular structures: an impediment to efficient data mining. IUCrJ. 2014;1:179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Tanley SWM, Schreurs AMM, Kroon-Batenburg LMJ, et al. Re-refinement of 4g4a: room-temperature X-ray diffraction study of cisplatin and its binding to His15 of HEWL after 14 months chemical exposure in the presence of DMSO. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016;72:253–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Helliwell JR, McMahon B, Guss JM, et al. The science is in the data. IUCrJ. 2017;4:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Grabowski M, Langner KM, Cymborowski M, et al. A public database of macromolecular diffraction experiments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2016;72:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Meyer PA, Socias S, Key J, et al. Data publication with the structural biology data grid supports live analysis. Nat. Commun. 2016;7:10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Minor W, Dauter Z, Helliwell JR, et al. Safeguarding Structural Data Repositories against Bad Apples. Structure. 2016;24:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kluza A, Niedzialkowska E, Kurpiewska K, et al. Crystal structure of thebaine 6-O-demethylase from the morphine biosynthesis pathway. J. Struct. Biol. 2018;202:229–235. [DOI] [PubMed] [Google Scholar]