

Abstract
Background
Homologous recombination deficiency (HRD) is shown to predict response to DNA-damaging therapies in patients with high-grade serous ovarian cancer (HGSOC); however, changes in HRD during progression remains unknown.
Methods
HRD scores were evaluated in paired primary and/or recurrent HGSOC samples (N = 107) from 54 patients with adjuvant platinum-based chemotherapy. BRCA1/2 mutation, BRCA1 methylation, loss of heterozygosity (LOH), and HRD scores were characterised using tumour DNA-based next-generation sequencing assays.
Results
Among 50 evaluable pairs (N = 100 samples), high intra-patient correlation in HRD score was observed (r2 = 0.93). BRCA1/2 mutations, BRCA1/2 LOH, and HRD were maintained between primary and recurrent samples, except for one pair in which a BRCA1 reversion mutation was identified in the recurrent sample. Despite the reversion, both samples were classified as having high HRD scores ( ≥ 42). All samples with BRCA1/2 mutations exhibited high HRD scores; however, high HRD scores were more prevalent than BRCA1/2 mutations (55% vs. 30%, respectively).
Conclusion
Markers of HRD were maintained between the primary and recurrent samples, regardless of other genomic changes that occurred during recurrence. HRD score/markers in primary tumours may be valuable and adequate for selection of platinum-based therapy and/or poly-ADP-ribose-polymerase (PARP) inhibitors in recurrent HGSOC.
Introduction
Optimal treatment for patients diagnosed with advanced ovarian cancer is platinum–taxane chemotherapy and surgical debulking. Despite initial response rates of more than 80%, tumour recurrence develops in approximately 70–80% of patients, with a median progression-free survival of 18 months.1 Identification of genomic alterations in BRCA1 and BRCA2 that render ovarian cancer deficient in the homologous recombination (HR) DNA repair pathway has made a major impact in development of novel therapeutic strategies.2,3 Of particular significance is the development of poly-ADP-ribose-polymerase (PARP) inhibitors, which induce synthetic lethality via inhibition of DNA single-stranded break repair mechanisms, and are useful in maintenance therapy of recurrent tumours that carry BRCA1 or BRCA2 mutations, or are HR deficient.4–12 Other germline mutations can exist in HR pathway genes (e.g., RAD51 and ATM) and patients without germline mutations can also exhibit somatic mutations of genes associated with HR deficiency (HRD). Data from The Cancer Genome Atlas suggests that approximately 50% of high-grade serous ovarian cancers (HGSOCs) harbour mutations within the HR pathway.13
To allow for the identification of HRD in a larger subset of patients beyond those with BRCA1 or BRCA2 mutations, an HRD assay was developed, which assesses three metrics of genomic instability: number of telomeric allelic imbalances (TAI), loss of heterozygosity (LOH), and large-scale state transitions (LSTs).14–16 In addition to limited heterogeneity observed with intratumour HRD in primary breast cancer,17 analysis of TAI, LOH, and LST in patients with triple-negative breast cancer on neoadjuvant chemotherapy has allowed for identification of tumours likely to respond to platinum-based chemotherapy.18 Somatic mutations in BRCA1/2 that can impact response have been reported in ovarian carcinoma following adjuvant chemotherapy.19–21 Mutations in HR pathway genes and BRCA1/BRCA2 mutations have been evaluated in primary-recurrent pairs of HGSOC.20,21 Given the high recurrence rate in HGSOC, early information on HRD status could be potentially useful for implementation of novel therapeutic modalities, even in the recurrent setting. In the current study, we sought to analyse HRD and BRCA status in paired primary and recurrent HGSOC samples, to better understand molecular changes during recurrence and the potential value of the HRD score for selecting first-line and subsequent treatments.
Materials and methods
Patients and treatment
Tumour tissue was obtained at primary cytoreductive surgery and first recurrence from HGSOC patients providing complete informed consent from 2001–2012 according to an Institutional Review Board approved protocol. Site for tumour collection was Charité Medical University of Berlin, Berlin, Germany. All patients underwent primary cytoreductive surgery followed by first-line platinum-based chemotherapy, except for one patient who received immunotherapy. The patients were from a sequential series that had surgically resected primary and first recurrent tumours, except for one set of paired patient tumours that comprised a primary and a sample at second recurrence. Recurrent specimens were from surgical resection. Germline sequencing was not performed as part of this study and the mutations identified in the tumour samples may be either germline or somatic.
Molecular analysis
A detailed description of the molecular analysis methods used during this study is provided in the Supplementary Information as Supplementary Methods.
DNA extraction
Tissue sections from formalin-fixed paraffin-embedded tumour samples were used for DNA extraction.
Promoter methylation analysis
Isolated genomic DNA was subjected to bisulfite conversion followed by two rounds of PCR amplification using primers specific for bisulfite converted DNA. The resulting PCR products were size fractionated and then sequenced using a MiSeq sequencer (Illumina, San Diego, CA). Analysis of the methylation assay sequence data was as described in ref. 18 (Supplementary Methods).
Mutation analysis and whole-genome SNP analysis
BRCA1/2 gene mutation detection and single-nucleotide polymorphism (SNP) whole-genome analysis was performed using a custom hybridisation capture method. A detailed description of the SNP selection process is described in ref. 22 (Supplementary Methods). Whole-exome libraries were created from isolated DNA. DNA from 16 samples was pooled and target enrichment performed by hybridisation using 1 of 2 hybridisation capture methods. Post-capture libraries were amplified, quantified, and sequenced using a HiSeq2500 (Illumina).
Data analysis
The SNP analysis and methods used to calculate the three individual component scores (LOH, TAI, and LST) and the final HRD score, and a description of the quality control metrics for the assay are described in detail in ref. 22 (Supplementary Methods). Identification of BRCA1- and BRCA2-deficient tumours has been described previously;22 only deleterious and suspected deleterious mutations were included in the analysis.23,24 HRD status was defined as HRD positive for tumours with HRD scores ≥ 42 or a BRCA1/2 mutation or HRD negative for tumours with HRD scores < 42 and a BRCA1/2 wild type.
To assess whether there was an association between time to first recurrence and the amount of genomic rearrangement that had occurred between the primary and recurrent tumour samples, correlation of allele dosage profiles was assessed at the individual SNP level. A description of how allele dosage profiles are calculated is provided in Supplementary Methods. A predefined HRD score threshold at 42 (fifth percentile of HRD scores observed in BRCA1 or BRCA2 mutant tumours) was used.18 Tumours were considered HR deficient (HRD positive) if they have an HRD score greater than or equal to the threshold or a tumour had BRCA1 or BRCA2 (tBRCA) mutation. HR non-deficient (HRD negative) tumours have a score below the threshold of 42.
Statistical analysis
The analyses described here were performed using R version 3.1.1 or higher. Pearson’s correlation coefficient was used to evaluate the intra-patient association between paired primary and recurrent samples. A bivariate linear regression model was constructed with correlation of paired allele dosage profiles as the response variable, time from surgery to first recurrence, and HRD score of the primary tumour as the covariates using the lm function from the R stats package. All P-values were reported as two-sided. P-values < 0.05 were considered to indicate statistically significant results.
Results
A total of 107 samples from 54 primary and/or recurrent pairs were analysed for HRD, BRCA1 and BRCA2 mutation status, and BRCA1 promoter hypermethylation. Two pairs were excluded due to uncertainty regarding the tumour site. Only primary tissue data was obtained from a third subject and one pair was excluded because the patient had clear cell carcinoma, resulting in 50 pairs, or 100 samples from 50 unique patients. Table 1 describes the patient demographics. The median age was 55 (range 23–74 years) and 96% received standard adjuvant platinum plus paclitaxel chemotherapy. One patient received neoadjuvant platinum plus paclitaxel chemotherapy. The median time to disease recurrence was 12 months (range 1–55 months).
Table 1.
Patient characteristics
| Patients and paired primary-recurrent samples | N = 50 |
|---|---|
| Total number of unique samples | N = 100 |
| Median age (range) | 55 (23–74) |
| Stage | |
| II | 3 |
| III | 42 |
| IV | 5 |
| Tumor histology | |
| Serous | 46a |
| Undifferentiated | 4 |
| Treatment | |
| Carboplatin-paclitaxel | 48 |
| Other platinum | 2 |
| Neoadjuvant treatment | |
| No | 49 |
| Yes | 1 |
| Median time to first recurrence (range) | 12 months (1–55 months) |
aOne patient with peritoneal cancer
HRD score analysis was successful on 88/100 samples (88%), with 78% of primary/recurrent pairs yielding evaluable HRD scores from both samples (N = 39 pairs). The remaining samples failed quality control analysis. Failures were primarily due to low tumour DNA content (N = 10; median non-tumour content of 72%; range 62–95%), with DNA degradation resulting in high assay noise failure of the remaining two samples. HRD status positive (HRD score ≥ 42 or BRCA1/2 mutant) or negative (HRD score < 42 and BRCA1/2 wild type) was concordant for all 39-evaluable primary-recurrent pairs (Fig. 1). There was high intra-patient correlation between the HRD score in the primary and paired recurrent sample (r2 = 0.93, Fig. 1), despite evidence of additional acquired genomic rearrangements in the recurrent sample (Fig. 2). The Median (interquartile range, IQR) scores were 1.0 (− 3.5, 3.5) for BRCA1/2 wild-type samples and 5.0 (1.5, 8.3) for BRCA1/2 mutant samples (p = 0.075). The Median (IQR) scores were 2.0 (−1.0, 8.0) for BRCA1 unmethylated and 0.5 (−8.3, 7.8) for BRCA1 methylated samples (p = 1). The Median (IQR) scores were 2.0 (−1.0, 4.0) for samples with low HRD scores and 2.0 (−1.8, 8.0) for samples with high HRD scores (p = 0.34).
Fig. 1.
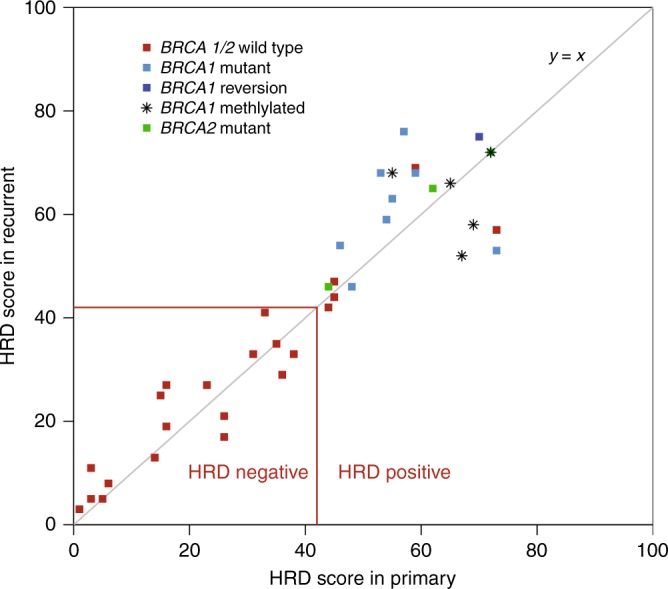
Correlation between HRD scores in primary (x axis) and recurrent (y axis) tumour samples. BRCA1/2 mutation status is shown by the colour of the data points. Tumours positive for BRCA1 promoter methylation are indicated by asterisks. The red horizontal and vertical lines indicate the cut-off (42) between HRD-positive and HRD-negative tumours for both the primary and recurrent tumours
Fig. 2.

Genomic profiles of primary and recurrent tumour samples with low and high HRD. Examples are shown for tumours with similar (a) and very divergent (b) primary and recurrent pairs. The yellow line indicates regions of LOH (allele dosage of 0) and non-LOH (allele dosage of 1). Areas of divergence (red arrows) and similarity (green arrows) are indicated. HRD scores are in the bottom right of each profile
Table 2 summarises the tumour HRD characteristics of individual samples and primary-recurrent pairs. Of 39 pairs with evaluable HRD scores, 22 (56%) had high HRD ( ≥ 42). Mutations in BRCA1 were observed in 13/50 pairs (26%), of which nine were evaluable for locus-specific LOH and 9/9 (100%) had BRCA1 LOH. Mutations in BRCA2 were observed in 3/50 pairs (6%), all of which had BRCA2 LOH. All BRCA1 or BRCA2 mutated pairs that passed HRD score analysis (N = 13) had high HRD scores ( ≥ 42). BRCA1 promoter methylation was observed in 10/100 tumour samples from 6/50 pairs. In two pairs, the results from primary-recurrent tumour pairs were discordant with methylation of only the primary tumour in one case and the recurrent tumour in the other. HRD scores were available for six of the pairs, which contained at least one tumour sample positive for BRCA1 promoter methylation. All tumour pairs positive for BRCA1 promoter methylation had high HRD scores ( ≥ 42). Twenty-two percent (5/22) of primary-recurrent tumours pairs with high HRD scores did not have a BRCA1/2 defect and 10/22 (45%) were BRCA1/2 non-mutant. HRD scores in tumours with BRCA1 or BRCA2 mutations tended to be higher (p = 0.075) in the recurrent than in the primary sample (Supplementary Figure 1).
Table 2.
Tumor HRD score charactersistics
| Samples | Primary-recurrent pairs | |
|---|---|---|
| Total tumor samples | 100 | 50 |
| Evaluable HRD scores | 88% (88/100) | 78% (39/50) |
| High HRD score ( ≥ 42) | 57% (50/88) | 56% (22/39) a |
| BRCA1 mutation | 26% (26/100) | 26% (13/50) |
| BRCA2 mutation | 6% (6/100) | 6% (3/50) |
| BRCA1 methylation | 10% (10/100) | 12% (6/50) b |
aPositive in both the primary and recurrent specimen
bPositive in the primary and/or recurrent specimen (two pairs were discordant)
A potential reversion mutation in BRCA1 was observed in one recurrent tumour sample (Fig. 3 and Supplementary Figure 2). The original mutation was a 1 bp insertion (c.5266dupC (p.Gln1757Profs*74) and was the only mutation observed in the primary tumour sample. This mutation shifts the transcript out of frame due to a one base pair shift in the location of a splice junction, and results in a premature stop codon downstream. The original mutation was observed in 285/363 forward and 295/383 reverse reads from the primary tumour sample, and in 512/599 forward and 508/604 reverse reads from the recurrent tumour sample. This mutation is a frequently observed BRCA1 deleterious Ashkenazi founder mutation. In the recurrent tumour sample, sequence analysis confirmed the presence of the original mutation and additionally detected two substitutions located 10 and 12 bp downstream. The reversion mutation was not observed in the primary tumour sample and was observed in 47/649 forward and 60/657 reverse reads in the recurrent tumour sample. These additional substitutions move the splice donor site back in frame. The resulting transcript is predicted to be full length with three amino acid changes (Gln → Pro, Asp → Gly, Arg → Gln) due to the three single base substitutions. Based on in silico analyses it is unlikely that any of these three amino acids changes are pathogenic; however, all three are classified as of uncertain clinical significance as insufficient information is available for a definitive classification. In addition, the new splice donor site that is present is predicted to be functional; however, functional assays to confirm this were beyond the scope of this study. Both the primary and recurrent tumour samples had high HRD scores. In all other primary-recurrent pairs, BRCA1 and BRCA2 mutation status in the primary tumour was preserved in the paired recurrent sample.
Fig. 3.
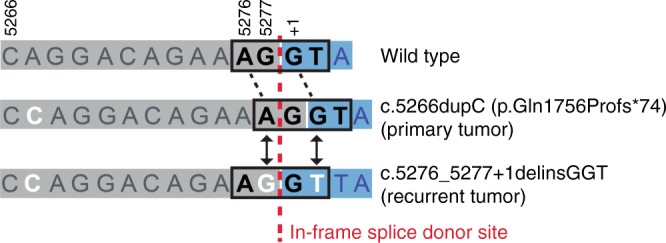
Schematic illustration of BRCA1 reversion mutation. Alignment of sequences of a region in wild-type BRCA1, mutated BRCA1 in the primary tumour, and reversion BRCA1 mutant in the recurrent tumour. The mutated bases are shown in white. Exonic sequence is shaded in grey and intronic sequence is shaded in blue. The splice donor site bases are shown by the boxed region on each sequence. The vertical red line indicates the position in the sequence where the in-frame splice donor site would be located. The numbers above the wild-type sequence read indicate the codon position within the wild-type transcript. The second line shows the sequence observed in the primary tumour. The third line shows the sequence observed in the recurrent tumour
We assessed how well allele dosage was correlated within each primary-recurrent tumour pair and between unrelated primary and recurrent samples (Fig. 4a, b). This analysis showed a high degree of correlation within the pairs (r2 ≥ 0.89), but not between unrelated primary and recurrent tumour samples (r2 ≤ 0.19). If the degree of divergence between the genomic profiles of the primary and recurrent pairs was associated with time to first recurrence, there should be a correlation between the allele dosage for each pair and time to first recurrence. No such association was observed (Fig. 4c; correlation coefficient − 0.062), indicating that accumulation of genomic rearrangements is not associated with time, and that the rate at which an individual tumour accumulates genomic rearrangements varies.
Fig. 4.
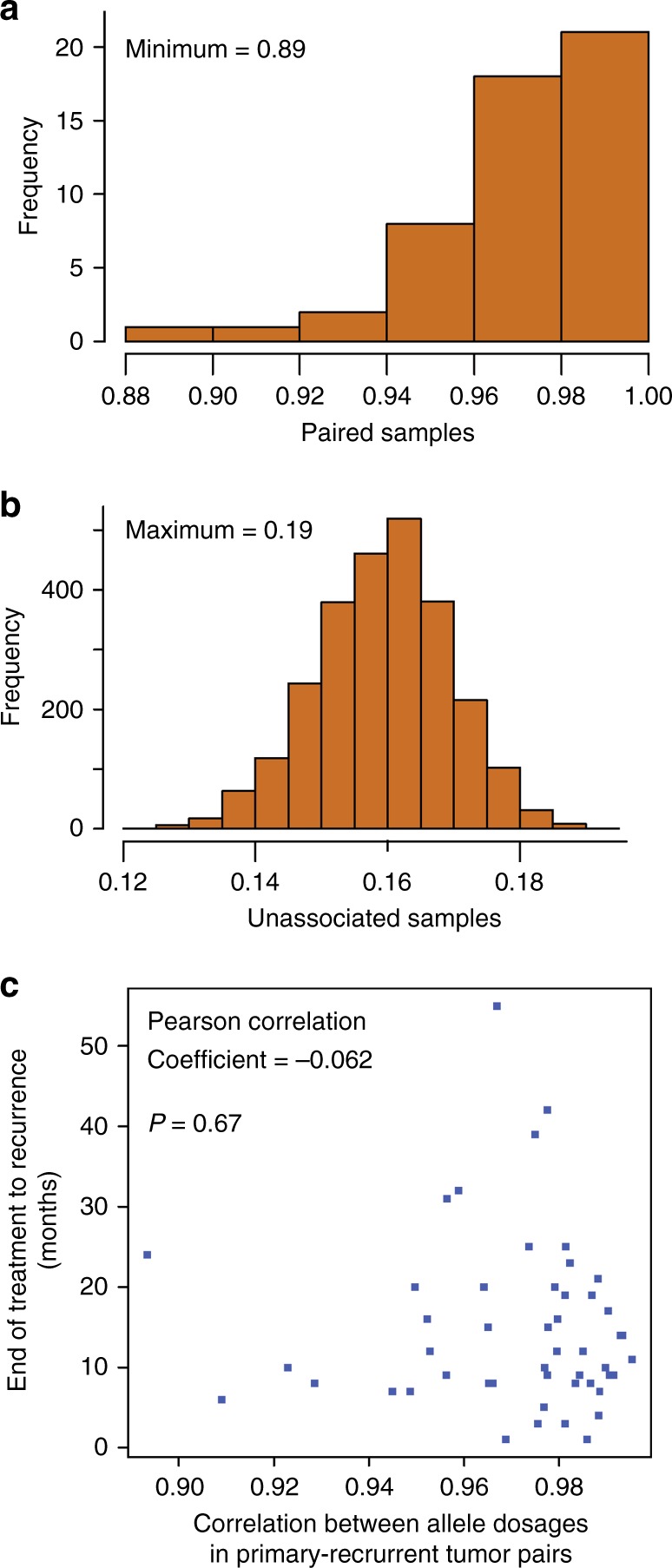
a Correlations between allele dosages in primary-recurrent tumour pairs. The histogram represents the frequency of Pearson’s correlation coefficients between paired allele dosages for matched primary-recurrent tumour pairs. b Correlations between unrelated primary and recurrent tumour samples. The histogram represents the frequency of Pearson’s correlation coefficients between paired allele dosages for unrelated primary-recurrent tumour pairs. c association between time to first recurrence and change in genomic profile. The scatter plot shows the relationship between Pearson’s correlation coefficients between allele dosage in matched primary-recurrent tumour pairs and time from end of treatment to first recurrence in months
Lastly, the more genomic instability observed in the primary tumour sample the greater the likelihood that the tumour has a defect in the HRD pathway. Therefore, we assessed whether the amount of divergence in the genomic profiles between the primary and recurrent samples is related to HRD status and time to first recurrence. Using a linear regression of correlation of paired allele dosages as a function of time to first recurrence and the HRD score of the primary tumour, no significant association was identified (p = 0.62). The coefficients associated with time to first recurrence and HRD score of the primary are − 1.2 × 10−4 (p = 0.70) and − 1.2 × 10−4 (p = 0.44), respectively.
A summary of time to recurrence following first and second treatment, BRCA1/2 status (mutation, methylation, and reversion) and HRD score for the paired primary and recurrent samples from 50 patients is outlined in Fig. 5a. All 50 patients were treated with first-line platinum-based chemotherapy and in 35 patients with available data, 26/35 patients were treated at recurrence with second-line platinum-based chemotherapy. The majority of patients in this group had response lasting > 6 months. The remaining 9/35 patients were treated with second-line topotecan at recurrence and responses in most patients was < 6 months. An exploratory analysis of primary tumours only for association between HRD status and recurrence-free survival (RFS) following first-line platinum and/or taxane chemotherapy is outlined in Fig. 5b. The median survival observed in the HRD-negative samples (N = 17) was 10 months (95% confidence interval (CI): 6–14) compared with 12 months in the HRD-positive group (N = 27; 95% CI: 9–20). No significant association between HRD status and RFS (P = 0.56) was observed.
Fig. 5.

a Summary of time to recurrence after 1st and 2nd line chemotherapy, BRCA1/2 status (mutation, methylation and reversion), and HRD score for the paired primary and recurrent samples from 50 patients. Samples with available data is represented by %. Patient 516688 received 1 course of neoadjuvant carboplatin-paclitaxel chemotherapy followed by surgery and subsequent carboplatin-paclitaxel, other platinum-based chemotherapy/immunotherapy when recurrent tumour was obtained. Tumour at 2nd recurrence was obtained for patient 516682. b Kaplan–Meier curve of recurrence-free survival (RFS) and HRD status of primary tumours following first-line adjuvant platinum and/or taxane chemotherapy
Discussion
To our knowledge, this study is the first to report changes in characteristics of HRD between paired primary and recurrent HGSOC samples. Markers of HRD examined, including mutations in BRCA1 and BRCA2, LOH in BRCA1 and BRCA2, BRCA1 promoter methylation, and HRD score, were maintained between the primary and recurrent tumour samples, regardless of other genomic changes that occurred during recurrence. High HRD scores ( ≥ 42) were observed in all tumour samples with mutations in BRCA1 or BRCA2; however, high HRD scores were more prevalent than mutations in BRCA1 or BRCA2 (55% vs. 30%, respectively). More than one-quarter of primary-recurrent pairs had high HRD scores but intact BRCA1 or BRCA2, suggesting that HRD testing via the proposed methods used in this study15,19 allows for the identification of more tumours with possible deficiencies in the HR pathway vs. BRCA testing alone. This relative frequency of BRCA1/2 mutant vs. non-mutant HRD scores is consistent with previously published studies in both triple-negative breast18 and ovarian cancers.25 There was one possible reversion mutation in a recurrent sample; however, both the primary and recurrent tumour samples had high HRD scores and were classified as HR deficient. One likely explanation is that the genomic scarring present in the tumour is permanent, and therefore the HRD score will reflect the tumour’s prior HR deficient state. Although the primary intent of this study was a comparison of HRD characteristics between paired primary and recurrent tumours, an exploratory analysis revealed no significant association between HRD status and RFS in the primary tumours following first-line adjuvant chemotherapy. This observation is not surprising given the small sample size. A recent report in a larger HGSOC cohort treated with carboplatin monotherapy25 has shown a significant association between HRD status and improved progression-free survival and overall survival.
Significant variation was observed in the degree of divergence of genomic profiles in the primary-recurrent pairs included in this study. Time from end of treatment to first recurrence varied from 1 to 55 months, with a mean of 14 months. The three biomarker components of the HRD score each count the presence of different types of genomic rearrangements. To be counted towards any one biomarker score a genomic region or breakpoint must meet certain pre-specified criteria. For example, breakpoints that are included in the LST score count must be at least 10 Mb from the next closest breakpoint. As additional genomic rearrangements develop they may be located more than 10 Mb from a preexisting breakpoint and therefore both breakpoints will be counted and the LST score will increase, or conversely the appearance of a new breakpoint closer than 10 Mb to a previously existing breakpoint would result in neither breakpoint being counted and the LST score would decrease. Therefore, the accumulation of new genomic rearrangements does not necessarily result in an increase in the HRD score as new rearrangements may either create or destroy regions or breakpoints that were previously counted. As a result, the direction and degree of change in HRD score between the primary and recurrent tumour samples does not necessarily reflect the amount of new genomic rearrangements that have occurred between the samples.
The present study did not directly address the role of heterogeneity, as a single primary or paired recurrent tumour was analysed. To possibly understand the heterogeneity of BRCA1/BRCA2 mutations, analysis of multiple tumours retrieved from different sites at primary surgery in a single patient could be useful. Indeed, at recurrence five independent reversion events in a patient with germline BRCA2 mutation has been reported following rapid autopsy and collection of multiple samples.19 The present results suggest concordance of HRD and BRCA1/2 mutations between the paired primary and recurrent tumours; however, analysis of these events in multiple tumours at primary surgery could be important to provide more comprehensive information in decisions for second-line treatment at recurrence. Although the present study did not address the implications of germline BRCA1/2 mutations in primary-recurrent paired samples, in patients with germline BRCA1/2 mutations, reversions that lead to platinum resistance have been reported only following chemotherapy.20
Extensive characterisation of the molecular events contributing to drug resistance or disease relapse have largely been hindered by the failure to acquire biopsies from the same ovarian cancer patient throughout the course of their disease. Investigation of large cohorts of paired samples are critical to uncover mechanisms of acquired resistance, which may present further opportunities for therapeutic intervention. This study is the first to report high intra-patient correlation in HRD between paired primary and recurrent HGSOC samples. Although BRCA1 and BRCA2 mutations are well recognised as positive-predictive biomarkers to DNA-damaging therapies, such as platinum analogs, and PARP inhibitors,4–12 the development of robust platforms to analyse additional defects in the HR pathway allows for greater selection of patients potentially eligible to receive these therapies.
Overall, high intra-patient correlation of HRD scores between paired primary and recurrent HGSOC samples suggest that characterisation of HRD in the primary sample may be valuable for selection of DNA-damaging therapies, including PARP inhibitors, as maintenance or in recurrent HGSOC. Further, additional prospective studies should investigate whether selection of DNA-damaging therapies based on HRD score from the primary tumour can improve clinical outcomes in the maintenance and/or recurrent setting.
Electronic supplementary material
Acknowledgements
We gratefully acknowledge the patients who consented to donate tissue specimens and the Charité Universitätsmedizin Berlin—Tumour Bank Ovarian Cancer Network and Rare Gynecological Tumours.
Author contributions
Study concept development: JNP, IB, KMT, JSL, MKG, JS, and RNG. Data/specimen collection: JNP, IB, KMT, JSL, SDE, MKG, JS, and RNG. Data analysis: all authors. Manuscript preparation: all authors.
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki. The study was approved by Ethics Commission Professor Dr H. Eichstädt, Charité Campus Virchow-Klinikum.
Consent for publication:
No additional consent for publication was required for this manuscript.
Competing interests
KMT, CS, PT, JR, and JS were employed by Myriad Genetics, Inc. at the time of this study. JNP, IB, SDE, MKG, JS, and RNG do not have any disclosures.
Funding
This study was funded in part by an investigator-initiated grant by Myriad Genetics.
Footnotes
Availability of data and material: Not applicable.
These authors contributed equally: Jai N. Patel and Ioana Braicu
Contributor Information
Jalid Sehouli, Email: jalid.Sehouli@charite.de.
Ram N. Ganapathi, Phone: +(704) 355-9759, Email: ram.ganapathi@carolinashealthcare.org
Electronic supplementary material
Supplementary information is available for this paper at 10.1038/s41416-018-0268-6.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham JM, et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014;4:4026. doi: 10.1038/srep04026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsop K, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012;30:2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer. 2016;60:49–58. doi: 10.1016/j.ejca.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Yates MS, et al. Evaluation of BRCA1/2 and homologous recombination defects in ovarian cancer and impact on clinical outcomes. J. Clin. Oncol. 2017;35:5511. doi: 10.1200/JCO.2017.35.15_suppl.5511. [DOI] [Google Scholar]
- 6.Konecny GE. Understanding exceptional responses to poly (ADP-ribose) polymerase inhibition in sporadic ovarian cancer. J. Clin. Oncol. 2017;35:1151–1153. doi: 10.1200/JCO.2016.72.0052. [DOI] [PubMed] [Google Scholar]
- 7.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 8.Mirza MR, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016;375:2154–2164. doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 9.Ledermann JA, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17:1579–1589. doi: 10.1016/S1470-2045(16)30376-X. [DOI] [PubMed] [Google Scholar]
- 10.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukhopadhyay A, et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012;72:5675–5682. doi: 10.1158/0008-5472.CAN-12-0324. [DOI] [PubMed] [Google Scholar]
- 12.Swisher EM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 13.Iijima M, et al. Genome-wide analysis of gynecologic cancer: The Cancer Genome Atlas in ovarian and endometrial cancer. Oncol. Lett. 2017;13:1063–1070. doi: 10.3892/ol.2017.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birkbak NJ, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abkevich V, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Popova T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 17.von Wahlde MK, et al. Intratumor heterogeneity of homologous recombination deficiency in primary breast cancer. Clin. Cancer Res. 2017;23:1193–1199. doi: 10.1158/1078-0432.CCR-16-0889. [DOI] [PubMed] [Google Scholar]
- 18.Telli ML, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin. Cancer Res. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patch AM, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 20.Norquist B, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pennington KP, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timms KM, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475. doi: 10.1186/s13058-014-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eggington JM, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin. Genet. 2014;86:229–237. doi: 10.1111/cge.12315. [DOI] [PubMed] [Google Scholar]
- 24.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stronach EA, et al. Biomarker assessment of HR deficiency, tumor BRCA1/2 mutations and CCNE1 copy number in ovarian cancer: associations with clinical outcome following platinum monotherapy. Mol. Cancer Res. 2018;16:1103–1111. doi: 10.1158/1541-7786.MCR-18-0034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.