

Summary
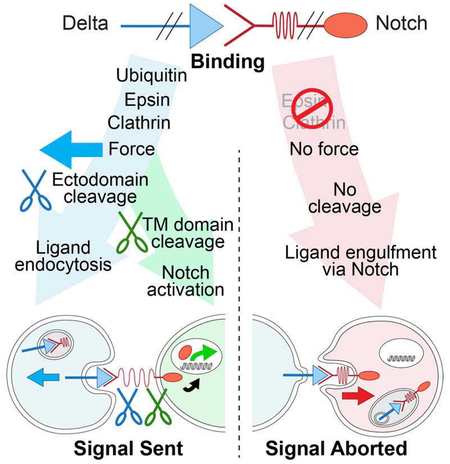
DSL ligands activate Notch by inducing cleavage and shedding of the receptor ectodomain—an event that requires ligand to be endocytosed in signal-sending cells by the adaptor protein Epsin. Two classes of explanation for this unusual requirement are: (i) recycling models, in which ligand must be endocytosed to be modified or repositioned before it binds Notch, and (ii) pulling models, in which ligand must be endocytosed after it binds Notch to exert force that exposes an otherwise buried cleavage site. We demonstrate in vivo that ligands that cannot enter the Epsin pathway nevertheless bind Notch but fail to activate the receptor because they cannot exert sufficient force. This argues against recycling models and in favor of pulling models. Our results also suggest that once ligand binds receptor, activation depends on a competition between Epsin-mediated ligand endocytosis, which induces cleavage, and transendocytosis of ligand by receptor, which aborts the incipient signal.
Graphical Abstract
Summary
Force exerted by endocytosis induces ectodomain cleavage of Notch to initiate signaling.
Introduction
Notch proteins are single-pass transmembrane receptors for cell surface ligands of the Delta/Serrate/Lag2 (DSL) superfamily (reviewed in Kovall et al., 2017). DSL ligands induce extracellular cleavage of the juxtamembrane “Negative Regulatory Region” (NRR) of Notch. This “S2” cleavage is the key ligand-dependent event responsible for activating Notch signal transduction as it renders the remainder of the receptor subject to intramembrane “S3” cleavage that allows the cytosolic domain—a transcription factor—to activate target genes.
How DSL ligands induce the S2 cleavage remains unsolved. Crucially, DSL ligands must be internalized by the endocytic adaptor Epsin in signal-sending cells to activate Notch in signal-receiving cells (Overstreet et al., 2004; Wang and Struhl, 2004; 2005; Chen et al., 2009). Epsin binds Ubiquitinated transmembrane proteins and targets them for Clathrin-mediated endocytosis (Chen et al., 1998; Wendland, 2002). Remarkably, in the Drosophila wing only a small fraction of the prototypical ligand Delta (Dl) is endocytosed via Epsin—yet, only this fraction activates Notch (Wang and Struhl, 2004; 2005). Thus, a central challenge is to determine why DSL ligands must enter the Epsin pathway to induce S2 cleavage.
Two classes of explanation have been proposed (Wang and Struhl, 2004; 2005; Weinmaster and Fischer, 2011; Musse et al., 2012). “Recycling” models posit that Epsin is required before ligand engages Notch, either to modify ligand from an inactive to active state, or to reposition it to a surface domain where it can gain access to Notch. “Pulling” models posit that Epsin is required after DSL ligands bind Notch, to allow endocytosis of Dl to exert force on the NRR exposing the S2 site for cleavage. Diverse experiments have been taken as evidence for or against these models (Weinmaster and Fischer, 2011; Musse et al., 2012; Kovall et al., 2017). However, this evidence has been more circumstantial than compelling.
For recycling models, recent findings now provide the basis for incisive tests of both modification and repositioning mechanisms. Specifically, S2 cleavage can now be induced in cell culture and in vivo using chimeric DSL/Notch pairs in which the native, extracellular domains are replaced by heterologous domains of other ligand/receptor pairs that are unlikely to require Epsin-dependent modifications (Gordon et al., 2015; Morsut et al., 2016; Roybal et al., 2016). If DSL ligands normally need to be modified via Epsin, such chimeric ligand/receptor pairs should bypass this requirement. Conversely, if recycling via Epsin is required to reposition ligand, the chimeric ligands should still require Epsin to access their receptors. Hence, a key question is whether such ligands require Epsin in vivo, either to engage or activate their receptors.
For pulling models, structural studies indicate that the S2 site is normally buried within the NRR unless exposed by an allosteric change induced by ligand binding (Gordon et al., 2007; 2008; Kovall et al., 2017) and atomic force microscopy indicates that force can expose the S2 site for cleavage (Stephenson and Avis, 2012). Also, the NRR of Notch on cultured cells can be cleaved when mechanical tension is applied to ligand bound to the receptor (Ahimou et al., 2004; Gordon et al., 2015). However, these experiments do not test if force is the operative agent in vivo, or if Epsin-mediated ligand endocytosis exerts the necessary force.
Experiments using optical tweezers are also consistent with Epsin-mediated endocytosis exerting force, as knock-down of Epsin reduced the capacity of ligand-expressing cells to displace Notch-coated beads away from the tweezers (Meloty-Kapella et al., 2012). However, Epsin impaired cells also exhibit abnormally high pushing forces on beads, challenging whether the diminished displacement was due to reduced pulling activity by ligand versus an increase in pushing activity by the cell. The beads in these experiments were also ~100 fold larger than endocytic pits and assayed under conditions in which many molecules of ligand on the cell can bind to the bead, consistent with displacement of the bead reflecting general forces exerted by the cell, rather than specific forces exerted by individual ligands or pits.
Here, we ask whether Epsin mediates “recycling” or “pulling” mechanisms in vivo by manipulating the structure and function of chimeric ligand/receptor pairs that reconstitute Epsin-dependent DSL/Notch signaling. Our results argue against recycling and in favor of pulling mechanisms by showing that ligand that cannot enter the Epsin pathway nevertheless binds receptor, but cannot exert sufficient force to cleave the NRR. Unexpectedly, they also suggest that once the intercellular ligand/receptor bridge forms, it is normally resolved by a competition between Epsin-mediated endocytosis of ligand, which induces S2 cleavage of the receptor, and engulfment of the ligand by the receiving cell, which terminates the prospective signal.
Results
Overview
To determine why DSL ligands must be endocytosed via Epsin to activate Notch, we manipulated the structures of Dl and Notch and monitored their signaling capacities and endocytic fates in the developing Drosophila wing. As summarized in the vade mecum in Figure 1, diverse manipulations yielded outcomes that fall mostly into two simple classes. If ligand can enter the Epsin pathway and exert sufficient force on the receptor, we observe productive signaling, as indicated by target gene expression in the receiving cell, and when assayed (†), transendocytosis of the cleaved ectodomain of the receptor into the sending cell (Figure 1A). If it cannot, we observe no signaling, and when assayed (†), transendocytosis of the entire ligand in the opposite direction, into the receiving cell (Figure 1B).
Figure 1. Signaling and endocytic fates of chimeric DSL/Notch pairs.

A) Productive ligand/receptor pairs. Binding of Dl to Notch induces S2 cleavage of the Negative Regulatory Region (NRR), S3 cleavage of the transmembrane (TM) domain, nuclear access of the intracellular domain (ICD) and activation of target genes (e.g., cut); the shed ectodomain is transendocytosed (TE) into the signal sending cell († = ectodomain TE confirmed by experiment).
FSH-Dl/FSHR-N.
The Dl extracellular domain (ECD) was replaced by the β subunit of Follicle Stimulating Hormone (FSHβ) and FSHα was expressed to reconstitute the composite FSH ligand (FSH). Reciprocally, the ligand-binding (EGF) portion of the Notch ectodomain was replaced by the FSH receptor ectodomain (FSHR; see STAR Methods and Figure S1 for composition of all proteins).
FSH-Dl ICD variants.
The Dl ICD was replaced by either (i) an unrelated 38 aa peptide bearing two K’s that target ligand to the Epsin pathway (FSH-Dl-K*; Wang and Struhl, 2004), or (ii) a Myc epitope that includes a LI dipeptide that comprises a Clathrin internalization signal that bypasses the requirement for Epsin (FSH-Dl-myc).
Other ligand/receptor pairs.
Alternative ligand/receptor pairs were generated by swapping the FSH and FSHR domains (FSHR-Dl/FSH-N) or replacing these domains with the Tropomyosin receptor kinase C and Neurotrophin-3 ectodomains (TrkC-Dl/NTF-N), or with GFP and an anti-GFP nanobody (GFP-Dl/Nano-N).
B) Non-productive ligand/receptor pairs. Preventing ligand entry into the Epsin/Clathrin pathway (Ø) by removing Epsin (epsin¯) or by altering the ligand ICD blocks S2 cleavage and results in transendocytosis of ligand into the signal-receiving cell (TE; † = transendocytosis of the ligand ectodomain confirmed by experiment).
FSH-Dl and other chimeric ligand ICD mutants
FSH-Dl, FSH-Dl-K* and FSH-Dl-myc were blocked from entering the Epsin/Clathrin pathway by mutating the cytosolic K’s to R’s (FSH-Dl-K>R, FSH-Dl-R*), or the LI internalization signal to AI (FSH-Dl-mycmut).
C-F) FSH-Dl/FSHR-N signaling and TE require FSHα to reconstitute the functional FSHαβ heterodimer (C), and Kuz and Net to execute the S2 and S3 cleavages (F). FSH-Dl, and not FSH-Dl-K>R, can activate FSHR-A2-N chimeric receptors if they carry any of three disease-associated A2 variants (D,E). FSH-Dl does not activate receptors carrying A2 domains that are cleaved less readily in response to mechanical tension (wildtype, WT, and MV1528; D).
Critical to our analysis, we devised a genetic strategy, Mosaic Analysis by Promoter Swap (MAPS), to subdivide the developing wing epithelium into mutually exclusive subpopulations of chimeric ligand and receptor expressing cells, such that ligand and receptor interact only in trans wherever the two subpopulations abut. We outline this strategy first, and then present our results as they discriminate between the proposed roles of Epsin in recycling (modification/repositioning) versus pulling models.
Mosaic Analysis by Promoter Swap
In essence, we use heat shock induced, Flp/FRT-mediated mitotic recombination (Golic, 1991) to generate clones of cells that express one of the two proteins, e.g. the receptor, in a background of cells that express the other, e.g. the ligand (Figures 2A, S2A; STAR Methods). The resulting, mutually exclusive subpopulations of receptor and ligand expressing cells are distinguished by epitope tagging either the ligand or receptor (Figure banners and/or legends; STAR Methods). Finally, both proteins are expressed in the prospective wing under Gal4/UAS control, using nubbin.Gal4 (nub.Gal4) or rotund.Gal4 (rn.Gal4) transgenes. Peak Notch activation in this tissue is normally restricted to a thin stripe of “border” cells flanking the dorso-ventral (D/V) compartment boundary (Blair, 1997; Figure 2B): this allows us to monitor signaling between UAS>ligand and UAS>receptor cells by assaying for ectopic expression of Notch target genes, such as cut or wingless (wg; Figure 2C,D).
Figure 2. FSH-Dl/FSHR-N signaling in the developing wing.

A) Mosaic Analysis by Promoter Swap (MAPS). This strategy relies on (i) the use of ligand and receptor encoding transgenes, each carrying an FRT (“>”) immediately upstream of the coding sequence, (ii) a UAS promoter in front of one coding sequence (e.g., the ligand) and the absence of a functional promoter (Ø) in front of the other (e.g., the receptor), and (iii) the insertion of both transgenes at the same genomic docking site. Heterozygous UAS>ligand/Ø>receptor cells express only the ligand (blue). However, Flp-mediated mitotic recombination (red “X”) generates two daughter cells, one of which now expresses only the receptor (black) whilst the other continues to express only the ligand (blue). The resulting, mutually exclusive subpopulations of receptor and ligand expressing cells are distinguished by epitope tagging either the ligand or receptor, in this case HRP-tagged Dl, stained blue (see Figure S2 and STAR Methods).
B) The wing primordium comprises a circular domain of cells (blue) within the wing imaginal disc (grey), which is subdivided into dorsal (D) and ventral (V) compartments (D/V boundary in black); the middle panel shows HRP-tagged Dl expression in the wing. D cells express the DSL ligand Serrate as well as a glycosyl-transferase Fringe, whereas V cells express Dl: Fringe biases Notch to respond to Dl whereas absence of Fringe biases Notch to respond to Serrate; Notch target genes (e.g., cut, yellow) are induced on both sides of the boundary. Here and in the remaining Figures, UAS transgenes are expressed under the wing specific driver nub.Gal4 (or similarly rn.Gal4), and only the epitope tags relevant to the experiment are indicated.
C) UAS>Dl cells (blue) induce ectopic Cut (yellow) in abutting UAS>Notch cells (black) in the D but not the V compartment; coexpression of Neur overcomes the Fringe-dependent bias and results in ectopic Cut in both compartments.
D) FSH-Dl/FSHR-N signaling induces ectopic Cut in both compartment, up to ~10-20 cell diameters from the D/V boundary in wildtype discs, and up to ~30 or more cell diameters in Neur coexpressing discs.
E) FSH-Dl/FSHR-N signaling requires FSHα, even when Neur is coexpressed. Scale bars: 50μm.
To introduce this approach as well as DSL/Notch signaling in the wing, we present a MAPS experiment in which we generated mutually exclusive subpopulations of UAS>Dl and UAS>Notch cells, the former encoding HRPDl, a biologically active, HRP tagged form of Dl (Wang and Struhl, 2004), to distinguish UAS>Dl from UAS>Notch cells (blue versus black in Figures. 2B,C). Normally, Notch in dorsal (D) compartment cells responds preferentially to Dl from ventral (V) compartment cells, whereas Notch in V compartment cells responds preferentially to the other fly DSL ligand Serrate (Ser) from D compartment cells (Blair, 1997; Figure 2B). This bias can, however, be overcome by coexpressing Dl with the E3 ubiquitin ligase Neuralized (Neur), which increases Dl signaling activity by driving its ubiquitination and recruitment into the Epsin pathway (Deblandre et al., 2001; Lai et al., 2001; Wang and Struhl, 2004; 2005). In accord, UAS>Dl cells induce ectopic Cut in abutting UAS>Notch cells in the D compartment of wild type discs, and in both compartments of discs that coexpress Neur (Figure 2C).
Evidence against ligand modification models
To test if nascent DSL ligands must be modified via Epsin-dependent recycling, we replaced the native ligand/receptor interaction domains of Dl and Notch with the corresponding domains of mammalian Follicle Stimulating Hormone (FSH) and its receptor (FSHR) to create a chimeric FSH-Dl/FSHR-N ligand/receptor pair that should bypass any such requirement (Figure 2D). We chose the ectodomains of FSH and FSHR because (i) FSH is a secreted signaling molecule, so unlikely to require Epsin-dependent modification to bind FSHR (Fox et al., 2001), (ii) FSH is heterodimer composed of distinct α and β subunits, allowing us to reconstitute a functional FSH-Dl ligand in a conditional manner by using the β subunit in place of the Dl ectodomain and providing or withholding expression of the α subunit, and (iii) FSH and the ectodomain of FSHR interact as monomers (Fan and Hendrickson, 2005), providing a one-to-one ligand/receptor interaction. As shown below, the FSH-Dl/FSHR-N pair recapitulates native DSL/Notch signaling independent of endogenous DSL ligands and Notch, obviating confounding interactions between the chimeric proteins and their native counterparts.
Activation of FSHR-N by FSH-Dl
We first asked if FSH-Dl can activate FSHR-N by using MAPS to generate mutually exclusive subpopulations of UAS>FSHR-N and UAS>FSH-Dl cells. We found that FSH-Dl cells can induce abutting FSHR-N cells to ectopically express Cut, but only when FSHα is supplied to reconstitute the composite FSH ligand (Figures. 2D,E). Notably, FSH-Dl/FSHR-N signaling induced ectopic Cut in both the D and V compartments, as expected given that the biased response observed for native Dl/Notch signaling (Figure 2C) depends on a special attribute of the Notch ectodomain, namely sugar modification by the glycosyl-transferase Fringed (Blair, 1997).
FSH-Dl/FSHR-N signaling does, however, appear weaker than Dl/Notch signaling. Here, and previously (Wang and Struhl, 2004), the ability of ectopic Dl/Notch signaling to induce cut and wg expression peaks in cells closest to the D/V boundary and declines further away. Hence, ligands with reduced signaling activity induce ectopic Cut and Wg only when the receiving cells are located close to the boundary. For FSH-Dl/FSHR-N signaling, ectopic Cut is limited to within ~10-20 cell diameters of the boundary (Figure 2D); by contrast, UAS>N cells respond to UAS>Dl cells in the D compartment, 30 or more cell diameters away (Figure 2C). However, FSH-Dl/FSHR-N signaling, like Dl/Notch signaling, is enhanced by Neur coexpression, resulting in ectopic Cut 30 or more cell diameters away in both compartments—an output that is still strictly dependent on FSHα (Figures 2D,E).
Importantly, FSH-Dl does not require endogenous Dl or Ser to signal. Instead, UAS>FSH-Dl Dl¯ Ser¯ cells, which are devoid of both ligands, still induce ectopic Cut in abutting UAS>FSHR-N cells (Figures. S2C, S3C). Likewise, coexpression of Scabrous, which inhibits the response of the ligand-binding domain of Notch to Dl (Lee et al., 2000), does not reduce activation of FSHR-N by FSH-Dl (Figure S4A). Thus, FSH-Dl has the intrinsic ability to activate FSHR-N, albeit less potently than native Dl/Notch signaling.
FSH-Dl/FSHR-N signaling recapitulates the basic parameters of Dl/Notch signaling
Notch activation normally depends on Kuz (S2) and γ-secretase (S3) cleavages. Similarly, UAS>FSHR-N cells that lack either Kuz or the γ-secretase component Nicastrin (Net) fail to express Cut in response to UAS>FSH-Dl cells (Figure S3A,B).
Further, FSH-Dl/FSHR-N signaling, like Dl/Notch signaling, exhibits “cis-inhibition”. When Dl is coexpressed, it binds Notch non-productively in cis, reducing or abolishing the ability of Notch to be activated in trans by Dl on neighboring cells (del Álamo et al., 2011). Likewise, when we generated homozygous UAS>FSH-Dl and UAS>FSHR-N twin clones in a background of heterozygous UAS>FSH-Dl/UAS>FSHR-N cells, we found that the heterozygous cells are refractory to FSH-Dl signal from abutting, UAS>FSH-Dl cells (Figure S3D).
FSH-Dl/FSHR-N signaling requires Epsin-dependent ligand endocytosis
If Epsin is normally required to modify the ectodomains of native DSL ligands, FSH-Dl should escape this requirement. However, two independent approaches indicate that signaling by FSH-Dl still depends on Epsin.
First, FSH-Dl clones that are concomitantly null for liquid facets (Iqf), the gene encoding the sole Drosophila Epsin (henceforth, epsin) did not induce Cut expression in abutting FSHR-N cells, in contrast to control FSH-Dl clones generated in the same discs that retain Epsin function (Figure 3A). Moreover, we obtained the same result even when FSH-Dl signaling was boosted by Neur coexpression (Figure S5). Hence, FSH-Dl signaling depends strictly on Epsin activity, like native DSL signaling (Overstreet et al., 2004; Wang and Struhl, 2004; 2005).
Figure 3. Signaling by FSH-Dl requires access to the Epsin/Clathrin endocytic pathway.

A) epsin¯ clones coinduced in wing discs composed of mutually exclusive subpopulations of FSH-Dl and FSHR-N expressing cells (epsin¯ clones are marked “black” by the absence of anti-Epsin staining and outlined in yellow; FSHR-N cells are marked red by a Cherry tag in FSHR-N; FSH-Dl cells are marked black by the absence of Cherry; here and in subsequent Figures, the relevant clonal genotypes are outlined and color coded as in the banners, and boxed regions are shown at higher magnification). UAS>FSH-Dl epsin¯ cells do not induce Cut in abutting FSHR-N cells (empty arrow heads), in contrast to UAS>FSH-Dl cells that retain wild type epsin function (filled arrow heads; the white asterisk marks the loss of Cut expression where the epsin¯ clone abuts the D/V boundary; see Figures S3-S5). Scale bar: 10μm.
B) Signaling by FSH-Dl variants requires that they access the Epsin/Clathrin pathway. FSH-Dl cells (blue) induce ectopic Wg (yellow) in adjacent FSHR-N cells (black) when the ligand has access to the Epsin pathway or can be targeted directly to Clathrin, bypassing the requirement for Epsin (FSH-Dl, FSH-Dl-K*, and FSH-Dl-myc; Figure 1A). In contrast mutated forms of these ligands that cannot access the Epsin/Clathrin route (FSH-Dl-K>R, FSH-Dl-R* and FSH-Dl- mycmut; Figure 1B) do not. Scale bars: 50μm.
C) FSHR-Dl/FSH-N, TrkC-Dl/NTF-N, and GFP-Dl/Nano-N chimeric ligand pairs (Figure 1A) all signal, albeit weakly in the case of GFP-Dl/Nano-N, in response to their corresponding ligand, but not the K>R variant of that ligand. Scale bars: 50μm.
Second, we used cytosolic domain variants of FSH-Dl (Figures 1, S1) to manipulate targeting of ligand to the Epsin or Clathrin endocytic pathway as previously described (Wang and Struhl, 2004; 2005), and found that access to both pathways is essential for signaling. Specifically, we (i) allowed or blocked ubiquitination of the Dl cytosolic domain by leaving all 12 Lysines intact or mutating them to Arginine (FSH-Dl versus FSH-Dl-K>R) or by removing the entire domain (FSH-Dl-AC); (ii) replaced the cytosolic domain with wild type or K-to-R mutant versions of a heterologous peptide that independently targets ligand to the Epsin pathway via ubiquitination (FSH-Dl-K* versus FSH-Dl-R*); and (iii) replaced the cytosolic domain with wild type or mutant versions of the classic Myc epitope (FSH-Dl-myc and FSH-Dl-rnycmut), which serendipitously contains a LI dipeptide internalization signal for Clathrin-mediated endocytosis (Letourneur and Klausner, 1992) and is sufficient to bypass the requirement for native Dl to enter the Epsin pathway (Wang, 2006). We find that the wild type versions of all of these ligands activate FSHR-N whereas their mutant derivatives do not (Figure 3B).
To further assess the dependence of chimeric ligands on Epsin, we tested three additional chimeric ligand/receptor pairs for which the heterologous ligand ectodomain is just as unlikely as FSH to require an Epsin-dependent modification: (i) we swapped the FSH and FSHR domains of FSH-Dl and FSHR-N, to create the reciprocal FSHR-Dl/FSR-N pair; (ii) we used the ligand/receptor binding domains of Neurotrophin-3 (NTF) and the Tropomyosin receptor kinase C (TrkC; Ultsch et al., 1999) to create a TrkC-Dl/NTF-N pair, and (iii) we used GFP and a single chain anti-GFP nanobody (Rothbauer et al., 2008) to create a GFP-Dl/Nano-N pair (Gordon et al., 2015; Figures. 1A, S1). In all three cases, we again observed productive signaling, albeit only weakly so for the GFP-Dl/Nano-N pair, but only in response to wild type, and not K>R, versions of each ligand (Figure 3C). We note (i) that the TrkC and NT3 ectodomains differ from those of the FSH/FSHR and GFP/Nano pairs in functioning as obligate homodimers, but this did not affect signaling, and (ii) that all four ligand/receptor pairs exhibit cis-inactivation (as shown for FSH-Dl/FSRH-N; Figure S5), suggesting that cis-inactivation is not due to any special property of the native ligand/receptor interaction aside from the capacity of their ectodomains to bind.
In sum, all four chimeric ligands, like native Dl, must enter the Epsin pathway to activate their cognate receptors, arguing against Epsin-dependent recycling being required to modify DSL ligands so that they can activate Notch.
Evidence against ligand repositioning models
Repositioning models, like modification models, posit that Dl must undergo Epsin-mediated recycling before it encounters Notch, albeit to be relocated to a position on the cell surface where it can access receptor on neighboring cells rather than to be modified so that it can bind receptor. Hence, one can test repositioning models by asking if Epsin is required for ligand to gain access to the receptor. Since ligand/receptor bridges that cannot undergo S2 cleavage should remain at the cell surface until cleared by uptake into either the sending or receiving cell, assaying for transendocytosis of either ectodomain into the opposing cell provides a way to determine if ligand can access receptor in the absence of Epsin.
To monitor transendocytosis of either domain, we used MAPS to generate mutually exclusive subpopulations of UAS>FSH-Dl and UAS>FSHR-N cells in which one of the two subpopulations also carries a UAS.YFP-Rab5CA transgene (Figures S6A, S1). YFP-Rab5CA is a constitutively active form of Rab5 that impairs the maturation of early endosomes resulting in wing cells that contain enlarged endosomes that accumulate cargo proteins but appear otherwise to develop normally (Rink et al., 2005; Zhang et al., 2007). Hence, when YFP-Rab5CA expression is restricted to one of the two subpopulations, the enlarged, YFP-tagged endosomes accumulate any protein they have transendocytosed from cells of the abutting subpopulation (as validated for transendocytosis of the cleaved ectodomain of FSHR-N by FSH-Dl; Figure S6B).
Ligand that cannot enter the Epsin/Clathrin pathway does not transendocytose receptor
To test if FSH-Dl ligands that cannot enter the Epsin/Clathrin pathway form uncleaved ligand/receptor bridges that are transendocytosed into the sending cell, we generated interfaces between cells that express a Cherry tagged form of FSHR-N (FSHR-CherryN) and cells that express YFP-Rab5CA plus FSH-Dl or a cytosolic domain variant thereof (Figures 1, 3B). All three ligands that have access to the Epsin/Clathrin pathway (FSH-Dl, FSH-Dl-K* and FSH-Dl-myc) serve as controls, since they induce S2 cleavage, and, as expected, transendocytose the ectodomain of the receptor in an FSHα-dependent fashion (Figure 4A, box #1; Figure S7C). By contrast, all of the mutated forms that cannot enter the Epsin/Clathrin pathway fail to show detectable transendocytosis of the receptor ectodomain (Figure 4B, box #3 and Figure S7A). Thus, FSH-Dl that cannot enter the Epsin/Clathrin pathway either does not have access to FSHR-N, or if it does, forms ligand/receptor bridges that are not cleared by uptake into the ligand expressing cells.
Figure 4. Transendocytosis of the FSH-Dl/FSHR-N ectodomain bridge depends on ligand entry into the Epsin/Clathrin pathway.

A) FSH-Dl variants that can access the Epsin/Clathrin pathway induce S2 cleavage of FSHR-N and transendocytose the S2-cleaved ectodomain of the receptor into the signal-sending cell, as indicated by accumulation of the Cherry tag (red) in YFP-Rab5CA endosomes (endosome #1). No transendocytosis of the ligand ectodomain is detected in the other direction, into YFP-Rab5CA endosomes in the signal-receiving cell (endosome #2), as indicated by the absence of accumulation of the HRP tag (blue). Here, and in (B), accumulation of the Cherry and HRP tags is assayed in separate experiments in which YFP-Rab5CA is expressed either in the sending or receiving cell (see Figures S6, S7).
Box #1) Images show abutting populations of UAS>FSH-Dl, UAS.YFP-Rab5CA cells (YFP labeled endosomes, green) and UAS>FSHR-N cells (red), for the three FSH-Dl variants that can enter the Epsin/Clathrin pathway (FSH-Dl, FSH-Dl-K* and FSH-Dl-myc). The magnified images show Cherry accumulation (red) inside YFP-Rab5CA endosomes in the ligand-expressing cells for all three ligands (middle column), as well as grey scale images of the Cherry signal (right column).
Box #2) Similar to box 1, except that YFP-Rab5CA is coexpressed with FSHR-N and the staining is for the HRP-tagged ectodomain of the ligand (blue). No transendocytosed HRP-tagged ligand is detectable in the YFP-Rab5CA endosomes.
B) All three FSH-Dl variants that are excluded from the Epsin/Clathrin pathway (FSH-Dl-K>R, FSH-Dl-R* and FSH-Dl-mycmut) do not induce S2 cleavage or transendocytose the receptor ectodomain into the sending cell, as indicated by the absence of Cherry accumulation in YFP-Rab5CA endosomes (endosome #3). Instead, the ectodomains of all three ligands are transendocytosed in the opposite direction, into the receiving cell, as indicated by accumulation of the HRP tag (blue; endosome #4).
Boxes #3 and #4) Labeled and presented as in boxes #1 and #2, but with opposite results. Scale bars: 5μm.
Ligand that is excluded from the Epsin/Clathrin pathway is transendocytosed by receptor
To test if FSH-Dl ligands that cannot enter the Epsin/Clathrin pathway form ligand/receptor bridges that are cleared by uptake into receiving cells, we used the same strategy, except expressing YFP-Rab5CA in the FSHR-N rather than the FSH-Dl expressing cells and assaying for transendocytosis of the HRP tagged ligand. In this case we obtained a positive result, namely that all four mutated forms of the ligand, which cannot enter the Epsin/Clathrin pathway, were transendocytosed into the receptor expressing cell in an FSHα-dependent fashion (Figures 4B, box #4; Figures S7B, S7D). By contrast, we failed to detect evidence for transendocytosis of the wild type forms of these ligands, which can enter the Epsin/Clathrin pathway and induce S2 cleavage of the receptor (Figures 4A, box #2).
To determine if abolishing Epsin activity also results in the unidirectional transendocytosis of the ligand ectodomain into receiving cells, we concomitantly induced clones of epsin null cells while using MAPS to generate mutually exclusive subpopulations of FSH-Dl and FSHR-N expressing cells that also express YFP-Rab5CA (Figures 5A; S2B; STAR Methods). For FSH-Dl expressing clones that lack Epsin (box #1), we observe transendocytosis of the HRP tagged ectodomain of the ligand into abutting receptor expressing cells but no evidence for transendocytosis of the Cherry-tagged receptor ectodomain into the ligand expressing cells—corroborating the results obtained with mutant variants of FSH-Dl that are excluded from the Epsin/Clathrin pathway. In contrast, for FSH-Dl expressing clones that retain Epsin function (box #2), we obtain the opposite result: the receptor ectodomain accumulates in endosomes of the sending cells, whereas no accumulation of the ligand ectodomain is detected in the receiving cells.
Figure 5. FSHR-N versus FSH-Dl transendocytosis depends on Epsin.

A, top) Transendocytosis of the extracellular FSH-Dl/FSHR-N bridge was assayed using HRP and Cherry extracellular tags, as in Figure 4, except that YFP-Rab5CA is expressed in both ligand and receptor expressing cells. Two independent types of clones were induced within the same disc. First, epsin¯ clones, outlined in yellow and marked “black” by the absence of Epsin (green, middle panel). Second, UAS>FSH-Dl clones (HRP, blue) generated by MAPS in a background of UAS>FSHR-N cells (Cherry, red), shown outlined in red (right panel). Some UAS>FSH-Dl clones are null for epsin (box #1); others are wildtype for epsin (box #2).
Box #1). FSH-Dl clone that is epsin¯ (blue, in the cartoon) in a background of FSHR-N cells (pink). Grey scale images of HRP and Cherry are shown in the middle and right panels. The FSH-Dl ectodomain accumulates in puncta in the abutting FSHR-N expressing cells (e.g., white arrowhead), whereas no accumulation of the FSHR-N ectodomain is detected in abutting FSH-Dl expressing cells (empty arrowhead).
Box #2). FSH-Dl expressing clone that is wildtype for epsin depicted and imaged as in the middle row. The results are reciprocal: the FSHR-N ectodomain accumulates in puncta in the neighboring FSH-Dl cells (e.g., white arrowhead right panel). In contrast, little or no accumulation of the FSH-Dl ectodomain is detected in puncta in the abutting FSHR-N cells (empty arrowhead, middle panel). Scale bar: 50μm.
B) FSH-Dl carrying an intracellular HA tag (FSH-DlHA) was used to monitor the fate of the Dl ICD (blue) following transendocytosis of the ligand from epsin¯ cells into FSHR-N receiving cells. As in A, two independent types of clones were induced within the same disc, namely, (i) epsin¯ clones (labelled as in A), and (ii) UAS>FSH-DlHA clones (blue) generated by MAPS in a background of UAS>FSHR-N cells (red). HA accumulation is apparent in puncta of FSHR-N cells that abut FSH-Dl epsin¯ cells (white arrowhead; grey scale image), but not in FSHR-N cells that abut wildtype FSH-Dl cells (empty arrow head). Taken together with the evidence of ligand transendocytosis in box #1 in (A), this indicates that the entire ligand has been internalized by the receiving cell. Concordant with the results shown in (A), transendocytosis of the receptor ectodomain in the opposite direction depends on whether the signal-sending cell is wildtype or mutant for epsin (middle panel): Cherry labeled puncta are evident in abutting FSH-Dl cells that retain epsin activity (red arrowhead), but are absent from FSH-Dl epsin¯ cells (empty red arrowhead). Scale bar: 10μm.
Thus, FSH-Dl that cannot gain entry to the Epsin/Clathrin pathway nevertheless has access to FSHR-N, contradicting the model that Epsin/Clathrin-dependent recycling is normally required to reposition ligand so that it can engage receptor.
Ligand that cannot enter the Epsin/Clathrin pathway is transendocytosed, intact, by receptor
Receptor-dependent transendocytosis of ligand that cannot enter the Clathrin/Epsin pathway could occur via an S2-like cleavage of the ligand, allowing receptor to internalize the severed ligand ectodomain, or by uptake of the entire ligand, e.g., by engulfment of a patch of the sending cell surface. To distinguish between these possibilities, we repeated the experiment above (Figure 5A), to assay for unidirectional transendocytosis of the ligand ectodomain into receiving cells, only this time using FSH-HRPDlHA, which carries a cytosolic HA tag as well as an extracellular HRP tag. Under these conditions, we detect accumulation of the intracellular HA tag in FSHR-N expressing cells that abut FSH-Dl expressing epsin null cells (Figure 5B). In contrast, no such accumulation is observed when the abutting FSH-Dl expressing cells retain epsin function; instead, the ligand expressing cells transendocytose the Cherry tagged ectodomain of the receptor.
Thus, the capacity of ligand to enter the Epsin/Clathrin pathway dictates whether ligand binding induces S2 cleavage and transendocytosis of the severed receptor ectodomain into the sending cell, or alternatively, results in the non-productive transendocytosis of the ligand, in its entirety, into the receiving cell.
Evidence for pulling models
Both structural and biophysical studies indicate that the S2 site is buried within the NRR and is exposed for cleavage by ligand binding to the amino-terminal EGF-repeat containing portion of Notch (Kovall et al., 2017). The capacity of all four chimeric ligands (FSH-Dl, FSHR-Dl, TrkC-Dl and GFP-Dl) to recapitulate Epsin/Clathrin-dependent activation of their corresponding receptors argues for an allosteric change that is intrinsic to the NRR as a physical link between the ligand-bound ectodomain and transmembrane domain of the receptor. Specifically, as posited in “pulling” models, the NRR could function as a force sensor that is unfolded by a threshold level of mechanical tension generated across the ligand/receptor bridge. If so, a heterologous force sensor that can be cleaved in response to a similar threshold of mechanical tension should be able to substitute for the NRR.
We have tested this using the A2 domain of von Willibrand Factor (vWF), a well-characterized force sensor. The A2 domain requires a defined threshold of mechanical tension of ~ 8pN to render an otherwise hidden target site subject to cleavage by ADAM proteolysis (Tsai et al., 1994; Tsai, 1996; Zhang et al., 2009). This is significantly higher than the threshold of 3.5 – 5.4 pN for the NRR determined by comparable experiments (Gordon et al., 2015). However, several disease-related variants of the A2 domain have lower force thresholds in blood (Hassenpflug, 2006; Xu and Springer, 2013) and kinetic analysis of one such variant, R1597W, suggests that it is cleaved at a threshold ~ 2 pN lower than wild type A2 (Xu and Springer, 2013), close to if not overlapping the range of the NRR. However, even if the NRR functions, in vivo, as a force sensor, the capacity of variants such as R1597W to substitute for it would require that (i), Drosophila cells have an endogenous protease, whether Kuz or some other, that can cleave the exposed A2 site, and (ii) the resulting cleaved form of the receptor has a sufficiently small ectodomain stub to be subject to S3 cleavage (Struhl and Adachi, 2000). Nevertheless, we find that some disease-related A2 variants, including R1597W, can indeed substitute for the NRR in mediating Epsin-dependent FSH-Dl/FSHR-N signaling, indicating that these requirements are met.
We first tested FSHR-A2WT-N, a form of FSHR-N that contains the wild type A2 domain in place of the NRR. We failed to detect ectopic Cut expression induced by UAS>FSH-Dl cells in abutting UAS>FSHR-A2WT-N cells, even when the UAS>FSH-Dl and UAS>FSHR-A2WT-N transgenes were homozygous and the experiment performed at 29°C—both conditions that should optimize expression of the two proteins (Figure 6A; STAR Methods).
Figure 6. Signal transduction by FSHR-N receptors containing the force sensing A2 domain of von Willibrand Factor in place of the NRR.

A) UAS>FSH-Dl sending cells (blue) fail to induce UAS>FSHR-A2-N receiving cells (black, outlined in red) to ectopically express Cut (white in upper panels, yellow in the lower panels) when the A2 domain is wildtype or carries the disease associated M1528V mutation, which modestly elevates its potential to be cleaved by mechanical stress in blood. B,C) FSHR-A2-N receptors that contain any one of three other disease-associated mutant A2 domains that are more readily cleaved in blood are activated by FSH-Dl, as indicated by ectopic Cut expression (B, left); the response is limited to 5-10 cell diameters of the D/V boundary indicating that it is weaker than canonical FSH-Dl/FSHR-N signaling. Activation of all three receptors requires Epsin-mediated ligand endocytosis, as indicated by their failure to respond to FSH-Dl-K>R (B, right), and by the requirement for FSHα (C). Scale bar: 10μm.
We next tested, FSHR-A2R1597W-N, using the same optimized conditions as for FSHR-A2WT-N, and obtained a positive result: ectopic expression of Cut (Figure 6B). The response was confined to within 5-10 cell diameters of the D/V compartment boundary, rather than within 10-20 cell diameters, as observed for FSHR-N. This more restricted response could reflect less efficient S2 or S3 cleavage, as noted above, and/or a modest difference in the tuning of the R1597W A2 domain relative to the native NRR.
Further corroborating this result, two other disease variants of the A2 domain, E1638K and I1628T, that result in similarly elevated levels of proteolysis in blood (Hassenpflug, 2006), and hence are likely cleaved in response to a similar force threshold, behaved like R1597W when used in place of the NRR (Figure 6B). Importantly, for all three of these A2 variant receptors, signaling was FSHα dependent (as shown for FSHR-A2E1638K-N; Figure 6C), and was only observed in response to wild type FSH-Dl but not its FSH-Dl-K>R mutant derivative (Figure 6B). Thus, all three respond in a manner that depends on ligand binding as well as ligand entry into the Epsin pathway.
Finally, we tested a fourth A2 variant, M1528V, that is associated with a markedly weaker effect on vWF cleavage in blood than the first three, and hence appears to be tuned to a higher force threshold (Hassenpflug, 2006). The resulting FSHR-A2M1528V-N receptor, like the wildtype FSHR-A2-N receptor, appears refractory to signaling by FSH-Dl (Figure 6A), reinforcing the correlation between the force necessary to render the different A2 domains subject to proteolysis in blood and their capacity to function in place of the NRR.
We conclude that Epsin-dependent ligand endocytosis exerts a specific level of force that is sufficient to render the first three A2 variants—but neither the M1528V nor the wild type A2 domain—subject to an S2-like cleavage. They thus provide in vivo evidence that the native NRR need only function as an equivalent force sensor to the R1597W, E1638K and I1628T variants to mediate activation of the receptor by ligand.
Discussion
The pivotal regulated event in Notch signal transduction is S2 cleavage of the receptor, but the mechanism by which ligand binding exposes the S2 cleavage site in vivo has remained unsolved. The absolute requirement for ligand to be endocytosed by Epsin provides a challenge as well as a potential key to elucidating this mechanism.
Evidence against “recycling” models and for “pulling” models
To distinguish between recycling and pulling models—the two major classes proposed to explain Epsin-dependent Notch activation—we reconstituted Dl/Notch signaling in vivo using chimeric ligands and receptors that allow us to test both models by altering the structural domains on which they depend. This strategy allowed us to dissect the basic requirements for S2 cleavage independent of any special attributes of the native proteins and under all of the normal constraints that operate in intact epithelia in vivo.
First, we negated models in which ligand has to be modified via Epsin-dependent recycling. We showed that four different chimeric ligand/receptor pairs in which the extracellular binding domains of native Dl and Notch have been replaced with those of unrelated ligands and receptors still require Epsin. This finding indicates that signaling does not depend on any special property of the native ectodomains other than their ability to bind to each other. More incisively, it argues against modification models as all four chimeric ligands are unlikely to require Epsin-dependent modification, yet all four still depend on Epsin to signal. This finding corroborates biophysical evidence that DSL ligands bind to Notch with similar affinity whether or not they have undergone endocytic recycling (Shergill et al., 2012). We note that our results do not rule out an auxiliary role of recycling-dependent modification in maximizing native Dl signaling in some contexts (e.g., Benhra et al., 2010); however, any such role is distinct from the requirement for Epsin, which is fundamental to the activation mechanism.
Second, we refuted repositioning models by showing that ligand can still bind receptor in vivo, even when it cannot undergo Epsin-dependent endocytosis. Further, ligands that are targeted directly for Clathrin-mediated endocytosis, bypassing the normal requirement for Epsin, can still bind receptor when precluded from the Clathrin pathway. Again, these results do not exclude repositioning as a means to augment signaling in some contexts, but any such provision would be supplemental to the basic activation mechanism.
Third, and in contrast, we obtained positive evidence for pulling models by showing that the A2 domain from von Willibrand Factor—a bona-fide force sensor (Tsai et al., 1994; Zhang et al., 2009)—can substitute for the NRR in mediating Epsin-dependent activation of our canonical FSHR-N chimera. Importantly, signaling was only observed when we used disease-related A2 variants that are more readily cleaved in blood than wildtype A2, correlating with biophysical data that such variants, as well as the native NRR, are tuned to a lower force threshold (Hassenpflug, 2006; Xu and Springer, 2013; Gordon et al., 2015). We conclude that Epsin-mediated ligand endocytosis exerts a distinct level of mechanical tension on the ligand/receptor bridge that is both necessary and sufficient to induce S2 cleavage in vivo.
The requirement for a distinct level of force may explain why all four chimeric ligand/receptor pairs signal less strongly than the native DSL/Notch pair. If binding of such chimeric pairs is of lower affinity than DSL/Notch binding, which employs a specialized catch bond mechanism (Marshall et al., 2003) to stabilize and prolong binding (Luca et al., 2017), the resulting bridges might be more likely to dissociate before the S2 site is cleaved. Conversely, disease associated mutations of the NRR that cause adventitious receptor activity (Malecki et al., 2006) may lower the force-threshold necessary for S2 cleavage. Indeed, a chimeric FSHR-N receptor with a mutation in the NRR that confers hyper-sensitivity to ligand (Lieber et al., 1993) shows a more sensitive response to FSH-Dl (Figure S4B).
Why does ligand that is internalized by non-Epsin mediated endocytosis fail to activate receptor?
A striking aspect of the requirement for Epsin in the Drosophila wing is that only a small fraction of Dl appears to be ubiquitinated, allowing it to enter the Epsin/Clathrin pathway and signal, whereas the larger fraction fails to signal despite being efficiently endocytosed by other mechanisms (Wang and Struhl, 2004; 2005). However, we found that ligand that cannot be ubiquitinated or that cannot enter the Epsin/Clathrin pathway can still bind receptor on neighboring cells, posing the question of why Dl fails to induce S2 cleavage when it is internalized by other means.
Our evidence suggests that once Dl binds Notch, both the sending and receiving cell compete for uptake of the bridge, with the outcome being determined by whether the ligand can enter the Epsin pathway (Figure 7). If it can, the activating S2 cleavage occurs and the severed bridge is internalized by the sending cell. If not, the bridge remains intact and is engulfed, together with the entire receptor-bound ligand by the receiving cell. Importantly, we do not detect evidence of ligand engulfment by the receiving cell under normal conditions (e.g., Figure 4A box #2), even though most Dl may not be ubiquitinated when it first engages receptor (Wang and Struhl, 2004; 2005). Hence, we infer that Dl that is not ubiquitinated when it first binds receptor is rapidly induced to become ubiquitinated. Such receptor-dependent ubiquitination should target most if not all Dl that binds Notch to the Epsin/Clathrin pathway, activating S2 cleavage and leaving few if any non-productive bridges behind for engulfment by the receiving cell (Figure 7). Conversely, Dl that has not yet encountered receptor or is bound in cis, would not be in a position to signal, and may comprise the majority fraction that is internalized non-productively by non-Epsin pathways.
Figure 7. Dl/Notch signaling and competition between sending and receiving cells for the ligand/receptor bridge.

A) Prior to engagement with Notch in trans, Dl exists in two forms: free, and sequestered in cis with Notch; both forms can be internalized via non-Epsin routes. Binding to Notch in trans induces a race between ubiquitination of Dl in the sending cell (B) and uptake of Dl into the receiving cell (C).
B) Sending cell wins: Epsin targets ubiquitinated Dl for Clathrin mediated endocytosis, applying force across the ligand receptor bridge that opens up the NRR (depicted as a spring) to uncover the S2 site for cleavage. Ectodomain shedding renders the remainder of the receptor subject to S3 cleavage, allowing the cytosolic domain to enter the nucleus and activate target genes. The available evidence suggests that Dl ubiquitination is normally induced by receptor binding (see Discussion).
C) Receiving cell wins: Ligand is internalized in its entirety by receptor-mediated ligand transendocytosis, possibly by engulfment of a patch of the sending cell surface in which the ligand is embedded, as depicted. Under normal conditions, receptor induced ubiquitination of ligand triggers Epsin-dependent S2 cleavage of the receptor before the receptor can transendocytose the ligand. However, manipulations or natural processes that compromise access of ligand to the Epsin/Clathin pathway, tip the competition in favor of the receiving cell, quenching the signal.
Fate of the intercellular ligand/receptor bridge and implications for DSL/Notch signaling
Our analysis suggests that Dl binding to Notch in trans initiates a race between a productive interaction (receptor-induced ubiquitination, Epsin-mediated ligand endocytosis and S2 cleavage) versus a non-productive interaction (receptor-mediated uptake of the ligand into the receiving cell; Figure 7). This scenario raises three intriguing questions.
First, how does contact with Notch induce ubiquitination of Dl? Because the phenomena we describe are observed for chimeric ligands in which the entire ectodomain is replaced by heterologous ligand domains, we posit that the influence of Notch on the ubiquitination of Dl does not depend on any special quality of the native ligand/receptor interaction, but instead results from recruitment of ligand into an intercellular bridge. Such recruitment might limit the movement and possibly cluster Dl on the surface of the sending cell, providing a cue that induces ubiquitination of the ligand. Notably, Mind bomb (Mib), the conserved E3 ligase that ubiquitinates Dl in the developing Drosophila wing, is expressed in all wing cells but appears to ubiquitinate Dl only in sending cells that are engaged in signaling (Wang and Struhl, 2004; 2005). Hence, ubiquitination of Dl by Mib may be induced by Notch binding to Dl. This contrasts with Neuralized (Neur), a structurally distinct E3 ligase that is regulated transcriptionally and may act constitutively to ubiquitinate Dl ligands in other contexts (e.g., Bang et al., 1995). Experiments in mammalian cell culture have shown that intermixing DSL ligand and Notch expressing cells modestly increases ligand ubiquitination (Hansson et al., 2010; Meloty-Kapella et al., 2012), although the functional significance of this finding has been unclear. In contrast, our results suggest that the control of Dl ubiquitination by Notch, whether via the regulation of Mib activity or Neur expression, may be essential for Dl signaling activity.
Second, how does Epsin/Clathrin-dependent endocytosis exert force across the Dl/Notch bridge? One possibility is that translocation of ligand in the plane of the membrane (e.g., clustering in Clathrin-coated pits) is opposed by a restriction on the lateral movement of the receptor. Such spatio-mechanical regulation has been suggested for the S2-like cleavage of EphrinA1 ligands by EphA2 receptors (Salaita et al, 2010). Alternatively, internalization of the ligand via invagination of Clathrin pits could be opposed by the intrinsic stiffness of the abutting cell membrane or by a counter force mediated by receptor. The actin nucleation activity of the Arp2/3 complex can contribute to both the lateral movement and invagination of Clathrin coated pits (Merrifield et al., 2002; 2004; Yarar et al., 2005). However, abolishing Arp2/3 function using mutations in any of the genes encoding the Arpc1, Arp2 and Arp3 subunits has no apparent effect on Notch activation along the DV border (Legent et al., 2012, our unpublished observation). A future challenge will be to identify the molecular machines that generate the requisite force.
Third: what governs the competition between the signal-sending and signal-receiving cell for S2 cleavage versus engulfment of the Dl/Notch bridge? We observed that the receiving cell “wins” when ligand cannot be targeted for Epsin/Clathrin mediated endocytosis. Accordingly, we suggest that engulfment may reflect a constitutive mechanism that is initiated whenever Dl engages Notch unless ubiquitination occurs quickly enough to target Dl into the Epsin/Clathrin pathway. We note that receptor-mediated engulfment of ligand has been observed previously, e.g., for Boss, the ligand for the Drosophila receptor tyrosine kinase Sevenless (Sev) (Cagan et al., 1992), and for Ephrin family ligands for Eph family receptors (Marston et al., 2003; Zimmer et al., 2003). However, uptake of these other transmembrane ligands is associated with, and possibly essential for, signal transduction, as is also observed for receptor-mediated endocytosis of several kinds of soluble ligands, e.g., Wnts (Seto and Bellen, 2006). By contrast, an essential property of pulling models is that receptor activation depends on the generation of force across the ligand/receptor bridge. Notch-mediated engulfment of Dl would alleviate any such force and hence abort, rather than facilitate, the incipient signal.
Recently, the capacity of the NRR to mediate ligand-dependent cleavage and nuclear import of the Notch cytosolic domain has been harnessed to engineer synthetic signaling systems that allow chosen transmembrane ligands to induce specific target genes in neighboring cells (Morsut et al., 2016; Roybal et al., 2016). Our present results suggest both constraints and opportunities for optimizing such “syn-Notch” systems, as informed by the basic requirements for ligand ubiquitination, Epsin-dependent endocytosis and force-dependent S2 cleavage
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to the Lead Contact, Gary Struhl (gs20@columbia.edu).
Experimental Model and Subject Details
In all experiments, we studied both male and female Drosophila melanogaster with no detectable difference between sexes. Animals were cultured at 25°C for all experiments except for those assaying the response of FSH-A2-N receptors (Figure 6), which were performed at 29°C to increase UAS transgene expression. To induce Flp-mediated mitotic recombination across FRTs, first or second instar larvae of the appropriate genotype were heat shocked at 36°C for one hour and wing discs from mature third instar larvae were dissected, fixed (2% formaldehyde and 0.1% Triton for 30 minutes; room temperature) washed three times in PBT (PBS, 0.1% Triton, 1% bovine serum albumin) and, if required, incubated with primary antibody in PBT, washed as before, incubated in secondary antibody, before a final wash prior to mounting (as in Wang and Struhl, 2004; 2005). Antibody incubation was carried out either at room temperature for 2 hours or overnight at 4°C. Protein expression was visualized by confocal microscopy. Cut and Wg expression were monitored using mouse monoclonal antisera (2B10 and 4D4 respectively); HA and HRP epitopes were visualized using rabbit polyclonal antisera (Santa Cruz 805 and Abcam ab34885 respectively; Wang and Struhl, 2004; 2005), Cherry and GFP were detected by native fluorescence, and all cells were counterstained by DAPI.
The complete genotypes of animals used in this study are shown in Table S1.
Mutations and transgenes used are as follows:
kuz e29-4 (BDSC Stk# 5804, Flybase ID: FBal0051471), NctR46 (Flybase ID: FBal0129200), lqf1227 (Flybase ID: FBal0191203), DlRevF10 SerRx82 (BDSC Stk# 6300, Flybase ID: FBst0006300), hsp70.flp (BDSC Stk# 23649, Flybase ID: FBtp0001101), arm.lacZ (BDSC Stk# 7371, Flybase ID: FBti0023290), UAS.CD2 (BDSC Stk# 9906, Flybase ID: FBtp0019068), UAS.neur (Flybase ID: FBtp0013307), UAS.sca (Ellis et al., 1994), nub.Gal4 (BDSC Stk# 42699, Flybase ID: FBtp0009119), rn.Gal4 (Stk# 7405, Flybase ID: FBti0023720), and a genomic rescuing lqf (epsin) transgene P[lqf+](Flybase ID: FBtp0012394) (http://flybase.org;http://flystocks.bio.indiana.edu/).
Method Details
Transgenes
All ligand and receptor coding sequences, with the exception of FSHα, were inserted into a modified form of pUAST-attB (www.flyc31.org) that contains a single Flp Recombinase Target (FRT, ‘>’) positioned between the UAS promoter and the coding sequence, and the resulting UAS>ligand and UAS>receptor transgenes were introduced at a single genomic docking site, attP-86Fb located on the right arm of the third chromosome (http://flybase.org/reports/FBst0024749.html), oriented so that the promoter is centromere proximal to the coding sequence. As required, a “no promoter” (Ø) element consisting of the transcriptional terminating 3’UTR of the hsp70 gene was swapped for the UAS promoter element in vivo, via Flp-mediated mitotic recombination with a Ø>CD2 transgene inserted at the same docking site to generate Ø>ligand and Ø>receptor transgenes. The FSHα coding sequence was inserted into pUAST and introduced into the genome by conventional P-element mediated transformation; a single UAS.FSHα transgene inserted on the X chromosome was used in all experiments.
The various tagged and chimeric forms of Dl are depicted in Figure S1A, which shows the amino-acid sequences of the relevant joins between native Dl, the Horse Radish Peroxidase (HRP) and Haemagglutinin (HA) tags, and the heterologous extracellular and intracellular domains. The native extracellular domain of Dl was replaced in its entirety by (i) the β subunit of human Follicle Stimulation Hormone (FSHβ, Fan and Hendrickson, 2005), (ii) the ectodomain of FSH Receptor (FSHR; Fan and Hendrickson, 2005), (iii) the ectodomain of Tropomyosin receptor kinase C (TrkC; Ultsch et al., 1999), or (iv) Green Fluorescent Protein (GFP) preceded by the FSHβ signal-peptide and followed by the ectodomain of CD4; in the case of FSH-Dl only an HRP tag was inserted immediately downstream of the heterologous ligand domain, immediately upstream of the Dl transmembrane domain. Signaling of FSH-Dl with or without the HRP tag is indistinguishable, and for most of the work the tagged form was used (Figure S1A). Specifically Figure S3B uses the untagged version of FSH-Dl, designated as FSH-Dl (no HRP). The Dl intracellular domain was deleted (∆C), mutated so that all Lysines were changed to Arginine (K>R), C-terminally tagged with HA, or replaced just after the stop transfer sequence downstream of the transmembrane domain by wildtype or mutant versions of (i) a small heterologous peptide containing two Lysines that are sufficient to mediate Epsin-dependent endocytosis (Wang and Struhl, 2004; 2005), and (ii) six repeats of a K>R form of the classic Myc epitope tag of which either five (myc) or six (mycmut) are mutated to change the LI dipeptide to Al. To reconstitute the composite FSHα/FSHβ ligand domain, secreted FSHα was co-expressed from a UAS.FSHα transgene. Note that the extracellular, juxta-membrane portion of Dl has been reported to contain a sequence that is subject to proteolytic cleavage by the Kuz-related ADAM protease Kuz-like (Sapir, 2004). This domain is not present in our chimeric FSH-, FSHR-, TrkC- or GFP-Dl ligands; hence, its role in the mechanism of native DSL/Notch signaling is not assessed in our experiments.
The various tagged and chimeric forms of Notch are similarly depicted in Figure S1B. All versions of FSHR-N in this work carried the extracellular Cherry tag (Figure S1B) and for simplicity this is omitted from their designation, except where necessary to ensure clarity. The amino-terminal Epidermal Growth Factor (EGF) Repeat containing portion of the native extracellular domain of Dl was replaced by the ectodomain of FSHR, Neurotrophin-3 (NTF) or a single chain anti-GFP nanobody (Nano, Rothbauer et al., 2008) preceded by the signal-peptide from FSH; the extracellular, juxta-membrane NRR was replaced by the wildtype and mutant forms of the A2 domain of von Willibrand Factor (Hassenpflug, 2006; Xu and Springer, 2013). The extracellular domain was tagged by the insertion of Cherry just upstream of the juxtamembrane NRR or A2 domain; the intracellular domain was tagged by a centrally located insertion of GFP, as previously described for the NiGFP transgene (Flybase: FBtp0072075).
Complete DNA sequences are available on request.
Analysis at the interface of ligand and receptor cells
Signaling between dedicated ligand and receptor cells was analyzed using Mosaic Analysis by Promoter Swap (MAPS), as outlined in Figures 2A and S3D and depicted in full detail in Figure S2. In essence, mitotic recombination across the FRTs in cells transheterozygous for UAS> and Ø> transgenes is induced in the presence of a Gal4 driver that acts in the developing wing (nub.Gal4 or rn.Gal4). This subdivides the prospective wing into mutually exclusive ligand and receptor expressing subpopulations, allowing signaling to be monitored by assaying the ectopic induction of Notch target genes (cut and wg) wherever the two subpopulations abut.
As depicted in Figures 2A and S2, gene functions can also be selectively abolished or activated in either of the two sub-populations by introducing the appropriate transgenes and mutations, and in a further permutation, both sub-populations can be generated in a background of cells that co-express both proteins (as in Figure S3D, to assay the potential for cis-inhibition of receptor by co-expressed ligand, and Figure 6, to render the FSHR-A2-N expressing cells homozygous for the UAS>FSHR-A2-N transgene)
To induce mosaics by promoter swap, first or second instar larvae of the appropriate genotype were heat shocked at 36°C for one hour and wing discs from mature third instar larvae were dissected and processed as described above.
For assaying transendocytosis, we performed maximum projections on Z stacks of images planes collected at 1 μm intervals. Importantly, ubiquitous expression of the UAS.YFP-Rab5CA transgene under nub.Gal4 control has no apparent effect on normal wing development consistent with native DSL/Notch signaling, as well as all other signaling events controlling wing growth and pattern, functioning as in wild type animals. In all transendocytosis experiments presented here (Figure 4,5,S6,S7), UAS.Nintra (BDSC Stk# 52008) was coexpressed throughout the prospective wing. This negates possible confounding effects of ectopic FSHR-N activation by abutting FSH-Dl cells and helps keep the wing epithelium flat, aiding visualization of tagged early endosomes.
Quantification and Statistical Analysis
In all experiments, most if not all of the imaginal wing discs contained several mutually exclusive subpopulations of ligand and receptor expressing cells within each wing primordium. In all cases, the images shown in the Figures are representative, and the outcome of the experiments qualitatively apparent (e.g., in showing the presence or absence of ectopic Cut or Wg expression, or in showing the presence or absence of transendocytosis of either the ligand or receptor ectodomain).
For simple MAPS experiments in which mutually exclusive ligand and receptor expressing subpopulations were generated in otherwise wild type discs, at least 20, and usually more than 50, discs were scored. For more complex MAPS experiments in which mutant clones were coinduced with receptor or ligand expressing clones, at least 10, and usually more than 25, discs were examined.
Supplementary Material
A) Chimeric ligands
Top. Each ligand contains a receptor-binding, extracellular domain (ECD), a transmembrane domain (TM) and an intracellular domain (ICD). In addition, most FSH-Dl ligands carry an HRP extracellular tag and in one case an intracellular HA tag (tagged and untagged forms of FSH-Dl activated FSHR-N indistinguishably).
Bottom. The peptide sequences flanking the joins between each of these domains, as well as the variant forms of the intracellular domains, are shown color-coded as indicated under the ligand designation (left). The K>R variants are identical to the wildtype FSH-Dl ligand except that all 12 lysines downstream of the stop transfer peptide following the transmembrane domain are replaced by Arginine. The full sequence of the K* peptide is:
GSWIPSFYNVVTGKTLALPNLIALQHIPLSPAGVIAKRPAPIALPNSCAA (the R* peptide is identical except for the two, underlined lysines, which are replaced by arginines). The myc variants contain 6 tandem repeats of a modified form of the classic Myc epitope tag, EQRLISEEDLN, in which the sole lysine in the original is replaced by arginine. For the wild type variant, the last repeat retains the LI dipeptide, whereas the preceding five have the dipeptide mutated to Al. In the mycmut variant, all six repeats contain the mutated (AI) dipeptide. Uncolored sequences indicate linker peptides introduced to accommodate restriction sites. All DNA coding sequences are available on request.
B) Chimeric receptors.
Top. Each receptor contains a ligand-binding extracellular domain (ECD), a juxtamembrane NRR or A2 domain, a transmembrane domain (TM), and an intracellular domain (ICD). In addition, some FSHR-N receptors carry a Cherry extracellular tag and either a v5 or GFP intracellular tag (tagged and untagged forms of FSHR-N were activated indistinguishably by FSH-Dl ligands).
Bottom. The peptide sequences flanking the joins between each of these domains are shown, color-coded as indicated under the receptor designation (left). Uncolored sequences indicate linker peptides introduced to accommodate restriction sites. All DNA coding sequences are available on request.
A) MAPS for subdividing a tissue into mutually exclusive subpopulations of cells expressing either of two different UAS> transgenes, in this case expressing either a DSL ligand or Notch receptor, so that the two proteins only encounter each other in trans, wherever cells from the different subpopulations abut. The mother cell carries a UAS>ligand transgene in trans to a Ø>receptor transgene (Ø = a “no promoter” element) inserted at the same attB docking site, and oriented in the same direction [the centromere is located to the left (not shown), with the 5’ end of each coding sequence positioned centromere proximal to the 3’ end]. As a consequence, the mother cell expresses the ligand but not the receptor (blue). Flp mediated recombination (red X) across the FRT (>) at the four strand stage, followed by chromatid exchange (not shown) and either of the two possible chromosome segregations (Seg. 1 or Seg. 2), produces one daughter that expresses only the ligand (blue) and a sibling daughter (red) that expresses only the receptor. The result is subdivision of the tissue into mutually exclusive UAS>ligand cells (derived from UAS>ligand daughter cells as well as transheterozygous mother cells) and UAS>receptor cells (cartoon and image on the right).
B) An elaboration of MAPS to generate clones that are homozygous for a genetic element “X” in one of the two mutually exclusive subpopulations (in this case, the UAS>ligand daughter cells; blue, yellow outline) together with sibling clones that are homozygous for the absence of X in the other subpopulation (the UAS>receptor daughter cells; red, black outline). X represents the general case, and can be either (i) a recessive mutant condition (e.g., Nct¯, Figure S3, the interface of interest being between UAS>receptor cells that are homozygous for X and UAS>ligand cells that are heterozygous or homozygous for the wild type state of X), or (ii) a transgene (e.g., UAS.YFP-Rab5CA in Figure S6, the interface of interest being between UAS>receptor daughter cells homozygous for the absence of X and UAS>ligand cells carrying one or two copies of X). As depicted, the mother cell carries a UAS>ligand transgene in cis with X and in trans to a Ø>receptor transgene, and hence expresses the ligand (blue; heterozygosity for X is depicted by the dotted yellow and black outline): if X is a recessive mutation, the mother cell will be phenotypically wildtype; if X is a transgene, it will contain one copy. Flp mediated recombination across the FRT at the four strand stage, followed by chromatid exchange (not shown) results in either of the two possible chromosome segregations (Seg. 1 or Seg. 2). Seg. 1 yields one ligand expressing X/X daughter that is either mutant or expresses two copies of the UAS transgene (blue, with yellow outline) and a sibling receptor expressing +/+ daughter that is wildtype or carries no copies of the UAS transgene (red, with black outline). Seg. 2 yields ligand and receptor expressing twin cells (blue and red, respectively), both of which remain heterozygous for X (remain wildtype for gene function and express one copy of the UAS transgene; dotted yellow and black outline). Since multiple recombination events are induced in each wing disc, the resulting tissue is a mosaic of the four cell subpopulations from the four possible daughter cells, as well as a fifth population derived from mother cells in which recombination has not occurred. These can be distinguished by assaying for expression of the ligand or receptor, the UAS transgene, and a marker for the presence or absence of X (e.g., as in the case of Nct¯, Figure S3B).
C) An elaboration of MAPS to generate subpopulations of cells that express two UAS> transgenes in a mutually exclusive manner in a background of cells that coexpress both transgenes (e.g., to analyze cis-inactivation of receptor by expression of ligand; Figure S3D). The depiction shows the general case in which a genetic element, X, is heterozygous in the mother cell, and mitotic recombination results in X becoming homozygous in one of the two daughter cells (e.g., as in the analysis of Dl Ser loss of function; Figure S3C), and the other daughter cell becoming homozygous for the absence of X. As depicted, the mother cell carries a UAS>ligand transgene in cis with X and in trans to a UAS>receptor transgene. Hence, the mother cell expresses both the ligand and receptor, as well as X, if X is a UAS transgene (purple with dotted yellow outline). Flp mediated recombination across the FRT at the four strand stage, followed by chromatid exchange and one of the two possible segregations results in one daughter that expresses ligand and is homozygous for X (blue, with yellow outline), and a sibling daughter that expresses receptor and homozygous for the lack of X (red, with black outline). The alternative segregation generates daughter cells that have the same genotype as the mother cell and is not shown.
A) Kuz is required in receiving cells for FSH-Dl/FSHR-N signaling. kuz¯ clones have been induced in wing discs composed of mutually exclusive FSH-Dl and FSHR-N expressing cells generated by MAPS [the relevant clonal genotypes are outlined and color coded as depicted in the banners; kuz¯ clones are marked “black” by the absence of expression of an arm-lacZ transgene in the left panel, and in the remaining two panels, the FSHR-N protein is visualized by the presence of an extracellular Cherry tag (red); See Figure S2 for a detailed depiction of the use of MAPS for this experiment as well as the experiments shown in C and D]. In the boxed region shown at higher magnification (right), FSHR-N expressing kuz¯ cells do not express Cut in response to abutting FSH-Dl expressing cells (empty green arrow head; the FSH-Dl cells are marked “black” by the absence of FSHR-N), in contrast to FSHR-N expressing cells that retain wild type kuz function, which do (filled arrow heads) even when located further away from the DV boundary than the kuz¯ clones (yellow outlined filled arrowhead). Similarly, kuz¯ FSHR-N cells close to the D/V boundary fail to express Cut when confronting FSH-Dl cells (empty yellow arrowhead), in contrast to their wild type neighbors. The white asterisk marks the loss of normal Cut expression associated with a small kuz¯ clone that blocks native DSL/N signaling across the D/V boundary.
B) Nct is required in receiving cells for FSH-Dl/FSHR-N signaling (color coded as in A, Nct¯ clones are marked “black” by the absence of expression of a UAS.CD2 transgene).
C) Native Dl and Ser are not required in sending cells for FSH-Dl/FSHR-N signaling (color coded as in A). Dl¯ Ser¯ clones that express FSH-Dl have been induced in a background FSHR-N expressing cells: Dl¯ Ser~ FSH-Dl expressing cells in such clones can induce ectopic Cut expression in abutting FSHR-N cells (filled arrowheads for the boxed region shown at higher magnification; the white asterisk indicates clones associated with a local loss of normal Cut expression along the D/V boundary).
D) FSH-Dl/FSHR-N signaling is subject to cis-inactivation. Top) Generation of homozygous UAS>FSH-Dl and UAS>FSHR-N expressing twin-clones in a background of FSH-Dl/FSHR-N cells by MAPS (depicted as in Figure 2; see also Figure S2C). Bottom) Boxed region of a wing primordium containing homozygous UAS>FSH-Dl, homozygous UAS>FSHR-N and transheterozygous UAS>FSH-Dl/ UAS>FSHR-N cell populations, color coded as in the banner. FSH-Dl expressing cells (blue) induce Cut in abutting FSHR-N cells (red; filled arrow heads), but not in abutting FSHR-N cells that express FSH-Dl (purple; empty arrow heads). Scale bars: 10μm.
A) Expression of UAS.scabrous in a wing disc subdivided into mutually exclusive subpopulations of FSH-Dl (blue) and FSHR-N (black) by MAPS. UAS.scabrous expression blocks the capacity of native Notch to respond to DSL ligands (as indicated by the absence of Cut expression along the DV border; white asterisk), but does not interfere with FSH-Dl/FSHR-N signaling, as indicated by the capacity of UAS>FSH-Dl cells to induce ectopic Cut expression (yellow) in adjacent UAS>FSHR-N cells.
B) Introduction of the NRRSC mutation, which confers hypersensitivity in the context of the native receptor, renders the resulting FSHR-N receptor (FSHR-NRRSC-N) hypersensitive to FSH-Dl, as indicated by the capacity of FSH-Dl to induce ectopic Cut in adjacent UAS>FSHR-NRRSC-N cells that are located ~30 or more cell diameters from the D/V border (using MAPS, as in A; compare with Figure 2D). For this Figure as well as Figure S5, see STAR Methods and Table S1 for exact genotypes. Scale bars: 100μm.
epsin¯ clones have been induced in wing discs that are subdivided using MAPS into mutually exclusive populations of FSH-Dl and FSHR-N expressing cells, both of which express UAS.Neur. As in Figure 3A, and as depicted in the banners, the epsin¯ clones are marked by the absence of Epsin protein expression (grey scale image), and FSHR-N and FSH-Dl cells are marked by the presence of FSHR-N (red), or its absence (black). Neur expression promotes FSH-Dl signaling capacity by driving ubiquitination of the ligand, directing it to the Epsin/Clathrin pathway (Figure 2D). Nevertheless, FSH-Dl expressing cells (black) still require Epsin function to be able to activate FSHR-N in abutting, receptor expressing cells (red), as indicated by the ectopic induction of Cut (green, arrowheads); no response is elicited when the FSH-Dl expressing cells lack Epsin (open arrowheads; the white asterisk marks the loss of normal Cut expression where the epsin¯ clone abuts the D/V boundary). Scale bar: 20μm.
A) Generating mutually exclusive subpopulations of ligand and receptor expressing cells by MAPS as in Figure 2, but with UAS.YFP-Rab5CA located distal to the UAS>FSH-Dl transgene, resulting in populations of UAS>FSH-Dl, UAS.YFP-Rab5CA cells that abut reciprocal populations of UAS>FSHR-N cells. See STAR Methods and Table S1 for exact genotypes.
B) Top. FSHR-N with an extracellular Cherry tag and an intracellular GFP tag was used to assay the transendocytosis of the receptor into the neighboring FSH-Dl expressing cell (ligand structure as in Figure 1). To allow the visualization of endocytosed cargo proteins and to restrict analysis to endosomes within the sending cells, YFP-Rab5CA was coexpressed with FSH-Dl. YFP-Rab5CA expression blocks the maturation of early endosomes, resulting in abnormally enlarged endosomes with YFP at their perimeter and trapped cargo proteins in their interior.
Bottom. Abutting populations of UAS>FSH-Dl, UAS.YFP-Rab5CA sending cells and UAS>FSHRCherry-NGFP receiving cells (genotypes and color codes as in the banner). The UAS>FSHRCherry-NGFP cells appear yellow due to the coincidence of GFP and Cherry. Where UAS>FSH-Dl, UAS. YFP-Rab5CA cells abut UAS>FSHRCherry-NGFP cells, the YFP-Rab5CA endosomes of the signal-sending cells accumulate predominantly Cherry (appear red, rather than yellow). In contrast, as shown in the higher magnification images to the right, the YFP/GFP signal is apparent at the perimeter of Cherry positive endosomes but not in their interior (asterisks), indicating little or no transendocytosis of the GFP-tagged intracellular domain of FSHRCherry-NGFP. Note that the fluorescence intensity of Cherry relative to GFP/YFP is high inside the endosomes, owing to the restricted accumulation YFP-Rab5CA at the endosomal perimeters as well as its relatively weak fluorescence compared to the Cherry signal. Scale bar: 5μm.
A) Images show abutting populations of UAS>FSH-DlΔC, UAS.YFP-Rab5CA cells and UAS>FSHRCherry-N cells (labeled as in the banner and in Figure 4A box#1). No Cherry accumulation is visible in the YFP-Rab5CA tagged endosomes, as also observed for the other versions of FSH-Dl that cannot access the Epsin/Clathrin pathway (Figure 4B).
B) The images show abutting populations of UAS>FSH-DlΔC cells and UAS>FSHR-N, UAS.YFP-Rab5CA cells (labeled as in banner and Figure 4A box#2). HRP accumulates inside the YFP-Rab5CA endosomes, as shown for other versions of FSH-Dl that cannot access the Epsin/Clathrin pathway (Figure 4B).
C) No Cherry accumulates within the endosomes of UAS>FSH-Dl, UAS.YFP-Rab5CA cells abutting UAS>FSHR-N cells when FSHα is not expressed to reconstitute the composite FSHα/β ligand domain (labeled and depicted as in Figure 4).
D) No HRP accumulates within the endosomes of UAS>FSHR-N, UAS.YFP-Rab5CA cells abutting UAS>FSH-Dl-K>R cells when FSHα is not expressed (labeled and depicted as in Figure 4). Scale bars: 5μm.
Highlights.
In vivo dissection of the mechanism of Notch activation by ligand endocytosis
Epsin/Clathrin-mediated endocytosis of ligand exerts force on Notch
Force induces ectodomain cleavage of Notch to initiate signal transduction
Ligand engulfment by receiving cell in the absence of force aborts incipient signal
ACKNOLWEDGEMENTS
We thank T. Chunyao, J. Recio, Y. Chen, G. Saharia and R. De Luca for assistance; T. Springer Q. Fan and B. Zhang for DNAs and antisera; the Bloomington Drosophila Stock Center (NIH P400D018537) for stocks; I. Greenwald, A. Tomlinson, S.Blacklow, M. Zecca, R. Coleman and J. Parker for advice; and the NIH (R01 GM109183) and HHMI for support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahimou F, Mok L, Bardot B, and Wesley C (2004). The adhesion force of Notch with Delta and the rate of Notch signaling. J. Cell Biol. 167, 1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang AG, Bailey AM, and Posakony JW (1995). Hairless promotes stable commitment to the sensory organ precursor cell fate by negatively regulating the activity of the Notch signaling pathway. Dev. Biol. 172, 479–494. [DOI] [PubMed] [Google Scholar]
- Benhra N, Vignaux F, and Dussert A (2010). Neuralized promotes basal to apical transcytosis of Delta in epithelial cells. Mol. Biol. Cell 21, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair SS (1997). Limb development: marginal fringe benefits. Curr. Biol. 7, R686–R690. [DOI] [PubMed] [Google Scholar]
- Cagan RL, Krämer H, Hart AC, and Zipursky SL (1992). The bride of sevenless and sevenless interaction: internalization of a transmembrane ligand. Cell 69, 393–399. [DOI] [PubMed] [Google Scholar]
- Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, and De Camilli P (1998). Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 394, 793–797. [DOI] [PubMed] [Google Scholar]
- Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, et al. (2009). Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc. Natl. Acad. Sci. USA 106, 13838–13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblandre GA, Lai EC, and Kintner C (2001). Xenopus neuralized is a ubiquitin ligase that interacts with XDelta1 and regulates Notch signaling. Dev. Cell 1, 795–806. [DOI] [PubMed] [Google Scholar]
- del Álamo D, Rouault H, and Schweisguth F (2011). Mechanism and significance of cis- Inhibition in Notch Signalling. Curr. Biol. 21, R40–R47. [DOI] [PubMed] [Google Scholar]
- Ellis MC, Weber U, Wiersdorff V, and Mlodzik M (1994). Confrontation of scabrous expressing and non-expressing cells is essential for normal ommatidial spacing in the Drosophila eye. Development 120, 1959–1969. [DOI] [PubMed] [Google Scholar]
- Fan QR, and Hendrickson WA (2005). Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KM, Dias JA, and Van Roey P (2001). Three-dimensional structure of human follicle-stimulating hormone. Mol. Endocrinol. 15, 378–389. [DOI] [PubMed] [Google Scholar]
- Golic KG (1991). Site-specific recombination between homologous chromosomes in Drosophila. Science 252, 958–961. [DOI] [PubMed] [Google Scholar]
- Gordon WR, Arnett KL, and Blacklow SC (2008). The molecular logic of Notch signaling - a structural and biochemical perspective. J. Cell Sci. 121, 3109–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WR, Zimmerman B, He L, Miles LJ, and Huang J (2015). Mechanical allostery: Evidence for a force requirement in the proteolytic activation of Notch. Dev. Cell 33, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, and Blacklow SC (2007). Structural basis for autoinhibition of Notch. Nat. Struct. Mol. Biol. 14, 295–300. [DOI] [PubMed] [Google Scholar]
- Hassenpflug WA (2006). Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood 107, 2339–2345. [DOI] [PubMed] [Google Scholar]
- Kovall RA, Gebelein B, Sprinzak D, and Kopan R (2017). The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 41, 228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Deblandre GA, Kintner C, and Rubin GM (2001). Drosophila neuralized is a ubiquitin ligase that promotes the internalization and degradation of Delta. Dev. Cell 1, 783–794. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu SY, and Baker NE (2000). The Scabrous protein can act as an extracellular antagonist of Notch signaling in the Drosophila wing. Curr. Biol. 10, 931–934. [DOI] [PubMed] [Google Scholar]
- Legent K, Steinhauer J, Richard M, and Treisman JE (2012). A screen forX-linked mutations affecting Drosophila photoreceptor differentiation identifies casein kinase 1α as an essential negative regulator of Wingless signaling. Genetics 190, 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F, and Klausner RD (1992). A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell 69, 1143–1157. [DOI] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Alcamo E, Corbin V, and Young MW (1993). Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes Dev. 7, 1949–1965. [DOI] [PubMed] [Google Scholar]
- Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, Haltiwanger RS, Zhu C, Ha T, and Garcia KC (2017). Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 355, 1320–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, and Blacklow SC (2006). Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol. Cell. Biol. 26, 4642–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsall BT, Long M, Piper JW, Yago T, McEver RP, and Zhu Cheng. (2003). Direct observation of catch bonds involving cell-adhesion molecules. Nature, 423, 190–193. [DOI] [PubMed] [Google Scholar]
- Marston DJ, Dickinson S, and Nobes CD (2003). Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nat. Cell Biol. 5, 879–888. [DOI] [PubMed] [Google Scholar]
- Meloty-Kapella L, Shergill B, Kuon J, Botvinick E, and Weinmaster G (2012). Notch ligand endocytosis generates mechanical pulling force dependent on Dynamin, epsins, and actin. Dev. Cell 22, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, and Almers W (2002). Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4, 691–698. [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Qualmann B, Kessels MM, and Almers W (2004). Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur. J. Cell Biol. 83, 13–18. [DOI] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, and Lim WA (2016). Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musse AA, Meloty-Kapella L, and Weinmaster G (2012). Notch ligand endocytosis: Mechanistic basis of signaling activity. Semin. Cell Dev. Biol. 23, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet E, Fitch E, and Fischer JA (2004). Fat facets and Liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development 131, 5355–5366. [DOI] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, and Zerial M (2005). Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, and Leonhardt H (2008). A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell Proteomics 7, 282–289. [DOI] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016). Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapir A (2004). Unidirectional Notch signaling depends on continuous cleavage of Delta. Development 132, 123–132. [DOI] [PubMed] [Google Scholar]
- Seto ES, and Bellen HJ (2006). Internalization is required for proper Wingless signaling in Drosophila melanogaster. J. Cell Biol. 173, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shergill B, Meloty-Kapella L, Musse AA, Weinmaster G, and Botvinick E (2012). Optical tweezers studies on Notch: single-molecule interaction strength is independent of ligand endocytosis. Dev. Cell 22, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson NL, and Avis JM (2012). Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc. Natl. Acad. Sci. USA 109, E2757–E2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, and Adachi A (2000). Requirements for Presenilin-dependent cleavage of Notch and other transmembrane proteins. Mol. Cell 6, 625–636. [DOI] [PubMed] [Google Scholar]
- Tsai HM (1996). Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood 87, 4235–4244. [PubMed] [Google Scholar]
- Tsai HM, Sussman II, and Nagel RL (1994). Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood 83, 2171–2179. [PubMed] [Google Scholar]
- Ultsch MH, Wiesmann C, Simmons LC, Henrich J, Yang M, Reilly D, Bass SH, and de Vos AM (1999). Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. Journal of Molecular Biology 290, 149–159. [DOI] [PubMed] [Google Scholar]
- Wang W, and Struhl G (2004). Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development 131, 5367–5380. [DOI] [PubMed] [Google Scholar]
- Wang W, and Struhl G (2005). Distinct roles for Mind bomb, Neuralized and Epsin in mediating DSL endocytosis and signaling in Drosophila. Development 132, 2883–2894. [DOI] [PubMed] [Google Scholar]
- Wang W (2006). Ligand endocytosis and activation of DSL-Notch signal transduction pathway. Ph.D. Thesis, Columbia University. [Google Scholar]
- Weinmaster G, and Fischer JA (2011). Notch Ligand Ubiquitylation: What Is It Good For? Dev. Cell 21, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B (2002). Epsins: adaptors in endocytosis? Nat. Rev. Mol. Cell Biol. 3, 971–977. [DOI] [PubMed] [Google Scholar]
- Xu AJ, and Springer TA (2013). Mechanisms by which von Willebrand disease mutations destabilize the A2 Domain. J. Biol. Chem. 288, 6317–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, and Schmid SL (2005). A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Molecular Biology of the Cell 16, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Schulze KL, Hiesinger PR, Suyama K, Wang S, Fish M, Acar M, Hoskins RA, Bellen HJ, and Scott MP (2007). Thirty-one flavors of Drosophila Rab proteins. Genetics 176, 1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Halvorsen K, Zhang CZ, Wong WP, and Springer TA (2009). Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science 324, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M, Palmer A, Köhler J, and Klein R (2003). EphB-ephrinB bi-directional endocytosis terminates adhesion allowing contact mediated repulsion. Nat. Cell Biol. 5, 869–878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) Chimeric ligands
Top. Each ligand contains a receptor-binding, extracellular domain (ECD), a transmembrane domain (TM) and an intracellular domain (ICD). In addition, most FSH-Dl ligands carry an HRP extracellular tag and in one case an intracellular HA tag (tagged and untagged forms of FSH-Dl activated FSHR-N indistinguishably).
Bottom. The peptide sequences flanking the joins between each of these domains, as well as the variant forms of the intracellular domains, are shown color-coded as indicated under the ligand designation (left). The K>R variants are identical to the wildtype FSH-Dl ligand except that all 12 lysines downstream of the stop transfer peptide following the transmembrane domain are replaced by Arginine. The full sequence of the K* peptide is:
GSWIPSFYNVVTGKTLALPNLIALQHIPLSPAGVIAKRPAPIALPNSCAA (the R* peptide is identical except for the two, underlined lysines, which are replaced by arginines). The myc variants contain 6 tandem repeats of a modified form of the classic Myc epitope tag, EQRLISEEDLN, in which the sole lysine in the original is replaced by arginine. For the wild type variant, the last repeat retains the LI dipeptide, whereas the preceding five have the dipeptide mutated to Al. In the mycmut variant, all six repeats contain the mutated (AI) dipeptide. Uncolored sequences indicate linker peptides introduced to accommodate restriction sites. All DNA coding sequences are available on request.
B) Chimeric receptors.
Top. Each receptor contains a ligand-binding extracellular domain (ECD), a juxtamembrane NRR or A2 domain, a transmembrane domain (TM), and an intracellular domain (ICD). In addition, some FSHR-N receptors carry a Cherry extracellular tag and either a v5 or GFP intracellular tag (tagged and untagged forms of FSHR-N were activated indistinguishably by FSH-Dl ligands).
Bottom. The peptide sequences flanking the joins between each of these domains are shown, color-coded as indicated under the receptor designation (left). Uncolored sequences indicate linker peptides introduced to accommodate restriction sites. All DNA coding sequences are available on request.
A) MAPS for subdividing a tissue into mutually exclusive subpopulations of cells expressing either of two different UAS> transgenes, in this case expressing either a DSL ligand or Notch receptor, so that the two proteins only encounter each other in trans, wherever cells from the different subpopulations abut. The mother cell carries a UAS>ligand transgene in trans to a Ø>receptor transgene (Ø = a “no promoter” element) inserted at the same attB docking site, and oriented in the same direction [the centromere is located to the left (not shown), with the 5’ end of each coding sequence positioned centromere proximal to the 3’ end]. As a consequence, the mother cell expresses the ligand but not the receptor (blue). Flp mediated recombination (red X) across the FRT (>) at the four strand stage, followed by chromatid exchange (not shown) and either of the two possible chromosome segregations (Seg. 1 or Seg. 2), produces one daughter that expresses only the ligand (blue) and a sibling daughter (red) that expresses only the receptor. The result is subdivision of the tissue into mutually exclusive UAS>ligand cells (derived from UAS>ligand daughter cells as well as transheterozygous mother cells) and UAS>receptor cells (cartoon and image on the right).
B) An elaboration of MAPS to generate clones that are homozygous for a genetic element “X” in one of the two mutually exclusive subpopulations (in this case, the UAS>ligand daughter cells; blue, yellow outline) together with sibling clones that are homozygous for the absence of X in the other subpopulation (the UAS>receptor daughter cells; red, black outline). X represents the general case, and can be either (i) a recessive mutant condition (e.g., Nct¯, Figure S3, the interface of interest being between UAS>receptor cells that are homozygous for X and UAS>ligand cells that are heterozygous or homozygous for the wild type state of X), or (ii) a transgene (e.g., UAS.YFP-Rab5CA in Figure S6, the interface of interest being between UAS>receptor daughter cells homozygous for the absence of X and UAS>ligand cells carrying one or two copies of X). As depicted, the mother cell carries a UAS>ligand transgene in cis with X and in trans to a Ø>receptor transgene, and hence expresses the ligand (blue; heterozygosity for X is depicted by the dotted yellow and black outline): if X is a recessive mutation, the mother cell will be phenotypically wildtype; if X is a transgene, it will contain one copy. Flp mediated recombination across the FRT at the four strand stage, followed by chromatid exchange (not shown) results in either of the two possible chromosome segregations (Seg. 1 or Seg. 2). Seg. 1 yields one ligand expressing X/X daughter that is either mutant or expresses two copies of the UAS transgene (blue, with yellow outline) and a sibling receptor expressing +/+ daughter that is wildtype or carries no copies of the UAS transgene (red, with black outline). Seg. 2 yields ligand and receptor expressing twin cells (blue and red, respectively), both of which remain heterozygous for X (remain wildtype for gene function and express one copy of the UAS transgene; dotted yellow and black outline). Since multiple recombination events are induced in each wing disc, the resulting tissue is a mosaic of the four cell subpopulations from the four possible daughter cells, as well as a fifth population derived from mother cells in which recombination has not occurred. These can be distinguished by assaying for expression of the ligand or receptor, the UAS transgene, and a marker for the presence or absence of X (e.g., as in the case of Nct¯, Figure S3B).
C) An elaboration of MAPS to generate subpopulations of cells that express two UAS> transgenes in a mutually exclusive manner in a background of cells that coexpress both transgenes (e.g., to analyze cis-inactivation of receptor by expression of ligand; Figure S3D). The depiction shows the general case in which a genetic element, X, is heterozygous in the mother cell, and mitotic recombination results in X becoming homozygous in one of the two daughter cells (e.g., as in the analysis of Dl Ser loss of function; Figure S3C), and the other daughter cell becoming homozygous for the absence of X. As depicted, the mother cell carries a UAS>ligand transgene in cis with X and in trans to a UAS>receptor transgene. Hence, the mother cell expresses both the ligand and receptor, as well as X, if X is a UAS transgene (purple with dotted yellow outline). Flp mediated recombination across the FRT at the four strand stage, followed by chromatid exchange and one of the two possible segregations results in one daughter that expresses ligand and is homozygous for X (blue, with yellow outline), and a sibling daughter that expresses receptor and homozygous for the lack of X (red, with black outline). The alternative segregation generates daughter cells that have the same genotype as the mother cell and is not shown.
A) Kuz is required in receiving cells for FSH-Dl/FSHR-N signaling. kuz¯ clones have been induced in wing discs composed of mutually exclusive FSH-Dl and FSHR-N expressing cells generated by MAPS [the relevant clonal genotypes are outlined and color coded as depicted in the banners; kuz¯ clones are marked “black” by the absence of expression of an arm-lacZ transgene in the left panel, and in the remaining two panels, the FSHR-N protein is visualized by the presence of an extracellular Cherry tag (red); See Figure S2 for a detailed depiction of the use of MAPS for this experiment as well as the experiments shown in C and D]. In the boxed region shown at higher magnification (right), FSHR-N expressing kuz¯ cells do not express Cut in response to abutting FSH-Dl expressing cells (empty green arrow head; the FSH-Dl cells are marked “black” by the absence of FSHR-N), in contrast to FSHR-N expressing cells that retain wild type kuz function, which do (filled arrow heads) even when located further away from the DV boundary than the kuz¯ clones (yellow outlined filled arrowhead). Similarly, kuz¯ FSHR-N cells close to the D/V boundary fail to express Cut when confronting FSH-Dl cells (empty yellow arrowhead), in contrast to their wild type neighbors. The white asterisk marks the loss of normal Cut expression associated with a small kuz¯ clone that blocks native DSL/N signaling across the D/V boundary.
B) Nct is required in receiving cells for FSH-Dl/FSHR-N signaling (color coded as in A, Nct¯ clones are marked “black” by the absence of expression of a UAS.CD2 transgene).
C) Native Dl and Ser are not required in sending cells for FSH-Dl/FSHR-N signaling (color coded as in A). Dl¯ Ser¯ clones that express FSH-Dl have been induced in a background FSHR-N expressing cells: Dl¯ Ser~ FSH-Dl expressing cells in such clones can induce ectopic Cut expression in abutting FSHR-N cells (filled arrowheads for the boxed region shown at higher magnification; the white asterisk indicates clones associated with a local loss of normal Cut expression along the D/V boundary).
D) FSH-Dl/FSHR-N signaling is subject to cis-inactivation. Top) Generation of homozygous UAS>FSH-Dl and UAS>FSHR-N expressing twin-clones in a background of FSH-Dl/FSHR-N cells by MAPS (depicted as in Figure 2; see also Figure S2C). Bottom) Boxed region of a wing primordium containing homozygous UAS>FSH-Dl, homozygous UAS>FSHR-N and transheterozygous UAS>FSH-Dl/ UAS>FSHR-N cell populations, color coded as in the banner. FSH-Dl expressing cells (blue) induce Cut in abutting FSHR-N cells (red; filled arrow heads), but not in abutting FSHR-N cells that express FSH-Dl (purple; empty arrow heads). Scale bars: 10μm.
A) Expression of UAS.scabrous in a wing disc subdivided into mutually exclusive subpopulations of FSH-Dl (blue) and FSHR-N (black) by MAPS. UAS.scabrous expression blocks the capacity of native Notch to respond to DSL ligands (as indicated by the absence of Cut expression along the DV border; white asterisk), but does not interfere with FSH-Dl/FSHR-N signaling, as indicated by the capacity of UAS>FSH-Dl cells to induce ectopic Cut expression (yellow) in adjacent UAS>FSHR-N cells.
B) Introduction of the NRRSC mutation, which confers hypersensitivity in the context of the native receptor, renders the resulting FSHR-N receptor (FSHR-NRRSC-N) hypersensitive to FSH-Dl, as indicated by the capacity of FSH-Dl to induce ectopic Cut in adjacent UAS>FSHR-NRRSC-N cells that are located ~30 or more cell diameters from the D/V border (using MAPS, as in A; compare with Figure 2D). For this Figure as well as Figure S5, see STAR Methods and Table S1 for exact genotypes. Scale bars: 100μm.
epsin¯ clones have been induced in wing discs that are subdivided using MAPS into mutually exclusive populations of FSH-Dl and FSHR-N expressing cells, both of which express UAS.Neur. As in Figure 3A, and as depicted in the banners, the epsin¯ clones are marked by the absence of Epsin protein expression (grey scale image), and FSHR-N and FSH-Dl cells are marked by the presence of FSHR-N (red), or its absence (black). Neur expression promotes FSH-Dl signaling capacity by driving ubiquitination of the ligand, directing it to the Epsin/Clathrin pathway (Figure 2D). Nevertheless, FSH-Dl expressing cells (black) still require Epsin function to be able to activate FSHR-N in abutting, receptor expressing cells (red), as indicated by the ectopic induction of Cut (green, arrowheads); no response is elicited when the FSH-Dl expressing cells lack Epsin (open arrowheads; the white asterisk marks the loss of normal Cut expression where the epsin¯ clone abuts the D/V boundary). Scale bar: 20μm.
A) Generating mutually exclusive subpopulations of ligand and receptor expressing cells by MAPS as in Figure 2, but with UAS.YFP-Rab5CA located distal to the UAS>FSH-Dl transgene, resulting in populations of UAS>FSH-Dl, UAS.YFP-Rab5CA cells that abut reciprocal populations of UAS>FSHR-N cells. See STAR Methods and Table S1 for exact genotypes.
B) Top. FSHR-N with an extracellular Cherry tag and an intracellular GFP tag was used to assay the transendocytosis of the receptor into the neighboring FSH-Dl expressing cell (ligand structure as in Figure 1). To allow the visualization of endocytosed cargo proteins and to restrict analysis to endosomes within the sending cells, YFP-Rab5CA was coexpressed with FSH-Dl. YFP-Rab5CA expression blocks the maturation of early endosomes, resulting in abnormally enlarged endosomes with YFP at their perimeter and trapped cargo proteins in their interior.
Bottom. Abutting populations of UAS>FSH-Dl, UAS.YFP-Rab5CA sending cells and UAS>FSHRCherry-NGFP receiving cells (genotypes and color codes as in the banner). The UAS>FSHRCherry-NGFP cells appear yellow due to the coincidence of GFP and Cherry. Where UAS>FSH-Dl, UAS. YFP-Rab5CA cells abut UAS>FSHRCherry-NGFP cells, the YFP-Rab5CA endosomes of the signal-sending cells accumulate predominantly Cherry (appear red, rather than yellow). In contrast, as shown in the higher magnification images to the right, the YFP/GFP signal is apparent at the perimeter of Cherry positive endosomes but not in their interior (asterisks), indicating little or no transendocytosis of the GFP-tagged intracellular domain of FSHRCherry-NGFP. Note that the fluorescence intensity of Cherry relative to GFP/YFP is high inside the endosomes, owing to the restricted accumulation YFP-Rab5CA at the endosomal perimeters as well as its relatively weak fluorescence compared to the Cherry signal. Scale bar: 5μm.
A) Images show abutting populations of UAS>FSH-DlΔC, UAS.YFP-Rab5CA cells and UAS>FSHRCherry-N cells (labeled as in the banner and in Figure 4A box#1). No Cherry accumulation is visible in the YFP-Rab5CA tagged endosomes, as also observed for the other versions of FSH-Dl that cannot access the Epsin/Clathrin pathway (Figure 4B).
B) The images show abutting populations of UAS>FSH-DlΔC cells and UAS>FSHR-N, UAS.YFP-Rab5CA cells (labeled as in banner and Figure 4A box#2). HRP accumulates inside the YFP-Rab5CA endosomes, as shown for other versions of FSH-Dl that cannot access the Epsin/Clathrin pathway (Figure 4B).
C) No Cherry accumulates within the endosomes of UAS>FSH-Dl, UAS.YFP-Rab5CA cells abutting UAS>FSHR-N cells when FSHα is not expressed to reconstitute the composite FSHα/β ligand domain (labeled and depicted as in Figure 4).
D) No HRP accumulates within the endosomes of UAS>FSHR-N, UAS.YFP-Rab5CA cells abutting UAS>FSH-Dl-K>R cells when FSHα is not expressed (labeled and depicted as in Figure 4). Scale bars: 5μm.