

Summary
IDH1 mutations are common in low-grade gliomas and secondary glioblastomas and cause overproduction of (R)-2HG. (R)-2HG modulates the activity of many enzymes, including some that are linked to transformation and some that are probably bystanders. Although prior work on (R)-2HG targets focused on 2OG-dependent dioxygenases, we found that (R)-2HG potently inhibits the 2OG-dependent transaminases BCAT1 and BCAT2, likely as a bystander effect, thereby decreasing glutamate levels and increasing dependence on glutaminase for the biosynthesis of glutamate and one of its products, glutathione. Inhibiting glutaminase specifically sensitized IDH mutant glioma cells to oxidative stress in vitro and to radiation in vitro and in vivo. These findings highlight the complementary roles for BCATs and glutaminase in glutamate biosynthesis, explain the sensitivity of IDH mutant cells to glutaminase inhibitors, and suggest a strategy for maximizing the effectiveness of such inhibitors against IDH mutant gliomas.
In Brief –
Gliomas with IDH mutations show increased sensitivity to radiation in concert with glutaminase inhibition offering a new approach towards treating these tumors.
Graphical Abstract
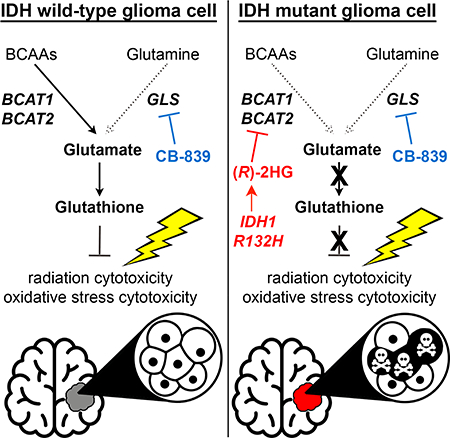
Introduction
IDH1 and IDH2 mutations occur in various cancers, including gliomas and leukemias (Losman and Kaelin, 2013). Tumor-associated IDH mutants convert 2-oxoglutarate (2OG) to the (R) enantiomer of 2-hydroxyglutarate, (R)-2HG, which accumulates to mM levels in tumors (Losman and Kaelin, 2013). (R)-2HG structurally resembles 2OG and competitively inhibits many 2OG-dependent dioxygenases, including JmjC histone demethylases and TET DNA hydroxylases (Losman and Kaelin, 2013). The challenge has been to determine which dioxygenases play causal roles in transformation by (R)-2HG in a given tumor type, and which are bystanders. In this regard, inhibition of TET2 promotes leukemic transformation and is also suspected of playing a role in IDH mutant gliomas (Flavahan et al., 2016; Losman and Kaelin, 2013).
Although studies of (R)-2HG targets have focused on 2OG-dependent dioxygenases, cells have other 2OG-dependent enzymes, including various transaminases that reversibly couple nitrogen transfer from their specific amino acid substrates to the interconversion of 2OG and glutamate. In this regard, Reitman and colleagues reported that the conditioned media of IDH mutant cells had decreased levels of branched chain α-ketoacids (BCKAs) compared to control cells (Reitman et al., 2011). BCKAs are made from the branched chain amino acids (BCAAs) leucine, isoleucine, and valine by the BCAA transaminases BCAT1 and BCAT2 (Figure 1A). Based on these findings and the 2OG dependence of BCAT1 and BCAT2, we hypothesized that the BCKA depletion observed by Reitman and coworkers was due to direct, competitive inhibition of BCAT activity by (R)-2HG and that this would create metabolic vulnerabilities that could be exploited therapeutically.
Figure 1. (R)-2HG Directly Inhibits BCAT1 and BCAT2.

(A) BCAT reactions. (B and C) Immunoblots of HOG cells expressing HA-tagged wild-type (WT) or mutant IDH1 (R132H) (n = 3) (B) or the indicated shRNAs (n = 2) (C). EV = empty vector. (D) Heatmap of metabolites in HOG cells as in (B) (n = 3) and (C) (n = 4) or HOG cells treated with the BCAT inhibitors Compound 2 (Cmpd 2) or gabapentin (gaba) (n = 3) relative to DMSO, shControl, and EV controls. Numbers after metabolites indicate m/z. (E) In vitro BCAT1 assays. Data were normalized to 0 mM (R)-2HG sample at each concentration of 2OG. For 150 μM 2OG samples, n = 4. For all other samples, n = 3. (F) Lineweaver-Burk plot of data in (E). (G) In vitro BCAT1 assays with 50 μM 2OG and (R)-2HG (n = 3) or (S)-2HG (n = 2). (H to K) In vitro transaminase assays with 50 μM 2OG. For panels (H), (I), (J), and (K), n = 3, 4, 8, and 4, respectively. (L to N) In silico modeling of 2OG, (R)-2HG, and (S)-2HG binding to human BCAT2 in the PMPbound conformation. Dashed lines = predicted hydrogen bonds. Asterisks = 2 additional hydrogen bonds predicted to be formed by (R)-2HG relative to (S)-2HG. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. See also Figure S1.
Results
(R)-2HG Inhibits BCAT1 and BCAT2
We infected IDH1/2 wild-type HOG human glioma cells (Reitman et al., 2011) with a lentivirus encoding the canonical R132H IDH1 mutant, wild-type IDH1, or with the empty vector (EV) (Figure 1B). We then did GC-MS-based metabolomic profiling of the resulting stable cell lines, as well as parental HOG cells that were treated with BCAT inhibitors (Compound 2 or gabapentin) (Hu et al., 2006) or were infected to produce one of two BCAT1 shRNAs or a control shRNA (Figures 1C and 1D). IDH1 R132H increased the abundance of the BCAAs and depleted glutamate and the BCKAs α-keto-β-methylvalerate (KMV) and α-ketoisocaproate (KIC), as did pharmacologically or genetically inhibiting BCAT1 (Figure 1D). Similarly, glioma stem-like cell (GSC) lines derived from IDH1 mutant gliomas (Figure S1B) had elevated BCAAs and lower levels of KIC and glutamate compared to IDH1 wild-type GSC lines (Figures S1A and S1C). Therefore, both engineered and patient-derived IDH1 mutant cells exhibit metabolic changes consistent with impaired BCAT activity.
We next developed and validated a recombinant BCAT1 activity assay (Figures S1D to S1G). (R)-2HG competitively inhibited BCAT1, which is a cytosolic enzyme, with respect to 2OG over a physiologically-relevant range of 2OG concentrations (Figures 1E to 1H) (Koivunen et al., 2012). (R)-2HG also inhibited the mitochondrial BCAT1 paralog, BCAT2 (Figure 1I), but minimally affected the GOT1 and GOT2 aspartate transaminases (Figures 1J and 1K). The alternative enantiomer, (S)-2HG, was a less effective BCAT inhibitor (Figure 1G), consistent with molecular docking studies that predict more extensive hydrogen bonding between (R)-2HG and BCAT2 (Figures 1L to 1N, S1H, and S1I) relative to (S)-2HG.
IDH Mutations Impair BCAA Catabolism and Glutamate Biosynthesis
To ask if mutant IDH inhibits BCAT in cells, we assayed the conversion of 15N-leucine to 15N-glutamate in immortalized human astrocytes (NHA) and HOG cells infected to produce the IDH1 R132H mutant, wild-type IDH1, or with the EV (Figures 2A and 2C). BCAT1 protein levels were lowered in late passage IDH1 mutant NHA cells (Figure S2C), consistent with a prior study (Tonjes et al., 2013) and with reduced BCAT1 mRNA levels in IDH1 mutant gliomas (Figure S2D), but were not downregulated in late passage HOG cells (Figures S2A and S2B). We therefore studied early passage NHA and HOG cells to avoid potential confounding effects in late passage NHA cells caused by decreased BCAT1 protein levels. Both the IDH1 mutant NHA and HOG cells had tumor-relevant (R)-2HG concentrations (Figure 2B) (Dang et al., 2009). Leucine transamination was impaired in IDH1 mutant NHA and HOG cells (Figures 2D, 2E, and S2F), mirroring the effects of Compound 2 (Figure S2E).
Figure 2. Mutant IDH Suppresses Glutamate Biosynthesis by Inhibiting BCAA Transamination.

(A and B) Immunoblots (A) and 2HG levels (B) in NHA and HOG cells expressing HA-tagged wild-type (WT) or mutant IDH1 (R132H) or EV (n = 3). (C) Schema of BCAT-dependent nitrogen transfer in 15N-leucine tracing assays. (D and E) 15N-leucine tracing assays in NHA (n = 3) (D) and HOG (n = 2) (E) stable cell lines. (F and G) 15N-leucine tracing assays in HOG cells pretreated 18 hr with 1 mM (R)- or (S)-2HG-TFMB esters (n = 3) (F) or for 3 days with 3 μM AGI-5198 or DMSO (n = 2) (G). (H) Metabolite levels in HOG stable cell lines treated as in (G) (n = 7). TIC = total ion counts. (I) In vitro OAT assay with 250 μM 2OG (n = 3). (J and K) Immunoblots (J) and glutamate levels (K) in HOG stable lines infected to produce OAT or EV (n = 3). Cells were given 300 μM dm2OG and/or 500 μM L-ornithine for 8 hr as indicated. (L) Immunoblots of GSC lines (n = 3). (M and N) Steady-state (M) and BCAAderived (N) glutamate in GSC lines (n = 4). In (N) cells were given 15N-BCAAs for 6 hr. (O and P) 2HG levels (n = 3) (O) and BCAA-derived glutamate (P) after 3 day treatment with 3 μM AGI-5198 or DMSO and 6 hr incubation with 15N-BCAAs. In (P), n = 3 for BT054 cells and n = 7 and 6, respectively, for DMSO- and AGI-5198-treated MGG152 cells. For panel (B), data presented are means ± SEM. For other panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. See also Figure S2.
A cell-permeable version of (R)-2HG, (R)-2HG-TFMB (Losman et al., 2013), but not (S)-2HG-TFMB, inhibited leucine transamination in parental HOG cells (Figure 2F). Conversely, leucine transamination in IDH1 mutant cells was rescued by AGI-5198, which blocks (R)-2HG production (Losman et al., 2013) (Figure 2G). Metabolic evidence of suppressed BCAT activity was also observed in HCT116 colorectal cancer cells treated with (R)-2HG or engineered to have heterozygous IDH1 or IDH2 mutations (Figure S2G), while BCAT activity was restored in HT1080 IDH1 +/R132C fibrosarcoma cells treated with AGI-5198 (Figure S2H). Therefore, loss of BCAT activity in IDH mutant cells is caused by (R)-2HG. Mutant IDH2, like mutant IDH1, also potently repressed BCAT activity when introduced into NHA and HOG cells (Figures S2I to S2K).
BCAA catabolism yields glutamate and BCKAs. We found that HOG cells avidly consume BCAAs from culture media (Figure S2L) and that the resulting BCKAs KMV and KIC are secreted by these cells rather than being further metabolized (Figures S2M and S2N). We therefore focused on glutamate, which is used to make many nitrogenous metabolites and acts as a neurotransmitter.
Glutamate does not efficiently cross the blood-brain barrier (BBB) and brain interstitial glutamate concentrations are ~100-fold lower than in serum (Espey et al., 1998; Smith, 2000). Neural cells rely heavily on transaminases, particularly BCATs, to sustain intracellular glutamate levels (Cooper and Jeitner, 2016). Moreover, we found that imported glutamate makes a negligible contribution to intracellular glutamate levels in HOG cells grown ex vivo, reaffirming the importance of glutamate synthesis to maintain the relatively large intracellular glutamate pool (Figure S2O).
To ask whether glutamate suppression by mutant IDH (Figures 1D and S1C) can be linked to loss of BCAT activity, we quantified steady-state 2HG, glutamate, and KIC levels in IDH1 mutant and wild-type HOG cell lines that were treated with AGI-5198 or DMSO (Figure 2H) while grown in cell culture media that contained glucose, glutamate, pyruvate, and amino acid levels based on physiological concentrations in the brain (Espey et al., 1998; Schug et al., 2015; Tisdall and Smith, 2006). AGI-5198 lowered (R)-2HG and normalized intracellular glutamate and KIC levels in IDH1 mutant cells, with the latter change indicative of restored BCAT activity (Figure 2H). Overexpressing an (R)-2HG-insensitive transaminase, ornithine aminotransferase (OAT), and supplementing media with the OAT substrates 2OG (in a cellpermeable form) and L-ornithine substantially reversed mutant IDH-induced glutamate depletion (Figures 2I to 2K).
IDH1 mutant GSC lines, like our engineered cell lines, also displayed reduced glutamate levels and BCAA catabolism relative to IDH1 wild-type lines despite comparable BCAT1 and BCAT2 protein levels (Figures 2L to 2N). Lowering (R)-2HG in MGG152 and BT054 cell lines with AGI-5198 potently stimulated BCAT activity and restored glutamate levels without affecting BCAT1 or BCAT2 protein levels (Figures 2O, 2P, S2P, and S2Q).
BCAT Inhibition Induces Compensatory Glutamine Metabolism
Cells can also make glutamate from glutamine using glutaminase (GLS). Interestingly, GLS is highly expressed in the brain compared to most normal tissues (Figures S3A and S3B) and low-grade gliomas upregulate a more active GLS splice isoform (Cassago et al., 2012) (Figures S3C and S3D). Furthermore, human brain tumors are highly glutamine avid (Venneti et al., 2015) (Tardito et al., 2015). We hypothesized that decreased BCAT function in IDH mutant cells would increase their reliance on GLS activity for glutamate production. Indeed, the conversion of glutamine to glutamate was increased in IDH1 mutant NHA and HOG cells compared to their wild-type counterparts (Figures 3A to 3C and 3E). This was not due to increased protein levels of GLS or c-Myc, which regulates GLS expression (Figures 3D and S3E) (Gao et al., 2009). Moreover, expression of mutant IDH1 in NHA and HOG cells cooperated with the GLS inhibitor CB-839 (Gross et al., 2014) to deplete intracellular glutamate levels >75% in vitro (Figure 3F) and the glutamate pools in IDH1 mutant GSC lines were hypersensitive to CB-839 (Figure 3G).
Figure 3. BCAT Repression Promotes Glutamine-dependent Glutamate Synthesis.

(A) Schema of GLS-dependent nitrogen transfer in α-15N-glutamine tracing assays. (B and C) α-15Nglutamine tracing assays (n = 3). (D) Immunoblot analysis (n = 2). (E) 15N-(amide)-glutamine tracing in HOG stable lines (n = 4). Fractional labeling of ammonia 8 hr after tracer addition is shown. (F) Glutamate levels in NHA and HOG stable cell lines treated with 100 nM CB-839 or DMSO for 2 hr (n = 4). (G) Glutamate levels in GSC lines treated with CB-839 or DMSO for 2 hr (n = 3). (H and I) Immunoblots (H) and 15N-leucine and α-15N-glutamine tracing assays (I) in HOG stable cells expressing the indicated sgRNAs (together with Cas9) and, where indicated, sgRNA-insensitive BCAT1 and BCAT2 cDNAs. n = 3. (J) Schema depicting the major routes of glutamate synthesis in glial cells. (K and L) 15N-BCAA and α-15N-glutamine tracing assays of NHA (K) and HOG (L) cells (n = 3). Fractional labeling of the glutamate pool at 6 hr is shown. (M) α-15N-glutamine (n = 6) and 15N-leucine (n = 3) tracing assays in HOG cells treated with 100 μM Compound 2 or 100 nM CB-839, respectively. Cells were given 15N-labeled amino acids for 20 minutes. (N) Immunoblot analysis of HOG stable lines infected to produce the GLS Y394L mutant or EV. KGA and GAC splice isoforms are indicated. n = 3. (O) Glutamate levels in HOG stable cell lines as in (N) (n = 3). For panels (N) and (O), see also Figures S3P to S3R. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. See also Figure S3.
To model this further we inactivated BCAT1 and BCAT2 in HOG cells with CRISPR/Cas9, leading to an on-target loss of leucine transamination (Figure 3H and 3I, left panel). 15N-glutamine tracing confirmed that, similar to IDH mutant cells, glutamine catabolism was induced in BCAT null cells (Figure 3I, right panel). In contrast to IDH1 mutant HOG cells, however, glutamate levels normalized over time, associated with dramatic 2OG upregulation, in BCAT null cells (Figures S3F to S3I). We suspected that 2OG induction in BCAT null cells drove other transamination reactions and that ongoing 2OG consumption by mutant IDH prevented this adaptation in the IDH1 mutant cells. Indeed, BCAT null cells potently increased alanine transamination, an effect that was only observed in IDH1 mutant cells when supplemented with 2OG (Figures S3J to S3N). Therefore, both loss of BCAT activity and increased 2OG consumption contribute to decreased transamination-dependent glutamate synthesis in IDH mutant cells (Figure S3O), leading to increased reliance on GLS (Figure 3J).
15N-BCAAs and 15N-glutamine tracer studies in NHA and HOG cells showed that these amino acids supply 80–90% of the nitrogen in the glutamate pool, with about half derived from BCAAs and half from glutamine (Figures 3K and 3L). To ask whether BCATs and GLS compensate for one another, we treated parental HOG cells with Compound 2 or CB-839 and performed 15N-glutamine and 15N-leucine tracing. As predicted, Compound 2 increased the conversion of glutamine to glutamate, while CB-839 increased flux through the BCATs (Figure 3M). Furthermore, expressing a hypermorphic GLS variant (DeLaBarre et al., 2011) rescued glutamate production in IDH mutant cells (Figures 3N, 3O, and S3P to S3R). Therefore, BCATs and GLS reciprocally support glutamate synthesis and GLS can be engaged to overcome the loss of BCAT activity caused by mutant IDH.
To address the significance of these findings in vivo we fed mice an 15N-leucine-containing diet and observed higher glutamate labeling in the brain as compared with muscle, liver, or adipose tissues or plasma (Figures 4A and 4B), consistent with an important role for BCATs in the CNS (Hull et al., 2015). We next infected HOG cells expressing IDH1 R132H or the EV (hereafter called IDH1 wild-type cells) with a virus encoding both luciferase and GFP (Figure S4A) and grew them orthotopically into nude mice. The GFP reporter enabled us to rapidly isolate high-purity tumor tissue from surrounding normal brain tissue for metabolic analyses, which confirmed clinically-relevant concentrations of 2HG in the IDH1 mutant tumors (Figures 4C and 4D). We then fed the tumor-bearing mice an 15N-leucine-containing diet and harvested tumor tissue 4 days later. We observed robust suppression of glutamate labeling, and its downstream metabolite aspartate, in IDH mutant gliomas relative to wild-type tumors despite similar plasma leucine tracer accumulation (Figures 4E to 4G). The 15N-glutamate levels in tumors vastly exceeded those in plasma, consistent with intracellular BCAT-dependent 15Nglutamate synthesis (Figure S4B).
Figure 4. Mutant IDH1 Reprograms Nitrogen Metabolism In Vivo.

(A and B) In vivo 15Nleucine tracing assay in naïve mice. Mice were fed an 15N-leucine-containing diet or standard diet (Unlabeled) and plasma and tissues were harvested 4 days later. Fractional labeling of plasma leucine (A) and the ratio of fractional glutamate labeling to fractional leucine labeling in mice fed an 15N-leucine diet (B) are shown (n = 4 mice per diet). (C and D) Absolute quantification of 2HG (C) and ex vivo fluorescence imaging (D) of isogenic orthotopic xenograft tumors established from mutant IDH- or EV-expressing HOG stable cell lines co-expressing GFP and firefly luciferase (FLuc) reporters. In (C), n = 3 mice per cohort and in (D) a representative image is shown. (E to G) In vivo 15N-leucine tracing assay in mice bearing IDH1 mutant and wild-type gliomas as in (C) and (D). Fractional labeling of plasma leucine (E) and the ratios of fractional glutamate (F) and aspartate (G) labeling to fractional leucine labeling in tumors are shown. In (E), n = 10 and 9 mice bearing IDH1 wild-type and mutant tumors, respectively. In (F) and (G), n = 14 and 18 IDH1 wild-type and mutant tumor samples, respectively. (H) Immunoblot analysis of HOG stable cell lines expressing Cas9 and the indicated sgRNAs (n = 3). KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. (I to L) 2HG (I), glutamine (J), glutamate (K), and aspartate (L) levels in xenografts formed by cells in (H). n = 6, 5, 5, and 9 mice in EV-sgCntl, EV-sgGLS, R132H-sgCntl, and R132H-sgGLS cohorts, respectively. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant. See also Figure S4.
Inactivation of GLS in the IDH1 mutant and wild-type HOG cells using CRISPR/Cas9 (Figure 4H) did not decrease their ability to form brain tumors (Figure S4C), but did, as expected based on our in vitro findings, cooperate with mutant IDH to lower glutamate and aspartate levels (Figures 4I to 4L and S4D). The effects of the single GLS sgRNA used in these experiments were on-target because they could be rescued by an sgRNA-insensitive GLS cDNA (Figures S4E and S4F) and were corroborated with CB-839 (Figures S4G to S4J). In contrast, glutamate levels in the IDH wild-type gliomas were impervious to GLS ablation (Figure 4K), consistent with sustained BCAT activity in these tumors (Figure 4F).
Consistent with prior cell culture studies (Emadi et al., 2014; Seltzer et al., 2010), neither genetic nor pharmacologic GLS inhibition compromised (R)-2HG synthesis in IDH1 mutant HOG tumors (Figures 4I and S4G). Moreover, U-13C-glutamine tracing in mice bearing orthotopic IDH1 mutant HOG tumors showed that the tumor 2HG pool was not labeled significantly (Figure S4L) despite substantial enrichment of labeled glutamine in plasma (Figure S4K) and robust labeling of tumor glutamate and 2OG pools. Therefore, GLS inhibition does not impede (R)-2HG synthesis in our IDH1 mutant glioma model.
Inhibition of Glutamate Synthesis Causes Glutathione Depletion
Glutamate is needed to make glutathione, which is decreased in IDH mutant brain tumors (Bisdas et al., 2016; Pope et al., 2012). We confirmed that glutathione levels are decreased in IDH1 mutant orthotopic xenograft gliomas (Figure 5A) and increased by AGI-5198 in both engineered (HOG) and endogenous (MGG152) IDH1 mutant glioma cell lines (Figure 5B). Decreased levels of reduced glutathione (GSH) in IDH1 mutant gliomas could reflect glutamate depletion caused by BCAT inhibition, consumption of NADPH by mutant IDH1, or both (Figure 5C). Arguing against a prominent role for NADPH consumption, the reduced/oxidized glutathione ratio was not impacted by mutant IDH in vitro or in vivo (Figures 5A and S5A) and, consistent with findings in IDH1 mutant macrophages (Sasaki et al., 2012), NADPH levels were not suppressed in IDH1 mutant HOG tumors (Figure S5B).
Figure 5. Mutant IDH Activity and GLS Inhibition Cooperate to Deplete Glutathione by Lowering Glutamate.

(A) GSH levels and reduced/oxidized glutathione ratios in HOG orthotopic gliomas. n = 8 and 6 IDH1 wild-type and mutant tumors, respectively. (B) Total glutathione levels in IDH1 mutant HOG (n = 3) and MGG152 (n = 5) cells treated with 3 μM AGI-5198 or DMSO for 3 days. (C) Schema for regulation of glutathione synthesis by mutant IDH. (D and E) Volcano plots of metabolites in NHA cells treated with 500 μM (R)-2HG-TFMB for 24 hr (D) or expressing a BCAT1 shRNA (E) relative to DMSO and a control shRNA, respectively. p values were determined via t-test using MetaboAnalyst. n = 3. (F) Venn diagram of metabolites co-regulated by (R)-2HG and a BCAT1 shRNA. (G) 15N-BCAA tracing assays of HOG stable lines (n = 3). Fractional labeling of the glutamate and GSH pools is shown at 6 and 12 hr, with values normalized to HOG EV cells at 6 hr. (H and I) Total glutathione levels in HOG and NHA cells treated for 8 hr with Compound 2 (n = 3) (H) or CB-839 (I). In (I), n = 3 for cells treated with 100 nM CB-839 and n = 4 for all others. (J) Total glutathione levels in HOG (n = 3) and NHA stable lines treated for 8 hr with 100 nM CB-839, 250 μM BSO, or DMSO. For NHA lines, n = 3 for BSO-treated IDH1 mutant cells and n = 5 for all others. (K) Total glutathione levels in HOG stable cell lines as in Figure 3N (n = 3). See also Figure S5H. (L to N) Total glutathione levels in GSC lines that were untreated (L) or treated for 8 hr with 1 μM CB-839 (M) or 250 μM BSO (N). n = 3. (O) LC-MS/MS-based GSH quantification in IDH1 mutant and wild-type HOG orthotopic gliomas treated with 7 doses of CB-839 (200 mg/kg BID) or vehicle control. n = 8, 9, 8, and 6 mice in EV-Vehicle, EV-CB-839, R132H-Vehicle, and R132H-CB-839 cohorts, respectively. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant. See also Figure S5.
To address the contribution of BCAT inhibition to this phenotype, we profiled NHA cells treated with (R)-2HG-TFMB or with a BCAT1 shRNA compared to controls using LC-MS/MS. Both (R)-2HG and the BCAT1 shRNA decreased both glutamate and glutathione levels (Figures 5D to 5F) without significantly reducing the levels of the glutathione precursors serine, which gives rise to glycine, and cysteine (Figures S5C and S5D). We then performed 15N-BCAA tracing in isogenic HOG cell lines and found that mutant IDH expression impedes the flow of nitrogen from BCAAs to glutamate and ultimately to glutathione (Figure 5G). Glutamate is limiting for glutathione synthesis because glutathione levels were decreased in HOG and NHA cells treated with Compound 2 (Figure 5H) or with the GLS inhibitors CB-839 and BPTES (Figures 5I and S5G), all of which lower glutamate levels (see Figures 1D, 3F, and S5F). The effects of BPTES were on-target because they were reversed by BPTES-resistant GLS mutants (DeLaBarre et al., 2011; Ferreira et al., 2013) (Figures S5E to S5G). The effects of CB-839 and mutant IDH on glutathione levels, like their effects on glutamate, were additive in NHA and HOG cells (Figures 5J and 3F). Conversely, expressing the hypermorphic GLS variant (DeLaBarre et al., 2011) that rescued glutamate levels in IDH mutant HOG cells (Figures 3O and S3Q) also rescued glutathione levels (Figures 5K and S5H). IDH mutant GSC lines display lower glutathione levels relative to IDH wild-type lines and a trend toward greater susceptibility to CB-839-induced glutathione depletion (Figures 5L and 5M). This did not reflect a general hypersensitivity to glutathione synthesis inhibitors because the glutamate-cysteine ligase inhibitor buthionine sulfoximine (BSO) suppressed glutathione levels to a similar extent in both genotypes (Figure 5N). TS516 cells displayed the greatest sensitivity to glutathione depletion by CB-839 amongst the IDH1 wild-type GSC lines tested and had the highest basal expression of GLS (Figure S5I) and c-Myc (Figure S3E), which increases GLS expression and dependence. Treatment of tumor-bearing mice with CB-839 depleted glutathione in IDH mutant HOG gliomas, but not in isogenic wild-type tumors (Figure 5O), mirroring its effects on glutamate (Figure S4I). Therefore, BCAT inhibition and resulting glutamate depletion decrease glutathione levels and induce reliance on GLS for glutathione synthesis in IDH mutant glioma cells.
Mutant IDH and GLS Inhibition Are Synthetic Lethal Under Oxidative Stress
GLS inhibitors selectively inhibit the proliferation of IDH mutant cells ex vivo, but the mechanism underlying this selectivity remains unknown (Emadi et al., 2014; Seltzer et al., 2010). Moreover, the effect sizes in these studies were small, possibly because they used standard cell culture media, which typically contains high levels of the ROS scavenger pyruvate. We postulated that the loss of glutathione observed after inhibiting GLS in IDH1 mutant cells would impair resistance to oxidative stress. In cell culture media with physiological metabolite levels, CB-839 cooperated with mutant IDH to profoundly inhibit HOG cell proliferation, mirroring their effects on glutamate and glutathione (Figures 6A, 3F, and 5J, note log scale in Figure 6A). This was due to redox stress because CB-839-induced growth inhibition was rescued by AGI-5198 and by media supplementation with glutamate or the antioxidant N-acetylcysteine (NAC) (Figures 6B to 6D).
Figure 6. Mutant IDH and GLS Inhibition are Synthetic Lethal during Oxidative Stress.

(A to D) Proliferation assays of HOG stable cell lines treated with 100 nM CB-839 or DMSO (n = 6) (A) and, where indicated, 3 μM AGI-5198 (n = 3) (B), 1 mM L-glutamate (n = 3) (C), or 1 mM NAC (n = 3) (D). Fold changes in cell numbers relative to Day 0 are displayed. (E to G) Cell death quantification (AnnexinV or DAPI positivity) in NHA and HOG stable lines that were treated for 18 hr, where indicated, with 100 nM CB-839 or 2.5 mM esterified reduced glutathione (eGSH) prior to and during a 5 hr treatment with tBH. In (E), n = 4 and 3 for cells treated with 100 μM and 150 μM tBH, respectively, and n = 5 for cells not treated with tBH. In (F), n = 2 for cells treated with 300 μM tBH and n = 3 for all others. In (G), n = 3 for cells treated with eGSH in the absence of CB-839 and n = 4 for all others. (H) Cell death quantification in HOG stable cell lines as in Figures 2J and S3P and treated as in (F). Cells were supplemented with 300 μM dm2OG and/or 500 μM L-ornithine starting 5 hours before CB-839 or DMSO pretreament, as indicated. tBH was added at 250 μM where indicated. In the left panel, n = 4 for cells treated with DMSO or cotreated with tBH and CB-839 and n = 3 for cells treated with tBH or CB-839 alone. In the right panel, n = 3. (I) Quantification of cell death in GSC lines pretreated with DMSO, 1 μM CB-839, or 250 μM BSO for 18 hr prior to and during treatment with tBH for 5 hr. Each cell line was treated with tBH at a dose empirically determined to cause 10–50% cell death without a second agent (50 μM for TS603, 75 μM for TS516, 100 μM for BT260/TS676, and 200 μM for BT054/MGG152) (see Figures S6I and S6J). n = 3, 2, 3, 4, 8, and 3 for TS516, TS676, BT260, MGG152, TS603, and BT054 cell lines, respectively. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant. See also Figure S6.
Exposure to 21% oxygen can, itself, induce oxidative stress (Parrinello et al., 2003) and steady-state ROS levels, as determined by an ROS-sensitive dye, were indeed higher in IDH1 mutant HOG cells under 21% oxygen conditions relative to IDH1 wild-type controls (Figures S6C to S6E). In contrast to the effects of CB-839 under 21% oxygen, we noted that GLS inhibition did not significantly impair the proliferation of IDH1 mutant HOG cells at 3% oxygen ex vivo or in orthotopic tumor assays (Figures S6A, S6B, and S4C). Nonetheless, we hypothesized that inhibiting GLS, by lowering glutathione levels (Bump and Brown, 1990), would preferentially sensitize IDH1 mutant cells to radiation and ROS-inducing drugs. Indeed, IDH1 mutant NHA and HOG cells were highly sensitive to the combination of CB-839 and the oxidant tert-butyl hydroperoxide (tBH) compared to their IDH wild-type counterparts and their effects were reversed by a cell-permeable form of GSH (Figures 6E to 6G). Synergistic cytotoxicity of tBH and CB-839 in IDH mutant HOG cells was also rescued by stimulating glutamate synthesis via ornithine transamination (Figure 6H, left panel) or glutamine catabolism using the hypermorphic Y394L GLS variant (Figure 6H, right panel). Note that Y394L GLS is resistant to BPTES (DeLaBarre et al., 2011) and CB-839 (data not shown). Despite their substantially lower basal glutathione content and higher ROS levels (Figures 5L, S6F, and S6G), IDH mutant GSC lines did not undergo apoptosis upon CB-839- or BSO-induced glutathione depletion (Figure S6H), nor did they display hypersensitivity to tBH treatment alone (Figures S6I and S6J). They were, however, hypersensitive to the combination of tBH and CB-839, consistent with our engineered cell culture models (Figure 6I). Whereas only 1 of 3 IDH wild-type GSC lines was susceptible to synergistic cell killing by tBH and CB-839 treatments (the c-Mychigh/GLShigh TS516 line), all 3 of the IDH1 mutant lines were sensitized to tBH by CB-839. In contrast, BSO synergized with tBH to kill all cell lines independently of IDH1 genotype. This suggests that CB-839’s ability to preferentially deplete glutathione in IDH mutant GSC lines underlies its selective potentiation of oxidative stress-induced cytotoxicity in these cells. Although mutant IDH increases the sensitivity of hematopoietic cells to oxidative DNA damage by downregulating ATM (Inoue et al., 2016), ATM levels are not suppressed IDH1 mutant glial cells compared to controls (Figures S6K to S6M).
CB-839 also enhanced the effectiveness of radiation against IDH1 mutant HOG cells, but not IDH1 wild-type HOG cells, in vitro, unless the cells were cotreated with NAC (Figures 7A and 7B). We next established isogenic orthotopic xenograft gliomas from IDH1 mutant and wildtype HOG cells engineered to co-express luciferase and GFP, as in Figure 4. Although CB-839 accumulates poorly in normal mouse brains, it accumulated in the orthotopic HOG cell tumors to levels comparable to those seen in orthotopic breast cancer xenografts (Figure S7A) (Gross et al., 2014), possibly due to local breakdown of the BBB. In IDH mutant gliomas, but not wild-type gliomas, radiation and CB-839 strongly inhibited tumor growth only when given in combination (Figures 7C to 7F, S7B, and S7C), extending median survival from 13 days in untreated mice to 30 days. These selective effects correlated with preferential depletion of glutamate (Figure S4I) and glutathione (Figure 5O) by CB-839 in the IDH1 mutant tumors. Similar results were observed when subcutaneous tumors formed by the TS603 IDH1 mutant GSC line were treated with radiation and CB-839 (Figures 7G, S7D, S7E, and S7K to S7M), associated with reduced proliferation, reduced angiogenesis and modestly increased apoptosis (Figures S7F to S7J).
Figure 7. GLS Inhibition Selectively Radiosensitizes IDH1 Mutant Glioma Cells.

(A and B) Clonogenic survival of irradiated HOG stable cell lines after pretreatment for 24 hr with 100 nM CB-839, 1 mM NAC, or DMSO. In panel (A), n = 4. In panel (B), cells received a radiation dose of 2 Gy (RT) or were not irradiated (Sham); n = 2 and 3 for HOG-EV and HOG-R132H cells, respectively. Data are normalized to DMSO-treated cells for each stable line. (C) Representative bioluminescence images of orthotopic gliomas formed by the indicated luciferase-positive HOG cells before and after treatment, where indicated, with CB-839 (200 mg/kg BID for 9 days) and radiation (RT, 3 fractions of 5 Gy). (D) Growth rates of tumors represented in panel (C). n = 11, 4, 6, and 4 mice bearing IDH1 mutant gliomas treated with both CB-839 and RT, CB-839 alone, RT alone, and neither, respectively. n = 11, 9, 13, and 11 mice bearing IDH1 wild-type gliomas treated with both CB-839 and RT, CB-839 alone, RT alone, and neither, respectively. (E and F) Kaplan-Meier survival curves of mice bearing orthotopic xenografts treated as in (C). Red arrows = radiation dosing; yellow shading = period of CB-839 treatment. (G) Volumes of TS603 subcutaneous xenografts treated with CB-839 (200 mg/kg BID for 9 days) and/or radiation (RT, 3 fractions of 3 Gy) as indicated. In panels (A) and (B), data presented are means ± SD, and twotailed p values were determined by unpaired t-test. For panels (C) to (G), results are from two experiments. In panel (D), data presented are means ± SEM and two-tailed p value was determined by unpaired t-test. In panels (E) and (F), two-tailed p values were determined by logrank test. In panel (G), data presented are means ± SEM and statistical analysis of tumor volumes at day 21 was performed. Two-tailed p values were determined by unpaired t-test. For all panels, *p < .05, **p < .01, ***p < .001; n.s. = nonsignificant. See also Figure S7.
Discussion
Our findings help explain why IDH1 mutant gliomas convert 2OG to glutamate more slowly than IDH1 wild-type tumors and have low glutamate and glutathione content (Bisdas et al., 2016; Chaumeil et al., 2014; Choi et al., 2012; Izquierdo-Garcia et al., 2015; Jalbert et al., 2017; Pope et al., 2012; Reitman et al., 2011). We show that (R)-2HG directly inhibits BCAT1 and BCAT2. Consistent with earlier analyses of IDH1 mutant brain tumors (Tonjes et al., 2013), our late-passage IDH1 mutant NHA cells also had low BCAT1 protein levels. However, IDH1 mutant early passage NHA and HOG cells and IDH1 mutant GSC lines had low BCAT activity despite preserved BCAT1 expression. Therefore, BCAT1 silencing is not required for the loss of BCAT activity displayed by IDH1 mutant glioma cells.
Glutamate is an abundant intracellular molecule that is used as a nitrogen donor to make many metabolites required for cell growth and proliferation, including non-essential amino acids, nucleotides, and glutathione. Many tumor cells display net glutamate secretion (Sharma et al., 2010), indicating a need for robust glutamate synthesis. Glioma cells might be especially reliant on intracellular glutamate biosynthetic pathways because glutamate, unlike other amino acids, does not efficiently cross the BBB into the CNS (Smith, 2000). Our study shows that glutamine (via GLS) and BCAAs (via the BCATs) are the primary nitrogen donors used to make glutamate in glial cells.
Many lines of evidence suggest that IDH mutational status dictates the relative contributions of glutamine versus BCAAs to glutamate synthesis in brain tumors. A prior in vivo study using 13C-labeled glucose and glutamine tracers showed that patient-derived xenografts made from primary GBMs, ~95% of which are IDH wild-type, consume glutamine, but do not readily convert it to glutamate (Marin-Valencia et al., 2012). Instead, they oxidize glucose to make glutamate, consistent with a biochemical mechanism wherein TCA cycle-derived 2OG is used by transaminases to make glutamate. Our in vivo 15N-leucine tracing studies bolster this earlier study by showing that IDH wild-type orthotopic HOG xenograft gliomas engage in significant BCAA transamination to make glutamate. Moreover, we found that the glutamate pool in these tumors is unaffected by GLS inactivation. In our isogenic, IDH1 mutant HOG tumors, however, we observed lower BCAT activity, as evidenced by reduced flux of leucinederived nitrogen into glutamate, decreased basal glutamate levels, and hyperdependence on GLS for the production of glutamate and its downstream product, glutathione. These findings were corroborated in vitro and in vivo with our isogenic models and with patient-derived GSC lines. Therefore, IDH mutations govern the selection of glutamate synthetic routes in glioma cells.
We observed minimal labeling of the (R)-2HG pool in our U-13C-glutamine in vivo tracing studies of IDH1 mutant orthotopic HOG tumors, consistent with earlier in vitro studies (Emadi et al., 2014; Seltzer et al., 2010). In contrast, a recent study found that glutamine contributes to (R)-2HG synthesis in an IDH1 mutant chondrosarcoma model (SalamancaCardona et al., 2017). Although there are many technical differences between our study and their study that could account for this discrepancy, even beyond tumor cell of origin and implantation site, both their and our in vivo tracing studies found that less than 3% of the 2HG pool is labeled in the first 30–45 minutes after U-13C-glutamine administration.
We show that genetic ablation of BCAT1 and BCAT2 causes cells to engage GLS to make glutamate, recapitulating the effects of mutant IDH. Inhibiting GLS in IDH1 mutant cells, in which BCAT is suppressed by (R)-2HG, blocks this adaptation and dramatically impairs their fitness by lowering glutamate, glutathione, and resistance to oxidative stress. Glutamate loss is key in this context because we could rescue the effects of mutant IDH1 on redox homeostasis by restoring glutamate levels using a GLS hypermorph or OAT, which is (R)-2HG-insensitive.
Chronic oxidative stress might contribute to the indolent clinical behavior of IDH mutant brain tumors (Cohen et al., 2013). Our findings bear on the recent observations that mutant IDH confers radiosensitivity in an (R)-2HG-dependent manner in preclinical models (Molenaar et al., 2015) and suggest that blocking (R)-2HG production could antagonize radiation and radiomimetic drugs.
Blocking (R)-2HG production causes regressions in IDH1 mutant AML (Waitkus et al., 2018). There are early indications, however, that this strategy will be less effective against IDH mutant gliomas, possibly because mutant IDH acts through a ‘hit-and-run’ mechanism in this setting (Waitkus et al., 2018). If so, it could be useful to devise therapeutic strategies that seek to exploit, rather than inhibit, (R)-2HG.
We showed that mutant IDH and GLS inhibition have a synthetic lethal relationship under conditions of oxidative stress because they both converge on glutamate production and redox homeostasis. Our findings were concordant using multiple cell lines, including engineered isogenic cells and patient-derived non-isogenic GSC lines, and were observed in both subcutaneous and orthotopic tumor models. Nonetheless, our work was limited by the dearth of available tumorigenic IDH1 mutant glioma lines (and no low-grade glioma lines) and by CB839’s poor BBB penetration. Indeed, it is conceivable that decreased BCAT activity and altered redox homeostasis contributes to the difficulty in growing low-grade IDH1 mutant glioma cells ex vivo and in mice. It will be important to study the interaction of redox stress and GLS inhibition in additional models of IDH mutant glioma as they become available, such as a recently described genetically engineered mouse model of this disease (Philip et al, 2018). It will also be important to further study the ability of GLS inhibitors to sensitize IDH mutant glioma cells to oxidative stress versus radiation, given that these treatments can induce distinct cellular responses, especially with respect to DNA damage pathways. Nonetheless, the National Cancer Institute Cancer Therapy Evaluation Program (CTEP) is now testing the combination of CB-839 with radiation and temozolomide for the treatment of IDH1 mutant glioma patients (ClinicalTrials.gov Identifier NCT03528642) based on our findings.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William G. Kaelin, Jr. (william_kaelin@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Puromycin-resistant NHA cells (human astrocytes immortalized with HPV E6 and E7 and hTERT, gender unknown) (Sonoda et al., 2001) were a kind gift of Dr. Russell Pieper (UCSF) or were made by infecting human astrocytes (Life Technologies N7805200) with retroviruses made with pLXSN-E6/E7 (see below) and pWZL-Blast-Flag-HA-hTERT (Addgene 22396). The latter cells were immortalized without a puromycin resistance marker to permit rapid puromycin selection in the RNAi experiment depicted in Figure 5E. The NHA cells from the Pieper lab were used in all data figures that used NHA cells except Figures 5D to 5F, S5C, and S5D. HT1080 (American Type Culture Collection, ATCC, male) and NHA cells were cultured in DMEM (Gibco 11995–065) containing 10% FBS and 1% penicillin/streptomycin. The early and late passage IDH1 mutant and wild-type NHA stable cell lines were previously generated (Koivunen et al., 2012). HOG cells (human oligodendroglioma line, kind gift of Dr. Pablo Paez, SUNY Univ. at Buffalo, male) were cultured in IMDM (Gibco 12440–053) containing 10% FBS and 1% penicillin/streptomycin. Wild-type, IDH1 R132H mutant, and IDH2 R172K mutant HCT116 isogenic clones (Horizon Discovery Ltd, male) were cultured in McCoy's 5A Modified Medium containing 10% FBS and 1% penicillin/streptomycin. NCI-H82 cells (male) and HEK293T cells (gender unknown) were originally obtained from ATCC and cultured in RPMI1640 (Gibco 11875–119) and DMEM media, respectively, supplemented with 10% FBS and 1% penicillin/streptomycin added to both. GSC lines TS603, TS516, TS676 (from I. Mellinghoff at MSKCC, genders unknown) (Rohle et al., 2013), BT054 (from S. Weiss at Univ. of Calgary, female) (Kelly et al., 2010), and BT260 (gender unknown) were cultured in NeuroCult NS-A Basal Medium with Proliferation Supplement (StemCell Technologies) supplemented with EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), 1% penicillin/streptomycin, Fungizone (250 ng/ml), and Plasmocin (2.5 μg/ml). GSC line MGG152 (male) (Wakimoto et al., 2014) was cultured in Neurobasal Medium supplemented with 3 mM glutamine, 1X B27, 0.25X N2, EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), 1% penicillin/streptomycin, and Fungizone (250 ng/ml). GSC lines were passaged as neurospheres and dissociated 1–2 times per week. All cell lines were maintained in the presence of 5% CO2 at 37 °C. Each cell line for which gender is listed as ‘unknown’ above was not annotated for gender in the publication originally describing its derivation or, if acquired from a commercial source, in the accompanying product literature.
Animal models
All care and treatment of experimental animals were carried out in strict accordance with Good Animal Practice as defined by the US Office of Laboratory Animal Welfare and approved by the Dana-Farber Cancer Institute (protocol 04–019) Institutional Animal Care and Use Committee. Animal welfare assessments were carried out daily during treatment periods. Female mice were housed together (2–5 mice per cage) and provided free access to standard diet and water. In order to house mice in groups according to treatment cohort after randomization, only female mice were used in this study. Male mice frequently engage in fighting when grouped with non-littermates, thus posing an animal health and welfare risk. Bioluminescence imaging of tumor-bearing mice was performed by technicians who were blinded to treatment assignments. Tumor size measurements and survival analyses were performed by researchers who were unblinded to treatment assignments.
Orthotopic xenograft glioma model
Orthotopic xenograft tumors were established by intracranial injection of 3×105 HOG cells (GFP-positive, luciferase-positive, HOG-EV and HOG-R132H stable cell lines) in a total volume of 3 μl (2% FBS in PBS) into the brains of naïve 6–10 week old female NCr nude mice (Taconic). Animals were anesthetized by IP injection of a ketamine/xylazine solution and a stereotactic frame was used to drill a hole 1 mm anterior and 2 mm lateral to the bregma. Cells were implanted at a depth of 3 mm using a 5 μl syringe (Hamilton). Wound clips were applied to close the incision and animals were provided buprenorphine for analgesia. Mice were bioluminescently imaged 10 days after tumor cell implantation. To minimize variance in tumor size at the time of treatment initiation, only mice with tumor signals ranging from 7×105 to 2×107 photons/sec (values unadjusted for background luminescence) were randomized to receive 200 mg/kg CB-839 or vehicle control twice daily via oral gavage, alone or in combination with whole-brain irradiation (15 Gy total administered in 3 fractions of 5 Gy each) (4 treatment arms total). CB-839 treatment began 12 days after surgery and continued for 8 more days. Mice were irradiated on days 14, 16, and 18 after surgery. Prior to irradiation, mice were anesthetized via IP injection of ketamine/xylazine and placed in a lead shield with a cutout centered over their heads. Radiation doses were administered using a Gammacell-40 irradiator (Nordion). Mice were subjected to bioluminescence imaging once per week and tumor growth rates were calculated by subtracting pretreatment signal from posttreatment signal and dividing by the number of intervening weeks. Mice were monitored daily and euthanized when they displayed symptoms of neurological distress or became moribund, whichever occurred first.
Primary xenograft model of IDH1 mutant glioma
TS603 neurospheres were dissociated and cells were resuspended in a 1:1 mixture of Neurobasal media supplemented with 2 mM glutamine and Matrigel Membrane Matrix (Corning 354234) at a final concentration of 1.5×107 cells/mL. 1.5×106 cells were injected into the flanks of naïve 612 week old female ICR SCID mice in a total volume of 100 μL. For tumor growth assays, tumor diameters were measured using calipers and tumor volumes were calculated based on the following formula: tumor volume (mm3) = width2 × length/2. Mice were monitored for tumor development and randomized to distinct treatment cohorts 30–45 days post-injection, once tumors had reached 50–350 mm3 in size. Mice received 200 mg/kg CB-839 or vehicle control twice daily via oral gavage, alone or in combination with flank irradiation (9 Gy total administered in 3 fractions of 3 Gy each) (4 treatment arms total). CB-839 and vehicle treatments began 1 day after randomization and enrollment and continued for 9 days. Mice were irradiated on days 3, 5, and 7 after randomization and enrollment. Prior to irradiation, mice were anesthetized via IP injection of ketamine/xylazine and placed in a lead shield with a cutout centered over their flanks. Radiation doses were administered using a Gammacell-40 irradiator (Nordion). After treatment initiation, tumor volumes were measured twice per week over the course of the 21-day growth assay.
METHOD DETAILS
Chemicals
TFMB esters of (R)- and (S)-2-hydroxyglutarate enantiomers were generated as previously described (Losman et al., 2013). Briefly, i-Pr2NEt and 3-(trifluoromethyl)benzyl bromide was added to a solution of (R)- or (S)-5-oxotetrahydrofuran-2-carboxylic acid in acetonitrile and the mixture was heated for 10 minutes and stirred at RT overnight. (R)- and (S)-2HG-TFMB esters were purified from the reaction mixture and concentrated. The BCAT inhibitor Compound 2 (N'-(5-chlorobenzofuran-2-carbonyl)-2-(trifluoromethyl)benzenesulfonohydrazide) (Hu et al., 2006) was produced via custom synthesis (Cayman Chemical). Where indicated, cell culture media also contained the following additives: gabapentin (Sigma), AGI-5198 (Xcessbio), CB839 (Calithera), BPTES (Sigma), tert-butyl hydroperoxide (Sigma), ethyl-GSH (Sigma), Lglutamic acid (Sigma), L-ornithine (Sigma), N-acetylcysteine (Sigma), dimethyl 2-oxoglutarate (Sigma), L-S,R-buthionine sulfoximine (BSO, Sigma).
Vectors for genetically manipulating cell lines
shRNA expression plasmids
To make the shRNA expression vectors, complementary oligonucleotides corresponding to BCAT1 shRNAs (#1: TRCN0000005907, #2: TRCN0000005909) and a non-targeting shRNA (target sequence: GCAAGCTGACCCTGAAGTTCA) were designed to generate 5’ EcoRI- and 3’ AgeI-compatible overhangs when annealed. The pLKO.1-puro cloning vector (Addgene 10878) was digested with EcoRI and AgeI, gel-purified, and ligated with annealed oligonucleotides.
CRISPR/Cas9 plasmids
The vectors lentiCRISPR_v2 (Addgene plasmid 52961) and lentiGuide-blast were used to express sgRNAs with and without Cas9, respectively. lentiGuide-blast was generated by replacing the EF1α-puro cassette in lentiGuide-puro (Addgene plasmid 52963) with the PGKblast cassette from pLKO.1-blast (Addgene 26655). lentiGuide-puro and pLKO.1-blast were digested with XmaI and MluI and the lentiGuide-puro backbone and PGK-blast cassette (released from pLKO.1-blast) were gel-purified and ligated to produce lentiGuide-blast. The BsmBI site present in the PGK-blast cassette was disrupted by site-directed mutagenesis (Quikchange II XL, Agilent) to facilitate cloning into the BsmBI site in the lentiGuide-blast sgRNA template cloning site. GLS (GCACGCATCCGCAGCCCG), BCAT1 (TATTAGGTCTTTAGCCTG), and non-targeting control (#1: GGAGGCTAAGCGTCGCAA) sgRNA templates were cloned into lentiCRISPR_v2 and BCAT2 (TCCACTACTCCCTGCAGG) and non-targeting control (#2: CTTCCGCGGCCCGTTCAA) sgRNA templates were cloned into lentiGuide-blast. BCAT1 and BCAT2 sgRNA templates were designed to target exon-intron boundaries to prevent editing of ectopically-expressed BCAT1 and BCAT2 cDNAs. Complementary oligonucleotides corresponding to each sgRNA were designed to generate overhangs compatible with sequences flanking BsmBI sites in the lentiCRISPR_v2 and lentiGuide-blast vectors when annealed. lentiCRISPR_v2 and lentiGuide-blast vectors were digested with BsmBI, gel-purified, and ligated with annealed oligonucleotides.
cDNA expression plasmids
pLXSN-E6/E7 was generated by PCR amplifying the E6/E7 bicistronic expression cassette from p1321 HPV-16 E6/E7 (Addgene 8641) with primers that introduced 5’ EcoRI and 3’ BamHI restriction sites. The pLXSN (Clontech) retroviral vector and the E6/E7 insert were digested with EcoRI and BamHI, gel-purified, and ligated.
IDH1 wild-type (NM_001282386.1) and R132H cDNAs were cloned into the lentiviral vectors plenti-Ubc-IRES-hygro (used to generate HOG stable cell lines) and plenti-EF1α-IRES-hygro (used to generate NHA and HOG stable cell lines in Figure 1). plenti-Ubc-IRES-hygro was made by ligating the plenti-Ubc-PGK-hygro vector (from Dr. Gang Lu) with an IRES-hygro cassette (pLL3.7-tTA-IRES-hygro, also from Dr. Gang Lu) after cutting both with XhoI and BsrGI followed by gel purification. plenti-EF1α-IRES-hygro was made by ligating pLL3.7EF1α-PGK-puro (from Dr. Gang Lu) with the IRES-hygro cassette from plenti-Ubc-IRES-hygro after digesting both with NotI and BsrGI followed by gel purification. IDH1 cDNAs were PCR amplified from pBABE-HA-hygro vectors (Koivunen et al., 2012) using a 5’ primer that introduced a 5’ NotI site and a 3’ primer that encoded an HA epitope tag and that introduced an XhoI site. The plenti-Ubc-IRES-hygro vector and PCR products were digested with NotI and XhoI, gel-purified, and ligated. IDH1 cDNAs were also PCR amplified using a 5’ primer that introduced a 5’ NheI site and a 3’ primer that encoded an HA epitope tag and introduced a BamHI site. The plenti-EF1α-IRES-hygro vector and PCR products were digested with NheI and BamHI, gel-purified, and ligated.
An IDH2 R172K-FLAG cDNA (Addgene 66807) was cloned into the lentiviral vectors plentiUbc-IRES-hygro (used to generate the HOG stable cell line) and plenti-EF1α-IRES-hygro (used to generate the NHA stable cell line). The IDH2 R172K-FLAG cDNA was PCR amplified with primers that introduced 5’ and 3’ homology arms targeted to regions of the plenti-Ubc-IREShygro and plenti-EF1α-IRES-hygro vectors between the promoter and IRES sequences. Primers corresponding to these homology arms were used in inverse PCR reactions with the vectors as templates. The IDH2 cDNA PCR products were gel-purified and used in InFusion exchange reactions with the inverse PCR products to generate the vectors plenti-Ubc-IDH2-R172K-FLAGIRES-hygro and plenti-EF1α-IDH2-R172K-FLAG-IRES-hygro.
A BCAT1 cDNA was amplified via nested PCR using a cDNA library prepared from NHA cells and the following primers:
Outer forward: TCGTGGCCGACGGAACAATG
Outer reverse: GTAGCATACAGTTGGTATCC
Inner forward: GACGTGGAATTCATGAAGGATTGCAGTAACGG
Inner reverse: AAGGCTCTCGAGGGATAGCACAATTGTCCAG
The PCR product recovered with the outer primers was PCR amplified with a 5’ primer that introduced a 5’ EcoRI site and a 3’ primer that introduced an XhoI site and eliminated the BCAT1 stop codon. This PCR product and pTRIPZ-C3F-neo vector (from Dr. Gang Lu) were digested with EcoRI and XhoI, gel-purified, and ligated. The stop codon from the endogenous cDNA was removed to allow read-through to the 3X-FLAG tag encoded by the pTRIPZ-C3Fneo vector. The amplified BCAT1 cDNA was confirmed by sequencing to match the reference sequence NM_005504.6. The BCAT1–3X-FLAG cDNA was PCR amplified using primers that introduced 5’ NheI and 3’ BamHI sites. This PCR product and plenti-EF1α-IRES-hygro vector were digested with NheI and BamHI, gel-purified, and ligated. In order to avoid promoter interference when simultaneously ectopically expressing BCAT1 and BCAT2 (see below) cDNAs, the human EF1α promoter driving BCAT1 expression was changed to a mouse PGK promoter. The mouse PGK promoter from pMSCV (Clontech) was PCR amplified with primers that introduced 5’ SwaI and 3’ NheI restriction sites. The PCR product and plenti-EF1αBCAT1-FLAG-IRES-hygro vector were digested with NheI and SwaI, gel-purified, and ligated. The resulting vector, plenti-mPGK-BCAT1-FLAG-IRES-hygro, was used in conjunction with plenti-EF1α-BCAT2-FLAG-IRES-neo (described below) to express both transaminases together.
A BCAT2 cDNA (NM_001190.3, Dharmacon, Mammalian Gene Collection, IMAGE Id 3537603) was PCR amplified using a 5’ primer that introduced a NotI site and a 3’ primer that encoded a 3X-FLAG epitope tag and that introduced a PacI site. The PCR product and plentiEF1α-IRES-neo vector (from Dr. Gang Lu) were digested with NotI and PacI, gel-purified, and ligated.
A GLS cDNA (Dharmacon, Mammalian Gene Collection, IMAGE Id 5263220) corresponding to the KGA splice isoform (NM_014905.4) was PCR amplified with primers that introduced 5’ NotI and 3’ PacI restriction sites. The PCR product and plenti-EF1α-IRES-neo vector were digested with NotI and PacI, gel-purified, and ligated. The resulting vector, plenti-EF1α-GLSIRES-neo, was mutagenized via In-Fusion cloning (Clontech) to introduce the K320A or Y394L mutations. Wild-type, K320A, and Y394L GLS expression vectors were also mutagenized via InFusion cloning to introduce silent mutations disrupting the binding site and PAM sequence targeted by the GLS sgRNA listed above (GCACGCATCCGCAGCCCGGGG > GtACaCAcCCaCAaCCaGGaG, changed nucleotides in lower case, PAM site in bold).
An OAT cDNA (Dharmacon, Mammalian Gene Collection, IMAGE Id 3913352) was PCR amplified with primers that introduced 5’ and 3’ homology arms targeted to regions of the pLVX-IRES-puro vector (Clontech) between the CMV promoter and the IRES sequence. Primers corresponding to these homology arms were used in an inverse PCR reaction with the pLVX-IRES-puro vector as template. The OAT cDNA PCR product was gel-purified and used in an InFusion exchange reaction with the inverse PCR product to generate the vector pLVX-OAT-IRES-puro.
A dual GFP and firefly luciferase reporter plasmid was generated by cloning a firefly luciferase cDNA into the LeGO-iG2 vector (from Dr. Kristoffer Weber, University Medical Center Hamburg-Eppendorf). Firefly luciferase cDNA was amplified from the pIRIGF vector (from Dr. Gang Lu) with primers that introduced 5’ BamHI and 3’ EcoRI restriction sites. The PCR product and LeGO-iG2 vector were digested with BamHI and EcoRI, gel-purified, and ligated. The resulting vector, LeGO-iG2-FLuc, encodes the bicistronic expression cassette FLuc-IRESGFP. All of the plasmids described above were authenticated by DNA sequencing.
The human ATM cDNA expression plasmid used to transfect HEK293T cells was obtained from Addgene (pcDNA3.1(+)Flag-His-ATM wt, Addgene 31985).
Transient transfection and viral transduction of cell lines
Lentiviral and retroviral particles were made by Lipofectamine 2000-based cotransfection of HEK293T cells with expression vectors and packaging plasmids psPAX2 (Addgene 12260) and pMD2.G (Addgene 12259) or gag/pol (Addgene 14887) and VSV.G (Addgene 14888), respectively, in a ratio of 8:3:1 (lentiCRISPR_v2 plasmids) or 4:3:1 (all others). Viruscontaining media was collected 48 and 72 hours after transfection, passed through a 0.45 μm filter, divided into 1 ml aliquots, and frozen at -80 °C until use.
For infections, NHA or HOG cells were plated at a density of 0.3×106 cells per well in 6-well plates. The next day, 0.1–1 ml of virus-containing media was added to each well in a final volume of 3 ml media with polybrene included at a final concentration of 8 μg/ml. Cells were centrifuged at 4000 × g for 30 min at room temperature to enhance infection efficiency. The next day, cells were expanded and allowed to recover for an additional 24 hours before being placed under drug selection. Stable cell lines were established by selection in media containing 500 μg/ml hygromycin, 2 μg/ml puromycin, 1 mg/ml G418, or 10 μg/ml blasticidin as appropriate for the drug resistance cassette present in each virus.
HEK293T cells were transiently transfected with 4 μg of a human ATM cDNA expression plasmid (Addgene 31985) or mock-transfected according to the protocol described above for lentivirus and retrovirus production but without inclusion of packaging plasmids. Cells were harvested 72 hours after transfection for immunoblot analysis of expression.
Immunoblot analyses of protein expression
Cells were lysed in EBC lysis buffer supplemented with a protease inhibitor cocktail (Roche). Protein lysates were resolved via SDS-PAGE and transferred to nitrocellulose membranes (BioRad). For immunoblots of GSC lines, membranes were stained with Ponceau S solution (Sigma P7170) for 5 minutes at RT to assess protein loading prior to blocking. Membranes were blocked with 5% non-fat milk in TBST and probed with the following antibodies, as indicated: anti-GLS (Abcam ab156876, rabbit monoclonal), anti-HA (Covance MMS-101P, mouse monoclonal), anti-FLAG (Sigma F1804, mouse monoclonal), anti-BCAT1 (BD 611270, mouse monoclonal), anti-BCAT2 (Abcam ab197917, rabbit polyclonal), anti-OAT (Abcam ab137679, rabbit polyclonal), anti-c-Myc (Cell Signaling 9402, rabbit polyclonal), anti-ATM (Cell Signaling 2873, rabbit monoclonal), anti-vinculin (Sigma V9131, mouse monoclonal). The following HRP-conjugated secondary antibodies were used: Goat anti-Mouse IgG (Thermo Fisher 31430, goat polyclonal) and Goat anti-Rabbit IgG (Thermo Fisher 31460, goat polyclonal).
IDH1 sequencing of GSC lines
Genomic DNA was extracted from GSC lines using the QIAamp DNA Blood Mini Kit (Qiagen 51104). 100 ng of genomic DNA from each cell line was used in separate PCR reactions to generate 500 bp amplicons containing exon 4 of the IDH1 gene. IDH1 sense and antisense primers were 5’-AATGAGCTCTATATGCCATCACTG-3’ and 5’-TTCATACCTTGCTTAATGGGTGT-3’. PCR conditions were as follows: 5 min at 95 °C, 40 cycles of 30 sec at 95 °C, 30 sec at 61 °C, and 30 sec at 72 °C, followed by a final extension step for 7 min at 72 °C. Amplicons were gel-purified and subjected to Sanger sequencing (GENEWIZ) using the IDH1 sense primer listed above.
Recombinant transaminase activity assays
Recombinant human BCAT1, BCAT2, GOT1, GOT2, and OAT enzymes were purchased from MyBioSource. Recombinant bovine GLUD1 enzyme was purchased from Sigma. Activity assays for each transaminase were performed in 96 well plates. Each well contained 200 μl of 100 mM Tris buffer, pH 7.4 with the following additives: 50 μM pyridoxal 5’-phosphate, 5 mM NAD+, 5 mM DTT, 5 mM amino acid substrate (L-leucine, L-aspartate, or L-ornithine), 100 μg GLUD1, and 50–250 ng recombinant transaminase. Where indicated, (R)-2-hydroxyglutarate or (S)-2-hydroxyglutarate (Sigma) was included in the reaction mixture. Transamination reactions were initiated by the addition of 2-oxoglutarate and immediately subjected to kinetic, spectrophotometric quantification of A340 to assess the rate of NADH formation. Readings were acquired every 78 sec for a duration of 11 minutes at 37 °C.
In silico docking studies with human BCAT2
Docking studies of the binding of 2OG and the (R) and (S) enantiomers of 2HG in the active site of human BCAT2 (RCSB codes 1KTA for the PMP form and 1EKF for the PLP form of the enzyme) were performed using the COOT program (Emsley and Cowtan, 2004). The positions, orientations, and interactions of 2OG and the 2HG enantiomers within the active site of human BCAT2 were modeled to be similar to the structure of BCAT from Geoglobus acetivorans complexed with 2OG (RCSB code 5E25). The resulting ligand-bound forms of human BCAT2 with 2OG, (R)-2HG, or (S)-2HG were energy-minimized using the UCSF Chimera software program (Pettersen et al., 2004). PyMOL (The PyMOL Molecular Graphics System Version 1.8, Schrödinger) was used to predict hydrogen bond interactions and generate molecular graphics.
Metabolite measurement by mass spectrometry
Metabolomic analyses of HCT116 and HT1080 cell lines were performed as previously described (Grassian et al., 2014). Briefly, extracts of polar metabolites were derivatized with methoxylamine hydrochloride and N-tert-3 butyldimethylsily-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane. Derivatized samples were analyzed by GC-MS using an Agilent 7890A gas chromatograph interfaced with an Agilent 5975C mass spectrometer. The GC-MS was operated in electron impact mode and scanned the 100–650 m/z range.
Tracing 15N-(amide)-glutamine into ammonia was performed as previously described (Spinelli et al., 2017). Briefly, HOG stable cell lines were given 15N-(amide)-glutamine for 8 hours before metabolites were extracted with 80:20 methanol:water and assessed for 15NH3. Ammonia was derivatized into indophenol with the Berthelot reaction, as previously described (Spinelli et al., 2017), and measured on an Agilent mass spectrometer coupled to reverse phase ion-pairing chromatography. Percent 15N-isotopologue enrichment was measured by dividing the ion counts of 15N-indophenol by the sum of 14N and 15N isotopologues of indophenol.
For steady-state metabolomics experiments involving HOG and NHA cell lines, cells were cultured in low glutamate Serum-like Modified Eagles Medium (SMEM) (1 μM glutamate, consistent with reported glutamate concentrations in the interstitial space of human brain tissue) (Espey et al., 1998; Smith, 2000) containing 10% dialyzed FBS, with noted exceptions below. In Figures 1D, 2B, S2L, and S2N, HOG cells were cultured in IMDM media containing 10% FBS. In Figures 5D to 5F, S5C, and S5D, NHA cells were cultured in DMEM media containing 10% FBS. For tracing assays of HOG cell lines, cells were cultured in IMDM lacking glutamate and pyruvate and supplemented with 10% dialyzed FBS. For tracing assays of NHA cell lines, cells were cultured in DMEM lacking glutamate and pyruvate and supplemented with 10% dialyzed FBS. Both glutamate/pyruvate-free IMDM and DMEM media for tracing assays were prepared from amino acid/pyruvate-free powdered media (US Biological D9800–13) and all components other than glutamate, pyruvate, and glutamine were added in concentrations consistent with the standard formulation of each media type. For tracing assays in DMEM and IMDM media, tracer concentrations were: 0.802 mM 15N-Leu/U-13C-Leu, 0.802 mM 15N-Ile/U-13C-Ile, 0.803 mM 15N-Val, 0.1 mM 1-13C-Glu, and 1 mMα- N-Gln/U- C-Gln. For alanine tracing assays in IMDM media, 15N-Ala tracer concentration was 0.281 mM. For steady-state metabolomics and tracing experiments in GSC lines with the exception of Figure S2Q, cells were cultured in low glutamate SMEM media containing EGF (20 ng/ml), bFGF (20 ng/ml), and heparin (2 μg/ml). In Figure S2Q, cells were cultured in NeuroCult NS-A Basal Medium with Proliferation Supplement (StemCell Technologies) supplemented with EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), 1% penicillin/streptomycin, Fungizone (250 ng/ml), and Plasmocin (2.5 μg/ml). For tracing assays in SMEM media, tracer concentrations were: 0.17 mM 15N-Leu, 0.14 mM 15N-Ile, and 0.23 mM 15N-Val. All labeled amino acids were purchased from Cambridge Isotope Laboratories. For all metabolomics experiments, cells were harvested no longer than 6 hours after switching to SMEM media in order to avoid nutrient depletion from the media and resulting cell starvation.
To quantify metabolites and metabolite labeling in HOG and NHA cell lines by GC-MS, cells were seeded in 6-well plates. To harvest samples, cells were washed with ice-cold saline and plates were snap frozen in liquid nitrogen. To quantify metabolites and metabolite labeling in GSC lines, neurospheres were plated in low-adherence 24-well plates. To harvest samples, metabolic activity was quenched by the addition of excess 4 °C saline and neurospheres were transferred to Eppendorf tubes and rapidly pelleted at 4 °C. The supernatant was aspirated and pellets were snap frozen in liquid nitrogen. All samples were stored at -80 °C prior to metabolite extraction. To extract metabolites from cultured cells, 350 μl -20 °C 70% MeOH was added to each well or tube. To quantify metabolites in tissue samples by GC-MS, tissues (3–30 mg) were quickly dissected from euthanized mice, weighed, and snap frozen in liquid nitrogen. To extract metabolites from tissues, samples were homogenized in -20 °C 70% MeOH using a TissueLyser II (Qiagen) in a volume that correlated with tissue sample size. To quantify metabolites in plasma samples or conditioned media samples, 10 μl plasma or 20 μl conditioned media was added to 350 μl -20 °C 70% MeOH. Methanol extracts from all cell culture, tissue, plasma, and conditioned media samples were combined with -20 °C chloroform (150 μl for cell culture samples, volumes for tissue samples that correlated with size) and vortexed for 20 min at 4 °C. Samples were then centrifuged (17,000 × g at 4 °C for 10 min) and the upper phase containing polar metabolites was collected and dried in a CentriVap vacuum concentrator (Labconco) overnight at 4 °C. Dried metabolite samples were stored at -80 °C before derivatization. For GC-MS analyses, dried metabolites were derivatized and analyzed using an Agilent 7890B GC/5977A MSD system. Polar metabolite extracts were derivatized with methoxyamine (Thermo Scientific) for 60 min at 37 °C and subsequently with N-(tert-butyldimethylsilyl)-Nmethyl-trifluoroacetamide/1% tert-butyldimethylchlorosilane (Sigma) for 90 min at 65 °C. 1 μl of each derivatized sample was injected onto the column and a m/z range of 100–650 was scanned in the mass spectrometer after electron impact ionization at 70 eV. Data analysis was performed using the Metran (Yoo et al., 2008) software tool. Mass isotopologues were quantified and corrected for natural isotope abundance. Our GC-MS platform is designed to detect the majority of proteinogenic amino acids as well as key intermediates in central carbon metabolism.
To quantify metabolites in cultured cells and tumor tissues by LC-MS/MS, samples were harvested as described above. Metabolites were extracted twice (tissues) or three times (cultured cells) in 80% MeOH and extracts were combined and dried using a SpeedVac or a CentriVap vacuum concentrator. Dried metabolites were re-suspended in 20 μL HPLC-grade water. 10 μL was injected and analyzed using a 5500 QTRAP hybrid triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) with selected reaction monitoring (SRM) with positive/negative polarity switching to detect a total of 263 water-soluble metabolites including amino acids and amino acid biosynthetic pathway intermediates, central carbon metabolism intermediates, nucleotides and their precursors, redox cofactors, and α-ketoacids (Yuan et al., 2012). Amide HILIC chromatography (Waters) at pH 9.0 was used for metabolite separation over a 15 min. gradient. Peak areas were integrated using MultiQuant 2.1 software and metabolite quantification was performed using Matlab.
Absolute quantification of 2HG by mass spectrometry
To quantify 2HG levels in cells grown in culture, cells were plated at a density of 0.5×106 per well in triplicate in 6-well plates. The following day, cells in one well were trypsinized and a ViCELL XR cell viability analyzer (Beckman Coulter) was used to record cell number and average cellular diameter. Cell sample volume was then calculated using the following formula: , where d is average cellular diameter, n is cell number, and V is cell sample volume. Polar metabolites were extracted from cells in the two remaining wells and 2HG was quantified in these experimental samples and in standards containing known quantities of pure (R)-2HG (20 ng – 6 μg range) by GC-MS as described above. The 2HG content of experimental samples was determined by interpolation using 2HG total ion counts measured in the standards. 2HG content was divided by cell sample volume to obtain cellular 2HG concentrations. For quantification of 2HG in tumors, sample mass was used to estimate sample volume based on a published value of brain tissue density (Bothe et al., 1984). 2HG content of tumor samples was measured via GC-MS as described above and tumor 2HG concentrations were calculated by dividing these values by tumor sample volume.
DNA methylation assay of HOG stable cell lines
DNA methylation was analyzed by whole genome bisulfite sequencing (WGBS). Briefly, genomic DNA was isolated from two replicates of HOG stable cell line samples collected at high and low passages using the QIAamp DNA Blood Mini kit (Qiagen 51104). 500 ng of gDNA were sheared to a fragment size around 200bp using a Covaris E220 sonicator. Whole genome bisulfite libraries were prepared with the NEXTflex Bisulfite-Seq Kit (Bioo Scientific 5119–02) according to the manufacturer’s instructions. Low coverage sequencing was conducted on the Illumina NextSeq 550. WGBS data was aligned using the BWA-MEM (Li and Durbin, 2009) and methylCtools (Hovestadt et al., 2014) software programs. Beta was plotted using all reads derived from CpG islands at BCAT1 Promoter 1 (chr12:24948674–24949139) and Promoter 2 (chr12:24902666–24903312).
In vivo 15N-leucine tracing in tumor-free mice
8 week old ICR SCID mice (Taconic) were randomized to standard diet or MouseExpress 15Nleucine-containing diet (50% labeled, 50% unlabeled, Cambridge Isotope Laboratories) 96 hours before organs and plasma were harvested. In the morning on the day of harvest, plasma and tissues samples were collected 4 hours after the dark to light transition in the animal facility. First, mice were deeply anesthetized with isoflurane and blood was collected via retroorbital sampling. Next, mice were euthanized quickly and epididymal adipose, liver, gastrocnemius muscle, and brain tissues were harvested and immediately snap-frozen in liquid nitrogen. Lastly, plasma was isolated from whole blood samples and frozen. Tissue and plasma samples were stored at -80 °C until metabolites were extracted for GC-MS analysis as outlined above.
Tumor visualization
In vivo imaging of orthotopic xenograft gliomas was based on expression of a firefly luciferaseIRES-GFP bicistronic expression cassette (LeGO-iG2-FLuc vector) in all implanted cell lines. For bioluminescence imaging studies, mice were given luciferin (50 mg/kg) by IP injection, anesthetized with isoflurane, and imaged using an IVIS camera (PerkinElmer). Bioluminescence images were obtained and analyzed using Living Image software (PerkinElmer). For data in Figures 7D, S7B, and S7C, values were corrected for background luminescence.
Ex vivo isolation of tumor tissue for metabolomics was accomplished by detection of GFP fluorescence. A head-mounted ‘miner’s lamp’ GFP detection apparatus, including an excitation lamp and an emission filter/magnification lens (BLS Ltd, Hungary), was used to rapidly distinguish tumor tissue from mouse brain tissue during dissection. Dissected tumor tissue was quickly weighed and snap frozen in liquid nitrogen. Tumor tissue was stored at -80 °C until metabolite extraction.
In vivo metabolite tracing studies in tumor-bearing mice
Orthotopic xenograft tumors were established in NCr nude mice using the GFP-positive, luciferase-positive, HOG-EV and HOG-R132H stable cell lines described above. For 15Nleucine tracing studies, mice were switched from standard chow to MouseExpress 15N-leucinecontaining diet (50% labeled, 50% unlabeled, Cambridge Isotope Laboratories) 3–4 weeks after tumor cell implantation and maintained on the diet for 96 hours. Tumor tissues (2 samples per mouse) and blood samples were collected 4 hours after the dark to light transition in the animal facility as described above. 3 mice bearing IDH wild-type HOG gliomas displayed small, diffuse tumors upon dissection that precluded recovery of sufficient tissue (<3 mg) for analysis. For U-13C-glutamine tracing studies, tracer was administered to mice bearing IDH1 mutant orthotopic HOG tumors 3–5 weeks after tumor cell implantation according to a published protocol (Lane et al., 2015). Briefly, mice were administered 200 μl of U-13C-glutamine (36.2 mg/mL in saline) via tail vein injection. Injections were performed a total of 3 times per mouse at 15 minute intervals. Mice were euthanized and blood and tumor tissues were collected 15 minutes after the 3rd injection. For both leucine and glutamine tracing studies, GFP-positive brain tumor tissue samples were collected using the head-mounted GFP detection apparatus detailed above. Plasma was isolated from whole blood samples and all samples were snap frozen in liquid nitrogen and stored at -80°C until they were processed for GC-MS analysis.
CB-839 PK and PD studies in HOG xenografts
Orthotopic xenograft tumors were established in NCr nude mice using the GFP-positive, luciferase-positive, HOG-EV and HOG-R132H stable cell lines as described above. 3–4 weeks after tumor cell implantation, mice were randomized to receive CB-839 (200 mg/kg BID via oral gavage) or vehicle control. For the PD study, 7 doses total were administered and mice were euthanized 4 hours after the final dose. For the PK study assessing CB-839 accumulation in IDH mutant HOG tumors, 4 doses total were administered and mice were euthanized 4 hours after the final dose. GFP-positive brain tumor tissue samples were collected using the head-mounted GFP detection apparatus detailed above. All samples were stored at -80 °C until they were processed for GC-MS or LC-MS/MS analysis. LC-MS/MS-based quantification of CB-839 in tumor samples was performed by Calithera Biosciences as previously described (Gross et al., 2014). Briefly, tumor samples were homogenized in an 80:20 mixture of methanol:water containing 50 nM carbamazepine as an internal standard and analyzed for CB-839 levels by LC-MS/MS using a SCIEX API 4000 instrument.
CB-839 PD study in TS603 xenograft gliomas
Subcutaneous xenograft tumors were established in ICR SCID mice using TS603 cells as described above. 7–9 weeks after tumor cell implantation, mice were randomized to receive CB839 (200 mg/kg BID via oral gavage) or vehicle control. 7 doses total were administered and mice were euthanized 4 hours after the final dose. Flank tumor samples were rapidly harvested, snap frozen in liquid nitrogen, and stored at -80 °C until they were processed for LC-MS/MS analysis.
Histology and immunohistochemistry analyses of TS603 xenografts
TS603 subcutaneous xenograft tumor tissues were harvested and immediately fixed for 24 hours in 10% formalin in PBS. Tissues were washed with and stored in 70% ethanol prior to paraffin embedding. Paraffin-embedded tissues were sectioned and stained with hematoxylin and eosin (H&E). Tumor sections stained with H&E or stained for immunohistochemistry (IHC) were reviewed by board-certified pathologist R.D.C. For IHC analyses, 4 μm tissue sections were incubated for 1 hour at 60°C, deparaffinized, and rehydrated. Sections were blocked with 3% H2O2 in methanol for 10 min, washed, and antigens were retrieved at 120°C in citrate buffer (DAKO S1699) using the Decloaking Chamber (Biocare Medical). Cooled slides were incubated with primary antibody for 40 minutes and secondary antibody (DAKO K4011/K4007) for 30 minutes at room temperature in a humidified chamber. Slides were visualized using 3,3′diaminobenzidine (DAB) and counterstained with Mayer’s Hematoxylin. Primary antibodies included anti-IDH1 R132H (Dianova DIA-H09, mouse monoclonal, 1:30), anti-Ki67 (DAKO M7240, mouse monoclonal, 1:100), anti-CD31 (DAKO M0823, mouse monoclonal, 1:30), and anti-Cleaved Caspase 3 (Cell Signaling Technology #9664, rabbit monoclonal, 1:500).
To quantify IHC stains, slides were scanned using the Vectra 3 Slide Analysis System (PerkinElmer). 20X multispectral regions of interest (ROIs) were selected and acquired from initial 10X brightfield overview scans. 4–12 ROIs were acquired per tumor. 29–40 ROIs derived from 4–6 tumors per stain per treatment group were used for IHC quantification. ROIs were processed and analyzed using inForm Tissue Finder software (PerkinElmer). Analysis algorithms were developed using select representative samples that span variation in tissue morphology, stain distribution, tissue quality, and other factors.
For Ki67 and cleaved caspase 3 quantification, analysis algorithms consisted of spectral unmixing, counterstain-based cell segmentation, and signal threshold-based binary scoring. The spectral library was unmixed from pure instances of the chromogen (DAB) and counterstain (hematoxylin) and the signal in each layer was converted to optical density values as counts per pixel. Cell segmentation was optimized by tuning inForm parameters to anticipate cell features from the counterstain and then visually assessing performance. The percentage of segmented cells meeting an assigned threshold of the reported optical density units for the DAB chromogen was scored in each ROI. The threshold was manually set by a pathologist as they would score the brightfield slides under a microscope. Scores for Ki67 and cleaved caspase 3 were assessed within nuclear compartments using a threshold of 0.04 counts per pixel.
For CD31 quantification, the analysis algorithm consisted of spectral unmixing followed by trainable tissue segmentation within inForm. The classifier for scoring CD31 positivity was manually trained to segment pixels based on DAB positivity. inForm software was used to develop a machine learning algorithm to predict labeled classes from annotated training regions. Once the algorithm was developed, it was applied on a batch of images and segmented CD31positive objects (microvessels) were summed in each ROI. The batch output was pathologyreviewed for segmentation and classification accuracy, then optimized via iterative re-training.
Glutathione quantification in tissue and cell samples
Glutathione quantification in tumor tissue samples and in cellular extracts from 15N-BCAA tracing experiments was performed via LC-MS/MS analysis as described above. In Figures 5A and S5B, cell lines used to establish orthotopic xenograft gliomas that were analyzed for GSH content also expressed Cas9 and a control sgRNA, as in Figure 4H.
The GSH-Glo Glutathione Assay and the GSH/GSSG-Glo Assay (both from Promega) were used to quantify total and reduced/oxidized glutathione, respectively, in cultured cells. Cells were seeded in white-walled, clear-bottom 96 well plates at a density of 2500–15000 cells/well 24 hours before analysis. After dissociating neurospheres, GSC lines were seeded into plates that had been coated with CTS CELLstart substrate (Life Technologies) to promote adherence. 16 hours later, cells were treated with the compound of interest for 8 hours in low glutamate SMEM containing 10% dialyzed FBS (NHA and HOG cells) or Neurobasal medium supplemented with 3 mM glutamine, 0.25X N2, EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), and 1% pen/strep (GSC lines, except for MGG152 cells in Figure 5B) before glutathione was quantified using the relevant kit listed above. In Figure 5B, MGG152 cells were cultured in low glutamate SMEM media containing EGF (20 ng/ml), bFGF (20 ng/ml), and heparin (2 μg/ml). For all glutathione quantification experiments involving SMEM media, cells were cultured for no longer than 8 hours before lysis in order to avoid nutrient depletion from the media and resulting cell starvation.
Intracellular ROS quantification in HOG cells and GSC lines
HOG stable lines or GSC lines were cultured in IMDM medium with 10% FBS or Neurobasal medium supplemented with 3 mM glutamine, 0.25X N2, EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), and 1% pen/strep, respectively, prior to ROS quantification. Cells were harvested, washed once with PBS, and resuspended in 300 nM chloromethyl-2',7'dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Life Technologies C6827) in PBS and stained at 37 °C for 10 minutes. Cells were centrifuged and H2DCFDA staining solution was aspirated. Cell pellets were resuspended in PBS with 5% FBS and DAPI and maintained on ice until analysis. Quantification of H2DCFDA fluorescence in the FITC channel in DAPI-negative viable cells was performed using an LSRFortessa flow cytometer (BD Biosciences). Median fluorescence intensity (MFI) ratios of H2DCFDA-stained to unstained cells were calculated. Data analysis was performed with FlowJo 10 software (FlowJo).
Cell death quantification in cultured cells
Cell death assays in NHA and HOG cells were performed in low glutamate SMEM media containing 10% dialyzed FBS. GSC lines were plated in 24-well plates pretreated with CTS CELLStart substrate in Neurobasal medium supplemented with 3 mM glutamine, 0.25X N2, EGF (20 ng/ml), bFGF (20 ng/ml), heparin (2 μg/ml), and 1% pen/strep. Where indicated, cells were cultured in the presence of 300 μM dm2OG and 500 μM L-ornithine and pretreated with CB-839 (100 nM for HOG and NHA cells, 1 μM for GSC lines), 250 μM L-S,R-buthionine sulfoximine, or 2.5 mM ethyl-GSH for 18 hours. Cells were then treated with tert-butyl hydroperoxide for 5 hours and subsequently co-stained with AnnexinV-FITC and DAPI to identify early and late apoptotic cells, respectively. Cell death quantification was performed using an LSR II flow cytometer (BD Biosciences). Data analysis was performed with FCS Express software (De Novo).
Clonogenic survival assay after irradiation
To assess radiation sensitivity of HOG stable cell lines in vitro, cells were pretreated with CB839, NAC, or DMSO for 19 hours overnight. The following morning, cells were seeded in 6well plates in low glutamate SMEM media containing 10% dialyzed FBS with CB-839, NAC, or DMSO at 50, 500, and 5000 cells per well. Cells were irradiated (0, 2, 4, 6 Gy) 5 hours later using a Gammacell-40 irradiator (Nordion) and incubated for 48 hours before changing media to IMDM media containing 10% FBS (without CB-839, NAC, or DMSO) to allow for colony formation. Colonies were stained with 0.4% Crystal violet in 20% MeOH 8–10 days after irradiation and counted manually to determine clonogenic survival rates.
Bioinformatics analyses of gene expression patterns in human tissue samples
Transcript-level GLS splice isoform expression in various normal human tissues was obtained from the BioGPS web portal (Su et al., 2004). Peptide-level GLS splice isoform expression in various normal human tissues was obtained from the Human Proteome Map web portal (Kim et al., 2014). Spectral counts for all GLS-specific peptides were summed to generate expression values used in heatmap construction. Oncomine™ (Compendia Bioscience) was used for analysis and visualization of transcript-level GLS splice isoform expression in human oligodendroglioma and astrocytoma patient samples relative to normal brain tissue samples compiled and profiled by Sun and colleagues (Sun et al., 2006).
The TCGA Brain Lower Grade Glioma (LGG) dataset was analyzed to quantify BCAT1 and ATM mRNA abundance in IDH1 mutant and wild-type glioma samples. Level 3 RSEM normalized (Li and Dewey, 2011) RNA-seq data were extracted from GDAC Firehose (http://gdac.broadinstitute.org/). IDH1 mutation annotations were found for 283 samples (n = 218 IDH1 mutant samples, n = 65 IDH1 wild-type samples). The R statistical software (https://www.r-project.org) was used to plot log2 transformed, RSEM normalized values for BCAT1 and ATM mRNAs. p values were calculated using Student’s t-test. The results from these analyses are based in part upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
2D proliferation assay of HOG stable cell lines
Where indicated, cells were treated with 3 μM AGI-5198 for 3 days prior to and during proliferation assay. To initiate growth assays, 5×104 cells were plated in 6 cm dishes in low glutamate SMEM media with 10% dialyzed FBS. Where indicated, CB-839 and NAC treatments were employed at concentrations of 100 nM and 1 mM, respectively. Hypoxic cell culture was conducted using a hypoxic chamber (Coy Lab Products) at 37 °C in 5% CO2 and 3% O2. Viable cell counts were recorded at the time of plating and 2, 4, and 6 days afterward using a Vi-CELL XR cell viability analyzer (Beckman Coulter). In order to avoid nutrient depletion from the media and resulting cell starvation, SMEM media was changed every 48 hours and the volume increased by 50% in all proliferation assays.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical information for individual experiments can be found in the corresponding figure legends. Statistical analyses were carried out using GraphPad Prism software or the web-based metabolomics data analysis tool MetaboAnalyst. No statistical method was used to predetermine sample size in animal studies. Significance of all comparisons other than those pertaining to survival data was calculated by unpaired two-tailed t-test using a cutoff of p < 0.05. For comparisons of two groups with significantly different variances, Welch’s t-test was used. For comparisons of two groups without significant differences in variances, Student’s t-test was used. Significance of survival data was determined by log-rank test using a cutoff of p < 0.05.
Supplementary Material
(A) Sanger sequencing of the IDH1 gene spanning nucleotide positions 391 through 399 in the indicated GSC lines (nucleotide positions are derived from the CDS of the transcript NM_005896.3). The nucleotide change G395A encodes the R132H mutation. Both G and A nucleotides are detected at position 395 in MGG152, TS603, and BT054 cell lines, indicating heterozygosity for the IDH1 R132H mutation. Sequencing was performed twice and representative chromatograms are shown. (B) 2HG measurements, normalized to lactate, in the indicated GSC lines (n = 4). (C) GC-MS metabolomic analysis of GSC lines from (A) and (B) (n = 4). Total ion counts for each metabolite in each of the 6 cell lines were normalized to protein content, summed with counts from lines of the same IDH1 genotype, and are depicted as a fraction of the total counts from all 6 cell lines. (D) Schema depicting basis for the BCAT1 assay. Glutamate generated by BCAT1 is a substrate for glutamate dehydrogenase. NADH generation by glutamate dehydrogenase can be easily measured via absorbance at 340 nm in a spectrophotometer over time. Recombinant BCAT1 and recombinant glutamate dehydrogenase are both present in each assay and the reaction is initiated by the addition of 2OG. (E) BCAT1 activity measured over a range of 2OG concentrations. For 150 μM 2OG, n = 4. For all other 2OG concentrations, n = 3. (F) BCAT1 activity measured in the presence of Compound 2 (n = 3). (G) Control experiment showing that (R)-2HG does not inhibit the glutamate dehydrogenase reaction required for the assay depicted in (D). Glutamate dehydrogenase activation by glutamate in the presence of (R)-2HG and absence of BCAT1 (n = 3). (H and I) In silico modeling of (R)-2HG (H) and (S)-2HG (I) binding at the active site of human BCAT2 in the PLP-bound conformation. Predicted hydrogen bond interactions are depicted by dashed lines. Four of five oxygen atoms in (R)-2HG form a total of five hydrogen bonds with residues comprising the BCAT2 active site while three of five oxygen atoms in (S)-2HG form four hydrogen bonds total. Asterisk denotes additional hydrogen bond predicted to be formed by (R)-2HG relative to (S)-2HG. For all panels, data presented are means ± SD.
(A and B) Immunoblot analysis (n = 3) (A) and fraction of sites methylated (Beta) as determined by whole-genome bisulfite sequencing (n = 2) (B) of early and late passage HOG stable cell lines. Methylation of the CpG islands surrounding the transcriptional start sites for BCAT1 Promoters 1 and 2 (chr12:24,949,459 and chr12:24,903,075, respectively, in human genome assembly hg38) is shown. These transcriptional start sites are identical to those studied by Tonjes and colleagues (Tonjes et al., 2013). Coordinates for the CpG islands associated with BCAT1 Promoters 1 and 2 are chr12:24,948,674–24,949,139 and chr12:24,902,666–24,903,312, respectively. We observed a pattern of BCAT1 promoter CpG island methylation in HOG cells that mirrors that seen in low-grade gliomas and secondary GBM patient samples analyzed by Tonjes and colleagues (i.e. low methylation of BCAT1 Promoter 1 and high methylation of BCAT1 Promoter 2). Expressing mutant IDH1 in HOG cells does not significantly impact methylation of either BCAT1 promoter at early or late passage. (C) Immunoblot analysis of early and late passage NHA stable lines (n = 3). 3DN signifies an enzymatically inactive IDH1 R132H variant in which three conserved aspartic acid residues within the IDH1 catalytic domain were replaced with asparagines. (D) BCAT1 mRNA levels in IDH1 mutant and wild-type glioma patient samples. Data are derived from analysis of 283 samples in the Brain Lower Grade Glioma TCGA dataset (n = 218 IDH1 mutant samples, n = 65 IDH1 wild-type samples). (E) 15N-leucine tracing assay in HOG cells pretreated with the indicated concentrations of Compound 2 for 17 hours (n = 3). (F) Labeling of intracellular leucine 10 minutes after 15Nleucine tracer administration in NHA (n = 3) and HOG (n = 2) stable cell lines confirming that the results in Figures 2D and 2E are not due to differential leucine tracer accumulation. (G) Ratios of the indicated metabolites in parental HCT116 cells treated with 10 mM (R)-2HG for 48 hours or isogenic HCT116 cells in which an IDH1 R132H or IDH2 R172K mutation was introduced into the endogenous IDH1 or IDH2 locus, respectively, by homologous recombination. n = 3. (H) Ratios of the indicated metabolites in HT1080 IDH1 R132C/+ fibrosarcoma cells cells treated with 1.5 μM AGI-5198 for 72 hours (n = 3). (I and J) Immunoblot analysis (I) and 15N-leucine tracing assay (J) of NHA and HOG stable cell lines infected to produce HA-IDH1 R132H, FLAG-IDH2 R172K, or with the empty vector (EV) (n = 3). (K) Steady-state 2HG and glutamate levels in HOG stable cell lines as in (I) (n = 3). TIC = total ion counts. Compared to mutant IDH1, mutant IDH2 more potently depletes glutamate and more potently inhibits BCAA catabolism. This effect correlates with higher (R)-2HG levels in IDH2 mutant cells relative to IDH1 mutant cells. (L) Levels of the indicated metabolites in conditioned media of HOG cells over time (n = 3). TIC are expressed relative to t = 0. Positive values indicate net secretion; negative values indicate net consumption. (M) U-13C-leucine, U-13C-isoleucine, and U-13C-glutamine tracing assays in HOG cells (n = 3). Fractional labeling of the citrate pool is shown. (N) Ratios of extracellular to intracellular TIC for the BCKAs KMV and KIC at the indicated time points (n = 3). Ratios are normalized to the 0 hour samples. (O) 1–13C-glutamate tracing assays in HOG cells (n = 3). Labeled and unlabeled glutamate isotopologues were quantified in both cellular and conditioned media extracts. (P) Immunoblot analysis of the indicated GSC lines treated with 3 μM AGI-5198 or DMSO for 3 days (n = 3). (Q) Glutamate levels in IDH1 wild-type TS516, TS676, and BT260 lines and in the positive control IDH1 mutant BT054 line treated with 3 μM AGI-5198 or DMSO for 3 days (n = 3). For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A and B) Heatmaps depicting relative abundance of GLS mRNA (KGA and GAC splice isoforms) (A) and GLS-derived peptides (B) in various normal human tissues and cell types. In panel (B), expression levels reflect cumulative abundance of both GLS splice isoforms and 52 GLS-derived peptides were used for quantification. Legend in (A) applies to both panels (A) and (B). (C and D) GLS mRNA abundance (GAC and KGA splice isoforms) in human astrocytomas, oligodendrogliomas, and normal brain tissue based on the study by Sun and colleagues (Sun et al., 2006) (NCBI GEO accession GSE4290) in the Oncomine database. Boxes represent median, 75th and 25th percentiles while whiskers represent the minimum and maximum expression values in the cohort. For normal brain, astrocytoma, and oligodendroglioma samples, n = 23, 26, and 26, respectively. (E) Anti c-Myc immunoblot analysis of NHA and HOG stable cell lines and GSC lines (n = 3). The MYC-amplified NCIH82 (H82) small cell lung cancer cell line was included as a positive control. (F to H) Steadystate glutamate (F), KIC (G), and 2OG (H) levels in HOG stable cell lines depicted in Figure 3H. n = 2. (I) Steady-state 2OG levels in the indicated HOG stable cell lines (n = 4). (J and K) 15Nalanine tracing assays in HOG stable cell lines as in Figure 3H (J) and 2A (K). n = 3. (L) Total ion counts (TIC) for labeled glutamate (M1) in the HOG-R132H stable cell line grown in the presence of 15N-alanine for 2 hours (n = 2). The indicated amounts of dimethyl-2OG (dm2OG) were added 1 hour before switching to 15N-alanine-containing media. (M and N) Steady-state levels of glutamate (M) and alanine (N) in the indicated HOG stable cell lines grown in the presence or absence of 3 mM dm2OG for 2 hours (n = 3). (O) Schema depicting mechanisms contributing to repression of transamination-dependent glutamate biosynthesis by the IDH1 R132H enzyme. (P to R) Immunoblots (n = 3) (P) and steady-state glutamate (Q) and 2HG levels (R) of the indicated HOG stable cell lines infected to produce GLS wild-type (WT) or Y349L enzymes (KGA splice isoform) or infected with an empty vector control (EV). In (P), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. In panels (Q) and (R), n = 3 for the EV-WT GLS line, n = 2 for the EV-GLS Y394L line, and n = 4 for all other lines. For all parts, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A) Flow cytometry-based quantification of GFP fluorescence in HOG-EV and HOG-R132H stable cell lines infected to produce a firefly luciferase-IRES-GFP bicistronic transcript. A representative histogram is shown. (B) In vivo 15N-leucine tracing assay in mice bearing IDH1 mutant and wild-type orthotopic xenograft gliomas as in Figures 4F and 4G. Ratio of fractional glutamate labeling to fractional leucine labeling in the plasma of tumorbearing mice is shown. n = 7 and 8 mice bearing IDH1 wild-type and mutant tumors, respectively. (C) Bioluminescence imaging of orthotopic tumors formed by HOG cell lines as in Figure 4H. Representative mice are shown. (D) Immunoblot analysis of isogenic orthotopic xenograft gliomas established from HOG stable cell lines (lanes 1–4), as in Figure 4H. Tumor growth was tracked by serial imaging, as depicted in (C), and tumors were harvested for metabolomics when they reached a comparable size. n = 4 tumor samples per cohort. (E and F) Immunoblot analysis (n = 2) (E) and steady-state glutamate levels (n = 3) (F) of HOG cells infected to produce an sgRNA-resistant (sgRNAR) GLS KGA splice isoform cDNA (containing a silent mutation to perturb the PAM sequence targeted by the GLS sgRNA) or an empty vector and then subjected to CRISPR/Cas9-based gene editing with a GLS sgRNA or a control sgRNA. (G to J) GC-MS-based quantification of 2HG (G), glutamine (H), glutamate (I), and aspartate (J) levels in isogenic IDH1 mutant and wild-type orthotopic xenograft gliomas derived from the indicated HOG stable cell lines following treatment with 7 doses of CB-839 (200 mg/kg BID) or vehicle control. n = 7, 8, 9, and 8 mice in EV-Vehicle, EV-CB-839, R132H-Vehicle, and R132H-CB-839 cohorts, respectively. (K and L) In vivo U-13C-glutamine tracing assay in mice bearing IDH1 mutant orthotopic HOG xenograft gliomas (n = 5 mice). Fractional labeling of plasma glutamine (K) and the ratios of fractional glutamate, 2OG, and 2HG labeling to fractional glutamine labeling in tumors (L) are shown. In (D) and (E), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. For panels (K) and (L), data presented are means ± SEM. For all other panels, data presented are means ± SD. *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A) Reduced to oxidized glutathione ratios in the indicated HOG (n = 3) and NHA (n = 5) stable cell lines as in Figure 2A. (B) NADPH levels in orthotopic xenografts formed by the indicated HOG stable cell lines and analyzed in Figure 5A. n = 8 and 6 mice bearing IDH1 wild-type and mutant tumors, respectively. (C and D) Steady-state levels of the indicated metabolites following (R)-2HG-TFMB treatment (C) or BCAT1 shRNA expression (D) in NHA cells relative to DMSO and a control shRNA, respectively. Data are derived from metabolomic profiling studies in Figures 5D to 5F. n = 3. (E to G) Immunoblots (n = 2) (E), glutamate levels (n = 3) (F), and glutathione levels (n = 4) (G) of HOG cells infected (or not) to produce an sgRNA-resistant GLS KGA splice isoform cDNA (containing a silent mutation to perturb the PAM sequence targeted by the GLS sgRNA) and subjected to CRISPR/Cas9-based gene editing with a GLS sgRNA or a control sgRNA. K320A and Y394L mutants are BPTES-resistant. In (F) and (G), cells were treated with 1 μM BPTES or DMSO for 2 hours and 8 hours, respectively. (H) Glutathione levels of the indicated HOG stable cell lines infected to produce GLS wild-type (WT) or Y349L enzymes (KGA splice isoform) or infected with an empty vector (EV) control, as in Figure S3P. n = 3. (I) Immunoblot analysis of GLS expression in GSC lines (n = 4). For panels (E) and (I), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A and B) Monolayer cell proliferation assays of the indicated HOG stable cell lines in the presence of 100 nM CB-839 or DMSO under 21% (A) or 3% O2 (B). Fold changes in cell numbers relative to day 0 are displayed. n = 4. (C to E) Flow cytometry-based quantification of staining with the ROS-sensitive dye H2DCFDA in the indicated HOG stable cell lines after 1 hour treatments with 200 μM tert-butyl hydroperoxide (tBH) or 1 mM NAC (C) or under basal conditions (D and E). In (C), representative histograms confirm that fluorescence of H2DCFDA-stained HOG-EV cells increases or decreases after tBH or NAC treatment, respectively. n = 3. The MFI ratio data (H2DCFDA-stained to unstained) from three independent replicates of the experiment depicted in (D) are quantified in (E). (F and G) Quantification of H2DCFDA staining in untreated GSC lines. The MFI ratio data from two independent replicates of the experiment depicted in (F) are quantified in (G). (H) Quantification of cell death (AnnexinV or DAPI positivity) in GSC lines treated with DMSO, 1 μM CB-839, or 250 μM BSO for 23 hours. Data are presented as percent change in cell death relative to DMSO control. n = 5, 3, 4, 5, 8, and 4 for TS516, TS676, BT260, MGG152, TS603, and BT054 cell lines, respectively. (I) Quantification of cell viability (AnnexinV and DAPI negativity) in GSC lines treated with the indicated concentrations of tBH for 5 hours. Data are presented as percent viable relative to DMSO-treated controls. For BT054 cells, n = 2 for 100 and 400 μM tBH treatments and n = 3 for all others. For MGG152 cells, n = 3 for 100 and 200 μM tBH treatments and n = 2 for all others. For all tBH doses in other GSC lines, n = 3. (J) tBH IC50 values in IDH mutant and wild-type GSC lines derived from panel (I). (K and L) AntiATM immunoblot analysis of NHA and HOG stable cell lines (K) and GSC lines (L). HEK293T (HEK) cells transiently transfected with an ATM cDNA expression plasmid and mocktransfected cells were included as controls. n = 3. (M) ATM mRNA levels in IDH1 mutant and wild-type glioma patient samples. Data are derived from analysis of 283 samples in the Brain Lower Grade Glioma TCGA dataset (n = 218 IDH1 mutant samples, n = 65 IDH1 wild-type samples). For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Twotailed p values were determined by unpaired t-test.
(A) Quantification of CB-839 accumulation in IDH mutant HOG orthotopic xenografts. Mice were treated with 200 mg/kg CB-839 twice daily. Mice were euthanized and brain tumor tissues were harvested 4 hours after the 4th dose of CB-839. n = 6 mice. (B and C) Sizes of orthotopic tumors formed by HOG cells stably expressing mutant IDH1 (B) or an empty vector (EV) control (C) and infected to produce firefly luciferase and GFP. Tumor bioluminescence is shown for mice in each treatment group in Figures 7C and 7D at the time of enrollment before treatment initiation. (D) Immunohistochemistry of TS603 subcutaneous xenograft tumors using antibodies specific for IDH1 R132H and CD31. Note that CD31-positive vascular endothelial cells (identified by red arrows) are highlighted as negative controls for staining with the anti-IDH1 R132H antibody. Specific detection of the IDH1 R132H protein is represented by intense cytoplasmic staining in tumor cells. Scale bars = 200 μm. n = 3 tumors, a representative field of one tumor is shown. (E) Volumes of TS603 subcutaneous xenograft tumors on day 0 (1 day before starting treatment) of the tumor growth assay from Figure 7G. (F to H) Histologic and immunohistochemical analysis of tumors from Figure 7G using antibodies against Ki67 and CD31. Representative images are shown in (F) and the data for all tumors are quantified in (G) and (H). Mice were euthanized and tumor tissues were harvested on day 21. For Veh+Sham, Veh+RT, CB839+Sham, and CB-839+RT cohorts, n = 5, 6, 6, and 6 tumors, respectively. For Ki67 quantification in Veh+Sham, Veh+RT, CB-839+Sham, and CB-839+RT cohorts, n = 29, 37, 33, and 40 representative fields, respectively. For CD31 quantification in Veh+Sham, Veh+RT, CB839+Sham, and CB-839+RT cohorts, n = 29, 37, 33, and 39 representative fields, respectively. 4–12 representative fields per tumor were used for quantification. Scale bars = 200 μm. (I and J) Histologic and immunohistochemical analysis of tumors using an antibody against cleaved caspase 3. Tumors were treated as in Figure 7G but harvested 24 hours after the last dose of radiation. Representative images are shown in (I) and the data for all tumors are quantified in (J). For each treatment cohort, n = 4 tumors. For cleaved caspase 3 quantification in Veh+Sham, Veh+RT, CB-839+Sham, and CB-839+RT cohorts, n = 30, 30, 35, and 31 representative fields, respectively. 4–12 representative fields per tumor were used for quantification. Scale bars = 200 μm. (K to M) LC-MS/MS-based quantification of glutamine (K), glutamate (L), and reduced glutathione (M) levels in TS603 subcutaneous xenografts following treatment with 7 doses of CB-839 (200 mg/kg BID) or vehicle control. For panels (B), (C), and (E) to (H), results are derived from two independent experiments. For panels (A) to (C) and (E), data presented are means ± SEM; for all other panels, data presented are means ± SD. For all panels, *p < .05, **p < .01, ***p < .001 and two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
Highlights.
(R)-2-hydroxyglutarate, produced by IDH1/2 mutants, inhibits the BCAT transaminases
IDH mutant gliomas display a transamination-dependent glutamate biosynthesis defect
BCAT loss increases reliance on glutaminase for glutamate and glutathione synthesis
Mutant IDH and glutaminase inhibition are synthetic lethal under oxidative stress
Acknowledgements
We thank C. Stiles and J. Alberta at Dana-Farber Cancer Institute (DFCI) for help with the stereotactic surgeries, Kaelin and Losman laboratory members for helpful discussions, I. Mellinghoff (MSKCC), S. Weiss (Univ. of Calgary), and R. Pieper (UCSF) for sharing cell lines, Calithera Biosciences for providing and quantifying CB-839, G. Lu (Celgene) for sharing plasmids, M. Yuan (Harvard) for help with LC-MS/MS, M. Treasure (UNC School of Medicine) for help with enzyme activity assays, DFCI Animal Resources Facility for help with mouse work, and Clyde Bango (DFCI) for help with IHC experiments. This work was supported by Brain SPORE grant P50CA165962 from NIH/NCI (K.L.L., D.P.C., M.G.V.H., W.G.K.) and by a grant from The Koch Institute/DFCI/Harvard Cancer Center Bridge Project (D.P.C., M.G.V.H., and W.G.K.). This work was also supported by the following NIH grants: 5R35CA210068 (W.G.K.), 5P01CA120964 and 5P30CA006516 (J.M.A.), R01CA188652 (C.M.M.), F30CA183474 and T32GM007753 (J.R.M). S.K.M. is supported by an American Cancer Society fellowship (PF-14–144-01-TBE) and by a Career Enhancement Project award from the DFCI/Harvard Cancer Center Brain SPORE. M.G.V.H. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar and acknowledges support from SU2C. W.G.K. is an HHMI Investigator.
Footnotes
Declaration of Interests
M.G.V.H. and W.G.K. are paid advisors to Agios Pharmaceuticals. W.G.K. receives compensation for his roles as an Eli Lilly and Company Board Director and Tango Therapeutics founder. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bisdas S, Chadzynski GL, Braun C, Schittenhelm J, Skardelly M, Hagberg GE, Ethofer T, Pohmann R, Shajan G, Engelmann J, et al. (2016). MR spectroscopy for in vivo assessment of the oncometabolite 2-hydroxyglutarate and its effects on cellular metabolism in human brain gliomas at 9.4T. J Magn Reson Imaging. [DOI] [PubMed] [Google Scholar]
- Bothe HW, Bodsch W, and Hossmann KA (1984). Relationship between specific gravity, water content, and serum protein extravasation in various types of vasogenic brain edema. Acta Neuropathol 64, 37–42. [DOI] [PubMed] [Google Scholar]
- Bump EA, and Brown JM (1990). Role of glutathione in the radiation response of mammalian cells in vitro and in vivo. Pharmacol Ther 47, 117–136. [DOI] [PubMed] [Google Scholar]
- Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, Pereira HM, Garratt RC, Dias SM, and Ambrosio AL (2012). Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 109, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil MM, Larson PE, Woods SM, Cai L, Eriksson P, Robinson AE, Lupo JM, Vigneron DB, Nelson SJ, Pieper RO, et al. (2014). Hyperpolarized [1–13C] glutamate: a metabolic imaging biomarker of IDH1 mutational status in glioma. Cancer Res 74, 4247–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, Yang XL, Mashimo T, Raisanen JM, Marin-Valencia I, et al. (2012). 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 18, 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AL, Holmen SL, and Colman H (2013). IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, and Jeitner TM (2016). Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, Wei W, and Hurov JB (2011). Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770. [DOI] [PubMed] [Google Scholar]
- Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, and Dang CV (2014). Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol 42, 247–251. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Espey MG, Kustova Y, Sei Y, and Basile AS (1998). Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. Journal of neurochemistry 71, 2079–2087. [DOI] [PubMed] [Google Scholar]
- Ferreira AP, Cassago A, Goncalves Kde A, Dias MM, Adamoski D, Ascencao CF, Honorato RV, de Oliveira JF, Ferreira IM, Fornezari C, et al. (2013). Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 288, 28009–28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, and Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. (2014). IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res 74, 3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. (2014). Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol Cancer Ther 13, 890–901. [DOI] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al. (2014). Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. [DOI] [PubMed] [Google Scholar]
- Hu LY, Boxer PA, Kesten SR, Lei HJ, Wustrow DJ, Moreland DW, Zhang L, Ahn K, Ryder TR, Liu X, et al. (2006). The design and synthesis of human branched-chain amino acid aminotransferase inhibitors for treatment of neurodegenerative diseases. Bioorg Med Chem Lett 16, 2337–2340. [DOI] [PubMed] [Google Scholar]
- Hull J, Patel VB, Hutson SM, and Conway ME (2015). New insights into the role of the branched-chain aminotransferase proteins in the human brain. J Neurosci Res 93, 987–998. [DOI] [PubMed] [Google Scholar]
- Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, Lemonnier F, Kron KJ, Cescon DW, Hao Z, et al. (2016). Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 30, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo-Garcia JL, Viswanath P, Eriksson P, Chaumeil MM, Pieper RO, Phillips JJ, and Ronen SM (2015). Metabolic reprogramming in mutant IDH1 glioma cells. PLoS One 10, e0118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalbert LE, Elkhaled A, Phillips JJ, Neill E, Williams A, Crane JC, Olson MP, Molinaro AM, Berger MS, Kurhanewicz J, et al. (2017). Metabolic Profiling of IDH Mutation and Malignant Progression in Infiltrating Glioma. Scientific reports 7, 44792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, Demetrick DJ, Steele L, Auer RN, Hader WJ, et al. (2010). Oligodendroglioma cell lines containing t(1;19)(q10;p10). Neuro Oncol 12, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. (2014). A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. (2012). Transformation by the (R)-enantiomer of 2hydroxyglutarate linked to EGLN activation. Nature 483, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, Yan J, and Fan TW (2015). (13)C Tracer Studies of Metabolism in Mouse Tumor Xenografts. Bio Protoc 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, and Kaelin WG Jr. (2013). What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27, 836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, and Kaelin WG Jr. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. (2012). Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar RJ, Botman D, Smits MA, Hira VV, van Lith SA, Stap J, Henneman P, Khurshed M, Lenting K, Mul AN, et al. (2015). Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res 75, 4790–4802. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, and Campisi J (2003). Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pope WB, Prins RM, Albert Thomas M, Nagarajan R, Yen KE, Bittinger MA, Salamon N, Chou AP, Yong WH, Soto H, et al. (2012). Non-invasive detection of 2hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol 107, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, and Yan H (2011). Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A 108, 3270–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. (2013). An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamanca-Cardona L, Shah H, Poot AJ, Correa FM, Di Gialleonardo V, Lui H, Miloushev VZ, Granlund KL, Tee SS, Cross JR, et al. (2017). In Vivo Imaging of Glutamine Metabolism to the Oncometabolite 2-Hydroxyglutarate in IDH1/2 Mutant Tumors. Cell Metab 26, 830–841 e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. (2012). IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. (2010). Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70, 8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MK, Seidlitz EP, and Singh G (2010). Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun 391, 91–95. [DOI] [PubMed] [Google Scholar]
- Smith QR (2000). Transport of glutamate and other amino acids at the blood-brain barrier. J Nutr 130, 1016S–1022S. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, and Pieper RO (2001). Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res 61, 49564960. [PubMed] [Google Scholar]
- Spinelli JB, Kelley LP, and Haigis MC (2017). An LC-MS Approach to Quantitative Measurement of Ammonia Isotopologues. Scientific reports 7, 10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. (2004). A gene atlas of the mouse and human proteinencoding transcriptomes. Proc Natl Acad Sci U S A 101, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey R, et al. (2006). Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9, 287–300. [DOI] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO, Weinstock A, Wagner A, et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol 17, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdall MM, and Smith M (2006). Cerebral microdialysis: research technique or clinical tool. Br J Anaesth 97, 18–25. [DOI] [PubMed] [Google Scholar]
- Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AH, Karra D, Piro RM, et al. (2013). BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 19, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, Carlin SD, La Rocca G, Lyashchenko S, Ploessl K, et al. (2015). Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7, 274ra217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitkus MS, Diplas BH, and Yan H (2018). Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, et al. (2014). Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 20, 2898–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H, Antoniewicz MR, Stephanopoulos G, and Kelleher JK (2008). Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J Biol Chem 283, 20621–20627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Sanger sequencing of the IDH1 gene spanning nucleotide positions 391 through 399 in the indicated GSC lines (nucleotide positions are derived from the CDS of the transcript NM_005896.3). The nucleotide change G395A encodes the R132H mutation. Both G and A nucleotides are detected at position 395 in MGG152, TS603, and BT054 cell lines, indicating heterozygosity for the IDH1 R132H mutation. Sequencing was performed twice and representative chromatograms are shown. (B) 2HG measurements, normalized to lactate, in the indicated GSC lines (n = 4). (C) GC-MS metabolomic analysis of GSC lines from (A) and (B) (n = 4). Total ion counts for each metabolite in each of the 6 cell lines were normalized to protein content, summed with counts from lines of the same IDH1 genotype, and are depicted as a fraction of the total counts from all 6 cell lines. (D) Schema depicting basis for the BCAT1 assay. Glutamate generated by BCAT1 is a substrate for glutamate dehydrogenase. NADH generation by glutamate dehydrogenase can be easily measured via absorbance at 340 nm in a spectrophotometer over time. Recombinant BCAT1 and recombinant glutamate dehydrogenase are both present in each assay and the reaction is initiated by the addition of 2OG. (E) BCAT1 activity measured over a range of 2OG concentrations. For 150 μM 2OG, n = 4. For all other 2OG concentrations, n = 3. (F) BCAT1 activity measured in the presence of Compound 2 (n = 3). (G) Control experiment showing that (R)-2HG does not inhibit the glutamate dehydrogenase reaction required for the assay depicted in (D). Glutamate dehydrogenase activation by glutamate in the presence of (R)-2HG and absence of BCAT1 (n = 3). (H and I) In silico modeling of (R)-2HG (H) and (S)-2HG (I) binding at the active site of human BCAT2 in the PLP-bound conformation. Predicted hydrogen bond interactions are depicted by dashed lines. Four of five oxygen atoms in (R)-2HG form a total of five hydrogen bonds with residues comprising the BCAT2 active site while three of five oxygen atoms in (S)-2HG form four hydrogen bonds total. Asterisk denotes additional hydrogen bond predicted to be formed by (R)-2HG relative to (S)-2HG. For all panels, data presented are means ± SD.
(A and B) Immunoblot analysis (n = 3) (A) and fraction of sites methylated (Beta) as determined by whole-genome bisulfite sequencing (n = 2) (B) of early and late passage HOG stable cell lines. Methylation of the CpG islands surrounding the transcriptional start sites for BCAT1 Promoters 1 and 2 (chr12:24,949,459 and chr12:24,903,075, respectively, in human genome assembly hg38) is shown. These transcriptional start sites are identical to those studied by Tonjes and colleagues (Tonjes et al., 2013). Coordinates for the CpG islands associated with BCAT1 Promoters 1 and 2 are chr12:24,948,674–24,949,139 and chr12:24,902,666–24,903,312, respectively. We observed a pattern of BCAT1 promoter CpG island methylation in HOG cells that mirrors that seen in low-grade gliomas and secondary GBM patient samples analyzed by Tonjes and colleagues (i.e. low methylation of BCAT1 Promoter 1 and high methylation of BCAT1 Promoter 2). Expressing mutant IDH1 in HOG cells does not significantly impact methylation of either BCAT1 promoter at early or late passage. (C) Immunoblot analysis of early and late passage NHA stable lines (n = 3). 3DN signifies an enzymatically inactive IDH1 R132H variant in which three conserved aspartic acid residues within the IDH1 catalytic domain were replaced with asparagines. (D) BCAT1 mRNA levels in IDH1 mutant and wild-type glioma patient samples. Data are derived from analysis of 283 samples in the Brain Lower Grade Glioma TCGA dataset (n = 218 IDH1 mutant samples, n = 65 IDH1 wild-type samples). (E) 15N-leucine tracing assay in HOG cells pretreated with the indicated concentrations of Compound 2 for 17 hours (n = 3). (F) Labeling of intracellular leucine 10 minutes after 15Nleucine tracer administration in NHA (n = 3) and HOG (n = 2) stable cell lines confirming that the results in Figures 2D and 2E are not due to differential leucine tracer accumulation. (G) Ratios of the indicated metabolites in parental HCT116 cells treated with 10 mM (R)-2HG for 48 hours or isogenic HCT116 cells in which an IDH1 R132H or IDH2 R172K mutation was introduced into the endogenous IDH1 or IDH2 locus, respectively, by homologous recombination. n = 3. (H) Ratios of the indicated metabolites in HT1080 IDH1 R132C/+ fibrosarcoma cells cells treated with 1.5 μM AGI-5198 for 72 hours (n = 3). (I and J) Immunoblot analysis (I) and 15N-leucine tracing assay (J) of NHA and HOG stable cell lines infected to produce HA-IDH1 R132H, FLAG-IDH2 R172K, or with the empty vector (EV) (n = 3). (K) Steady-state 2HG and glutamate levels in HOG stable cell lines as in (I) (n = 3). TIC = total ion counts. Compared to mutant IDH1, mutant IDH2 more potently depletes glutamate and more potently inhibits BCAA catabolism. This effect correlates with higher (R)-2HG levels in IDH2 mutant cells relative to IDH1 mutant cells. (L) Levels of the indicated metabolites in conditioned media of HOG cells over time (n = 3). TIC are expressed relative to t = 0. Positive values indicate net secretion; negative values indicate net consumption. (M) U-13C-leucine, U-13C-isoleucine, and U-13C-glutamine tracing assays in HOG cells (n = 3). Fractional labeling of the citrate pool is shown. (N) Ratios of extracellular to intracellular TIC for the BCKAs KMV and KIC at the indicated time points (n = 3). Ratios are normalized to the 0 hour samples. (O) 1–13C-glutamate tracing assays in HOG cells (n = 3). Labeled and unlabeled glutamate isotopologues were quantified in both cellular and conditioned media extracts. (P) Immunoblot analysis of the indicated GSC lines treated with 3 μM AGI-5198 or DMSO for 3 days (n = 3). (Q) Glutamate levels in IDH1 wild-type TS516, TS676, and BT260 lines and in the positive control IDH1 mutant BT054 line treated with 3 μM AGI-5198 or DMSO for 3 days (n = 3). For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A and B) Heatmaps depicting relative abundance of GLS mRNA (KGA and GAC splice isoforms) (A) and GLS-derived peptides (B) in various normal human tissues and cell types. In panel (B), expression levels reflect cumulative abundance of both GLS splice isoforms and 52 GLS-derived peptides were used for quantification. Legend in (A) applies to both panels (A) and (B). (C and D) GLS mRNA abundance (GAC and KGA splice isoforms) in human astrocytomas, oligodendrogliomas, and normal brain tissue based on the study by Sun and colleagues (Sun et al., 2006) (NCBI GEO accession GSE4290) in the Oncomine database. Boxes represent median, 75th and 25th percentiles while whiskers represent the minimum and maximum expression values in the cohort. For normal brain, astrocytoma, and oligodendroglioma samples, n = 23, 26, and 26, respectively. (E) Anti c-Myc immunoblot analysis of NHA and HOG stable cell lines and GSC lines (n = 3). The MYC-amplified NCIH82 (H82) small cell lung cancer cell line was included as a positive control. (F to H) Steadystate glutamate (F), KIC (G), and 2OG (H) levels in HOG stable cell lines depicted in Figure 3H. n = 2. (I) Steady-state 2OG levels in the indicated HOG stable cell lines (n = 4). (J and K) 15Nalanine tracing assays in HOG stable cell lines as in Figure 3H (J) and 2A (K). n = 3. (L) Total ion counts (TIC) for labeled glutamate (M1) in the HOG-R132H stable cell line grown in the presence of 15N-alanine for 2 hours (n = 2). The indicated amounts of dimethyl-2OG (dm2OG) were added 1 hour before switching to 15N-alanine-containing media. (M and N) Steady-state levels of glutamate (M) and alanine (N) in the indicated HOG stable cell lines grown in the presence or absence of 3 mM dm2OG for 2 hours (n = 3). (O) Schema depicting mechanisms contributing to repression of transamination-dependent glutamate biosynthesis by the IDH1 R132H enzyme. (P to R) Immunoblots (n = 3) (P) and steady-state glutamate (Q) and 2HG levels (R) of the indicated HOG stable cell lines infected to produce GLS wild-type (WT) or Y349L enzymes (KGA splice isoform) or infected with an empty vector control (EV). In (P), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. In panels (Q) and (R), n = 3 for the EV-WT GLS line, n = 2 for the EV-GLS Y394L line, and n = 4 for all other lines. For all parts, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A) Flow cytometry-based quantification of GFP fluorescence in HOG-EV and HOG-R132H stable cell lines infected to produce a firefly luciferase-IRES-GFP bicistronic transcript. A representative histogram is shown. (B) In vivo 15N-leucine tracing assay in mice bearing IDH1 mutant and wild-type orthotopic xenograft gliomas as in Figures 4F and 4G. Ratio of fractional glutamate labeling to fractional leucine labeling in the plasma of tumorbearing mice is shown. n = 7 and 8 mice bearing IDH1 wild-type and mutant tumors, respectively. (C) Bioluminescence imaging of orthotopic tumors formed by HOG cell lines as in Figure 4H. Representative mice are shown. (D) Immunoblot analysis of isogenic orthotopic xenograft gliomas established from HOG stable cell lines (lanes 1–4), as in Figure 4H. Tumor growth was tracked by serial imaging, as depicted in (C), and tumors were harvested for metabolomics when they reached a comparable size. n = 4 tumor samples per cohort. (E and F) Immunoblot analysis (n = 2) (E) and steady-state glutamate levels (n = 3) (F) of HOG cells infected to produce an sgRNA-resistant (sgRNAR) GLS KGA splice isoform cDNA (containing a silent mutation to perturb the PAM sequence targeted by the GLS sgRNA) or an empty vector and then subjected to CRISPR/Cas9-based gene editing with a GLS sgRNA or a control sgRNA. (G to J) GC-MS-based quantification of 2HG (G), glutamine (H), glutamate (I), and aspartate (J) levels in isogenic IDH1 mutant and wild-type orthotopic xenograft gliomas derived from the indicated HOG stable cell lines following treatment with 7 doses of CB-839 (200 mg/kg BID) or vehicle control. n = 7, 8, 9, and 8 mice in EV-Vehicle, EV-CB-839, R132H-Vehicle, and R132H-CB-839 cohorts, respectively. (K and L) In vivo U-13C-glutamine tracing assay in mice bearing IDH1 mutant orthotopic HOG xenograft gliomas (n = 5 mice). Fractional labeling of plasma glutamine (K) and the ratios of fractional glutamate, 2OG, and 2HG labeling to fractional glutamine labeling in tumors (L) are shown. In (D) and (E), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. For panels (K) and (L), data presented are means ± SEM. For all other panels, data presented are means ± SD. *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A) Reduced to oxidized glutathione ratios in the indicated HOG (n = 3) and NHA (n = 5) stable cell lines as in Figure 2A. (B) NADPH levels in orthotopic xenografts formed by the indicated HOG stable cell lines and analyzed in Figure 5A. n = 8 and 6 mice bearing IDH1 wild-type and mutant tumors, respectively. (C and D) Steady-state levels of the indicated metabolites following (R)-2HG-TFMB treatment (C) or BCAT1 shRNA expression (D) in NHA cells relative to DMSO and a control shRNA, respectively. Data are derived from metabolomic profiling studies in Figures 5D to 5F. n = 3. (E to G) Immunoblots (n = 2) (E), glutamate levels (n = 3) (F), and glutathione levels (n = 4) (G) of HOG cells infected (or not) to produce an sgRNA-resistant GLS KGA splice isoform cDNA (containing a silent mutation to perturb the PAM sequence targeted by the GLS sgRNA) and subjected to CRISPR/Cas9-based gene editing with a GLS sgRNA or a control sgRNA. K320A and Y394L mutants are BPTES-resistant. In (F) and (G), cells were treated with 1 μM BPTES or DMSO for 2 hours and 8 hours, respectively. (H) Glutathione levels of the indicated HOG stable cell lines infected to produce GLS wild-type (WT) or Y349L enzymes (KGA splice isoform) or infected with an empty vector (EV) control, as in Figure S3P. n = 3. (I) Immunoblot analysis of GLS expression in GSC lines (n = 4). For panels (E) and (I), KGA and GAC splice isoforms are indicated; n.s. denotes nonspecific band. For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.
(A and B) Monolayer cell proliferation assays of the indicated HOG stable cell lines in the presence of 100 nM CB-839 or DMSO under 21% (A) or 3% O2 (B). Fold changes in cell numbers relative to day 0 are displayed. n = 4. (C to E) Flow cytometry-based quantification of staining with the ROS-sensitive dye H2DCFDA in the indicated HOG stable cell lines after 1 hour treatments with 200 μM tert-butyl hydroperoxide (tBH) or 1 mM NAC (C) or under basal conditions (D and E). In (C), representative histograms confirm that fluorescence of H2DCFDA-stained HOG-EV cells increases or decreases after tBH or NAC treatment, respectively. n = 3. The MFI ratio data (H2DCFDA-stained to unstained) from three independent replicates of the experiment depicted in (D) are quantified in (E). (F and G) Quantification of H2DCFDA staining in untreated GSC lines. The MFI ratio data from two independent replicates of the experiment depicted in (F) are quantified in (G). (H) Quantification of cell death (AnnexinV or DAPI positivity) in GSC lines treated with DMSO, 1 μM CB-839, or 250 μM BSO for 23 hours. Data are presented as percent change in cell death relative to DMSO control. n = 5, 3, 4, 5, 8, and 4 for TS516, TS676, BT260, MGG152, TS603, and BT054 cell lines, respectively. (I) Quantification of cell viability (AnnexinV and DAPI negativity) in GSC lines treated with the indicated concentrations of tBH for 5 hours. Data are presented as percent viable relative to DMSO-treated controls. For BT054 cells, n = 2 for 100 and 400 μM tBH treatments and n = 3 for all others. For MGG152 cells, n = 3 for 100 and 200 μM tBH treatments and n = 2 for all others. For all tBH doses in other GSC lines, n = 3. (J) tBH IC50 values in IDH mutant and wild-type GSC lines derived from panel (I). (K and L) AntiATM immunoblot analysis of NHA and HOG stable cell lines (K) and GSC lines (L). HEK293T (HEK) cells transiently transfected with an ATM cDNA expression plasmid and mocktransfected cells were included as controls. n = 3. (M) ATM mRNA levels in IDH1 mutant and wild-type glioma patient samples. Data are derived from analysis of 283 samples in the Brain Lower Grade Glioma TCGA dataset (n = 218 IDH1 mutant samples, n = 65 IDH1 wild-type samples). For all panels, data presented are means ± SD; *p < .05, **p < .01, ***p < .001. Twotailed p values were determined by unpaired t-test.
(A) Quantification of CB-839 accumulation in IDH mutant HOG orthotopic xenografts. Mice were treated with 200 mg/kg CB-839 twice daily. Mice were euthanized and brain tumor tissues were harvested 4 hours after the 4th dose of CB-839. n = 6 mice. (B and C) Sizes of orthotopic tumors formed by HOG cells stably expressing mutant IDH1 (B) or an empty vector (EV) control (C) and infected to produce firefly luciferase and GFP. Tumor bioluminescence is shown for mice in each treatment group in Figures 7C and 7D at the time of enrollment before treatment initiation. (D) Immunohistochemistry of TS603 subcutaneous xenograft tumors using antibodies specific for IDH1 R132H and CD31. Note that CD31-positive vascular endothelial cells (identified by red arrows) are highlighted as negative controls for staining with the anti-IDH1 R132H antibody. Specific detection of the IDH1 R132H protein is represented by intense cytoplasmic staining in tumor cells. Scale bars = 200 μm. n = 3 tumors, a representative field of one tumor is shown. (E) Volumes of TS603 subcutaneous xenograft tumors on day 0 (1 day before starting treatment) of the tumor growth assay from Figure 7G. (F to H) Histologic and immunohistochemical analysis of tumors from Figure 7G using antibodies against Ki67 and CD31. Representative images are shown in (F) and the data for all tumors are quantified in (G) and (H). Mice were euthanized and tumor tissues were harvested on day 21. For Veh+Sham, Veh+RT, CB839+Sham, and CB-839+RT cohorts, n = 5, 6, 6, and 6 tumors, respectively. For Ki67 quantification in Veh+Sham, Veh+RT, CB-839+Sham, and CB-839+RT cohorts, n = 29, 37, 33, and 40 representative fields, respectively. For CD31 quantification in Veh+Sham, Veh+RT, CB839+Sham, and CB-839+RT cohorts, n = 29, 37, 33, and 39 representative fields, respectively. 4–12 representative fields per tumor were used for quantification. Scale bars = 200 μm. (I and J) Histologic and immunohistochemical analysis of tumors using an antibody against cleaved caspase 3. Tumors were treated as in Figure 7G but harvested 24 hours after the last dose of radiation. Representative images are shown in (I) and the data for all tumors are quantified in (J). For each treatment cohort, n = 4 tumors. For cleaved caspase 3 quantification in Veh+Sham, Veh+RT, CB-839+Sham, and CB-839+RT cohorts, n = 30, 30, 35, and 31 representative fields, respectively. 4–12 representative fields per tumor were used for quantification. Scale bars = 200 μm. (K to M) LC-MS/MS-based quantification of glutamine (K), glutamate (L), and reduced glutathione (M) levels in TS603 subcutaneous xenografts following treatment with 7 doses of CB-839 (200 mg/kg BID) or vehicle control. For panels (B), (C), and (E) to (H), results are derived from two independent experiments. For panels (A) to (C) and (E), data presented are means ± SEM; for all other panels, data presented are means ± SD. For all panels, *p < .05, **p < .01, ***p < .001 and two-tailed p values were determined by unpaired t-test. n.s. = nonsignificant.