

ABSTRACT
Salmonella and Citrobacter are gram negative, members of Enterobacteriaceae family that are important causative agents of diarrhea and intestinal inflammation. TGF-β1 is a pleiotropic multifunctional cytokine that has been implicated in modulating the severity of microbial infections. How these pathogens alter the TGF-β1 signaling pathways in the intestine is largely unknown. Streptomycin-pretreated C57BL/6J mouse model colonized with S. typhimurium for 8 hours (acute) and 4 days (chronic) infection and FVB/N mice infected with C. rodentium for 6 days were utilized. Results demonstrated an increase in TGF-β1 receptor I expression (p<0.05) in S. typhimurium infected mouse ileum at both acute and chronic post-infection vs control. This was associated with activation of Smad pathways as evidenced by increased phosphorylated (p)-Smad2 and p-Smad3 levels in the nucleus. The inhibitory Smad7 mRNA levels showed a significant up regulation during acute phase of Salmonella infection but no change at 4d post-infection. In contrast to Salmonella, infection with Citrobacter caused drastic downregulation of TGF receptor I and II concomitant with a decrease in levels of Smad 2, 3, 4 and 7 expression in the mouse colon. We speculate that increased TGF-β1 signaling in response to Salmonella may be a host compensatory response to promote mucosal healing; while C. rodentium decreases TGF-β1 signaling pathways to promote inflammation and contribute to disease pathogenesis. These findings increase our understanding of how enteric pathogens subvert specific aspects of the host-cellular pathways to cause disease.
KEYWORDS: Citrobacter, Salmonella, Smad
Introduction
TGF-β1 is an important multifunctional cytokine that is expressed in many different cell types in the intestine including epithelial, fibroblasts and mononuclear cells.1,2 It regulates a diverse set of cellular responses such as cell growth and proliferation, cellular differentiation, extracellular matrix production, tissue repair and immune-modulation.3 Because of its role in immune modulation, there has been a considerable interest in investigating the role of TGF-β1 in microbial infections. Indeed, HIV and many other pathogens such as M avium, L amazonensis, L brazilensis,4,5 S aureus,6 T cruzi7 increase TGF-β1 levels to cause immune suppression and progression of infection. However, other studies have shown that TGF-β1 may have a beneficial effect for host such as in L monocytogenes or C albicans infection. These conflicting reports of TGF-β1 either causing progression or resistance to infection could be due to distinct effects of different microbes on TGF-β1-induced signaling pathways in different cell types.
Indeed, TGF-β1signaling is very complex. The ligand TGF-β1 initiates signaling by binding to and bringing together type I (TGF-RI) and type II receptor (TGF-RII) serine/threonine kinases on the cell surface. This allows TGF-RII to phosphorylate the receptor I kinase domain, which then propagates the signal through phosphorylation of the canonical Smad proteins. R-Smads (Smad1, 2, 3, 5, and 8) are directly phosphorylated and activated by the type I receptor kinases and undergo formation of heteromeric complexes with the Co-Smad, Smad4. The activated Smad complexes are translocated into the nucleus and, in conjunction with other nuclear cofactors, regulate the transcription of target genes. The I-Smad, Smad7, keeps a check on TGF-β1 signaling by competing with R-Smads for receptor or Co-Smad interaction and by targeting the receptors for degradation.3,8 Eliminating TGF-β1 or its downstream signaling cascade (Smad2, 3 and 4) leads to inflammatory disease or cancer.9-11 For example, TGF-β-null mice develop systemic inflammation, whereas, transgenic mice expressing a dominant negative TGF-R2 are known to develop severe pulmonary and gut inflammation10,12,13 or tumor formation.14 On the other hand, activation of TGF-β1 signaling pathways leads to tissue repair or remodeling, although excessive signaling can have deleterious consequences such as fibrosis or tumor metastasis.14,15 Whether TGF-β1 signaling is exploited by enteric pathogens to contribute to disease pathogenesis is largely unknown.
We chose two important pathogens namely Salmonella typhimurium, a major cause of salmonellosis (characterized by diarrhea and inflammation in ileum and colon); and C. rodentium, a mouse pathogen that closely resembles clinically relevant pathogens Enteropathogenic E. coli (EPEC) and Enterohemorrhagic E. coli (EHEC) and causes colitis. Our novel findings demonstrated that Salmonella elicits alterations in TGF-β1 signaling with increased TGF-R abundance and activation of Smad2/3 signaling pathways in the mouse ileum. C. rodentium infection was associated with a downregulation in TGF-R I and II abundance and reduction in Smad gene expression. It is speculated that increased TGF-β1 signaling in response to Salmonella may be a host compensatory response to promote mucosal healing or pathogen survival; while C. rodentium decreases TGF-β1 signaling pathways to promote inflammation. These data indicate specific host-microbial interaction that enteric pathogens exploit to contribute to disease progression or survival in the host.
Results
-
(1)
TGF-1 receptor mRNA Expression is Increased by Salmonella TGF-β1 superfamily comprises of a family of secreted signaling molecules that regulate a number of cellular processes including growth, differentiation, adhesion and migration.3 Whether Salmonella infection alters TGF-β1 signaling pathways is not known. In a streptomycin-pretreated C57BL/6J mouse model colonized with wild-type pathogenic Salmonella ATCC 14028 for 8 h (acute) and 4 d (chronic), we found that the mRNA levels of TGF-RI were significantly increased (4–5 fold) by Salmonella infection in the ileum at 8 h and 4 d as compared to control (Fig. 1A). TGF-RII mRNA levels were also markedly increased (∼20 fold) after acute infection with Salmonella and remained increased, although to a lesser degree (8 fold) 4 days post-infection (Fig. 1B).
-
(2)
Salmonella Infection Increased TGF-RI Protein Abundance To examine whether increase in mRNA levels corresponded with protein abundance, western blot utilizing TGF-RI and TGF-RII specific antibodies was performed in mucosa isolated from ileum of uninfected and Salmonella colonized mice. A significant increase in TGF-RI (but not TGF-RII) protein expression in response to Salmonella infection was observed at 8 h and 4 day (Fig. 1C). Villin and β actin were used as an internal control markers for normalization. As shown in densitometric analysis, TGF-RI protein levels were significantly up regulated (∼2–3 fold) post-infection (Fig. 1D). However, the protein levels of TGF-RII did not change unlike the increase in its mRNA levels (Fig. 1E). Taken together, our data showed that TGF-RI is up regulated at both mRNA and protein levels in the ileum in the early as well as late stages of Salmonella infection.
-
(3)
Salmonella Activates Smad2 and Smad3 Pathways: Activation of Smad proteins by TGF-RI mediated phosphorylation is a principal event of TGF-β1 signaling in influencing gene transcription.8,16 To check whether Salmonella alters Smad signaling cascade, we first examined total cellular levels of R-Smads (Smad2 and Smad3) in response to infection with Salmonella. mRNA levels of Smad2 were unaltered (Fig. 2A), whereas, Smad3 mRNA levels were up regulated acutely post infection (Fig. 2B). Activation of Smad2/3 usually involves phosphorylation and translocation to the nucleus. Utilizing phosphospecific antibodies, increased staining of p-Smad2 in the nucleus was observed at both 8 h and 4 d post colonization with Salmonella in the ileal tissue, indicative of its activation (Fig. 3A). The p-Smad2 staining in the nucleus was distributed throughout the crypt-villus axis in response to infection compared to control with negligible p-Smad2 staining. Similarly, p-smad3 nuclear staining was also increased in Salmonella colonized samples at acute phase of infection compared to control (Fig. 3A). Immunostaining showed no significant alteration in total protein levels of Smad2 and 3 proteins as detected by antibody that cross-reacts with both Smad2 and Smad3 (Supplementary Fig. 1).
-
(4)
Salmonella effects on Smad4 and Smad7 Levels Following activation of Smad2 and 3, a heteromeric complex with co-Smad (Smad4) is formed which influences gene transcription of target genes. As shown in Fig. 4A, mRNA levels of Smad4 were acutely (8 h) up regulated post-infection (∼30–40 fold), which remained significantly increased at chronic phase (4 d), although to a lesser degree (∼5-fold). We further examined levels of Smad7, representing the inhibitory or “anti-Smad” because it interferes with the activation of R-Smads. Smad7 mRNA levels were increased at early stage (8 h) of infection (∼5 fold) with no change observed at 4 d (Fig. 4B).
-
(5)
TGF-β signaling pathways in response to C. rodentium: In order to determine whether the effects of Salmonella on TGF-β related pathways are specific, we investigated another related but distinct pathogen, Citrobacter rodentium. Our previous studies showed that there was substantial increase in mRNA levels of proinflammatory cytokines including IL-1β, CXCL1, and IFN-γ in the colonic mucosa of Citrobacter rodentium infected mice.17 However, effects of Citrobacter infection on the TGF-β signaling pathways have not been systematically investigated. A previous microarray analysis of differential gene expression in Citrobacter infected FVB/N mice showed a decreased abundance of TGF-R II at 9 days post-infection, which was not rigorously verified.18 In our streptomycin-pretreated FVB/N mouse model colonized with C. rodentium, the data demonstrated a significant decrease in TGF-RI mRNA levels (∼2 fold) in the colonic mucosa at 6 d post-infection as compared to control (Fig. 5A). TGF-RII mRNA was also markedly decreased (∼2 fold) after infection with Citrobacter (Fig. 5B). With respect to the downstream Smad pathways, there was a significant decrease in mRNA levels of both Smad 2 (Fig. 5C) and Smad 3 (Fig. 5D) post-infection. Interestingly, levels of the Smad 4 (Table 1) as well as the inhibitory Smad (Smad7) were downregulated in response to Citrobacter infection (Table 1). These data indicate that TGF-β signaling pathways are downregulated post C. rodentium infection and this decrease in anti-inflammatory pathways along with increase in pro-inflammatory cytokines may contribute to disease pathogenesis associated with Citrobacter infection.
-
(6)
C. rodentium decreases Smad2/3 immunostaining: To investigate whether mRNA levels of Smad2/3 correlate with protein expression, we examined cellular localization of Smad 2 and 3 by immunofluoresence staining utilizing an antibody that cross reacts with Smad2 and Smad 3. As shown in Fig. 6, in the control colon, Smad2/3 staining was localized to the crypts region, mostly in the nucleus with some cytoplasmic expression. However, infection with Citrobacter resulted in a drastic reduction in the Smad2/3 protein (Fig. 6). These data indicate that TGF-β signaling pathways are downregulated post C. rodentium infection and this decrease in anti-inflammatory pathways along with increase in pro-inflammatory cytokines may contribute to disease pathogenesis associated with Citrobacter infection.
Figure 1.

Effect of Salmonella infection on ileal TGF-R1 and TGF-RII mRNA and protein levels. Streptomycin-pretreated C57BL/6J mice were colonized with S. typhimurium for 8 hours (acute infection) and 4 days (chronic infection). RNA extracted from mouse ileal tissues was amplified using gene specific primers for real-time PCR quantification. Data represent the relative mRNA expression of TGF-R1 (A) and TGF-RII (B) normalized to the respective GAPDH mRNA (internal control) levels. To check protein expression, mouse ileal mucosal tissue lysates were prepared from control and Salmonella-infected mice (8 h and 4 d). Immunoblotting was carried out using receptor-specific antibodies (C). Densitometric analysis showing relative expression of TGF-RI (D) and TGF-RII (E) protein normalized to Villin. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 2.

The mRNA expression levels of Smad2 and Smad3 in response to Salmonella infection in vivo. RNA was extracted from mouse ileal tissues and amplified using gene specific primers for real-time PCR quantification. (A) Smad2 mRNA levels and (B) Smad3 mRNA levels at 8 h and 4 d post Salmonella infection. Data represent the relative expression of Smad2 and Smad3 normalized to the respective GAPDH mRNA (internal control) levels. Data are expressed as mean ± SEM ***p<0.001.
Figure 3.

Activation of Smad2/3 proteins in response to acute and chronic Salmonella infection. An increase in immunofluorescence staining levels of Smad2 and Smad3 phosphorylated proteins in the ileal tissues. p-Smad2 staining (alexa fluor 568, red), p-Smad3 staining (alexa fluor 568, red) (scale bar: 200 μm) in representative images.
Figure 4.

Smad4 and Smad7 expression in response to acute and chronic Salmonella infection. (A) Smad4 mRNA levels at 8 h and 4 d post infection. (B) Smad7 mRNA levels at 8 h and 4 d post infection. Data are expressed as mean ± SEM. **p<0.01; ****p<0.0001; #p<0.05; ####p<0.0001.
Figure 5.

Citrobacter rodentium infection decreases colonic mucosal TGF-R1 and TGF-R2 mRNA levels and downregulates Smad2 and Smad3 levels. Streptomycin-pretreated FVB/N mice were colonized with C. rodentium or control for 6 days. RNA extracted from mouse colonic mucosa was amplified using gene specific primers for real-time PCR quantification. Data represent the relative mRNA expression of TGF-R1 (A), TGF-RII (B), Smad2 (C) and Smad3 (D) normalized to the respective GAPDH mRNA (internal control) levels. Data are expressed as mean ± SEM. *p 0.05; **p<0.01.
Table 1.
Smad4 and Smad7 mRNA levels in Citrobacter rodentium infection to FVB/N mice. RNA was extracted from mouse colonic mucosa and amplified using gene specific primers for real-time PCR quantification. Data represent relative expression of Smad normalized to the respective GAPDH mRNA (internal control) levels. A.U: Arbitrary Units (A.U). Data are expressed as mean ± SEM. *p < 0.05.
| Infection Groups | Smad4 mRNA /GAPDH mRNA (A.U) | Smad7 mRNA /GAPDH mRNA (A.U) |
|---|---|---|
| Uninfected | 1.22 ± 0.09 | 1.95 ± 0.42 |
| Citrobacter rodentium (6 d) | * 0.79 ± 0.15 | * 0.40 ± 0.1 |
Figure 6.

Smad 2/3 Immunostaining in response to Citrobacter infection. OCT embedded tissue sections of representative region of the colon were fixed and stained for total Smad2/3 (alexa fluor 488, green) and Villin (alexa fluor 568, red). A decrease in immunofluorescence staining of Smad2 and Smad3 proteins in the distal colon tissues is shown following C. rodentium infection in representative images. (scale bar: 50 μm).
Discussion
Salmonella and Citrobacter belong to the pathogenic members of the Enterobacteriaceae family of bacteria, which utilizes a type 3-secretion system to cause disease. The major phenotype of the gastro-intestinal disease caused by these microbes is diarrhea and inflammation, although the underlying mechanisms are not fully understood. To examine how these pathogens exploit the host-signaling pathways to contribute to disease pathogenesis, our studies focused on TGF-β, a multifunctional cytokine with anti-inflammatory properties.
The results presented in this study show that both pathogens modulate TGF signaling pathways with an activation of the Smad signaling pathways in response to Salmonella and a decrease in TGFR abundance and Smad signaling pathways by Citrobacter. For these studies, we utilized the previously well-characterized streptomycin-pretreated mouse models of infection mice.17,18,19,20,21,54 Streptomycin-treated model of Salmonella infection primarily develops severe colitis, however, noticeable degree of inflammation is present in the distal ileum as well.22 Indeed, the Salmonella infected small intestine on C57/BL6J background showed erosion of the epithelial surface and polymorphonuclear cell infiltration into the lamina propria 4 days post infection (Supplementary Fig. 2) indicating an inflammatory response as judged by H&E staining. Mouse colon infected with Salmonella showed a marked increase in the pro-inflammatory response and epithelial damage compared to the infected small intestine (Supplementary Fig. 2). Since Salmonella preferentially invades the ileum by targeting the Peyer's patches, we utilized ileum for TGF-β signaling pathways analysis.
The mouse enteric bacterial pathogen, C. rodentium, causes diarrhea, transmissible colonic hyperplasia, and colitis in mice as a consequence of its ability to colonize murine large intestinal enterocytes using the histopathological attaching and effacing (A/E) lesion formation.23,24 The pathogenic features of EPEC or EHEC involve a disruption of the epithelial barrier, perturbation of ion transporters and stimulation of a mucosal immune-mediated extensive inflammatory response.25 FVB/N mice from Jackson Labs were used for Citrobacter infection based on a previous study comparing the susceptibility of different mouse strains (all bought from the same vendor) to C. rodentium infection.19 FVB/N mice showed more susceptibility to Citrobacter infection with development of diarrheal phenotype and severe intestinal inflammation as compared to the resistant C57/BL6J mice.19,26 Our previous studies have also shown increased myeloperoxidase activity, increase in mRNA levels of pro-inflammatory cytokines in streptomycin pre-treated FVB/N mice gavaged with Citrobacter.17 Histopathological analysis demonstrated neutrophil infiltration and crypt hyperplasia in response to Citrobacter as compared to uninfected mice given only streptomycin (Supplementary Fig. 3). Although, the response of different strains to infection by enteric pathogens could be variable,; it is unlikely to affect the outcome of our study. This notion is supported by the fact that TGF-beta dependent Smad2/3 signaling activity is similar in different strains of mice.27 In this regard, a previous study clearly showed that transgenic mice generated with the Smad responsive luciferase construct (SBE-luc) in a FVB/N or in a SJL/J × C57BL/6J mixed genetic background, showed a similar luciferase activity (representing Smad2/3 activation) in the intestine (and also in the brain, heart and skin) in these two different strains of mice.
Previous studies have shown that exogenous TGF-β1 confers resistance against Salmonella infection.28,29 The number of bacteria recovered in the spleens and in the livers of recombinant- TGF-β1-treated mice at 2 and 5 days after Salmonella administration (IP) have been shown to be significantly lower than that found in the same organs after phosphate-buffered saline (PBS) inoculation.30 Similar to Salmonella, C. difficile toxin A was recently shown to induce TGF-β1/Smad signaling pathways and addition of recombinant TGF-β1 showed protection from toxin induced damage to intestinal epithelial cells.31 It is possible that the pathogen-mediated induction in TGF-1 receptors may underlie protective effects against exogenously administered TGF-β1.
Evidence from a variety of in vivo animal models such as knock out mice shown that eliminating TGF-β1 or its downstream signaling cascade (Smad2, 3 and 4) leads to inflammatory disease or cancer.9-11 For example, Smad2 has been shown to be important for immune regulation and mediating signals during development.32,33 Smad3 is a critical mediator of TGF-β1 induced anti-inflammatory responses34 as targeted deletion of the Smad3 gene in mice show massive infiltration of T cells and multiple pyogenic abscesses in the stomach and intestine.35 Interestingly, disruption of Smad4 specifically in T cells resulted in colitis and an increased susceptibility to the spontaneous rectal tumorigenesis.36 Of note, diminished levels of p-Smad3 were reported in the gut mucosa of Crohn's disease.37,38 However, our studies showed that Salmonella is capable of evoking an anti-inflammatory response mediated by TGF-β. Our previous studies showed that wnt 11A, another pathway important for blocking pathogen invasion or suppression of inflammation was also induced after Salmonella colonization in both the ileum and colon.39 Our data indicate that TGF-β/Smad signaling pathways are protective against infection but may not be directly involved in major phenotype of Salmonella i.e diarrhea and inflammation. In this regard, previous studies have demonstrated that Salmonella can hijack host-cell machinery and manipulate the mucosal inflammatory responses. For example, our previous studies demonstrated perturbation of tight junctions, activation of inflammatory responses through NF-kB pathway and reduction in stem cell markers (Lgr5 and Bmi1) post-infection.40-44 Other factors that contributed to Salmonella associated inflammation and diarrhea may include dysregulation of epithelial transporters such as apical Cl−/HCO3− exchanger (SLC26A3).45 The role of T3SS effector molecules in the associated changes in TGF/Smad pathways remain to be investigated. Our previous studies have demonstrated that Salmonella effector protein AvrA dampens the inflammatory responses as a strategy to promote its survival within the host cells.43 Whether AvrA plays a role in increasing alterations in TGF-β/Smad signaling pathways remains to be investigated further.
Interestingly, chronic Salmonella infection of the murine gastrointestinal tract has been shown previously to lead to tissue fibrosis with increased levels of growth factors including TGF-β1 along with extensive type I collagen deposition in the cecal mucosa, submucosa, and muscularis mucosa of infected mice. Thus, it can be speculated that activation of TGF-β1 signaling during chronic inflammatory response is vital for epithelial-mesenchymal transition (EMT) essential for tissue remodeling and wound repair. Interestingly, although TGF-β levels were demonstrated to be higher in patients with IBD and necrotizing colitis model, there is increased Smad7 expression that may cause decrease in Smad2/3 phosphorylation or activation.46,47 These studies further emphasize that merely measuring changes in the levels of TGF-β during the progression of inflammatory and fibrotic disorders is inconclusive until actual continuous TGF-β-mediated signaling pathways are evaluated.48 Thus, exogenous administration of TGF-β1 to increase Smad2/3 activation or Smad7 inhibition have been shown to be protective in ameliorating inflammation.46 In our model of Salmonella infection, the inhibitory Smad7 was up regulated at mRNA level during early infection and was unaltered during chronic phase. The absence of inhibitory signaling during later phase of infection would result in sustained activation of Smad2/3.30 In fact, Smad3 has also been shown to be an important signaling molecule for promoting fibrosis. This is evident from Smad3 null mice that are protected from progressive fibrosis mediated by overexpression of TGF-β149,50 or bleomycin induced lung fibrosis.51
Our current studies showing that TGF receptor and Smad2/3 and Smad 4 pathways are markedly attenuated under these conditions suggest that Citrobacter may exploit TGF pathway as a potential mechanism to cause disease. Similarly, Smad3 and p-Smad3 were also shown to be decreased in viral infections including HSV1 contributing to pathology of human corneal diseases.52 However, Smad7 levels were found to be diminished in response to Citrobacter infection. Previous studies have shown that Smad7 levels are regulated by different stimuli, including TGF-β1, IFN-γ and TNF.53 The mechanism of decrease in Smad7 in response to Citrobacter is unclear at present, albeit this may be a compensatory response to mitigate further inhibition of R-Smads as their expression is already decreased in response to Citrobacter.
As shown in Fig. 7, our current study shows that Salmonella elicits alterations in TGF-β1 signaling with an increase in TGF-R1 receptor abundance and activation of Smad2/3 signaling in the mouse ileum during acute and chronic phases of infection. We speculate that induction of TGF-β1 signaling may be a host compensatory response to promote mucosal healing in response to Salmonella infection and may lead to fibrosis in chronic mouse model of Salmonellosis. On the other hand, our studies show that Citrobacter rodentium infection is associated with defective TGF-R1 and TGF-RII expression and decrease in expression of Smad pathways. These studies provide novel insights into the host-bacterial interactions that are important to understand disease mechanisms relevant to intestinal inflammation and infection.
Figure 7.
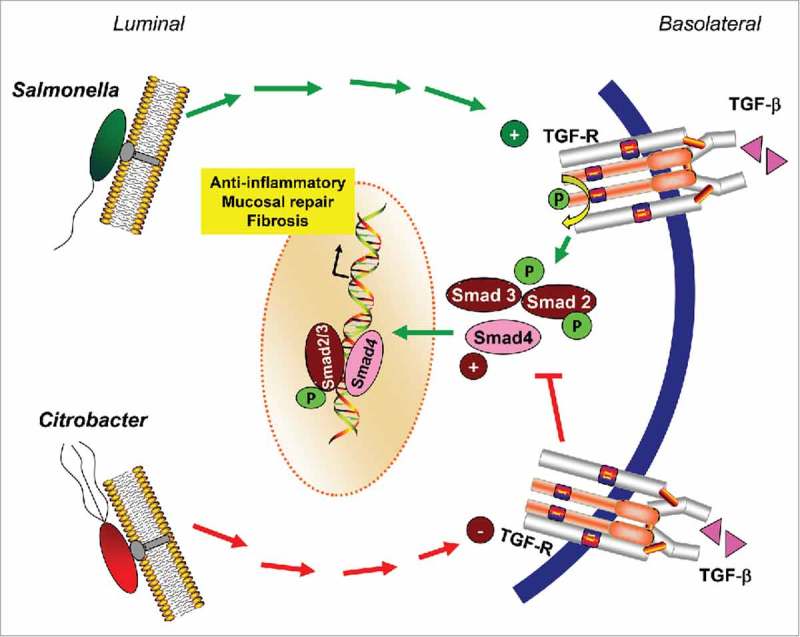
Model showing effects of Salmonella and Citrobacter on TGF-β1 signaling pathways in intestine.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains used in this study were wild-type Salmonella ATCC 14028 and Citrobacter rodentium (ATCC, DBS100/255). Non-agitated microaerophilic Salmonella cultures were prepared by inoculating 10 ml of Luria-Bertani broth with 0.01 ml of a stationary phase culture and were incubated overnight (∼18 h) at 37°C, as previously described.54-56 Citrobacter cultures were grown overnight with shaking in Luria-Bertani broth (LB) at 37°C, diluted (1:33) in serum-free and antibiotic-free tissue culture medium containing 0.5% mannose, and grown at 37°C with aeration to mid-log growth phase (5 × 108 cells/ml)17. Bacteria were spun down and resuspended in 1 ml of sterile 1 × PBS.17
Streptomycin pretreated mouse model of Salmonella Infection
Animal experiments were performed using specific pathogen-free female C57BL/6J mice (Taconic, Hudson, NY) that were 6–7 weeks old, as previously described.40 The protocol was approved by the Rush University Committee on Animal Resources. Water and food were withdrawn for 4 hours before oral gavage with 7.5 mg/mouse of streptomycin. After gavage, the animals were supplied with water and food ad libitum. Twenty hours after streptomycin treatment, water and food were withdrawn again for 4 hours before the mice were infected with 1 × 107 CFU of S. typhimurium (100 µl suspension in HBSS) or treated with sterile HBSS (control) by oral gavage, as previously described.41,54 Both the control and the infected mice received the same dose of streptomycin. At the indicated times after infection, the mice were sacrificed and tissue samples from the ileum were removed for analysis.
Streptomycin pretreated mouse model of Citrobacter Infection
FVB/N mice (male, age 4–6 weeks) obtained from Jackson Laboratories were maintained in Jesse Brown Veterans Affairs Medical Center. In vivo studies performed in these mice were approved by the Animal Care Committee of the University of Illinois at Chicago and Jesse Brown Veterans Affairs Medical Center. All mice were given (streptomycin, 5 g/l) treatments in drinking water for 24 h, followed by water alone for another 24 h. Citrobacter in I X PBS (∼1 × 109 CFU of bacteria/mouse/200 µl) were administered to susceptible FVB/N mice on day 1 by oral gavage.17 Control mice received 200 μl of sterile 1X PBS. Mice were euthanized on 6th day post infection. Intestines were resected, and mucosa was scraped for further analysis.
RNA Extraction and Real time PCR
Total RNA was extracted from mucosal scraping collected from ileum or colon using TRIzol reagent (Invitrogen, Grand Island, NY, USA). The RNA integrity was verified by gel electrophoresis. The RNA was subjected to quantitative real-time RT-PCR using the SYBR green master mix (Stratagene) according to the manufacturer's instructions.
Mouse Ileal or colonic mucosal Lysates
Lysates were prepared from the ileum or distal colon of the mouse. The scraped mucosa was sonicated in lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium ortho-vanadate, and protease inhibitor cocktail). The protein concentration was measured using the BioRad Reagent (BioRad, Hercules, CA, USA).
Immuno-blot analysis
Equal amounts of protein lysates were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and immunoblotted with primary antibodies specific to the protein of interest. Membranes that were probed with more than one antibody were stripped before reprobing. Bands were quantified using Kodak MI software (v.4.0.3).
Immunofluorescence
Intestinal tissues were freshly isolated and embedded in paraffin wax after fixation with 10% neutral buffered formalin or embedded in OCT. Immunofluorescence staining was performed on paraffin-embedded sections (1 µm) of mouse intestine. The slides were incubated in 3% hydrogen peroxide for 20 minutes at room temperature to block endogenous peroxidase activity and then incubated for 20 minutes in 5% BSA with 0.1% saponin in PBS to reduce nonspecific background. For OCT sections, staining was performed as previously described.17 The permeabilized tissue samples were incubated with the indicated antibodies, including Smad2/3 (3102), p-Smad2 (3101), p-Smad3 (9520), (Cell signaling technology), overnight at 4°C. Samples were incubated with goat anti-rabbit Alexa Fluor 594 or 488 (Molecular Probes, CA, USA) and DAPI (Molecular Probes) for one hour at room temperature. Tissues were mounted with SlowFade (SlowFade® AntiFade Kit, Molecular Probes), followed by a coverslip, and the edges were sealed to prevent drying. Specimens were examined with a Leica SP5 Laser Scanning confocal microscope.
Statistical analysis
All data are expressed as means ± SEM. Differences among three were analysed using one-way ANOVA with GraphPad Prism 5 (GraphPad Software, Inc La Jolla, CA, USA). P-Values of 0.05 or less were considered statistically significant.
Supplementary Material
Funding Statement
HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK098170) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK105118) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK71596) VA Merit Review (BX000152) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK92441) VA Merit Review (BX002687) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK105118) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK81858) HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK54016) Support for these studies was provided by the National Institute of Diabetes and Digestive and Kidney Diseases [Grants R01 DK098170 (to R. K. Gill); R01 DK54016, R01 DK81858, and R01 DK92441 (to P. K. Dudeja); and R01 DK109709 (to W. A. Alrefai) and R01 DK105118 (to Jun Sun] and the U.S. Department of Veterans Affairs (VA BX002867- 01A1, to S. Saksena; and BX000152, to W. A. Alrefai), VA Research Career Scientist Award (to W. A. Alrefai), and VA Senior Research Career Scientist Award (to P. K. Dudeja).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Barnard JA, Warwick GJ, Gold LI. Localization of transforming growth factor beta isoforms in the normal murine small intestine and colon. Gastroenterology. 1993;105:67–73. doi: 10.1016/0016-5085(93)90011-Z. PMID:8514063. [DOI] [PubMed] [Google Scholar]
- 2.Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989;108:661–9. doi: 10.1083/jcb.108.2.661. PMID:2645303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wharton K, Derynck R. TGFbeta family signaling: novel insights in development and disease. Development. 2009;136:3691–7. doi: 10.1242/dev.040584. PMID:19855012. [DOI] [PubMed] [Google Scholar]
- 4.Barral-Netto M, Barral A, Brownell CE, Skeiky YA, Ellingsworth LR, Twardzik DR, Reed SG. Transforming growth factor-beta in leishmanial infection: a parasite escape mechanism. Science. 1992;257:545–8. doi: 10.1126/science.1636092. PMID:1636092. [DOI] [PubMed] [Google Scholar]
- 5.Barral A, Barral-Netto M, Yong EC, Brownell CE, Twardzik DR, Reed SG. Transforming growth factor beta as a virulence mechanism for Leishmania braziliensis. Proc Natl Acad Sci U S A. 1993;90:3442–6. doi: 10.1073/pnas.90.8.3442. PMID:7682701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowrance JH, O'Sullivan FX, Caver TE, Waegell W, Gresham HD. Spontaneous elaboration of transforming growth factor beta suppresses host defense against bacterial infection in autoimmune MRL/lpr mice. J Exp Med. 1994;180:1693–703. doi: 10.1084/jem.180.5.1693. PMID:7964455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva JS, Twardzik DR, Reed SG. Regulation of Trypanosoma cruzi infections in vitro and in vivo by transforming growth factor beta (TGF-beta). J Exp Med. 1991;174:539–45. doi: 10.1084/jem.174.3.539. PMID:1908509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. PMID:22992590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahm KB, Im YH, Parks TW, Park SH, Markowitz S, Jung HY, Green J, Kim SJ. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–8. doi: 10.1136/gut.49.2.190. PMID:11454793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh CG, Gress RE, Karlsson S. Transforming growth factor-beta 1 null mice. An animal model for inflammatory disorders. Am J Pathol. 1995;146:264–75. PMID:7856732. [PMC free article] [PubMed] [Google Scholar]
- 11.Strober W, Fuss I, Kitani A. Regulation of experimental mucosal inflammation. Acta Odontol Scand. 2001;59:244–7. doi: 10.1080/00016350152509274. PMID:11570528. [DOI] [PubMed] [Google Scholar]
- 12.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al.. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. PMID:1436033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–8. doi: 10.1182/blood.V100.2.560. PMID:12091349. [DOI] [PubMed] [Google Scholar]
- 14.Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19:77–91. doi: 10.1517/13543780903382609. PMID:20001556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. PMID:21740331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–71. doi: 10.1038/37284. PMID:9393997. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Anbazhagan AN, Coffing H, Chatterjee I, Priyamvada S, Gujral T, Saksena S, Gill RK, Alrefai WA, Borthakur A, et al.. Lactobacillus acidophilus counteracts inhibition of NHE3 and DRA expression and alleviates diarrheal phenotype in mice infected with Citrobacter rodentium. Am J Physiol Gastrointest Liver Physiol. 2016;311:G817–G26. doi: 10.1152/ajpgi.00173.2016. PMID:27634011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borenshtein D, Fry RC, Groff EB, Nambiar PR, Carey VJ, Fox JG, Schauer DB. Diarrhea as a cause of mortality in a mouse model of infectious colitis. Genome Biol. 2008;9:R122. doi: 10.1186/gb-2008-9-8-r122. PMID:18680595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borenshtein D, Schlieper KA, Rickman BH, Chapman JM, Schweinfest CW, Fox JG, Schauer DB. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect Immun. 2009;77:3639–50. doi: 10.1128/IAI.00225-09. PMID:19546193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu R, Bosland M, Xia Y, Zhang YG, Kato I, Sun J. Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget. 2017;8:55104–15. PMID:28903406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu S, Liao AP, Xia Y, Li YC, Li JD, Sartor RB, Sun J. Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. Am J Pathol. 2010;177:686–97. doi: 10.2353/ajpath.2010.090998. PMID:20566739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garner CD, Antonopoulos DA, Wagner B, Duhamel GE, Keresztes I, Ross DA, Young VB, Altier C. Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar typhimurium murine model of infection. Infect Immun. 2009;77:2691–702. doi: 10.1128/IAI.01570-08. PMID:19433544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankel G, Phillips AD, Novakova M, Field H, Candy DC, Schauer DB, Douce G, Dougan G. Intimin from enteropathogenic Escherichia coli restores murine virulence to a Citrobacter rodentium eaeA mutant: induction of an immunoglobulin A response to intimin and EspB. Infect Immun. 1996;64:5315–25. PMID:8945583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schauer DB, Falkow S. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect Immun. 1993;61:4654–61. PMID:8406863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodges K, Gill R. Infectious diarrhea: Cellular and molecular mechanisms. Gut Microbes. 2010;1:4–21. doi: 10.4161/gmic.1.1.11036. PMID:21327112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borenshtein D, Nambiar PR, Groff EB, Fox JG, Schauer DB. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect Immun. 2007;75:3271–81. doi: 10.1128/IAI.01810-06. PMID:17470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin AH, Luo J, Mondshein LH, ten Dijke P, Vivien D, Contag CH, Wyss-Coray T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J Immunol. 2005;175:547–54. doi: 10.4049/jimmunol.175.1.547. PMID:15972691. [DOI] [PubMed] [Google Scholar]
- 28.Nakane A, Nishikawa S, Sasaki S, Miura T, Asano M, Kohanawa M, Ishiwata K, Minagawa T. Endogenous interleukin-4, but not interleukin-10, is involved in suppression of host resistance against Listeria monocytogenes infection in interferon-depleted mice. Infect Immun. 1996;64:1252–8. PMID:8606087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spaccapelo R, Romani L, Tonnetti L, Cenci E, Mencacci A, Del Sero G, Tognellini R, Reed SG, Puccetti P, Bistoni F. TGF-beta is important in determining the in vivo patterns of susceptibility or resistance in mice infected with Candida albicans. J Immunol. 1995;155:1349–60. PMID:7636200. [PubMed] [Google Scholar]
- 30.Galdiero M, Marcatili A, Cipollaro de l'Ero G, Nuzzo I, Bentivoglio C, Galdiero M, Romano Carratelli C. Effect of transforming growth factor beta on experimental Salmonella typhimurium infection in mice. Infect Immun. 1999;67:1432–8. PMID:10024591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tinoco-Veras CM, Santos A, Stipursky J, Meloni M, Araujo APB, Foschetti DA, López-Ureña D, Quesada-Gómez C, Leitão RFC, Gomes FCA, et al.. TGF-beta1/SMADs signaling pathway activation protects intestinal epithelium from Clostridium difficile toxin A-induced damage. Infect Immun. 2017;85(10):1–13. doi: 10.1128/IAI.00430-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–40. doi: 10.1016/S0092-8674(00)81696-7. PMID:9865691. [DOI] [PubMed] [Google Scholar]
- 33.Wakabayashi Y, Tamiya T, Takada I, Fukaya T, Sugiyama Y, Inoue N, Kimura A, Morita R, Kashiwagi I, Takimoto T, et al.. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-beta-Smad pathway. J Biol Chem. 2011;286:35456–65. doi: 10.1074/jbc.M111.236794. PMID:21862595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–91. doi: 10.1093/emboj/18.5.1280. PMID:10064594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owen CR, Yuan L, Basson MD. Smad3 knockout mice exhibit impaired intestinal mucosal healing. Lab Invest. 2008;88:1101–9. doi: 10.1038/labinvest.2008.77. PMID:18711354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szigeti R, Pangas SA, Nagy-Szakal D, Dowd SE, Shulman RJ, Olive AP, Popek EJ, Finegold MJ, Kellermayer R. SMAD4 haploinsufficiency associates with augmented colonic inflammation in select humans and mice. Ann Clin Lab Sci. 2012;42:401–8. PMID:23090737. [PMC free article] [PubMed] [Google Scholar]
- 37.Sedda S, Marafini I, Dinallo V, Di Fusco D, Monteleone G. The TGF-beta/Smad System in IBD Pathogenesis. Inflamm Bowel Dis. 2015;21:2921–5. doi: 10.1097/MIB.0000000000000542. PMID:26230862. [DOI] [PubMed] [Google Scholar]
- 38.Fiocchi C. TGF-beta/Smad signaling defects in inflammatory bowel disease: mechanisms and possible novel therapies for chronic inflammation. J Clin Invest. 2001;108:523–6. doi: 10.1172/JCI13863. PMID:11518725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Wu S, Xia Y, Li XE, Xia Y, Zhou ZD, Sun J. Wingless homolog Wnt11 suppresses bacterial invasion and inflammation in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G992–G1003. doi: 10.1152/ajpgi.00080.2011. PMID:21903761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. Beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest. 2007;87:613–24. doi: 10.1038/labinvest.3700545. PMID:17384665. [DOI] [PubMed] [Google Scholar]
- 41.Liao AP, Petrof EO, Kuppireddi S, Zhao Y, Xia Y, Claud EC, Sun J. Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLoS One. 2008;3:e2369. doi: 10.1371/journal.pone.0002369. PMID:18523661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Lu R, Wu S, Sun J. Salmonella regulation of intestinal stem cells through the Wnt/beta-catenin pathway. FEBS Lett. 2010;584:911–6. doi: 10.1016/j.febslet.2010.01.024. PMID:20083111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol. 2007;171:882–92. doi: 10.2353/ajpath.2007.070220. PMID:17690189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang YG, Wu S, Xia Y, Sun J. Salmonella-infected crypt-derived intestinal organoid culture system for host-bacterial interactions. Physiol Rep. 2014;2. doi: 10.14814/phy2.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, Klinkenberg R, Guiney DG, Fierer J, Barrett KE. Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella-induced enteritis. Gastroenterology. 2013;145:1358–68 e1-4. doi: 10.1053/j.gastro.2013.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monteleone G, Caruso R, Pallone F. Role of Smad7 in inflammatory bowel diseases. World J Gastroenterol. 2012;18:5664–8. doi: 10.3748/wjg.v18.i40.5664. PMID:23155305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Namachivayam K, Blanco CL, MohanKumar K, Jagadeeswaran R, Vasquez M, McGill-Vargas L, Garzon SA, Jain SK, Gill RK, Freitag N, et al.. Smad7 inhibits autocrine expression of TGF-beta2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G167–80. doi: 10.1152/ajpgi.00141.2012. PMID:23154975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosendahl A, Checchin D, Fehniger TE, ten Dijke P, Heldin CH, Sideras P. Activation of the TGF-beta/activin-Smad2 pathway during allergic airway inflammation. Am J Respir Cell Mol Biol. 2001;25:60–8. doi: 10.1165/ajrcmb.25.1.4396. PMID:11472976. [DOI] [PubMed] [Google Scholar]
- 49.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–108. doi: 10.4049/jimmunol.173.3.2099. PMID:15265946. [DOI] [PubMed] [Google Scholar]
- 50.Zanninelli G, Vetuschi A, Sferra R, D'Angelo A, Fratticci A, Continenza MA, Chiaramonte M, Gaudio E, Caprilli R, Latella G. Smad3 knock-out mice as a useful model to study intestinal fibrogenesis. World J Gastroenterol. 2006;12:1211–8. doi: 10.3748/wjg.v12.i8.1211. PMID:16534873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr., Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–93. doi: 10.1152/ajplung.00151.2001. PMID:11839555. [DOI] [PubMed] [Google Scholar]
- 52.Nie Y, Cui D, Pan Z, Deng J, Huang Q, Wu K. HSV-1 infection suppresses TGF-beta1 and SMAD3 expression in human corneal epithelial cells. Mol Vis. 2008;14:1631–8. PMID:18776948. [PMC free article] [PubMed] [Google Scholar]
- 53.Yan X, Liu Z, Chen Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai). 2009;41:263–72. doi: 10.1093/abbs/gmp018. PMID:19352540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu R, Wu S, Liu X, Xia Y, Zhang YG, Sun J. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PLoS One. 2010;5:e10505. doi: 10.1371/journal.pone.0010505. PMID:20463922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma J, Zhang YG, Xia Y, Sun J. The inflammatory cytokine tumor necrosis factor modulates the expression of Salmonella typhimurium effector proteins. J Inflamm (Lond). 2010;7:42. doi: 10.1186/1476-9255-7-42. PMID:20704730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. PMID:8227148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.