

ABSTRACT
The commensal microbiota influences many aspects of immune system regulation, including T cells, but molecular details of how this occurs are largely unknown. Here we review our findings that the microbiota regulates Erdr1, a secreted apoptotic factor, to control T cell survival. Erdr1 is highly upregulated in CD4+ T cells from germfree mice and antibiotic treated animals, and our study shows that Erdr1 is suppressed by the microbiota via Toll-like receptor signaling and MyD88 dependent pathways. Erdr1 functions in an autocrine fashion and promotes apoptosis through the FAS/FASL pathway. Suppression of Erdr1 leads to survival of autoreactive T cells and exacerbated autoimmune disease in the EAE model, and overexpression of Erdr1 results in lessened disease. This novel T cell apoptotic factor has implications for autoimmunity, cancer biology, and invasive pathogens and thus represents a novel therapeutic target in disease.
KEYWORDS: Erdr1, TLRs, autoimmunity, T cells, microbiota, Host-pathogen interactions
Introduction
Symbiotic bacteria that reside abundantly throughout the mammalian body are commonly referred to as the microbiota. These commensals have been shown to have beneficial effects on the host, and disruptions to the composition of the microbiota can be detrimental to the organism.1 One such deleterious result is the increasing prevalence of autoimmunity within industrialized civilizations that is beginning to extend across the globe. In most autoimmune diseases, T cells react against innocuous antigens such as environmental particles or self-tissue. Gut microbes are known to drive both inflammatory and regulatory T cell responses that influence autoimmune disease, however, the mechanisms by which the microbiota regulates these responses is still unclear.2,3
The commensal microbiota also has a role in protecting the body from pathogenic infections. Germfree (GF) and antibiotic-treated mice are more susceptible to enteric pathogens than mice with a diverse gut microbiota.4-7 Resident gut microbiota can provide competition against invading bacteria by nutrient acquisition, directed toxins, and by altering the host environment to favor commensals, and gut microbial signals work to sculpt induction of lymphoid tissue and activate immune cells capable of targeting and clearing invading pathogens.7,8
Recent studies have uncovered specific roles for the microbiota on CD4+ T cell differentiation and function at both extra-intestinal and mucosal locations. Systemically, it has been shown that germfree mice have a lower number of CD4+ T cells in their spleen than specific pathogen free (SPF) mice.9 After mono-colonization of GF mice with B. fragilis, a gut commensal, the mono-colonized mice had an expansion and restoration of CD4+ T cells to levels of SPF mice, showing that the proportion of systemic CD4+ T cells are directly related to microbial presence.9 The subsets and therefore function of those T cells can also change depending on what types of microbes are residing within the gastrointestinal tract. The presence of segmented filamentous bacteria (SFB) can drive Th17 expansion from CD4+ T cells in the lamina propria, leading to inflammation and boosting mucosal immunity.10 A human commensal, Bifidobacterium adolescentis, shows the same Th17 induction but without inflammation.11 In the colonic mucosa, Clostridium species and polysaccharide A (PSA) producing B. fragilis induce the production and differentiation of CD4+ T cells into inducible T regulatory cells (Tregs). A robust Treg presence was shown to induce IL-10 production, an anti-inflammatory cytokine, and provided resistance to colitis.12,13
Given the breadth of the types of microbes present in the mammalian microbiota, identifying how the immune system responds to different microbial products is crucial in understanding the complex relationship between microbiota-mediated diseases and resulting pathologies. To identify genes that are regulated by microbial signals, we screened for genes that were differentially regulated in splenic CD4+ T cells between GF and SPF animals. Genes involved in cellular maintenance, death, and survival had the greatest difference between GF and SPF mice and from that group Erythroid differentiation factor 1 (Erdr1) was one of the most highly up-regulated genes in T cells from GF mice.14
Erdr1 is a vesicle associated secreted protein that is ubiquitously expressed with multiple reported functions.15 Erdr1 was originally identified as hemoglobin synthesis factor in human cells, but has since been shown to also influence cellular survival, growth and motility in cancer cells16.17 However, how Erdr1 functions within T lymphocytes had yet to be explored. As there were multiple functions ascribed to Erdr1 within the literature we tested multiple hypotheses prior to uncovering a role for Erdr1 in T cell death and its importance during autoimmune disease.
Erdr1 is suppressed by the gut microbiota
Antibiotics are used heavily around the world, often prescribed without solid evidence of a bacterial infection and through different periods of development.18,19 The implications of the overuse of antibiotics in our society have become apparent through the selection of multi-drug resistant bacteria.18,20,21 In addition, disruptions to resident commensal microbes by antibiotic usage can have detrimental effects on host immunity and contribute to IBD.19,22 To mimic the use of antibiotics in our society and to test that the microbiota has a role in regulating Erdr1, a mixture of antibiotics was administered to adult SPF mice to deplete commensal bacteria. Reduction of the microbiota by antibiotics lead to elevated levels of Erdr1 expression within splenic CD4+ T cell populations, similar to what was observed in GF mice. That Erdr1 suppression can be disrupted by antibiotics in adult animals suggests that there is constant microbial pressure on Erdr1 regulation.
Toll-like receptors are a family of cellular receptors located on immune cells that respond to specific microbial patterns ranging from bacterial peptidoglycan, lipopolysaccharide, and flagella to non-self RNA and DNA.23 Most TLRs signal through the MyD88-dependent pathway and activate the transcription factor NFkB, resulting in production of pro-inflammatory cytokines and a robust immune response. As the microbiota is a source of TLR ligands, we tested whether Erdr1 could be regulated by TLR signaling. Indeed, animals deficient in Myd88 or TLR2 had elevated levels of Erdr1, and treatment of T cells in vitro with a TLR2 agonist suppressed Erdr1 expression. This suggests that TLR2 and MyD88 signaling downregulates Erdr1 (Fig. 1). Because most of the gut microbiota is present within the intestine, we wondered how TLR ligands from the gut could influence populations of T cells outside the intestine. Several reports have identified that TLR agonists can be found circulating within the intestine.24 Supporting this, we demonstrated that TLR2 signaling was detected in the serum of SPF but not GF animals. Moreover, feeding purified TLR2 ligands to GF animals potently suppressed Erdr1 expression. While this suggests that T cells at systemic sites might be directly influenced by circulating TLR ligands present within the blood, the possibility still exists that T cells traffic from gut to the spleen and therefore might encounter local TLR ligands. Future studies will be needed to differentiate between these possibilities.
Figure 1.
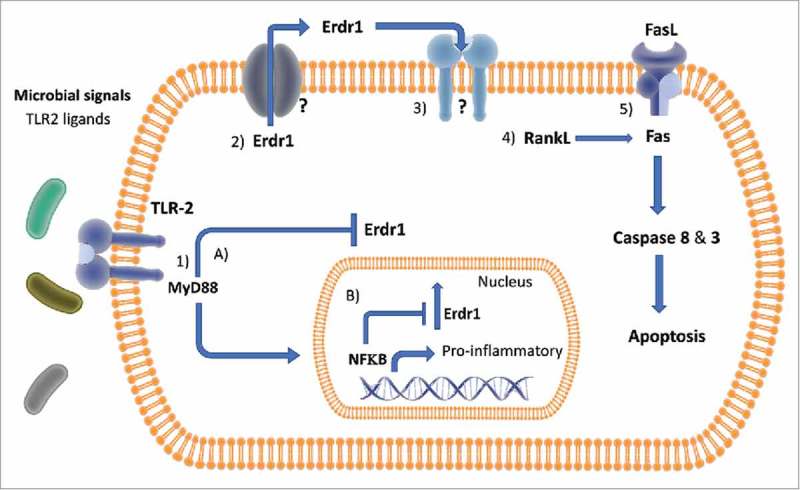
Model of Erdr1 Regulation and Downstream Pathways in CD4+ T Cells Figure 1: Erdr1 suppression is regulated by the microbiota via TLR signaling in CD4+ T cells. In the presence of commensal microbes, TLR2 signals through a MyD88 dependent pathway and suppresses Erdr1, resulting in RankL and Fas activation. Apoptosis is induced by Caspase 3 following Fas activation. The data suggests that the working model is 1) microbial ligands activate TLR2, which activates MyD88. MyD88 then either: A) suppresses Erdr1 independent of NFkB and outside the nucleus, or B) induces activated NFkB suppression of Erdr1 from inside the nucleus. 2) Free cytosolic Erdr1 is secreted by an unknown channel and works in an autocrine manner on the 3) currently unknown Erdr1 receptor, which upregulates RankL and Fas. 4) Activated RankL activates Fas/FasL pathway 5) Activation of FasL/FasR pathway leading to Caspase 8 and 3 activation and resulting apoptosis.
Uncovering a function for Erdr1 in T cells
Erdr1 was originally discovered in the supernatants of B-cell leukemic cells and was identified as a factor that induces hemoglobin production in erythroid progenitor lines.15 Since then, work has demonstrated that Erdr1 is downregulated in human cancer cell lines and contributes to metastasis.16,17 Its properties as an apoptosis factor had been suggested in other cell types, but a role in T cells had yet to be explored.15–17 As the microbiota has been shown to influence the differentiation of CD4+ T helper cell subsets, we initially performed a myriad of experiments to determine whether Erdr1 functioned in this process. However, neither over-expression nor knockdown of Erdr1 by shRNA had an effect on how T cells differentiated into various subsets.14 As Erdr1 had been reported to function in apoptosis and survival we began to further analyze these aspects of T lymphocyte biology. Supporting this, GF, MyD88 and TLR2 deficient mice had elevated levels of apoptotic splenic CD4+T cells. As these are the signals that suppress Erdr1 within T cells, these data suggested that Erdr1 expression might induce T cell apoptosis.14
We made several attempts to produce an Erdr1 knock-out mouse model. Initially, we created a whole-body knockout using Crispr/Cas9 technologies, however there were never any surviving animals indicating that it was embryonic lethal. Moreover, insertion of LoxP sites surrounding Erdr1 to create a conditional allele was difficult, potentially due to highly repetitive sequences surrounding the locus. In lieu of a knockout mouse, overexpression and shRNA silencing tools were used to manipulate Erdr1 expression and identify a function for Erdr1. Suppression of Erdr1 by shRNA led to a reduction in the proportion of pre-apoptotic cells, resulting in significant increases in the absolute numbers of live cells compared to the control. Forced over-expression of Erdr1 had the opposite effect, as it caused significant deficiencies in live T cells through apoptosis.
To investigate the mechanism responsible for Erdr1-mediated apoptosis, RNA-seq was performed on primary splenic Erdr1 overexpressing CD4+ T cells. The most significantly different genes and pathways were those involved in cell death and survival. One of the most significantly upregulated genes was RankL, which has previously been shown to be highly expressed in activated T cells. However, the function of RankL in T cells has been shown to be induction of cellular survival, therefore was unlikely to influence Erdr1 induced apoptosis on its own. Interestingly, RankL has been reported to enhance cell death in a collaboration with the Fas/FasL signaling pathway.25-27 Fas/FasL is a primary and well-studied pathway within T cells that leads to apoptosis through caspase 3 activity in T cells.28,29
Based on this, we first determined whether Erdr1 regulated the Fas pathway. Supporting this, suppression of Erdr1 expression by shRNA led to a reduction of both activated caspase 3 and surface expression of Fas. Moreover, chemical inhibition of caspase-3 or neutralization of Fas using a blocking antibody prevented Erdr1 induced apoptosis. How RankL might function in this pathway is still not fully understood. RankL has been reported to have many different functions including a report that it enhances T cell growth.25 However, RankL has also been shown to induce apoptosis in dendritic cells when in collaboration with the Fas/FasL signaling pathway that could drive tolerance.27 In T cells, it could be that when the Fas/FasL pathway is engaged, RankL functions to upregulate apoptosis, but in the absence of Fas/FasL it acts as a growth factor (Fig. 1).
Previous work has shown that Erdr1 can function in both an autocrine and paracrine manner, but this had not been tested in T cells.30 To test this, we overexpressed Erdr1 in CD45.2 T cells and incubating them with CD45.1 control cells. Within 48 hours of co-incubation the CD45.1 cells had outcompeted the CD45.2 T cells, demonstrating that Erdr1 likely acts in an autocrine manner to induce apoptosis. Taken together these data have continued to paint the picture of Erdr1 in T cells, regulated by the microbiota, as an inducer of apoptosis by way of the RankL and Fas/FasL signaling pathway affecting T cells in an auto-regulatory manner (Fig. 1).
Erdr1 mediated cell death affects antigen specific immune responses
Apoptosis, or programmed cell death, is an essential means by which the life of a cell can be regulated. In immune cells, if there are too many active cells, overly inflammatory reactions can cascade into autoimmune scenarios. Conversely, too little cells and an adequate response to an invading pathogen cannot be mounted.
Considering that Erdr1 induces T cell apoptosis, we thought it likely that the immune response would be affected by manipulation of Erdr1 expression. To test this, Erdr1 was overexpressed or silenced by shRNA in CD4+ OT-II TCR transgenic T cells, which were adoptively transferred into TCRβ-knockout mice (which have no T cells). The mice were then immunized with OVA and analyzed for their immune response to the OVA. Animals with suppressed Erdr1 had increases in spleen size with more total cells in them, increased numbers of OT-II cells and a reduced percentage of antigen-specific preapoptotic CD4+ T cells. Animals with suppressed Erdr1 also had a significant increase in inflammatory OT-II Th17 cells and enhanced germinal center B cells. The overexpression of Erdr1 had the converse result. The same experiment was repeated, but with Fas KO mice as a T cell donor. Without the presence of the Fas pathway, differential expression of Erdr1 did not lead to a difference in apoptotic cells. This strengthened the conclusion that Erdr1 enhances T cell apoptosis through the Fas pathway in vivo.
Erdr1 mediated cellular death dictates the severity of a multiple sclerosis model
Previous work has shown that GF mice are resistant to Experimental Autoimmune Encephalomyelitis (EAE), which is used as an animal model of multiple sclerosis (MS).3,31 In this model, immunization by an adjuvant containing myelin glycoprotein results in uncontrolled autoreactive T cells directed at myelin, which is a protective coating on nerves, leading to a loss of the myelin sheath and subsequent paralysis.32,33 Part of the reason that GF animals are resistant to EAE is likely due to a loss of inflammatory T cell lineages, leading to a decrease in an autoimmune phenotype. Given that the suppression of Erdr1 decreases T cell apoptosis, therefore increasing pro inflammatory T cells, we hypothesized that suppression of Erdr1 would lead to exacerbated autoimmune disease in the EAE model.
Indeed, suppression of Erdr1 within T cells enhanced cellular survival of autoreactive T cells during EAE, leading to increased paralysis and a worse outcome. Consistent with enhanced diseased, there were more infiltrating cells within the brains of animals with reduced Erdr1. Overexpression of Erdr1 showed the opposite effect: animals with elevated Erdr1 levels developed milder disease noted by partial loss of their hind limbs and no forelimb paralysis, fewer T cells infiltrated the brain, and T cells had an increased expression of Fas, again validating that Erdr1 leads to upregulation of apoptosis via Fas.
These experiments have shown that Erdr1 regulates T cell survival and its suppression can have detrimental effects in autoimmune disease progression. These experiments also show that Erdr1 could be used as a therapeutic target – inducing expression of Erdr1 in T cells could help alleviate MS symptoms.
Discussion and future directions
Future work is needed to identify Erdr1's receptor. While we have shown that Fas is a downstream target and is the apoptotic signaling pathway affected by Erdr1, Fas is not likely to be the primary receptor. It is also possible Erdr1 has no surface receptor, but rather is taken back into the cell by the vesicles it is contained within. More work is needed to tease apart one, or several, different molecules that Erdr1 is directly acting upon. Once a receptor is found, both Erdr1 and its receptor could be targeted as a therapeutic intervention both during autoimmune disease or during cancer progression.
We do not yet understand the specific interactions of Erdr1 with different bacteria, be they commensals, pathobionts or invasive pathogens. We know that the microbiota works to regulate Erdr1 expression in splenic CD4+ T cells; however, which bacteria influence Erdr1 and in what ways remain to be investigated. While gut commensals suppress Erdr1, other systemic inflammatory pathogens, such as Salmonella, might stimulate Erdr1 expression as a means of host evasion by limiting T cell responses. Investigations are needed to determine the roles of various invasive pathogen infections verses commensal interactions. Fig. 2 explores current models of Erdr1 regulation in T cells and resulting impacts on autoimmunity, invasive pathogens and cancer.
Figure 2.
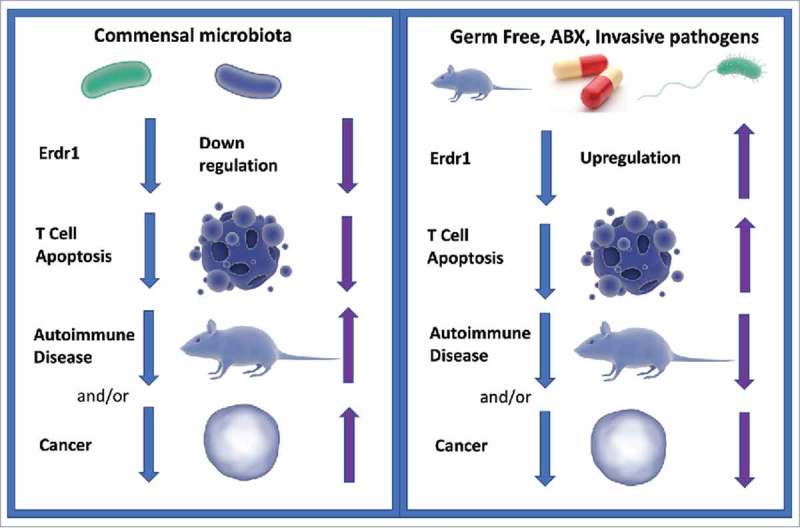
Working Model of Erdr1 Regulation and Resulting Disease Phenotypes Figure 2: In our current model of Erdr1 regulation, commensal microbes suppress Erdr1, which downregulates T cell apoptosis (left panel). Blue arrows indicate the order of events and purple arrows indicate Erdr1 expression level and the resulting phenotype (e.g. upregulation, more apoptosis, less autoimmune disease). Downregulation of Erdr1 leads to enhanced T cell survival, which in turn leads to greater autoimmune diseases and suppression of Erdr1 is noted in several cancer cell lines. In the absence of microbes via germfree or antibiotic treated animals, (right panel) Erdr1 expression is upregulated, T cell apoptosis is increased, and autoimmune disease severity is decreased. Upregulation of Erdr1 could lead to decreases in cancer. Invasive pathogens could exploit this pathway, upregulating Erdr1 and leading to immune cell apoptosis as a mechanism of immune evasion.
The onset and exacerbation of certain cancers often have a microbial component. For instance, H. pylori can cause gastric cancer and E. coli NC101 and F. nucleatum (among others) exacerbate colorectal cancer.34-36 Previous studies have showed an involvement of Erdr1 in cancer cells lines. In addition to its role as a pro-apoptotic factor during gastric cancer16 and melanoma,17 Erdr1 has been shown to have a pro-apoptotic role in keratinocytes during UV challenge.37 How Erdr1 might function in T cells during different types of cancer is yet to be determined, as is its role in certain cancers with closer proximity to the gut microbiota and with a known microbial component, such as colon cancer.
We know from this work that Erdr1 acts on T cells in an autocrine manner, but it is still unclear if Erdr1 from a T cell has a paracrine or autocrine effect on nearby epithelial cells, which could be critical in regulating cancer progression. More work is needed to investigate its potential involvement in these cancers, and how the microbiota might work to mediate suppression and induction of Erdr1 both in cancer cells and in lymphocytes. Its utility as a therapeutic target is reliant on understanding which nearby cell types become affected.
Further, we investigated mainly CD4+ T cells in this study without regard to specific subsets. It may be that Erdr1 is more actively expressed in certain populations of CD4+ T cells than others, which could have important effects on inflammation and disease progression. Additionally, the role of Erdr1 in other immune cells types such as CD8+ T cells, B cells, dendritic cells and macrophages have yet to be addressed.
There are also potential ways in which Erdr1 could be regulated by a viral infection. It would be interesting to assess the role of Erdr1 during HIV infection in CD4+ T cells. A T cell apoptosis factor could have a role in HIV progression or could be used as a therapy. For instance, during an HIV latency phase, if Erdr1 could be overexpressed in those cells, leading to specific apoptosis of HIV containing CD4+ T cells, perhaps the virus load in an individual could be lowered. During AIDS, perhaps suppression of Erdr1 could help to diminish the depletion of CD4+ T cells. Much more work would be needed to show these involvements and the potential for therapy.
All told, elucidating the role of Erdr1 in T cells has led to exciting insights in how the microbiota could be regulating autoimmune disease, and could be used as a therapeutic target alongside other treatments to help alleviate MS symptoms. It also has a role in different cancer types, where suppression of Erdr1 leads to increased oncogenesis, and induction of Erdr1 or stimulation of its receptor could be a future therapeutic target.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. doi: 10.1038/nri2515. PMID:19343057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–27. doi: 10.1016/j.immuni.2010.06.001. PMID:20620945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4615–22. doi: 10.1073/pnas.1000082107. PMID:20660719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osawa N, Mitsuhashi S. Infection of Germeree Mice with Shigella Flexneri 3a. Jpn J Exp Med 1964;34:77–80. PMID:14169586. [PubMed] [Google Scholar]
- 5.Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, Finlay BB. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun. 2008;76:4726–36. doi: 10.1128/IAI.00319-08. PMID:18678663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, et al.. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe. 2016;19:443–54. doi: 10.1016/j.chom.2016.03.004. PMID:27078066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14:685–90. doi: 10.1038/ni.2608. PMID:23778796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–23. doi: 10.1016/j.chom.2011.10.004. PMID:22018232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–18. doi: 10.1016/j.cell.2005.05.007. PMID:16009137. [DOI] [PubMed] [Google Scholar]
- 10.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al.. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. PMID:19836068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, Pasman L, Ortiz-Lopez A, Jupp R, Wu HJ, et al.. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. 2016;113:E8141–E50. doi: 10.1073/pnas.1617460113. PMID:27911839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al.. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41. doi: 10.1126/science.1198469. PMID:21205640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–9. doi: 10.1073/pnas.0909122107. PMID:20566854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soto R, Petersen C, Novis CL, Kubinak JL, Bell R, Stephens WZ, Lane TE, Fujinami RS, Bosque A, O'Connell RM, et al.. Microbiota promotes systemic T-cell survival through suppression of an apoptotic factor. Proc Natl Acad Sci U S A. 2017;114:5497–502. doi: 10.1073/pnas.1619336114. PMID:28487480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dormer P, Spitzer E, Frankenberger M, Kremmer E. Erythroid differentiation regulator (EDR), a novel, highly conserved factor I. Induction of haemoglobin synthesis in erythroleukaemic cells. Cytokine. 2004;26:231–42. [DOI] [PubMed] [Google Scholar]
- 16.Jung MK, Houh YK, Ha S, Yang Y, Kim D, Kim TS, Yoon SR, Bang SI, Cho BJ, Lee WJ, et al.. Recombinant Erdr1 suppresses the migration and invasion ability of human gastric cancer cells, SNU-216, through the JNK pathway. Immunol Lett. 2013;150:145–51. doi: 10.1016/j.imlet.2013.01.012. PMID:23370368. [DOI] [PubMed] [Google Scholar]
- 17.Jung MK, Park Y, Song SB, Cheon SY, Park S, Houh Y, Ha S, Kim HJ, Park JM, Kim TS, et al.. Erythroid differentiation regulator 1, an interleukin 18-regulated gene, acts as a metastasis suppressor in melanoma. J Invest Dermatol. 2011;131:2096–104. doi: 10.1038/jid.2011.170. PMID:21697887. [DOI] [PubMed] [Google Scholar]
- 18.Antibiotic resistance threats in the United States Centers for Disease Control and Prevention. 2013; http://www.cdc.gov/drugresistance/threat-report-2013/index.html
- 19.Hviid A, Svanstrom H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. PMID:20966024. [DOI] [PubMed] [Google Scholar]
- 20.Pasquale TR, Jabrocki B, Salstrom SJ, Wiemken TL, Peyrani P, Haque NZ, Scerpella EG, Ford KD, Zervos MJ, Ramirez JA, et al.. Emergence of methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of late-onset nosocomial pneumonia in intensive care patients in the USA. Int J Infect Dis. 2013;17:e398−403. doi: 10.1016/j.ijid.2012.12.013. PMID:23375542. [DOI] [PubMed] [Google Scholar]
- 21.Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al.. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–21. doi: 10.1016/j.cell.2014.05.052. PMID:25126780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leclercq S, Mian FM, Stanisz AM, Bindels LB, Cambier E, Ben-Amram H, Koren O, Forsythe P, Bienenstock J. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat Commun. 2017;8:15062. doi: 10.1038/ncomms15062. PMID:28375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. PMID:12524386. [DOI] [PubMed] [Google Scholar]
- 24.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16:228–31. doi: 10.1038/nm.2087. PMID:20081863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753. PMID:11861618. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Pan G, McKenna MA, Zayzafoon M, Xiong WC, McDonald JM. RANKL regulates Fas expression and Fas-mediated apoptosis in osteoclasts. J Bone Miner Res. 2005;20:107–16. doi: 10.1359/JBMR.041022. PMID:15619676. [DOI] [PubMed] [Google Scholar]
- 27.Izawa T, Ishimaru N, Moriyama K, Kohashi M, Arakaki R, Hayashi Y. Crosstalk between RANKL and Fas signaling in dendritic cells controls immune tolerance. Blood. 2007;110:242–50. doi: 10.1182/blood-2006-11-059980. PMID:17371940. [DOI] [PubMed] [Google Scholar]
- 28.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. PMID:17562483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41. doi: 10.1038/nrm2312. PMID:18073771. [DOI] [PubMed] [Google Scholar]
- 30.Dormer P, Spitzer E, Moller W. EDR is a stress-related survival factor from stroma and other tissues acting on early haematopoietic progenitors (E-Mix). Cytokine. 2004;27:47–57. doi: 10.1016/j.cyto.2004.03.014. PMID:15242693. [DOI] [PubMed] [Google Scholar]
- 31.Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–41. doi: 10.1038/nature10554. PMID:22031325. [DOI] [PubMed] [Google Scholar]
- 32.Bhat R, Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–32. doi: 10.1016/j.neuron.2009.09.015. PMID:19840554. [DOI] [PubMed] [Google Scholar]
- 33.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–9. doi: 10.1038/nprot.2006.285. PMID:17487163. [DOI] [PubMed] [Google Scholar]
- 34.Blaser MJ, Parsonnet J. Parasitism by the "slow" bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J Clin Invest 1994;94:4–8. doi: 10.1172/JCI117336. PMID:8040281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al.. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. PMID:22903521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. PMID:23954158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HJ, Song SB, Yang Y, Eun YS, Cho BK, Park HJ, Cho DH. Erythroid differentiation regulator 1 (Erdr1) is a proapototic factor in human keratinocytes. Exp Dermatol. 2011;20:920–5. doi: 10.1111/j.1600-0625.2011.01354.x. PMID:21995813. [DOI] [PubMed] [Google Scholar]