

ABSTRACT
Alcohol use disorder (AUD) results in increased intestinal permeability, nutrient malabsorption, and increased risk of colorectal cancer (CRC). Our understanding of the mechanisms underlying these morbidities remains limited because studies to date have relied almost exclusively on short-term heavy/binge drinking rodent models and colonic biopsies/fecal samples collected from AUD subjects with alcoholic liver disease (ALD). Consequently, the dose- and site-dependent impact of chronic alcohol consumption in the absence of overt liver disease remains poorly understood. In this study, we addressed this knowledge gap using a nonhuman primate model of voluntary ethanol self-administration where rhesus macaques consume varying amounts of 4% ethanol in water for 12 months. Specifically, we performed RNA-Seq and 16S rRNA gene sequencing on duodenum, jejunum, ileum, and colon biopsies collected from 4 controls and 8 ethanol-consuming male macaques. Our analysis revealed that chronic ethanol consumption leads to changes in the expression of genes involved in protein trafficking, metabolism, inflammation, and CRC development. Additionally, we observed differences in the relative abundance of putatively beneficial bacteria as well as those associated with inflammation and CRC. Given that the animals studied in this manuscript did not exhibit signs of ALD or CRC, our data suggest that alterations in gene expression and bacterial communities precede clinical disease and could serve as biomarkers as well as facilitate future studies aimed at developing interventions to restore gut homeostasis.
KEYWORDS: alcohol, bacterial 16S rRNA gene sequencing, colorectal cancer, gut microbiome, mucosal immunity, gut, rhesus macaque, RNA-Seq, gene expression
Introduction
The gastrointestinal (GI) tract is the largest mucosal surface composed of intestinal epithelial cells (IEC), immune cells, and microorganisms that together form the most critical barrier between the host and its environment.1,2 Interactions between these three components are vital to maintaining selective permeability and immune homeostasis in the gut.3,4 Each region of the GI tract is unique in its physiological function and microbial load5: the duodenum and jejunum are primarily responsible for absorption and digestion of dietary components; the ileum, the longest segment of the small intestine, harbors the majority of gut-associated lymphoid tissue (GALT); and the colon functions to reabsorb water, eliminate waste, and contains the highest bacterial content.
Alcohol use disorder (AUD) results in gut injury highlighted by increased permeability, perturbations of the gut microbiome, nutrient malabsorption, increased risk of colorectal cancer (CRC), and functional alterations in mucosal immune cells.6 Data from in vitro studies indicate that increased permeability is mediated by dysregulated expression and/or distribution of tight junction proteins, notably, zonula occludens (ZO)-1 and claudin-1 in ethanol or acetaldehyde-treated IEC lines.7-9 These in vitro findings are in line with clinical and animal model studies that showed reduced expression of claudin-1, occludin, and ZO-1 in the duodenum, ileum, and colon,10,11 in part due to increased expression of microRNA-212.8 Increased intestinal permeability coupled with translocation of luminal contents plays a critical role in the initiation and pathogenesis of alcoholic liver disease (ALD).12-14
AUD alters the fecal microbiome in both humans15-18 and rodent models.13,19 Early studies reported increased abundance of Gram-negative anaerobic bacteria and endospore-forming rods,15 as well as bacteria overgrowth in AUD subjects compared to healthy controls.16 Lower abundances of Bacteroidetes and higher abundances of Proteobacteria have been shown in colonic biopsies and fecal samples from patients17,18 as well as mice with ALD.13 Changes in microbial communities are believed to contribute to higher intestinal permeability since antibiotic treatment prevents the ethanol-induced increase in intestinal permeability in rats.20
Additionally, AUD affects nutrients absorption by decreasing secretion of digestive enzymes.21 This is further compounded by the fact that subjects with AUD often derive most of their caloric intake from alcohol limiting their supply of nutrients necessary for structural maintenance of IEC.22 Consequently, subjects with AUD are often deficient in essential vitamins and minerals that play a crucial role in the modulation of gut mucosal immunity.23 Furthermore, AUD is associated with a 70% greater risk for developing colorectal cancer (CRC).24-26 Finally, studies showed reduced cytokine production by lamina propria lymphocytes with ethanol consumption, suggestive of reduced immune surveillance.27,28
Despite these findings, several gaps in our understanding of the impact and mechanisms of ethanol-mediated GI injury persist. For instance, very few studies have examined the impact of AUD on GI tract segments other than the colon. Additionally, the impact of non-heavy alcohol consumption is largely understudied despite the fact it is widespread.29 Finally, mechanisms of reduced nutrient absorption and the increased risk for CRC remain unclear. Some of the major obstacles to addressing these questions are: 1) limited access to intestinal biopsies along the entire GI tract from human subjects; 2) ethical consideration in obtaining biopsies along the GI tract from subjects who are non-heavy consumers and don't exhibit disease; and 3) the short duration of ethanol consumption coupled with high ethanol concentration used in rodent studies.30
To address these questions, we used a nonhuman primate model of voluntary ethanol self-administration where following a 3-month ethanol induction period, animals are given “open-access” to both 4% ethanol and water for 22 h/day for up to 24 months.31,32 Because animals voluntarily consume varying doses of ethanol, they naturally segregate into non-heavy and heavy drinkers with intakes that closely mirror human data.31 After 18 months of chronic heavy ethanol consumption, animals develop ALD as evidenced by elevated liver enzymes and liver inflammation.33 Using this animal model, we recently reported robust gene expression changes and disruptions in innate immune pathways in peripheral blood mononuclear cells isolated from both male34 and female35 heavy drinkers. These data provide potential mechanisms to explain the higher incidence of infection, delay in wound healing, and the increased risk of cardiovascular disease, lung inflammatory disease, and cancer seen in subjects with AUD. We also previously showed that despite the lack of differences in the frequency of intestinal lymphocytes, chronic ethanol consumption resulted in a dose-dependent decreased production of IL-2, IL-17, TNFα, and IFNγ by gut-resident CD4+ and CD8+ T cells.28
In this study, we sought to uncover dose- and site-dependent ethanol-induced alterations in mucosal gene expression and bacterial composition of intestinal biopsies collected from all major gut sections from 8 ethanol-consuming and 4 control male rhesus macaques. Our RNA-Seq analysis revealed several gene expression changes associated with protein localization, metabolism, inflammation, and CRC in ethanol-consuming animals. We also found that chronic ethanol consumption led to a dose-dependent decrease in putatively beneficial bacteria and an increase in bacteria associated with inflammation and CRC along the GI tract. Additional in silico analyses suggest putative changes in bacterial metabolic pathways. Since none of our animals exhibited clinical signs of CRC or ALD, these data suggest that ethanol-mediated changes in gene expression and bacterial communities could serve as early biomarkers of GI disease.
Results
Chronic ethanol consumption modulates expression of genes critical for regulation of gene expression, metabolism, and cell adhesion in the jejunum.
Although it is believed most of the ethanol consumed is rapidly absorbed into circulation in the duodenum, we did not detect any differentially expressed genes (DEG) in the duodenal biopsies. In the jejunum, we detected 45 DEG between controls and ethanol-consuming male macaques (Fig. 1A). The 28 DEG with human homologs (Fig. 1B) play a role in the regulation of gene expression, protein trafficking, and metabolism (Fig. 1C). Upregulated DEG that play a role in regulating gene expression include DNA replication licensing factor (MCM6), histone H3 (H3F3A), and serine/arginine-rich splicing factor (SRSF11). Upregulated DEG that are important for protein trafficking include zinc finger DHHC-type-containing-6 (ZDHHC6; protein folding) and inner mitochondrial membrane peptidase-like-1 (IMMP1L; protein export). Finally, upregulated DEG with a role in cellular metabolism include those involved in digestion of carbohydrates such as sucrose-isomaltase (SI) and enolase-4 (ENO4), as well as fatty acid metabolism such as acyl-coenzyme-A synthetase medium chain-3 (ACSM3) and apolipoprotein B (APOB). Several of the downregulated DEG also played a role in regulation of: gene expression such as chromatin-modifying protein-1A (CHMP1A); metabolism such as the insulin-induced gene-2 (INSIG2); and cell movement such as the adhesion molecule CD109.
Figure 1.
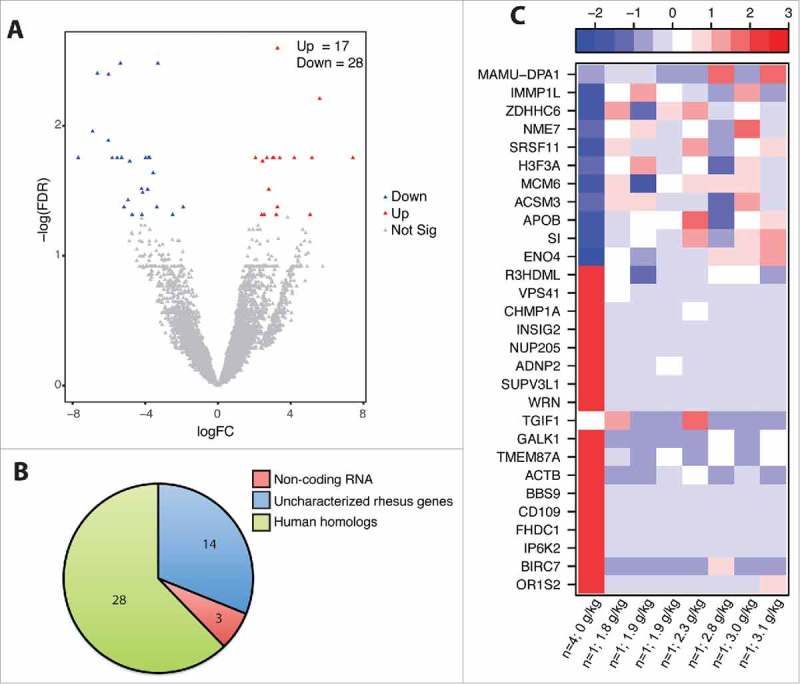
Chronic ethanol consumption modulates jejunum mucosal gene expression. DEG were identified using the generalized linear model likelihood ratio test method from the edgeR package as those with a fold change of ≥2 and a Benjamini-Hochberg- corrected false discovery rate (FDR) of <0.05. (A) Volcano plot summarizing the gene expression changes with red representing the upregulated DEG and blue representing the downregulated DEG. The number of up- and down-regulated genes is noted. (B) Pie chart representing the breakdown of DEG as: noncoding RNA, uncharacterized rhesus genes, and human homologs. (C) Heatmap representing gene expression (shown as absolute normalized RPKM values) of the genes listed; first column shows median RPKM values of the controls (n = 4), and subsequent columns show RPKM values of each individual ethanol-consuming animals ordered by alcohol dose (g of ethanol/kg/day); range of colors is based on scaled and centered RPKM values of the entire set of genes (red represents increased expression while blue represents decreased expression).
Chronic ethanol consumption modulates expression of genes important for immune processes in the ileum.
The ileum exhibited the most robust changes in gene expression with 510 DEG (Fig. 2A). Of the 454 DEG with human homologs (Fig. 2B), 363 were upregulated and 91 were downregulated. The upregulated DEG primarily enriched to gene ontology (GO) terms related to immunity and signal transduction (Fig. 2C). A nework analysis using the functional enrichment software Metacore™ showed that of the 136 upregulated DEG that functionally enriched to the GO process “immune system process”, 73 DEG directly interact with one another (Fig. 2D). These genes include T-cell markers (CD1C, CD40LG); B-cell markers (CD19, CD79B); antigen presenting cell markers (CD40, CD86); adhesion molecules (VCAM1, CD44); cytokines, chemokines, and their receptors (CCL19, CCR6, CCR7, TNFRSF13C); and Toll-like receptors (TLR1, TLR7, TLR10). The majority of the DEG that functionally enriched to “signal transduction” also enriched to “immune system process”. Those that uniquely enriched to “signal transduction” play a role in G-protein signaling e.g., regulator of G protein signaling RGS13, the signaling G protein RAC2, the guanine nucleotide exchange factor GIV/Girdin (CCDC88A), and the GTPase dynamin (DNM1).
Figure 2.
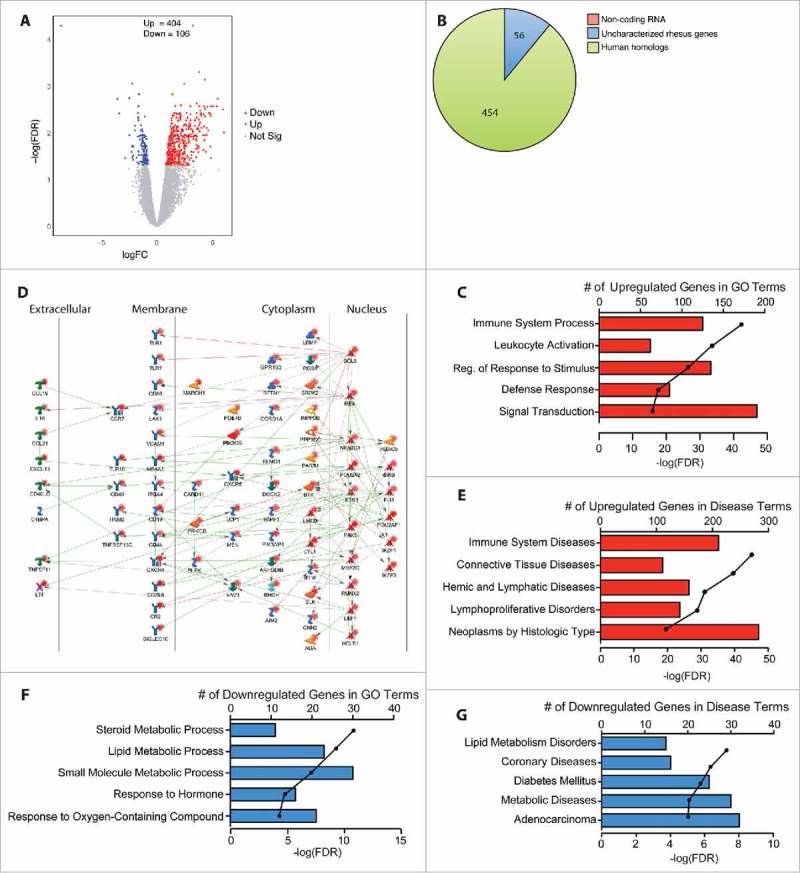
Chronic heavy ethanol consumption modulates expression of genes important for immune system processes in the ileum. DEG were identified using the generalized linear model likelihood ratio test method from the edgeR package as those with a fold change of ≥2 and a Benjamini-Hochberg- corrected false discovery rate (FDR) of <0.05. (A) Volcano plot summarizing the gene expression changes with red representing the upregulated DEG and blue representing the downregulated DEG. The number of up- and down-regulated genes is noted. (B) Pie chart representing the breakdown of DEG as noncoding RNA, uncharacterized rhesus genes, and human homologs. (C) Bar graphs displaying the number of DEG upregulated with ethanol consumption that enriched to the listed Gene Ontology (GO) terms; line represents the -log(FDR-adjusted P-values) associated with each enrichment to a GO term. (D) Network image of the upregulated DEG that enriched to “immune system process” and directly interact with one another. (E) Bar graphs displaying the number of DEG upregulated with ethanol consumption that enriched to the Disease terms; line represents the -log(FDR-adjusted P-values) associated with each Disease. (F) Bar graphs displaying the number of DEG downregulated with ethanol consumption that enriched to the listed GO terms; line represents the -log(FDR-adjusted P-values) associated with enrichment to each GO term. (G) Bar graphs displaying the number of DEG upregulated with ethanol consumption that enriched to the Disease terms; line represents the -log(FDR-adjusted P-values) associated with each Disease term.
Additional functional enrichment analysis using a disease database available on Metacore™ revealed significant enrichment to immune system diseases (Fig. 2E) including genes that are critical to the development and progression of various lymphomas such as BCL2A1, BCL6, Bruton's tyrosine kinase (BTK), B- and T-lymphocyte attenuator (BTLA), matrix metalloproteinases (MMP12), and mitogen-activated protein kinases (MAP4K1). Most of the 108 DEG that uniquely enriched to “neoplasms by histologic type” are associated with GI neoplasms including complement component-4 binding protein alpha (C4BPA), protein tyrosine phosphatase non-receptor type-7 (PTPN7), and transcription factor 4 (TCF4). Expression of genes involved in structural integrity such as gap junction protein (GJA5) was also upregulated.
The 91 DEG that were downregulated in the ileum with ethanol consumption enriched to GO processes related to steroid/lipid metabolism (Fig. 2F). Notable DEG include the signaling molecule phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type-2-gamma (PIK3C2G); transcription factors hepatocyte nuclear factor alpha 1 and 4 (HNFA1, HNFA4); and the low-density lipoprotein receptor (LDLR) and its ligand proprotein convertase subtilisin/kexin type-9 (PCSK9). Similarly, analysis using the diseases database showed enrichment to “lipid metabolism disorders” and “metabolic diseases”. Additionally, several of the 32 genes that enriched to the disease pathway “adenocarcinoma” (Fig. 2G) are associated with progression of CRC including caspase recruitment domain-containing protein (PYCARD), melanophilin (MLPH), transient receptor potential cation channel (TRPM6), and suppressor of cytokine signaling-6 (SOCS6).
Chronic ethanol consumption modulates expression of genes important for pathways involving cancer development and immunity in the colon mucosa.
In the colon, 3 genes were upregulated and 108 genes were downregulated with ethanol-consumption (Fig. 3A). Of the 107 downregulated DEG with human homologs (Fig. 3B), 67 enriched to the GO term “immune system process” (Fig. 3C and E) and 71 enriched to the disease term “immune system diseases” (Fig. 3D). These genes include B-cell markers (CD19, CD72, CD79B); co-stimulatory markers (CD83); as well as cytokines, chemokines, and their receptors (e.g. IL24, CCL19, IL21R, and CCR7). Several DEG also encode for T-cell markers (e.g. CD1C, CD2, CD3D, CD3E, CD5, CD8A) and enriched to GO terms “regulation of T-cell activation” and “T-cell differentiation”. Interestingly, several of these DEG were upregulated with ethanol consumption in the ileum (CD1C, CD19, CD22, CD72, CD79B, CCL19, CCR7, CXCR4, FCRL1, IL21R, TNFRSF13C). Additional downregulated immune genes include indoleamine-2,3-dioxygenase-1 (IDO1), which drives differentiation of regulatory T-cells; peptidase inhibitor (PI3); and defensin-6 (DEF6). Some downregulated genes are also critical for signal transduction including Janus kinase-3 (JAK3) and signal transducer and activator of transcription (STAT1). Several of the immune genes listed above enriched to the disease term “gastrointestinal neoplasms” and are also associated with CRC development including the T cell chemoattractant CXCL9, keratin-7 (KRT7), and IDO1.
Figure 3.
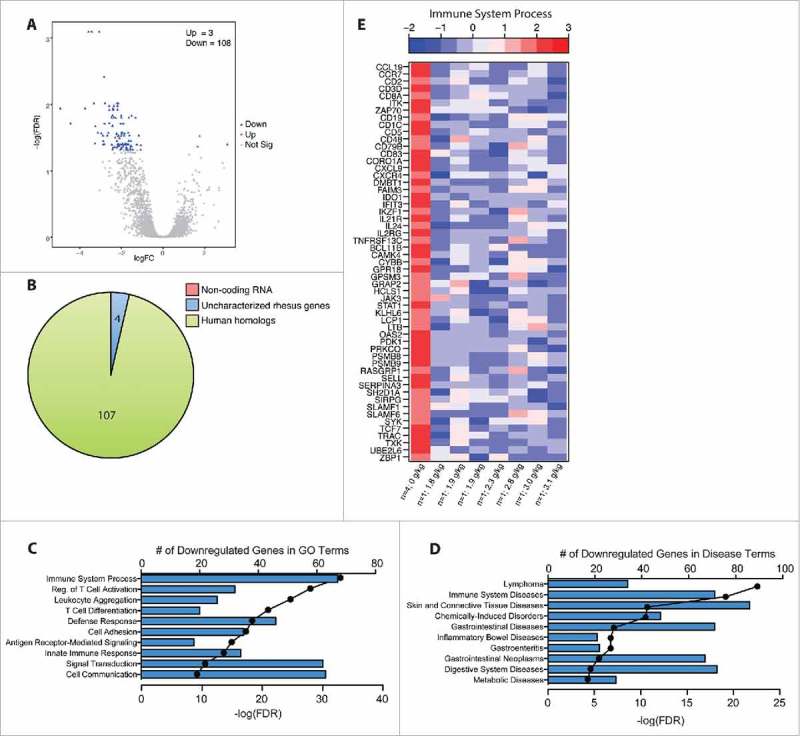
Chronic heavy ethanol consumption modulates expression of genes associated with cancer development and immune function in the colon. DEG were identified using the generalized linear model likelihood ratio test method from the edgeR package as those with a fold change of ≥2 and a Benjamini-Hochberg- corrected false discovery rate (FDR) of <0.05. n = 4/group. (A) Volcano plot summarizing the gene expression changes with red representing the upregulated DEG and blue representing the downregulated DEG. The number of up- and down-regulated genes is noted. (B) Pie chart representing the breakdown of DEG as noncoding RNA, uncharacterized rhesus genes, and human homologs. (C, D) Bar graphs displaying the number of DEG downregulated with ethanol consumption that enriched to GO and Disease terms respectively; line represents the -log(FDR-adjusted P-values) associated with enrichment to each GO/disease term. (E) Heatmap representing gene expression (shown as absolute normalized RPKM values) of DEG that enriched to the GO term “immune system process”; first column shows median RPKM values of the controls (n = 4), and subsequent columns show RPKM values of each individual ethanol-consuming animals ordered by alcohol dose (g of ethanol/kg/day); range of colors is based on scaled and centered RPKM values of the entire set of genes (red represents increased expression while blue represents decreased expression).
Ethanol consumption results in dynamic dose-dependent mucosal gene expression changes.
Since we collected samples from male macaques with varying ethanol intake, we next explored dose-dependent dynamic changes in gene expression that go beyond positive or negative correlations by utilizing Short Time-series Expression Miner (STEM). Animals were divided into 3 groups based on their ethanol consumption patterns: controls (n = 4), non-heavy drinkers (n = 4), and heavy drinkers (n = 4).36 We focused our analysis on highly expressed genes with an average RPKM value ≥50 (reads per kilobase of transcript per million mapped reads). In the duodenum, STEM analysis identified 2 clusters of genes the expression of which increases with non-heavy ethanol consumption then decreases to either control or near control levels with heavy drinking. Genes in cluster 1 play a role in protein transport and localization (Fig. 4A; Supplemental Table 1; Fig. S1A) and functionally enriched to GO terms such as “cotranslational protein targeting to membrane” (FDR-P = 1.15e-17; e.g ribosomal proteins RPL11 and RPS27) and “protein targeting to ER” (FDR-P = 2.66e-17). Genes also enriched to “transport” (FDR-P = 1.30e-10; e.g. solute carrier SLC26A3) and “immune response-regulating signaling pathway” (FDR-P = 8.11e-7; e.g. immunoglobulin genes IGHM and IGJ). Furthermore, 41 of these 47 genes enriched to the Disease term “digestive system diseases” (FDR-P = 4.95e-5) including those encoding for the transporter PIGR and mucin gene MUC6. Genes in cluster 2 (Fig. 4A; Supplemental Table 1) play a role in exocytosis and enriched to GO terms “secretion” (FDR-P = 1.92e-4) and “neutrophil degranulation” (FDR-P = 1.92e-4) such as heat shock protein HSPA8. All 13 genes within cluster 2 also enriched to “colonic diseases” (FDR-P = 3.00e-4).
Figure 4.
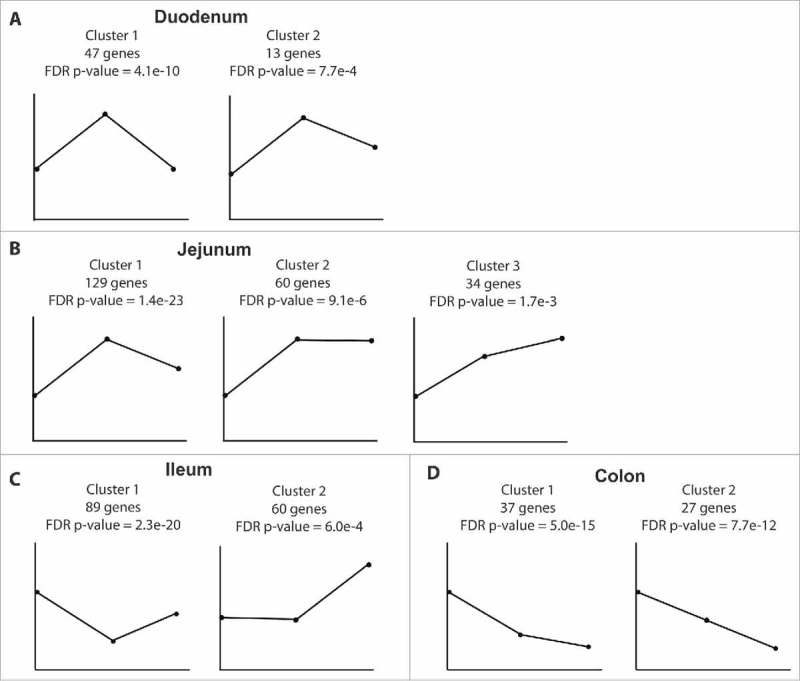
Dynamic gene expression changes mediated by ethanol consumption. Expression patterns of DEG clusters uncovered by STEM analysis in (A) duodenum, (B) jejunum, (C) ileum, (D) and colon with the number of genes and FDR p-value reported. First point represents controls (n = 4); second point represents non-heavy group (n = 4) and last point represents heavy group (n = 4).
Similarly, in the jejunum, STEM analysis revealed a cluster of genes whose expression increases with non-heavy ethanol consumption then returns to near control levels with heavy ethanol consumption (Fig. 4B; Supplemental Table 2). Majority of these 129 genes are important for cellular metabolism, and functionally enriched to the GO term “cellular metabolic process” (n = 80; FDR-P = 5.16e-3) including the Acetyl-CoA Acetyltransferase 1 ACAT1 and the ubiquitin ligase MARCH7. STEM also identified a second group of genes with expression that increases with non-heavy ethanol consumption and remains unchanged with heavy ethanol consumption (Fig. 4B; Supplemental Table 2) that also functionally enriched to GO term “metabolic process” (FDR-P = 8.42e-2; Fig. S1B) including the phosphofructokinase PFKM and the solute carrier SLC7A7. STEM further identified a third cluster of genes in the jejunum, with expression levels that drastically increase with non-heavy ethanol consumption then continue to increase with heavy ethanol consumption (Fig. 4B; Supplemental Table 2). These genes are critical for signal transduction and include natural killer cell receptor CD160 and the choline phosphotransferase CEPT1.
STEM analysis of the ileum transcriptome uncovered a cluster of 89 genes the expression of which decreases with non-heavy ethanol consumption and to a lesser extent with heavy ethanol consumption (FDR-P = 2.3e-20; Fig. 4C; Supplemental Table 3). These genes functionally enriched to GO terms “neutrophil mediated immunity” (FDR-P = 9.25e-6; e.g. chemokine CCL25 and innate immune gene PLAC8), “transport” (FDR-P = 9.25e-6; members of the solute carriers family SCL2, 5, 6 and 7), and “digestion” (FDR-P = 1.16e-5; mucin gene MUC2 and actin-binding protein VIL1). STEM analysis identified a second cluster of 60 genes the expression of which was unaltered with non-heavy ethanol consumption but greatly increased with heavy ethanol consumption (Fig. 4C; Supplemental Table 3). These genes enriched to the GO term “negative regulation of metabolic process” (FDR-P = 1.88e-25; Fig. S1C) including heat shock protein HSP90AB1 and ribosomal protein UBA52. Additionally, 38 other genes enriched to the Disease term “colonic diseases” (FDR-P = 1.00e-5) such as metastasis suppressor CD82, putative tumor suppressor TAGLN2, and actin-sequestering TMSB4X.
In the colon, STEM analysis identified 37 genes, the expression of which greatly decreased with non-heavy ethanol consumption, and even more with heavy ethanol consumption (Fig. 4D; Supplemental Table 4). These genes play a role in macromolecule transport and functionally enriched to GO terms such as “protein targeting” (FDR-P = 4.67e-20) and “macromolecule localization” (FDR-P = 6.20e-11) including cofilin gene CFL1 and carbohydrate binding galectin 3 LGALS3. Furthermore, 32 of these genes enriched to the Disease term “gastrointestinal neoplasms” (FDR-P = 1.88e-3) including gut permeability indicator FABP and signaling molecule RACK1. STEM identified an additional cluster of genes where expression negatively correlated with increasing ethanol dose in a linear fashion (Fig. 4D; Fig. S1D; Supplemental Table 4). Functional enrichment showed that 16 genes are associated with “localization” (FDR-P = 7.78e-12) and 19 of these genes mapped to the Disease term “colonic diseases” (FDR-P = 3.17e-4), notably adhesion molecule CD9, and components of the actin cytoskeleton such as ACTB.
Given the robust changes in gene expression observed along the GI tract, we next determined if these alterations were accompanied by an increased permeability of the GI barrier. We measured circulating endotoxin-core antibodies in plasma samples collected after 12 months of alcohol consumption and discovered a positive correlation between levels of anti-endotoxin IgM levels and ethanol intake (r = 0.8201; p = 0.0016) (Fig. S2G).
Alcohol consumption impacts mucosa-associated bacterial communities in both small and large intestine.
We next investigated ethanol-induced changes in gut mucosa-associated bacterial communities using 16S rRNA gene sequencing37 (Fig. 5A). Since we did not have access to luminal contents, we sequenced the second half of the mucosal biopsies used for RNA-Seq analysis. Although not ideal, this approach would provide insight into the microbial niche that is closely interacting with intestinal epithelial and immune cells. To assess alpha diversity, we calculated the Shannon diversity index, which revealed similarities in evenness among the intestinal regions and groups (Fig. S3A). The beta diversity analysis using weighted UniFrac values showed that biopsies from ethanol-consuming male macaques did not form a distinct cluster from those collected from controls (Fig. 5B and Fig. S3B-E). In line with these observations, only subtle differences were noted at the phyla level (Fig. 5A and Fig. S4A). In the jejunum and ileum, we observed a small decrease in relative abundance of Firmicutes in ethanol-consuming animals compared to controls (36.3% and 36.8% compared to 41.2% and 42.8%). In the colon, we detected a slight increase in Bacteroidetes (25.2% vs. 18.2%) that was accompanied by a decrease in Proteobacteria (33.2% vs. 38.3%) in ethanol-consumers compared to controls (Fig. 5A). A genus level analysis further revealed minimal changes in the colon, where levels of Flexispira slightly decreased while Clostridiales, S24-7, and Prevotella increased with ethanol consumption (Fig. S4B).
Figure 5.
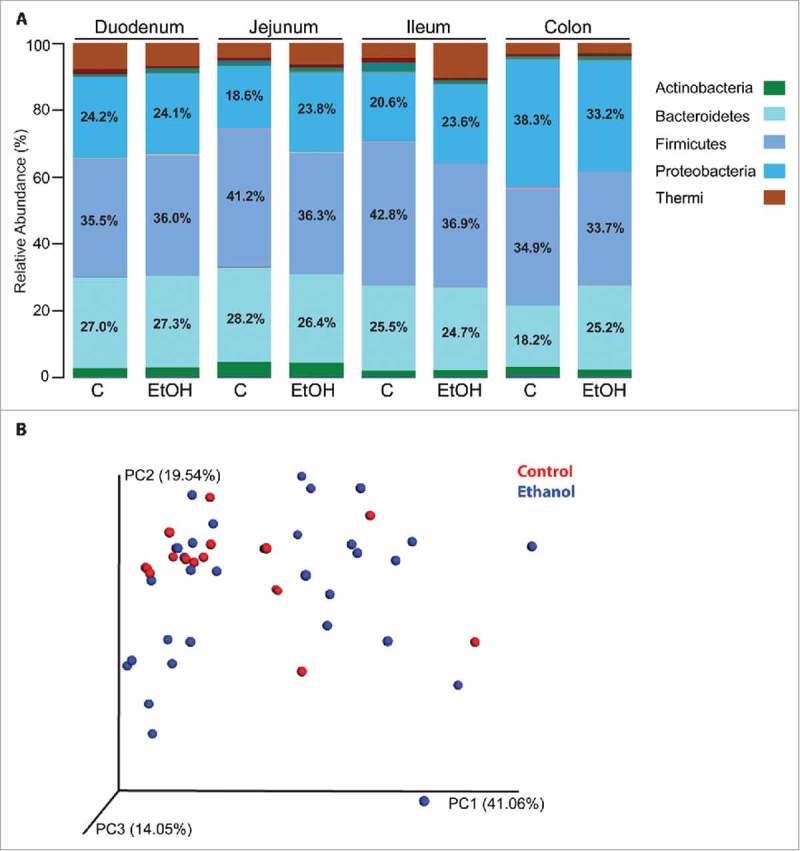
Chronic ethanol intake results in minor changes in mucosal microbial communities. Low quality, chimeric, and Cyanobacteria sequences were removed, reference OTU were selected at 97% similarity, and beta diversity calculations were performed using QIIME. n = 4/group. (A) The average abundance (% of total) of bacterial rRNA gene sequences at the phylum level in the duodenum, jejunum, ileum, and colon of controls and ethanol-consuming male macaques. The % abundance of Bacteroidetes, Firmicutes, and Proteobacteria are noted on the plots. (B) Principal component analysis (PCA) of weighted UniFrac values to examine beta diversity between controls and ethanol-consuming animals.
Since we did not detect significant differences in microbial communities at the group level, we decided to examine Spearman correlations between individual average daily ethanol consumption and operational taxonomic unit (OTU) abundance (Table S5-S8). We also utilized Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) to carry out an in silco analysis of the pathways associated with the bacterial communities using K numbers from the KEGG database and measured correlations with ethanol consumption (Table S9-S12). In the duodenum, these analyses revealed 19 OTU whose abundance positively correlated with ethanol consumption including several members of the Porphyromonadaceae (Table S5 and Fig. 6A). Only 4 OTU, which were members of the Proteobacteria and Bacteroidetes phyla, negatively correlated with ethanol consumption in the duodenum (Table S5 and Fig. 6A). PICRUSt analysis revealed 21 putative bacterial genes associated with the metabolism of carbon compounds, carbohydrates, and lipids that positively correlated with ethanol consumption (Table S9 and Fig. 6B). An additional putative 19 bacterial genes involved in metabolism of purines and carbohydrates negatively correlated with ethanol consumption (Table S9 and Fig. 6B).
Figure 6.
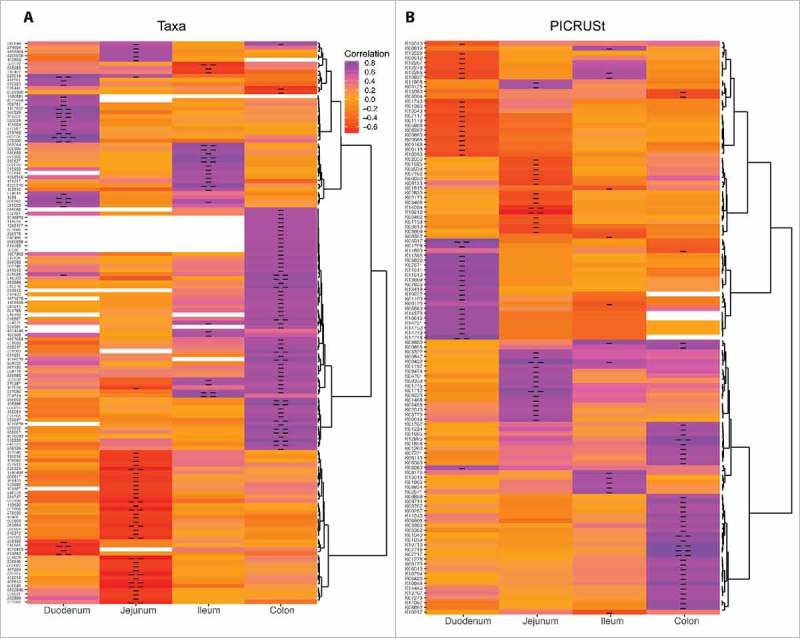
Chronic ethanol consumption alters bacterial communities and subsequently critical metabolic pathways in a dose-dependent manner. Heatmaps of (A) OTU (refer to Tables S5-S8) and (B) KEGG genes (refer to Tables S9-S12) that significantly correlated with average daily BEC either in a positive or negative manner as determined by the Spearman correlation coefficient (n = 12/gut region). Correlations were defined as those with an r-value ≥0.5 or ≤−0.5, and a p-value <0.05. Single and double hyphens in the cells indicate p < 0.05 and p < 0.01, respectively.
In the jejunum, 4 OTU positively correlated with ethanol consumption including the opportunistic pathogen Prevotella veroralis.38 Many of the 34 OTU that negatively correlated with ethanol consumption were members of Clostridiales including OTU in the Lachnospiraceae and Ruminococcaceae families (Table S6 and Fig. 6A). PICRUSt analysis revealed 18 and 16 putative bacterial genes in the jejunum that positively and negatively correlated with ethanol consumption (Table S10 and Fig. 6B), including 4′-phosphopantetheinyl transferase, which negatively correlated with ethanol consumption and is important for the metabolism of cofactors and vitamins.
In the ileum, 19 OTU positively correlated with BEC including several Prevotella species (P. copri, P. stercorea, and P. loescheii) that are known to be associated with inflammation (see Table S7 and Fig. 6A). The 3 OTU that negatively correlated with BEC included likely commensal bacteria such as Ruminococcus torques. PICRUSt analysis revealed 14 putative bacterial genes largely involved in sugar and fatty acid synthesis that positively correlated with ethanol consumption while 6 bacterial genes involved in amino acid, sulfur, and purine metabolism were negatively associated with ethanol consumption (Table S11 and Fig. 6B).
The most robust changes in bacterial communities following chronic ethanol consumption were detected in the colon where the abundance of 55 OTU positively correlated with ethanol consumption (Table S8 and Fig. 6A). These included Peptostreptococcaceae bacterium, Haemophilus parainfluenzae, and Selenomonas dianae, which are enriched in CRC patients39,40 and associated with worse prognosis.41 PICRUSt analysis suggests that these changes in relative OTU abundance could be associated with the upregulation of pathways involving biosynthesis of carbohydrates (glyoxylate metabolism, amino and nucleotide sugar metabolism); xenobiotics metabolism; and nutrient metabolism (Table S12 and Fig. 6B). In contrast, PICRUSt analysis indicated beta-ketoacyl-ACP synthase, critical for vitamin synthesis, was negatively correlated with ethanol intake.
Discussion
Among the different models used in alcohol research, nonhuman primates are critically important due to their genetic homology with humans, their propensity to voluntarily consume alcohol, and the similarity of their alcohol metabolism to that of humans.31 Here, we used a nonhuman primate model of voluntary ethanol self-administration to characterize the site- and dose-dependent impact of chronic ethanol consumption on intestinal mucosa gene expression and bacterial composition in male rhesus macaques.
We did not identify any gene expression differences at the group level associated with ethanol intake in the duodenum, possibly due to the rapid ethanol absorption, the scarcity of resident immune cells in this GI segment5, and/or the wide range of ethanol doses among the ethanol-consuming animals. However, additional analyses that leveraged the variable drinking patterns of the animals revealed that the expression level of 60 genes largely involved in protein localization, notably several ribosomal protein and solute carriers, increases with non-heavy ethanol consumption then returns to levels similar or slightly above control levels.
In the jejunum, a modest number of DEG that play a role in the regulation of gene expression, protein trafficking, and metabolism was identified between controls and ethanol-consuming animals. One of the most upregulated DEG in the jejunum is histone 3 (H3F3A), which is heavily phosphorylated and acetylated following acute ethanol administration in the liver of male rats.42 Future studies will determine whether increased expression of H3F3A in the jejunum of male ethanol-consuming macaques is also accompanied with increased protein expression and post-translational modifications that could in turn regulate gene expression in the jejunum. Similarly, STEM analysis revealed dose-dependent changes in the expression of genes involved in signal transduction, regulation of gene expression, and cellular metabolic processes including: several G-coupled protein receptors (GPR18, 22, 34, and 82), potassium channel family members (KCNIP4 and KCNJ13), transcription factors (EIF2B1, EIF2S2, and EIF4A1), and interestingly microRNA precursors (miR1-2, 133A, 181A, and 215). Collectively, these gene expression changes suggest that ethanol consumption disrupts metabolism, protein trafficking, and regulation of gene expression in the jejunum.
The most robust changes in gene expression at the group level were detected in the ileum, which harbors the majority of GALT.5 Several DEG upregulated with ethanol consumption in the ileum are important for lymphocyte function and trafficking such as CD19, CD27, CD37, CD40, CD44, CCL19, CCR6, and CCR7. These changes in gene expression occurred despite the lack of differences in T cell frequencies28 which suggests that ethanol regulates gene expression at the cell level rather than through changes in cell numbers. Moreover, we detected large increases in the expression of Toll-like receptors (TLR) that are critical in the detection of bacterial lipopeptides (TLR1), single-stranded RNA viruses (TLR7), and other unknown pathogen-associated molecular patterns (TLR10).43 STEM analysis also revealed increased levels of several genes associated with host defense and inflammation such as CCL25, ACE1, DGAT1, and CD13.
Interestingly, we found several DEG implicated in the initiation and/or progression of CRC in the ileum including the transcription factor lymphoid enhancer factor-1 (LEF1), which is highly expressed in human CRC cells.44 In addition, expression of NOS2, which plays a critical role in preventing tissue damage due to reactive oxygen species, was downregulated 6-fold with heavy drinking in the colon. Given that alcohol consumption increases production of reactive oxygen species45, loss of NOS2 expression can potentially exacerbate DNA damage and carcinogenesis. STEM also uncovered a cluster of 38 genes involved in “colonic diseases” that remain unchanged with non-heavy ethanol consumption but greatly increase with heavy ethanol consumption and included the metastasis suppressor gene CD82, and tumor protein translationally controlled-1 (TPT1), which protein levels have been shown to be drastically increased in the early stages of CRC development.46
We also report an increase in the expression of gap junction protein GJA5 in the ileum, which regulates the passage of ions and molecules between cells. Increased expression of GJA5 could lead to an increase in translocation of bacteria, their products, and dietary antigens, which in turn leads to increased inflammation and could explain the robust upregulation of immune genes in the ileum. This change is in line with the increased endotoxin-core IgM antibody titers observed, and is potentially indicative of decreased barrier function. Indeed, a clinical study showed that a one-time consumption of 20g ethanol (non-heavy ethanol consumption) is sufficient to cause an increase in both small and large intestinal permeability in healthy human volunteers.51
Fewer immune genes were dysregulated by ethanol consumption in the colon compared to the ileum, potentially due to reduced GALT in this compartment.5 Moreover, in contrast to the ileum, the expression of genes involved in immune function and regulation of inflammation were reduced in the colon following ethanol consumption including PI3, IFIT3, IDO1, CCL19, CCR7, and DEF6. The difference in direction of change could be due to the fact that immune cells in the colon have evolutionarily adapted to a more regulatory role due to the greater microbial load.47 Reduced expression of immune genes could also signal reduced immune surveillance in the colon with long-term ethanol consumption, which in turn could facilitate the development of malignancies. For instance, expression of IL21R, which plays an important role in anti-tumor responses,48 is reduced in the colon. Other genes that negatively correlated with alcohol dose are responsible for control of the cell cycle including CDC42, RAB1B, and RAB8A.
Similarly, STEM analysis identified 2 distinct clusters of genes the expression of which negatively correlated with ethanol consumption in the colon. These genes enriched to the disease term “gastrointestinal neoplasms” including CFL1, FABP, FTH1, and RACK1. STEM also revealed that expression of miR-663, which is known to be dysregulated in several types of cancer,50 decreased with ethanol consumption. Decreased expression of this microRNA was primarily driven by non-heavy ethanol consumption in the colon, and was only slightly further exacerbated following heavy ethanol consumption.
Interestingly, we did not detect a difference in microbial community diversity in ethanol-consuming animals compared to controls at the group level. This observation is in contrast to a recent study using a rodent model of acute ethanol liquid diet, which reported a decrease in bacterial diversity with alcohol exposure.13 The discrepancy between our data and the rodent studies could be due to differences in acute versus chronic consumption of ethanol, the total amount of ethanol consumed, the diet (our macaques have concurrent access to 4% ethanol as well as water versus the liquid chow diet for the rodent study), the variable dose of ethanol consumed by our animals (since it is a voluntary ethanol consumption model) and last but not least physiological and genetics differences between rodents and nonhuman primates.
Furthermore, previous studies showed decreased abundance of Bacteroidetes and Firmicutes and higher abundance of Proteobacteria and Actinobacteria in the sigmoid colon and feces of AUD patients with ALD compared to those without ALD.17,18 In contrast, data reported here show minor differences at the phyla level that reflected a slight increase in Bacteriodetes and a reduction of Proteobacteria. This difference is likely due to the lack of ALD in our animals and examination of mucosa-associated bacterial communities, which can greatly differ from stool communities.52 AUD has also been shown to result in an increase in the prevalence of families containing pathogenic bacteria including Enterobacteriaceae, Streptococcaceae, and Prevotellaceae as well as a reduction in beneficial families such as Lachnospiraceae in fecal material from patients with cirrhosis.18 Similarly, we report that chronic ethanol consumption was positively associated with Streptococcaceae, Prevotellaceae, as well as Lachnospiraceae in the duodenum, ileum, and colon.
In the duodenum, we uncovered a positive correlation between ethanol consumption and several OTU associated with inflammation such as Porphyromonadaceae.53 We also identified many OTU associated with inflammation that positively correlated with ethanol consumption in the ileum including major Prevotella species (P. copri, P. stercorea, and P. loescheii). Similarly, the frequency of opportunistic pathogens such as Prevotella copri, P. stercorea, P. loescheii, Clostridium perfrigens, C. celatum, C. clostridioforme, and Streptococcus suis were increased with daily ethanol dose in the colon. In contrast, chronic heavy ethanol consumption decreased putatively beneficial bacteria such as Blautia species in jejunum and Ruminococcus species in ileum. Interestingly, an expansion of P. copri in mice has been shown to exacerbate colitis.54 We also observed an increase in the genus Porphyromonas in the colon with heavy ethanol drinking compared to controls. Porphyromonas have been identified as a strong biomarker for CRC in humans.55 Decreases in Porphyromonadaceae have also been associated with inflammation in colorectal tumor-bearing mice.53 In the colon, we also found a positive correlation between ethanol consumption and frequency of Peptostreptococcaceae bacterium and Haemophilus parainfluenzae, which are enriched in CRC patients39 and associated with colorectal carcinoma-in-adenoma.41 Furthermore, Selenomonas dianae, which is enriched in proximal colon tumors,40 was also increased with ethanol consumption. These findings are particularly interesting given the increased incidence of CRC with AUD.56 Additionally, the abundance of Flexispira species, which are usually dominant in healthy macaques,57 was reduced with heavy ethanol consumption suggestive of a loss in commensal bacteria. Furthermore, PICRUSt analyses revealed several putative bacterial genes that positively correlated with ethanol consumption are involved in carbohydrate metabolism. We also uncovered a negative correlation between ethanol consumption and the putative bacterial gene 4′-phosphopantetheinyl transferase, which is involved in vitamin production and metabolism, a potential mechanism of alcohol-mediated malabsorption/malnutrition.
In summary, the studies presented in this manuscript show that ethanol consumption results in robust changes in gene expression, notably within the ileum where a large number of genes involved in host defense and inflammation were upregulated. Chronic ethanol consumption also leads to dysregulated expression of genes involved in GI cancers, especially CRC. Moreover, relative abundance of several bacterial taxa were altered throughout the GI tract including a dose-dependent decrease in the incidence of putatively beneficial bacteria as well as increased frequencies of taxa associated with inflammation and CRC. Similarly, in silico analysis suggests that several bacterial metabolic pathways may also be altered with ethanol consumption. It is important to note that animals used in our study lack liver damage, therefore the changes in gene expression and bacterial communities described precede overt clinical disease and could therefore potentially serve as prognostic biomarkers.
Limitations of our study include the use of full thickness biopsies, which precluded us from being able to trace changes in gene expression to specific cell types. Performing RNA-Seq on isolated cell populations (intestinal epithelial cells, lamina propria lymphocytes, intra-epithelial lymphocytes, and smooth muscle cells) may reveal additional gene expression changes within specific cell population that were diluted by the use of a full thickness biopsy. Moreover, the lack of access to biopsies before the animals were exposed to ethanol impeded our ability to conduct longitudinal studies. Access to fecal samples or luminal contents would have also allowed us to compare mucosa-associated versus luminal communities, and analyze microbial metabolites. Finally, we evaluated a small number of animals and only used male macaques. However, our study also has unique strengths, notably the use of an outbred nonhuman primate model, the analysis of biopsies from the 4 major gut sections, and the simultaneous measurement of changes in gene expression profiles and abundance of microbial communities with the same biopsy.
Future studies need to analyze bacterial communities using shotgun metagenomics and metabolomics. Additionally, the mechanisms underlying changes in gene expression will be explored by defining epigenetic changes such as histone modifications and alterations in microRNA profiles. The functional consequences of alterations in specific bacterial species will be determined by assessing their impact on barrier function, their ability to translocate, and invade and adhere to epithelial cells. Last but not least, these studies need to be extended to study female macaques in order to identify gender differences in the impact of ethanol consumption on GI health.
Materials and methods
Ethics approval
Tissues were acquired from the Monkey Alcohol Tissue Research Resource (MATRR/matrr.com). All animal work was was performed in strict accordance with the recommendations detailed in the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, the Office of Animal Welfare and the United States Department of Agricultures and approved by the Oregon National Primate Research Center (ONPRC) Institutional Animal Care and Use Committee (IACUC).
Rhesus macaque model of voluntary ethanol self-administration
We leveraged the Monkey Alcohol Tissue Research Resource (MATRR; http://www.matrr.com/) to conduct the experiments described in this manuscript. Samples from 12 male rhesus macaques 4–5 years of age were used in these studies (Table 1). Eight of these male rhesus macaques chronically consumed 4% ethanol in water for 12 months and segregated into two cohorts, n = 4 each, based on average daily ethanol intake values: non-heavy drinkers with an average daily blood ethanol concentration (BEC) of 22.3-48.8 mg/dl (or mean daily intake of 1.8-2.3 g/kg) and heavy drinkers with an average daily BEC of 90–126 mg/dl (or mean daily intake of 2.8-3.3 g/kg). Classification of the animals into non-heavy and heavy drinkers was previously described in31,36 and the Monkey Alcohol Tissue Research Resource (www.matrr.com). Four males served as controls and consumed a calorically-matched maltose dextrose solution. Although controls and ethanol-consuming animals consumed comparable numbers of total calories (Fig. S2A), heavy drinkers consumed a much higher number of ethanol-derived calories compared to non-heavy drinkers (Fig. S2B). Mean daily BEC and g/kg consumed (calculated over the entire 12-month open access period) significantly correlated with each other (Fig. S2C). All 12 animals were euthanized after 12 months of open access and biopsies were collected from duodenum, jejunum, ileum, and transverse colon, snap frozen, and stored at −80°C until analysis. Biopsies were cut in half: one half was used for RNA isolation and the other half was used for DNA isolation. Plasma samples were also collected at necropsy following 12 months of ethanol self-administration, however fecal samples and luminal contents were not available. Ethanol-consuming animals did not show overt signs of liver damage as assessed by measuring alanine transaminase (ALT) and aspartate transaminase (AST) enzyme levels and histological assessment of liver sections (Fig. S2D-F).
Table 1.
Summary of rhesus macaques used in this study.
| Animal ID | Mean daily ethanol intake (g/kg/day) | Blood EtOH Content (Avg mg%) | Drinking status |
|---|---|---|---|
| MC1 | 0 | 0 | Control |
| MC2 | 0 | 0 | Control |
| MC3 | 0 | 0 | Control |
| MC4 | 0 | 0 | Control |
| MM1 | 1.8 | 35.86 | Non-Heavy |
| MM2 | 1.9 | 32.68 | Non-Heavy |
| MM3 | 1.9 | 20.74 | Non-Heavy |
| MM4 | 2.3 | 33.89 | Non-Heavy |
| MH1 | 2.8 | 67.42 | Heavy |
| MH2 | 3 | 75.02 | Heavy |
| MH3 | 3.1 | 72.24 | Heavy |
| MH4 | 3.3 | 95.25 | Heavy |
Mean daily ethanol (EtOH) intake, as calculated during the period of 12-month EtOH self-administration is shown.
RNA sequencing (RNA-Seq)
Total RNA was isolated from duodenal, jejunal, ileal, and colonic biopsies using the Qiagen miRNeasy kit (Qiagen, Catalog #217004). Ribosomal RNA (rRNA) was depleted using the Epicentre Ribo-Zero rRNA Removal kit (Illumina, Catalog #MRZH11124). Libraries were constructed using the Bioo Scientific NEXTflex Rapid Directional RNA-seq kit (Bioo Scientific, Catalog #NOVA-5138-10). Briefly, rRNA-depleted RNA was fragmented and converted to double stranded cDNA. Adapters were ligated and ∼300 base pair fragments were amplified by PCR and selected by size exclusion. Each library was labeled with a unique barcode for multiplexing. To ensure proper sizing, quantitation, and quality prior to sequencing, libraries were analyzed on the Agilent 2100 Bioanalyzer. Multiplexed libraries were subjected to single-end 100 base pair sequencing using the Illumina HiSeq2500 platform.
RNA-Seq bioinformatic analysis
Data analysis was performed with the RNA-Seq workflow module of the systemPiperR package available on Bioconductor.58,59 Quality reports were generated with seeFastq. Reads were mapped with the splice-aware aligner suite Bowtie2/Tophat260,61 against the Macaca mulatta genome from Ensembl.62 Default parameters of Tophat2 optimized for mammalian genomes were used for alignment (allowing 2% nucleotide mismatches). Raw expression values in form of gene-level read counts were generated with summarizeOverlaps.63 Only reads overlapping exonic regions of genes were counted, discarding reads mapping to ambiguous regions of exons from overlapping genes.
Analysis of differentially expressed genes (DEG) was performed with the generalized linear model likelihood ratio test method from the edgeR package.64,65 To determine the impact of ethanol consumption on gene expression, we compared the transcriptome of controls versus that of ethanol-consuming animals. DEG were defined as those with a fold change of ≥2 and a Benjamini-Hochberg-controlled false discovery rate (FDR) of <0.05. To confirm the RNA-Seq results, expression levels of 8 DEG that play a role in ALD, CRC, and host defense/inflammation in the: 1) ileum [CD2 (Rh02839718_m1), CCL19 (Rh02621767_m1), CCR6 (Rh02788181_s1), CCR7 (Rh03985963_s1), and BCL6 (Rh02839507_m1)] (Fig. S5A); and 2) in the colon [CD2 (Rh02839718_m1), CCL19 (Rh02621767_m1), STAT1 (Rh02899274_m1), and ZAP70 (Rh02837378_m1)] (Fig. S5B) were selected for confirmation using Taqman probes and qRT-PCR. Changes in expression level of all 8 genes were confirmed. Functional enrichment analysis was performed to identify significant biological pathways including gene ontology (GO) terms and disease biomarkers using MetaCore™ software (GeneGo). Since this software requires the use of human gene ID, all rhesus macaque DEG were mapped to human homologs using annotations from ENSEMBL.
The software program Short Time-series Expression Miner (STEM; v1.3.11) was utilized to further identify clusters of genes that show similar patterns of expression as it relates to discrete doses of ethanol consumption: none, non-heavy, and heavy.66
16S rRNA gene library construction and sequencing
Total DNA was extracted from duodenal, jejunal, ileal, and colonic biopsies using PowerSoil DNA Isolation kit (MO BIO Laboratories, Inc., Catalog #12888-100). PCR and custom primers were used to amplify the V4-V5 region of the 16S rRNA gene as previously described.67 Libraries were sequenced (250 bases) using an Illumina MiSeq.
16S rRNA gene bioinformatics analyses
Low quality, chimeric, and Cyanobacteria sequences were removed using Quantitative Insights Into Microbial Ecology (QIIME).68 This process resulted in 2,820,185 total reads with per sample counts ranging from 19,579 to 128,131, with an average of 58,754 counts/sample. Reference operational taxonomic units (OTU) were selected at 97% similarity using QIIME and Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt). Taxonomic assignments were made using the 2013 Greengenes reference database.69 OTU with fewer than 100 sequences were removed from additional analyses. QIIME was used to calculate alpha diversity including OTU observed and Shannon diversity indices, beta diversity UniFrac values, and perform Adonis analyses. PICRUSt was used to adjust OTU abundances for their rRNA copy number and impute the predicted metagenome of the bacterial communities based on the 16S rRNA gene sequences.70 Spearman correlation analyses were performed in QIIME between 12-month average BEC and either the OTU or the Kyoto Encyclopedia of Genes and Genomes (KEGG) K numbers generated by the PICRUSt analyses. Correlations were defined as those with an r-value >0.5 or <−0.5, and a p-value <0.05. Heatmaps depicting these correlation values were created using custom R scripts and four libraries (ggplot2, ggdendro, reshape2, and grid).71-74 The 16S rRNA gene sequence data have been deposited in NCBI's SRA under the accession number SUB1983942 (http://www.ncbi.nlm.nih.gov/sra).
Determination of anti-endotoxin IgM antibodies
Endotoxin-core antibodies in plasma samples were measured using an enzyme-immunoassay technique (ELISA) after 12 months of alcohol consumption using EndoCab IgM ELISA kit (Hycult Biotech, Catalog# HK504-IgM). Plasma samples were diluted 50x.
Statistical analysis
Statistical significance of caloric intake, IgM, ALT, AST, values between controls, non-heavy drinkers, and heavy drinkers was assessed with a one-way ANOVA followed by Bonferroni's multiple comparison correction tests. Spearman correlation analyses were performed to define associations between anti-endotoxin IgM antibody titer and average 12 month BEC or daily g/kg ethanol consumed. Expression data for each Taqman probe was calculated relative to control RPL32 mRNA expression using ΔCt calculations and statistical significance was assessed using an unpaired T-test. These analyses were carried out by GraphPad Prism version 6 (GraphPad software).
Supplementary Material
Funding Statement
This work was supported by NIAAA grant AA021947, R21 AA024981, U01 AA013510, and R24 AA109431. Barr was supported by the NIEHS NRSA T32 ES018827 and NIAAA F31 AA025278 grants. The University of California Riverside Genomics Core was supported by NIH 1S10RR028934-01.
Availability of data and material
The RNA-Seq data have been deposited in NCBI's Sequence Read Archive (SRA) under the accession number SUB1885356 (http://www.ncbi.nlm.nih.gov/sra). The 16S rRNA gene sequence data have been deposited in NCBI's SRA under the accession number SUB1983942 (http://www.ncbi.nlm.nih.gov/sra).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Andrew Tremain and Michael George from University of California Davis Genomics core for the 16S rRNA gene sequencing. The authors declare that they have no competing interests.
Authors' contributions
KG and IM designed the study. TB, SS, JB, KG, and IM wrote the manuscript. TB and SS performed the RNA-Seq experiment and analyzed the RNA-Seq data. TB, PR, and JB analyzed the 16S rRNA gene sequence data. JZ and WM performed the multivariate analyses. TB and IM interpreted the data. All authors read and approved the final manuscript.
References
- 1.Kaiko GE, Stappenbeck TS. Host-microbe interactions shaping the gastrointestinal environment. Trends Immunol. 2014. doi: 10.1016/j.it.2014.08.002. PMID:25220948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sassone-Corsi M, Raffatellu M. No vacancy: how beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J Immunol. 2015;194:4081–7. doi: 10.4049/jimmunol.1403169. PMID:25888704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. doi: 10.1038/nri2515. PMID:19343057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–53. doi: 10.1038/nri3608. PMID:24566914. [DOI] [PubMed] [Google Scholar]
- 5.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–85. doi: 10.1038/nri3738. PMID:25234148. [DOI] [PubMed] [Google Scholar]
- 6.Molina PE, Gardner JD, Souza-Smith FM, Whitaker AM. Alcohol abuse: critical pathophysiological processes and contribution to disease burden. Physiology (Bethesda). 2014;29:203–15. PMID:24789985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Tong J, Chang B, Wang B, Zhang D, Wang B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep. 2014;9:2352–6. doi: 10.3892/mmr.2014.2126. PMID:24718485. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y, Banan A, Forsyth CB, Fields JZ, Lau CK, Zhang LJ, Keshavarzian A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Exp Res. 2008;32:355–64. doi: 10.1111/j.1530-0277.2007.00584.x. PMID:18162065. [DOI] [PubMed] [Google Scholar]
- 9.Elamin E, Jonkers D, Juuti-Uusitalo K, van Ijzendoorn S, Troost F, Duimel H, Broers J, Verheyen F, Dekker J, Masclee A. Effects of ethanol and acetaldehyde on tight junction integrity: in vitro study in a three dimensional intestinal epithelial cell culture model. PLoS One. 2012;7:e35008. doi: 10.1371/journal.pone.0035008. PMID:22563376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong W, Li Q, Sun Q, Zhang W, Zhang J, Sun X, Yin X, Zhang X, Zhou Z. Preventing Gut Leakiness and Endotoxemia Contributes to the Protective Effect of Zinc on Alcohol-Induced Steatohepatitis in Rats. J Nutr. 2015;145:2690–8. doi: 10.3945/jn.115.216093. PMID:26468492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elamin E, Masclee A, Dekker J, Jonkers D. Ethanol disrupts intestinal epithelial tight junction integrity through intracellular calcium-mediated Rho/ROCK activation. Am J Physiol Gastrointest Liver Physiol. 2014;306:G677−85. doi: 10.1152/ajpgi.00236.2013. PMID:24557761. [DOI] [PubMed] [Google Scholar]
- 12.Chen P, Schnabl B. Host-microbiome interactions in alcoholic liver disease. Gut Liver. 2014;8:237–41. doi: 10.5009/gnl.2014.8.3.237. PMID:24827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bull-Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, Gobejishvili L, Joshi-Barve S, Ayvaz T, Petrosino J, et al.. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS One. 2013;8:e53028. doi: 10.1371/journal.pone.0053028. PMID:23326376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canesso MCC LN, Ferreira CM, Gonçalves JL, Almeida D, Gamba C, Cassali G, Pedroso SH, Moreira C, Martins FS, Nicoli JR, et al.. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: the role of the gut microbiota. BMC Microbiol. 2014;14:240. doi: 10.1186/s12866-014-0240-4. PMID:25223989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bode JC, Bode C, Heidelbach R, Durr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31:30–4. PMID:6698486. [PubMed] [Google Scholar]
- 16.Bode C, Kolepke R, Schafer K, Bode JC. Breath hydrogen excretion in patients with alcoholic liver disease–evidence of small intestinal bacterial overgrowth. Z Gastroenterol. 1993;31:3–7. PMID:8447153. [PubMed] [Google Scholar]
- 17.Mutlu E, Gillevet P, Rangwala H, Sikaroodi M, Naqvi A, Engen P, Kwasny M, Lau C, Keshavarzian A. Colonic microbiome is altered in alcoholism. Am J physiol Gastrointest liver physiol. 2012;302:78. doi: 10.1152/ajpgi.00380.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–72. doi: 10.1002/hep.24423. PMID:21574172. [DOI] [PubMed] [Google Scholar]
- 19.Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33:1836–46. doi: 10.1111/j.1530-0277.2009.01022.x. PMID:19645728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–54. doi: 10.2353/ajpath.2006.050617. PMID:16565490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bode C, Bode JC. Alcohol's role in gastrointestinal tract disorders. Alcohol Health Res World. 1997;21:76–83. PMID:15706765. [PMC free article] [PubMed] [Google Scholar]
- 22.Lieber CS. The influence of alcohol on nutritional status. Nutr Rev. 1988;46:241–54. doi: 10.1111/j.1753-4887.1988.tb05443.x. PMID:3045703. [DOI] [PubMed] [Google Scholar]
- 23.Barr T, Helms C, Grant K, Messaoudi I. Opposing effects of alcohol on the immune system. Prog Neuropsychopharmacol Biol Psychiatry. 2016;65:242–51. doi: 10.1016/j.pnpbp.2015.09.001. PMID:26375241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su LJ, Arab L. Alcohol consumption and risk of colon cancer: evidence from the national health and nutrition examination survey I epidemiologic follow-up study. Nutr Cancer. 2004;50:111–9. doi: 10.1207/s15327914nc5002_1. PMID:15623458. [DOI] [PubMed] [Google Scholar]
- 25.Seitz HK, Czygan P, Simanowski U, Waldherr R, Veith S, Raedsch R, Kommerell B. Stimulation of chemically induced rectal carcinogenesis by chronic ethanol ingestion. Alcohol Alcohol. 1985;20:427–33. PMID:3910061. [PubMed] [Google Scholar]
- 26.Simanowski UA, Seitz HK, Baier B, Kommerell B, Schmidt-Gayk H, Wright NA. Chronic ethanol consumption selectively stimulates rectal cell proliferation in the rat. Gut. 1986;27:278–82. doi: 10.1136/gut.27.3.278. PMID:3699547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937−47. doi: 10.1152/ajpgi.00235.2001. PMID:12016118. [DOI] [PubMed] [Google Scholar]
- 28.Asquith M, Pasala S, Engelmann F, Haberthur K, Meyer C, Park B, Grant KA, Messaoudi I. Chronic Ethanol Consumption Modulates Growth Factor Release, Mucosal Cytokine Production, and MicroRNA Expression in Nonhuman Primates. Alcohol Clin Exp Res. 2013. doi: 10.1111/acer.12325. PMID:24329418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(SAMHSA) SAaMHSA National Survey on Drug Use and Health (NSDUH). 2015. [Google Scholar]
- 30.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8:627–37. doi: 10.1038/nprot.2013.032. PMID:23449255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker EJ, Farro J, Gonzales S, Helms C, Grant KA. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin Exp Res. 2014;38:2835–43. doi: 10.1111/acer.12547. PMID:25421519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grant KA, Leng X, Green HL, Szeliga KT, Rogers LS, Gonzales SW. Drinking typography established by scheduled induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin Exp Res. 2008;32:1824–38. doi: 10.1111/j.1530-0277.2008.00765.x. PMID:18702645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivester P, Roberts LJ 2nd, Young T, Stafforini D, Vivian J, Lees C, Young J, Daunais J, Friedman D, Rippe RA, et al.. Ethanol self-administration and alterations in the livers of the cynomolgus monkey, Macaca fascicularis. Alcohol Clin Exp Res. 2007;31:144–55. doi: 10.1111/j.1530-0277.2006.00276.x. [DOI] [PubMed] [Google Scholar]
- 34.Barr T, Girke T, Sureshchandra S, Nguyen C, Grant K, Messaoudi I. Alcohol Consumption Modulates Host Defense in Rhesus Macaques by Altering Gene Expression in Circulating Leukocytes. J Immunol. 2016;196:182–95. doi: 10.4049/jimmunol.1501527. PMID:26621857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sureshchandra S, Rais M, Stull C, Grant K, Messaoudi I. Transcriptome Profiling Reveals Disruption of Innate Immunity in Chronic Heavy Ethanol Consuming Female Rhesus Macaques. PLoS One. 2016;11:e0159295. doi: 10.1371/journal.pone.0159295. PMID:27427759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez VA, Grant KA. Studies using macaque monkeys to address excessive alcohol drinking and stress interactions. Neuropharmacology. 2017;122:127–35. doi: 10.1016/j.neuropharm.2017.03.027. PMID:28347838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto T, Sugano M. [16S rRNA gene sequence analysis for bacterial identification in the clinical laboratory]. Rinsho Byori. 2013;61:1107–15. PMID:24605544. [PubMed] [Google Scholar]
- 38.Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi: 10.1111/1574-6976.12075. PMID:24861948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. PMID:22761885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Z, Guo B, Gao R, Zhu Q, Qin H. Microbiota disbiosis is associated with colorectal cancer. Front Microbiol. 2015;6:20. doi: 10.3389/fmicb.2015.00020. PMID:25699023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasai C, Sugimoto K, Moritani I, Tanaka J, Oya Y, Inoue H, Tameda M, Shiraki K, Ito M, Takei Y, et al.. Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: Terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol Rep. 2016;35:325–33. doi: 10.3892/or.2015.4398. PMID:26549775. [DOI] [PubMed] [Google Scholar]
- 42.James TT, Aroor AR, Lim RW, Shukla SD. Histone H3 phosphorylation (Ser10, Ser28) and phosphoacetylation (K9S10) are differentially associated with gene expression in liver of rats treated in vivo with acute ethanol. J Pharmacol Exp Ther. 2012;340:237–47. doi: 10.1124/jpet.111.186775. PMID:22025646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002. 168:554–61. doi: 10.4049/jimmunol.168.2.554. PMID:11777946. [DOI] [PubMed] [Google Scholar]
- 44.Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF, Waterman ML. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–7. doi: 10.1038/ng0501-53. PMID:11326276. [DOI] [PubMed] [Google Scholar]
- 45.Wu D, Cederbaum AI. Alcohol, oxidative stress, and free radical damage. Alcohol Res Health. 2003;27:277–84. PMID:15540798. [PMC free article] [PubMed] [Google Scholar]
- 46.Bommer UA, Vine KL, Puri P, Engel M, Belfiore L, Fildes K, Batterham M, Lochhead A, Aghmesheh M. 2017. Translationally controlled tumour protein TCTP is induced early in human colorectal tumours and contributes to the resistance of HCT116 colon cancer cells to 5-FU and oxaliplatin. Cell Commun Signal. 15:9. doi: 10.1186/s12964-017-0164-3. PMID:28143584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mann ER, Bernardo D, English NR, Landy J, Al-Hassi HO, Peake ST, Man R, Elliott TR, Spranger H, Lee GH, et al.. Compartment-specific immunity in the human gut: properties and functions of dendritic cells in the colon versus the ileum. Gut. 2016;65:256–70. doi: 10.1136/gutjnl-2014-307916. PMID:25666191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. 2014;13:379–95. doi: 10.1038/nrd4296. PMID:24751819. [DOI] [PubMed] [Google Scholar]
- 49.Jin S, Mu Y, Wang X, Liu Z, Wan L, Xiong Y, Zhang Y, Zhou L, Li L. Overexpressed RACK1 is positively correlated with malignant degree of human colorectal carcinoma. Mol Biol Rep. 2014;41:3393–9. doi: 10.1007/s11033-014-3201-y. PMID:24504450. [DOI] [PubMed] [Google Scholar]
- 50.Zang W, Wang Y, Wang T, Du Y, Chen X, Li M, Zhao G. miR-663 attenuates tumor growth and invasiveness by targeting eEF1A2 in pancreatic cancer. Mol Cancer. 2015;14:37. doi: 10.1186/s12943-015-0315-3. PMID:25744894. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Elamin E, Masclee A, Troost F, Pieters HJ, Keszthelyi D, Aleksa K, Dekker J, Jonkers D. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: a combined in vivo and in vitro approach. PLoS One. 2014;9:e107421. doi: 10.1371/journal.pone.0107421. PMID:25226407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, Monteith P, Noble NA, Sikaroodi M, Gillevet PM. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol. 2012;303:G675−85. doi: 10.1152/ajpgi.00152.2012. PMID:22821944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692−13. doi: 10.1128/mBio.00692-13. PMID:24194538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, et al.. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. PMID:24192039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zackular JP, Rogers MA, Ruffin MTt, Schloss PD. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res (Phila). 2014;7:1112–21. doi: 10.1158/1940-6207.CAPR-14-0129. PMID:25104642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fedirko V, Tramacere I, Bagnardi V, Rota M, Scotti L, Islami F, Negri E, Straif K, Romieu I, La Vecchia C, et al.. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22:1958–72. doi: 10.1093/annonc/mdq653. PMID:21307158. [DOI] [PubMed] [Google Scholar]
- 57.Hensley-McBain T, Zevin AS, Manuzak J, Smith E, Gile J, Miller C, Agricola B, Katze M, Reeves RK, Kraft CS, et al.. Effects of Fecal Microbial Transplantation on Microbiome and Immunity in Simian Immunodeficiency Virus-Infected Macaques. J Virol. 2016;90:4981–9. doi: 10.1128/JVI.00099-16. PMID:26937040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, et al.. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–21. doi: 10.1038/nmeth.3252. PMID:25633503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Girke T. systemPipeR: NGS workflow and report generation environment, vR package version 1.2.4. 2015. https://github.com/tgirke/systemPipeR. [DOI] [PMC free article] [PubMed]
- 60.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. PMID:22388286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. PMID:23618408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al.. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–9. doi: 10.1093/nar/gku1010. PMID:25352552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. PMID:23950696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. PMID:19910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8:1765–86. doi: 10.1038/nprot.2013.099. PMID:23975260. [DOI] [PubMed] [Google Scholar]
- 66.Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191. doi: 10.1186/1471-2105-7-191. PMID:16597342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4516–22. doi: 10.1073/pnas.1000080107. PMID:20534432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. PMID:20383131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. doi: 10.1128/AEM.03006-05. PMID:16820507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al.. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. PMID:23975157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wickham H. Ggplot2: elegant graphics for data analysis. Springer, New York;2009. [Google Scholar]
- 72.Andrie de Vries BDR. ggdendro: Create Dendrograms and Tree Diagrams Using ‘ggplot2’. 2016. [Google Scholar]
- 73.Wickham H. Reshaping Data with the reshape Package. Journal of Statistical Software. 2007;21:1–20. doi: 10.18637/jss.v021.i12. [DOI] [Google Scholar]
- 74.Team RC. R: A language and environment for statistical computing. 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.