

Synapse formation relies on the coordination of dynamic pre- and postsynaptic structures during brain development. Liu et al. reveal that presynaptic terminal maturation of mossy fiber axons is retrogradely regulated by postsynaptic scaffold protein Lnx1 via stabilizing EphB receptor kinases.
Abstract
Neuronal connections are initiated by axon targeting to form synapses. However, how the maturation of axon terminals is modulated through interacting with postsynaptic elements remains elusive. In this study, we find that ligand of Numb protein X 1 (Lnx1), a postsynaptic PDZ protein expressed in hippocampal CA3 pyramidal neurons, is essential for mossy fiber (MF) axon targeting during the postnatal period. Lnx1 deletion causes defective synaptic arrangement that leads to aberrant presynaptic terminals. We further identify EphB receptors as novel Lnx1-binding proteins to form a multiprotein complex that is stabilized on the CA3 neuron membrane through preventing proteasome activity. EphB1 and EphB2 are independently required to transduce distinct signals controlling MF pruning and targeting for precise DG-CA3 synapse formation. Furthermore, constitutively active EphB2 kinase rescues structure of the wired MF terminals in Lnx1 mutant mice. Our data thus define a retrograde trans-synaptic regulation required for integration of post- and presynaptic structure that participates in building hippocampal neural circuits during the adolescence period.
Graphical Abstract
Introduction
Proper wiring of the developing brain relies on the dynamic formation of synapses (Cohen-Cory, 2002; Turrigiano and Nelson, 2004; Kolodkin and Tessier-Lavigne, 2011). The development of these specific synapses requires the accurate coordination of multiple developmental events, including axon targeting and pruning, dendritic growth, spinogenesis, and synapse formation (Ackley and Jin, 2004; Jüttner and Rathjen, 2005; Waites et al., 2005; Low and Cheng, 2006). Accumulating evidence has indicated that synapse formation and stabilization are dynamically modulated through the pre- and postsynaptic compartments in an anterograde, retrograde, or bidirectional way (Yuste and Bonhoeffer, 2004; Alvarez and Sabatini, 2007; McAllister, 2007; Bhatt et al., 2009; Shen and Scheiffele, 2010; Siddiqui and Craig, 2011; Sala and Segal, 2014). Extensive studies over the past decades both in vivo and in vitro have demonstrated that the presynaptic compartment plays a dominant role in initializing these processes, especially in the activity- or experience-dependent neuronal connection (Zucker, 1999; Hensch, 2005; Holtmaat and Svoboda, 2009; Kerschensteiner et al., 2009; Kozorovitskiy et al., 2012; Li et al., 2017).
The basic connectivity during development is likely through an integration of cell-intrinsic genetic programs and extrinsic influences of guidance cues, neurotrophic factors, and neuronal and synaptic adhesion systems (McAllister, 2007; Chen et al., 2008; Giagtzoglou et al., 2009; Shen and Cowan, 2010; Shen and Scheiffele, 2010; Siddiqui and Craig, 2011; Südhof, 2012; Bennett and Lagopoulos, 2014; Sala and Segal, 2014). Numerous molecules or molecular families, including receptors and adhesion proteins, kinases and small GTPases, and cytoskeletal regulators, interact with various scaffold proteins containing PDZ domains during the formation of functional synapses (Garner et al., 2000, 2002; Sheng and Sala, 2001; Kim and Sheng, 2004; Feng and Zhang, 2009; Sheng and Kim, 2011; Sala and Segal, 2014). Although a large number of PDZ proteins have been identified as participating in postsynaptic morphogenesis, including dendritic development and spinogenesis (El-Husseini et al., 2000; Penzes et al., 2001; Hoogenraad et al., 2005; Nakamura et al., 2011; Geiger et al., 2014; Heisler et al., 2014), these studies are largely restricted in postsynaptic compartments. Previous studies have shown that presynaptic structure and function are also regulated in a retrograde way (Contractor et al., 2002; Jüngling et al., 2006; Regalado et al., 2006; Futai et al., 2007; Hu et al., 2012; Orr et al., 2017), whereas the precise mechanism of how postsynaptic PDZ scaffolds participate in the maturation of presynaptic structure remains relatively less investigated.
The dentate mossy fiber (MF)-CA3 synapse in the hippocampus is an excellent model to study the dynamic formation of synaptic structures and neural circuits. The MF axons are composed of two distinct bundles, suprapyramidal bundle (SPB) and infrapyramidal bundle (IPB), which target CA3 neurons. The IPB undergoes a pruning process during the postnatal developing period (Bagri et al., 2003; Xu and Henkemeyer, 2009; Riccomagno et al., 2012) that make it easy to observe the coordinative change with postsynaptic remodeling on a large scale. The MF-CA3 synapses are represented as a large multiheaded morphology composed of highly plastic MF presynaptic terminals with massive separate vesicle release sites and thorny postsynaptic structures that are different from typical glutamatergic asymmetric synapses (Amaral and Dent, 1981; Chicurel and Harris, 1992; Nicoll and Schmitz, 2005; Rollenhagen et al., 2007). This specific axon structure is advantageous for the examination of terminal targeting and maturation with postsynaptic dynamics during postnatal development.
In this study, we identify a PDZ scaffold protein, ligand of Numb protein X (Lnx1), which is expressed specifically in the hippocampal CA3 neurons. Through gene targeting in mice, we demonstrate that Lnx1 is required for targeting and remodeling of presynaptic MF axon terminals that wire with postsynaptic spines to form efficient synapses. We further demonstrate that CA3-expressed EphB receptors serve as novel Lnx1-interacting proteins responsible for MF terminal refinement and maturation during MF-CA3 synapse formation. Constitutively active EphB2 receptor kinase in Lnx1−/− mice is sufficient to rescue the presynaptic structure of MF. Thus, our data indicate that presynaptic axon targeting and terminal maturation can be controlled by postsynaptic elements through a trans-synaptic regulation in hippocampus.
Results
Lnx1 is expressed in CA3 neurons and required for MF axon pruning
Lnx1 mRNA has been identified in hippocampal CA3 neurons (Rice et al., 2001), as confirmed by the Allen Brain Atlas (Fig. S1 A). To examine whether PDZ scaffold protein Lnx1 is important for the development of hippocampus in vivo, we generated a protein-null mutant in which the Lnx1 gene was knocked out through homologous recombination in embryonic stem cells. This results in deletion of coding exons needed for both P70 and P80 isoforms of Lnx1 and insertion of LacZ sequences to express β-galactosidase (β-gal), allowing for detection of Lnx1 expression (Fig. 1 A). The Lnx1 knockout was validated by Southern blot analysis using external 5′ and 3′ probes, PCR genotyping using gene-specific oligonucleotides, and protein detection using anti-Lnx1 antibodies (Fig. S1, B–D). Embryos containing the Lnx1 mutation were stained for β-gal expression using X-gal staining and showed strong expression in many tissues such as eyes, ears, and limbs (Fig. S1 E). The Lnx1 null homozygotes (Lnx1−/−) were viable at expected Mendelian ratios, appeared to be healthy, were fertile, and lived until adulthood. In view of the relatively low level of Lnx1 in brain compared with periphery organs (Lenihan et al., 2014), we precisely checked the Lnx1 expression from postnatal week 1 (PW1) until adulthood by immunoprecipitation of β-gal protein from hippocampal lysates of Lnx1 mutant mice (Fig. S2 A). Immunofluorescence with anti–β-gal antibodies in Lnx1 mutant mice indicated specific expression of Lnx1 in the hippocampal CA3 pyramidal neurons (Fig. 1 B). To examine the subcellular localization of Lnx1, we purified postsynaptic density (PSD) fractions from hippocampal tissues and immunoprecipitated with anti-Lnx1 to detect Lnx1 protein, and found that Lnx1 is expressed only in the postsynaptic fraction (Fig. S2 B). To validate the postsynaptic expression of Lnx1, we overexpressed Flag-Lnx1 into cultured hippocampal neurons and observed postsynaptic localization of Lnx1 in dendritic spines (Fig. S2 C). Our analysis thus identifies Lnx1 as a hippocampal CA3-specific postsynaptic protein in the adolescent brain.
Figure 1.

Defective MF axon pruning in Lnx1 null mice. (A) The schematic indicates the strategy for generating Lnx1 knockout by substituting Lnx1 with LacZ gene. 5′ untranslated sequences and exon coding sequences are shown as gray and dark boxes, respectively, and introns and 5′ and 3′ nontranscribed regions are shown as lines. EcoRI (E) and NcoI (N) restriction sites are indicated. The Lnx1-LacZ targeting vector, including a PGK-Neo cassette, was designed to replace Lnx1 exons including the p80 and p70 promoters after homologous recombination. The 5′ and 3′ external probes used to confirm the results by Southern blot are shown with the expected sizes indicated (see Fig. S1 B). (B) Immunofluorescence of β-gal in Lnx1 mutant mice indicated specific expression of Lnx1 in hippocampal CA3 pyramidal cell layer. Bars: 200 µm (left); 50 µm (right). (C) NeuroTrace dye–labeled CA3 pyramidal cells in 3-wk-old Lnx1−/− mice are loosely packed compared with WT littermates. Calbindin staining showed that the IPB axon–penetrated CA3 pyramidal neurons layer are much longer in Lnx1−/− mice compared with WT littermates. White brackets delineate IPB length, and distance between arrowheads delineates the IPB length in CA3 pyramidal neurons. Bars: 200 µm (left); 100 µm (right). (D) Quantification of the length ratio of IPB to CA3 area (from the hilus to the curvature) in Lnx1−/− mice and WT littermates during postnatal development. n = 5–7 mice. (E) Comparison of IPB length within CA3 pyramidal neurons in Lnx1−/− mice and WT littermates during postnatal development. n = 5–7 mice. (F) Diagram of coculture assay. (G) TdT+ primary hippocampal axons (arrowheads) cocultured with Lnx1−/− neurons showed fewer pruned axons than with WT neurons. Bar, 100 µm. (H) Comparison for neurite lengths in neuronal coculture with WT or Lnx1−/− neurons. (I) Comparison of primary neurite numbers in neuronal coculture with WT or Lnx1−/− hippocampal neurons. n = 72–403 neurons per group in H and I. (J) Comparison of longest neurite length (arrowheads) of calbindin-negative neurons and calbindin-positive neurons dissected from TdT+ hippocampi cocultured with WT or Lnx1−/− hippocampal neurons. n = 16–19 neurons per group. Bar, 100 µm. Means ± SEM; two-way ANOVA with Tukey’s post hoc test (J) or Student’s t test (D, E, and I); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The specific localization of Lnx1 prompted us to determine whether hippocampal morphogenesis is altered in juvenile Lnx1−/− mice. The absence of Lnx1 did not cause an obvious change in the overall structure of the hippocampus, as viewed by staining with the immunofluorescent dye NeuroTrace, to label neurons (Fig. 1 C). According to calbindin immunofluorescence in WT mice, most of the MF axons from the dentate gyrus were seen in the SPB above the CA3 pyramidal cell bodies, whereas a smaller group of IPB axons grew initially underneath the CA3 pyramidal cells but then were pruned back to their mature length and joined the SPB axons as the hippocampus developed in WT mice as reported previously (Bagri et al., 2003; Xu and Henkemeyer, 2009; Riccomagno et al., 2012). Strikingly, we observed that IPB axons in the Lnx1−/− mice grew into split CA3 pyramidal layers that were not shortened in a timely manner during development and instead maintained an inappropriate long and stable length comparable with the SPB until adulthood (Fig. 1, C–E), suggesting defective axon pruning and targeting during a critical development period.
Because Lnx1 was expressed restrictively in the CA3 pyramidal neurons but not in dentate granule (DG) cell neurons, the defective pruning and targeting of MF originating from granule cells in Lnx1−/− mice indicates a non–cell-autonomous mechanism. Two possibilities that account for the abnormalities could be considered: one could be that Lnx1 affects postsynaptic structure to mediate axon development upon axon–cell/dendrite contact; the other is that Lnx1 alters extracellular environmental factors that could affect axonal growth, targeting, and retraction in a secreted gradient manner. To test these possibilities, an in vitro coculture assay was developed. After a 12–14-d preculture of hippocampal neurons from Lnx1−/− or WT mice to allow for neuron distribution and contact on the dish, tdTomato–positive (TdT+) primary hippocampal neurons from TdT+ knock-in mice were plated on the culture by either direct addition or loading with coverslips onto the dish to prevent direct axon–cell contact (Fig. 1 F). The primary neurites with TdT fluorescence were analyzed for axon length at the indicated time points. We found that in the cultures of WT or Lnx1−/− hippocampal neurons, the plated TdT+ neurites underwent an initial fast growth by the first 3 d, resembling the early growth of MF axons, and then reached a relatively stable period for elongation and branching, which was followed by a later trimming back of most primary neurites (Fig. 1, G–I). However, neurons cocultured with Lnx1−/− neurons maintained obviously longer neurites than WT neurons from day 4 of coculture (Fig. 1, G–I), while neurons plated on coverslips showed no difference between the two groups (Fig. S3). We further compared the pruning of calbindin-negative and calbindin-positive neurons in the cocultured system (Fig. S4 A) and found that calbindin-positive neurons had even shorter neurites than the calbindin-negative neurons when cocultured with WT neurons, while both remained unchanged when cocultured with Lnx1−/− neurons (Fig. 1 J), indicating that the majority of DG cells become pruned in the coculture. These results suggest that the non–cell-autonomous regulation by Lnx1 on MF axon development is likely in a manner of axon–cell/dendrite contact in developing hippocampus.
Retrograde coordination of pre- and postsynaptic arrangement by Lnx1
We then investigated the development of MF axons projected from DG neurons to clarify whether the presynaptic structure was altered in Lnx1−/− mutants. To see with more detail about the morphological changes of axonal terminals in Lnx1−/− mice, brain slices from 3-wk-old WT mice or Lnx1−/− mice were immunostained with anti-ZnT3, a protein marker for the MF axon terminals. In WT brains, robust axon terminals formed along the SPB, while little was found in IPB adjacent to the CA3 pyramidal cell layer. In contrast, the MF axons in Lnx1−/− mice showed a dramatic increase in IPB axon terminals that had contact neurons within the CA3 pyramidal cell layer (Fig. 2 A). We next asked whether the increased MF axon terminals as we observed in Lnx1−/− mice are anatomically matched with the spine morphogenesis that led to functional synapses. By using a Thy1-GFP-M transgenic reporter (Feng et al., 2000), we observed increased spine density but reduced mushroom-shaped spines of CA3 neurons in PW3 Lnx1−/− mice compared with control littermates (Fig. S4 B). We then examined the MF axon terminals that contact CA3 neurons and analyzed the percentage of these terminals on spines (terminal-spine) and dendritic shaft (terminal-shaft; Fig. 2, B and C). We observed a decreased ratio of terminal-spine/terminal-shaft in Lnx1−/− mice compared with control littermates (Fig. 2 D). We then classified the spines in CA3 neurons into two categories: spines overlapped with ZnT3-labeled terminals (ZnT3+ synapse) and spines separated from the ZnT3+ terminals, and we observed a decreased ratio of ZnT3+ synapses/total synapses in Lnx1−/− mice (Fig. 2, B, C, and E).
Figure 2.

Defective MF terminal targeting in Lnx1-null mice. (A) ZnT3 and NeuN staining showed robust expanded IPB axon terminal field and more terminal-contacted CA3 pyramidal cells in Lnx1−/− mice compared with WT littermates. Bars: 200 µm (top); 30 µm (bottom). PCL, pyramidal cell layer. n = 7 mice per group. (B) Representative images showing spines of CA3 neuron (green) contacting terminals of MF axon (red) in PW3 Lnx1−/− mice and control littermates. The higher magnification deconvolved 3D-rendered views (right) are as shown from regions of white box (left arrowhead). The overlapped spines and ZnT3+ terminals are defined as ZnT3+ synapses. Arrows indicate terminals on shaft. Bars: 100 µm (left); 2.5 µm (right). (C) Schematic showing terminals on spine (Terminal-Spine) or shaft (Terminal-Shaft). (D and E) Quantification of ratio for terminals on spine or shaft and ZnT3+ synapses determined from 3D-rendered views. n = 14–16 neurons from three to four mice for per group. (F and G) Representative and percentage of four types of pre- and postsynaptic dynamics in coculture system. Red labels, presynaptic axons; green labels, postsynaptic dendritic branches or spines. Arrowheads indicate contacted boutons or spines. Dotted line marks the area of bouton. *, AI ≥ 0.2; ns, AI < 0.2. n = 13–17 neurons. (H) The areas of newborn boutons are smaller when cocultured with Lnx1−/− neurons (n = 17 boutons) than with WT neurons (n = 23 boutons). (I) Quantification of the dynamic areas of refined boutons in coculture system. n = 33–47 boutons per group. Means ± SEM; two-way ANOVA with Tukey’s post hoc test (I) or Student’s t test (A, D, E, G, and H); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further characterize the role of Lnx1 for axon targeting and maturation, we performed live imaging in a dual color–labeled coculture system to observe the temporal dynamics of the axon terminals and matched spines. We quantitated the morphological changes and calculated an area index (AI) to classify these changes into four types of alteration: presynaptic, postsynaptic, both, and none (Fig. 2 F and Videos 1, 2, 3, 4, and 5). Compared with WT neurons, we observed an increased rate in none type but a decreased rate in presynaptic type when cocultured with Lnx1−/− neurons, while the rates of postsynaptic and both types remained unchanged (Fig. 2, F and G). We further classified the presynaptic type into two subtypes: newborn bouton and refined bouton. We found that newborn boutons showed greater expanding terminals in contacted with WT spines than with Lnx1−/− spines (Fig. 2 H). In contrast, the refined boutons underwent rapid terminal shrinkage and split to match with the contacting spines when cocultured with WT neurons, and this effect was attenuated when cocultured with Lnx1−/− neurons (Fig. 2 I). These results indicate that axon targeting and maturation are coordinated with the spine structure by Lnx1 in a retrograde manner.
Lnx1 is essential for MF terminal maturation and release probability
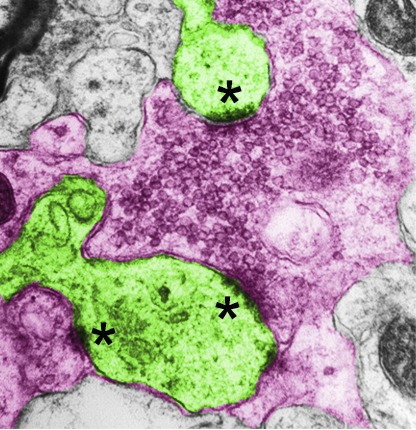
To determine the targeting specificity of MF axon terminals to spines of CA3 pyramidal neurons, we further validated the synapse formation by transmission EM to quantify the axon terminals and their matched spines. The unique morphological characteristics of the MF synapses consist of giant presynaptic boutons and multi-invaginating spines that are distinct from typical synapses (Fig. 3 A), the massive asymmetric synapses frequently observed in the adjacent area of projected MF axons and cell layer of CA3 pyramidal neurons, particularly the SPB region (∼90% of total synapses) and IPB region (∼20% of total synapses). We saw numerous large and complex presynaptic boutons containing massive vesicles and multiple vesicle release sites embracing the contacted spines in both strains of mice (Fig. 3 A). To evaluate the presynaptic terminal maturation that is associated with vesicle amount, we measured the numbers of vesicle release sites (indicated by PSDs) and docked vesicles in the presynaptic active zone, respectively. We found that both numbers were significantly reduced in Lnx1−/− mice compared with WT littermates (Fig. 3, A and B).
Figure 3.

Defective presynaptic maturation and function in Lnx1-null mice. (A) Schematic diagram shows synapse with giant bouton (left). Red, presynaptic terminal; green, postsynaptic spine. The docked vesicles are indicated as vesicle-contacted presynaptic membrane. Transmission electron micrographs of SPB regions from PW3 mice show MF terminals (red) and postsynaptic spines (green) in synapses with giant bouton (right). Asterisks mark PSD. The higher-magnification views show docked vesicles. Bars, 0.5 µm (left); 0.1 µm (right). (B) The PSD number per spine and docked vesicles per vesicle release site in MF synapses from SPB (301–312 synapses) or IPB (38–43 synapses) region of WT mice were more than in Lnx1−/− mice. n = 4 mice per group. (C) Schematic diagram shows typical synapse with regular bouton (left). Transmission electron micrographs from PW3 mice show axon terminals (red) and postsynaptic spines (green) in typical synapses (right). Asterisks mark PSD. Red dotted line indicates the presynaptic boutons without observed postsynaptic spine (nonsynaptic). Bars, 0.5 µm. (D) The number of vesicles per terminal (r2 = 0.62; P < 0.001) or areas of vesicles per terminal (r2 = 0.66; P < 0.001) in typical synapses were significantly correlated with PSD length in PW3 WT mice but not in Lnx1−/− mice. A dot presents a synapse. (E) The vesicle density in terminal that formed typical synapse in WT mice was more than that in Lnx1−/− mice. (F) A cumulative frequency plot of average vesicle distance from WT (n = 133 synapses) and Lnx1−/− (n = 165 synapses) mice with histogram distribution fit for the inset. n = 4 mice per group in D–F. (G) Schematic diagram in the upper panel shows EPSC recordings of the MF-CA3 pathway. Stimulating electrode was placed in the SPB layer, and recording pipette was placed in the CA3 area (between dotted line). Stim, stimulating electrode; Rec, recording pipette. Representative average traces (left) and summary graph (right) show PPRs at interstimulus intervals of 25, 50, 100, 200, and 400 ms in PW3 mice. Bar, 50 ms. n = 24–27 neurons from four mice per group. Means ± SEM; two-way ANOVA with Tukey’s post hoc test (E) or Student’s t test (B, F, and G); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In addition to synapses with giant boutons, we observed a number of typical synapses with regular boutons in the region (Fig. 3 C). We examined the maturation of these presynaptic compartments with the modification of postsynaptic structures in Lnx1−/− mice by measuring the number and distributed areas of vesicles in axon terminals as well as their connecting postsynaptic profile area, indicated by PSD length, with EM. We plotted the two presynaptic factors, respectively, versus PSD length and observed a strong positive correlation between the vesicle number/distribution area and PSD length in WT mice, while the correlation was diminished in Lnx1−/− mice (Fig. 3 D). We also examined the vesicle density in presynaptic terminals with (synaptic) or without (nonsynaptic) postsynaptic compartments and the distribution of these vesicles by calculating the average distance of vesicles to the presynaptic membrane, and observed a decreased vesicle density in synaptic terminals and an increased distance of the vesicles to membrane in Lnx1−/− mice (Fig. 3, C, E, and F). These results indicate that loss of Lnx1 causes abnormal development of presynaptic axon terminals projected to CA3 region.
We then assayed paired-pulse ratios (PPRs) of MF-CA3 synapses by stimulating SPB or IPB fibers that were measured as the amplitude ratio of the second to the first evoked excitatory postsynaptic current (EPSC) in CA3 neurons to evaluate function of presynaptic release. Compared with control mice, we observed an increased PPR upon either SPB or IPB stimulation in PW3 Lnx1−/− mice (Figs. 3 G and S4 C), indicating an impaired glutamate release probability. These results suggest that Lnx1 integrates postsynaptic structure dynamics and presynaptic terminal maturation for precise connection of functional synapses.
EphB receptors are stabilized on membrane through interaction with Lnx1
We then screened transmembrane molecules that might be involved upon MF-CA3 neuron contact and analyzed expression of numerous proteins that have been predicted to interact with Lnx1 (Wolting et al., 2011; Guo et al., 2012). These proteins involve families of EphBs, connexins, claudins, cadherins, and striatin, which were reported to mediate cell–cell interactions and spinogenesis (Söhl et al., 2005; Benoist et al., 2006; Giagtzoglou et al., 2009). We detected members of these proteins for each family that are highly expressed in the hippocampus and found a significant reduction of EphB receptor proteins, a family of tyrosine receptor kinases, in both total cell lysates and membrane components extracted from Lnx1−/− hippocampus (Figs. 4 A and S5 A). Both EphB1 and EphB2 have been demonstrated to express in CA3 neurons, but not in MF, and serve as binding receptors to their membrane-expressed ligand ephrin-B3, which is expressed specifically in MF axons (Xu and Henkemeyer, 2009). In contrast with EphBs, the amount of other transmembrane proteins was not affected by loss of Lnx1 (Figs. 4 A and S5 A).
Figure 4.

Lnx1 interacts with postsynaptic EphB receptors. (A) The expression of membrane proteins in hippocampus from PW3 Lnx1−/− and WT mice was detected by Western blot and quantified. n = 3 mice per group. (B) Coimmunoprecipitation of Lnx1 and EphB1/2 in synaptosomes of hippocampus. Lnx1 was pulled down by EphB2 antibody from PSD fraction of either WT or EphB2K661R/K661R mice but neither EphB2−/− nor EphB2ΔVEV/ΔVEV mice. ns, nonspecific band; IB, immunoblot. (C) Schematic representation of p70-Lnx1 and its mutant constructs. (D) Immunoprecipitation (IP) of Lnx1 mutants and EphB1 or EphB2 coexpressed in NG108 cells. Lnx1 interacted with EphB1 through the N terminus and with EphB2 through the second PDZ domain. (E) Colocalization of Lnx1 and EphB2 (arrowheads in the right panels) in CA3 pyramidal cell layer (arrow in the left panel) were determined by immunostaining in PW3 WT mice. The higher-magnification views in white boxes indicate superresolution imaging of Lnx1 and EphB2. White dotted lines indicate neuronal boundary. Bars: 250 µm (left); 5 µm (bottom right); 0.5 µm (top right). Means ± SEM. Student’s t test (A); ***, P < 0.001.
To determine whether Lnx1 directly interacts with EphB receptors, we pulled down Lnx1 with EphB1 or EphB2 antibody from protein lysates of WT hippocampal tissues compared with EphB1 or EphB2 mutant tissues. We found that Lnx1 could be pulled down by EphB1 or EphB2 antibodies from lysates of WT hippocampal tissues or by EphB2 antibody from EphB2K661R/K661R tissues, a kinase-dead EphB2 mutant (Genander et al., 2009), but not from tissues of EphB2−/−; EphB2LacZ/LacZ, a truncated mutant in which the intracellular domain was replaced with β-gal (Henkemeyer et al., 1996), or EphB2ΔVEV/ΔVEV, a mutant with PDZ domain-binding motif disrupted (Fig. S5 B; Genander et al., 2009). These results indicate that the C-terminal PDZ domain–binding motif is necessary for binding with Lnx1. We further purified the synaptosomes of hippocampal tissues and found that Lnx1 could be pulled down by EphB2 antibodies from PSD but not non-PSD fractions in WT and EphB2K661R/K661R tissues, indicating that Lnx1 interacts with EphB2 in postsynaptic compartments (Fig. 4 B). To clarify the binding sites of Lnx1, we generated Flag-tagged individual PDZ domain mutations of the P70-Lnx1 isoform, which was found to be expressed specifically in the brain (Dho et al., 1998; Xie et al., 2001), and cotransfected them with plasmids encoding HA-tagged EphB1/2 for immunoprecipitation. We found that different domains of Lnx1 were required for binding with the different subtypes of EphB receptors. Lnx1 interacted with EphB1 through the N terminus and with EphB2 through the second PDZ domain (Fig. 4, C and D). To further test colocalization of Lnx1 and EphB2 in CA3 pyramidal neurons, immunostaining was used in hippocampal sections and superresolution imaging with stimulated emission depletion (STED) techniques were performed to visualize the localization of these proteins. The STED imaging resolved discrete puncta containing both Lnx1 and EphB2 around the membrane of CA3 neurons (Fig. 4 E).
To figure out whether the decreased level of EphB proteins in Lnx1−/− mice was the result of mRNA reduction in the hippocampus, quantitative RT-PCR was performed, and no change was observed in Lnx1−/− mice (Fig. S5 C). To further determine whether Lnx1 influences the degradation of EphB receptors, we treated cultured Lnx1−/− hippocampal neurons with proteasome inhibitor or lysosomal inhibitors and observed that the decreased EphB1 and EphB2 in Lnx1−/− mutants were restored to the normal level after treatment of proteasome inhibitor MG132, while lysosomal inhibitor leupeptin or NH4Cl had no effect (Fig. 5 A), suggesting an involvement of the proteasome pathway in EphB degradation.
Figure 5.

Lnx1 sustains stability of EphB receptors. (A) Analysis of degradation of EphB1 and EphB2 after addition of inhibitors of proteasome or lysosome in primary hippocampal neurons from WT and Lnx1−/− mice. *, EphB1; #, EphB2. Quantitative results of three biological replicates are shown. (B) Analysis of endogenous EphB2 protein level in MEF cells infected with lentivirus containing mock, Lnx1, or Lnx1-△PDZ2 in the presence or absence of proteasome inhibitor MG132. Quantitative results of three biological replicates are shown. (C) Analysis of EphB protein levels in primary hippocampal neurons from Lnx1−/− mice infected with lentivirus containing mock, Lnx1, Lnx1-△NT, Lnx1-△PDZ1, or Lnx1-△PDZ2. Quantitative results of four biological replicates are shown. Means ± SEM; two-way ANOVA with Tukey’s post hoc test (A and C) or one-way ANOVA with Tukey’s post hoc test (B); *, P < 0.05; **, P < 0.01; ***, P < 0.001; ###, P < 0.001. IB, immunoblot.
To determine whether the binding with Lnx1 is essential for the stable expression of EphB receptors, we infected MEF cells that express endogenous EphB2 protein with lentivirus expressing Lnx1 or Lnx1 mutant lacking the second PDZ domain, which was necessary for Lnx1 binding with EphB2. We observed an obvious increased EphB2 protein level infected with Lnx1 compared with control virus, while deleting the second PDZ domain led to a decreased level of EphB2 protein, and this could be reversed by addition of MG132 (Fig. 5 B). Furthermore, the reduced EphB protein levels in Lnx1−/− primary neurons were also restored to normal levels by infecting with Lnx1 virus, while the protein levels of EphB1 or EphB2 were not rescued by the Lnx1 virus lacking the N terminus or the second PDZ domain, respectively (Fig. 5 C). Thus, these data suggest that Lnx1 stabilizes EphB receptors through specific binding sites to prevent their degradation in proteasome.
In contrast with the P70-Lnx1 isoform, the other P80-Lnx1 isoform expressed in periphery tissues has identical PDZ domains but an additional RING domain for E3 ligase activity (Dho et al., 1998; Xie et al., 2001). To investigate whether P80-Lnx1 has a similar role in the stability of the EphB receptor, we overexpressed P70-Lnx1 and P80-Lnx1 into MEF cells, respectively, and observed an increased level of EphB2 protein with P70-Lnx1 but a decreased level with P80-Lnx1. These results indicate that the two Lnx1 isoforms play opposite roles in the expression level of EphB2, owing to their difference in the RING domain (Fig. S5 D).
Activating EphB2 kinase promotes MF terminal maturation in Lnx1−/− mice
EphB receptors have been reported to be required for spine morphogenesis and synapse formation in CA3 pyramidal neurons in vivo (Henkemeyer et al., 2003). We thus analyzed EphB1−/− and EphB2−/− protein–null mutant mice for MF phenotype compared with Lnx1-null. We found that longer MF axons penetrated CA3 pyramidal cell layers in both mutants (Fig. 6, A–D), which resembled the phenotype observed in Lnx1−/− mice. To further validate the requirement of EphB2 forward signaling that was involved in spinogenesis (Henkemeyer et al., 2003), we examined the IPB morphology of EphB1LacZ/LacZ and EphB2LacZ/LacZ knock-in mice, in which the EphB intracellular segment is substituted with β-gal and the transmembrane and extracellular domains are left intact on the cell surface to activate ephrin-B3–mediated reverse signaling in MF axons (Henkemeyer et al., 1996; Chenaux and Henkemeyer, 2011). Interestingly, we found an opposite phenotype between EphB1LacZ/LacZ and EphB2LacZ/LacZ mice. Unlike the EphB1−/− mutant, EphB1LacZ/LacZ mutant mice showed no obvious change in IPB length compared with controls (Fig. 6, A and B), whereas EphB2LacZ/LacZ knock-in mice showed more serious defective CA3 cell patterning and axon shortening, including both IPB and SPB axons. We further checked the phenotype in EphB2K661R/K661R mice and observed a similar MF defect comparable with EphB2−/− or EphB2LacZ/LacZ mice (Fig. 6, C and D), suggesting that tyrosine kinase activity is required for MF axon pruning. This indicates that EphB1 and EphB2 transduce distinct signals in which the extracellular domain of EphB1 and the EphB2 intracellular kinase-dependent signaling are independently required for MF pruning.
Figure 6.

EphB receptors differ in signals required for MF pruning. (A) Neurotrace dye–labeled pattern of CA3 pyramidal cells in PW3 WT, EphB1−/−, and EphB1LacZ/LacZ mice. Calbindin staining showed that the IPB axon–penetrated CA3 pyramidal neurons layer are much longer in EphB1−/− mice compared with WT or EphB1LacZ/LacZ mice. White brackets delineate IPB length, and distance between white arrowheads delineates the IPB length in CA3 pyramidal neurons. (B) The ratio of IPB length to the length from hilus to curvature of CA3 area and IPB length within CA3 pyramidal neurons in WT, EphB1−/−, and EphB1LacZ/LacZ mice were quantified. n = 5–6 mice per group. (C and D) Calbindin staining for PW3 WT, EphB2−/−, EphB2LacZ/LacZ, and EphB2K661R/K661R mice. The length ratio of IPB and IPB length in CA3 pyramidal neurons are much longer in EphB2−/−, EphB2LacZ/LacZ, and EphB2K661R/K661R mice compared with WT mice. n = 4–5 mice per group. (E and F) Calbindin staining for PW3 Lnx1−/− and Lnx1−/−; EphB1LacZ/LacZ mice. No difference was observed in IPB axons between Lnx1−/− and Lnx1−/−; EphB1 LacZ/LacZ mice. n = 5 mice per group. (G and H) Calbindin staining for PW3 EphB2 F620D/F620D, Lnx1−/−, and Lnx1−/−; EphB2 F620D/F620D mice. The aberrant IPB axons in Lnx1−/− mice were rescued in Lnx1−/−; EphB2 F620D/F620D mice. n = 5–6 mice per group. Bars, 100 µm. Means ± SEM; one-way ANOVA with Tukey’s post hoc test (B, D, F, and H); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then crossed EphB1LacZ/LacZ, in which the intracellular segment is substituted with β-gal to prevent binding with intracellular partners for EphB1 protein degradation (Fig. S5 E), or EphB2F620D/F620D, a constitutively active form of EphB tyrosine kinase (Holmberg et al., 2006), with Lnx1−/−, to see whether the extracellular domain of EphB1 or constitutive catalytic activation of EphB2 is able to reverse the morphological defects caused by Lnx1 ablation. We saw that in Lnx1−/−; EphB1LacZ/LacZ mice, the morphological abnormalities remained unchanged compared with Lnx1−/− mice (Fig. 6, E and F). However, an obvious rescue in axon terminal targeting was observed in Lnx1−/−; EphB2F620D/F620D mice (Fig. 6, G and H; and Fig. 7, A and B). To test whether presynaptic signal transduction is involved, we first measured the expression level of ephrin-B3, the EphB binding ligand specifically expressed in MF axons (Xu and Henkemeyer, 2009), in Lnx1 mutants, and we did not see an obvious change (Fig. S5 F). We then crossed Efnb3−/− mice with Lnx1−/−; EphB2F620D/F620D mice and found that MF terminal targeting was disrupted in the Lnx1−/−; Efnb3−/−; EphB2F620D/F620D mice (Fig. 7, A and B), indicating that the presynaptic ephrin-B3 is also required for MF axon targeting. To figure out whether the postsynaptic or cell-autonomous changes of CA3 neurons secondarily contributed to the observed MF phenotypes, we carefully counted the CA3 cell number and cell density in each genetic condition. We did not observe an obvious change in cell number but found a decreased cell density in Lnx1−/− mice, which was exhibited as a loose pattern in CA3 cell layers (Fig. 7, A and C). As the loose cell layer was also observed in EphB mutants as shown previously (Bouché et al., 2013) and in our study (Fig. 6, A and C), the abnormal cell patterning observed in Lnx1−/− mice was likely attributable to the low level of EphBs comparable with that of EphB mutants.
Figure 7.

EphB kinase activation promotes MF terminal targeting. (A and B) ZnT3 and NeuN staining show the IPB axon terminal–wired CA3 pyramidal cells in different mice. The robust expanded IPB axon terminals in Lnx1−/− mice were rescued in Lnx1−/−; EphB2 F620D/F620D mice but were disrupted again in Lnx1−/−; Efnb3−/−; EphB2 F620D/F620D mice. Bars, 250 µm (top); 25 µm (bottom). n = 5–6 mice per group. (C) Quantification of number of NeuN-positive cells in CA3 region in PW3 different mice. The total number of NeuN+ cells of CA3 area per slice shows no difference among these mice, while the cell density of CA3 area in Lnx1−/− mice and Lnx1−/−; Efnb3−/−; EphB2 F620D/F620D mice showed a reduction compared with other groups. n = 18–19 slices from three mice per group. (D) Immunoprecipitation (IP) of EphB2 protein from hippocampal lysates of PW3 Lnx1−/− and WT mice. The EphB2 tyrosine phosphorylation level was slightly decreased in Lnx1−/− mice compared with WT mice (P = 0.095). n = 3 mice per group. (E) The expression of EphB proteins in hippocampus from PW3 mice was detected by Western blot and quantified. The decreased EphB2 protein level in Lnx1−/− mice was partly restored in Lnx1−/−; EphB2 F620D/F620D mice, while EphB1 remained unchanged. n = 3 mice per group. Means ± SEM; one-way ANOVA with Tukey’s post hoc test (B, C, and E) or Student’s t test (D); *, P < 0.05; **, P < 0.01, ***, P < 0.001. IB, immunoblot.
To clarify why constitutively activating EphB2 can reverse morphological defects caused by Lnx1 ablation, we studied the possible changes in tyrosine kinase activity of EphB2 in Lnx1−/− mice. By immunoprecipitation with EphB2 antibody from hippocampal lysates of WT or Lnx1−/− mice, a comparable total amount of EphB2 was extracted and loaded for detection of the tyrosine phosphorylation of EphB2. We observed a slight reduction in phosphorylated EphB2 level in Lnx1−/− mice compared with WT control mice, which suggests that Lnx1 plays a mild role in promoting or sustaining activation of EphB2 (Fig. 7 D). To further determine whether active EphB2 may be more resistant to the degradation, we detected EphB2 expression in Lnx1−/−; EphB2F620D/F620D mice and observed a partial restoration of EphB2 protein level compared with that in Lnx1−/− mice (Fig. 7 E).
Finally, we examined the terminal morphology visualized with EM in Lnx1−/−; EphB2F620D/F620D mice. In support of the diminished axon terminals in Lnx1−/−; EphB2F620D/F620D mice observed by ZnT3 staining, the number of release sites and docked vesicles in synapses with giant boutons, the vesicle properties, and their distance to the membrane in typical synapses were also restored to normal levels comparable with WT mice (Fig. 8, A–F). We further assayed the PPRs of MF-CA3 synapses and found that EphB2F620D/F620D mice per se showed no difference compared with the WT mice, while the increased PPR in Lnx1−/− mice was restored to a normal level in Lnx1−/−; EphB2F620D/F620D mice (Fig. 8 G).
Figure 8.

EphB kinase activation promotes MF terminal maturation. (A) Transmission electron micrographs from PW3 mice show MF terminals (red) and postsynaptic spines (green) in synapses with giant bouton. Asterisks mark PSD. Bars, 0.5 µm. (B) The decreased PSD number per spine and docked vesicles of MF synapses from SPB (281–312 synapses) or IPB (37–46 synapses) regions in Lnx1−/− mice were restored to normal level in Lnx1−/−; EphB2 F620D/F620D mice. n = 3–4 mice per group. (C) Transmission electron micrographs from PW3 mice show axon terminals (red) and postsynaptic spines (green) in typical synapses with regular bouton. Asterisks mark PSD. Bars, 0.5 µm. (D) The number of vesicles per terminal in typical synapses was significantly correlated with the PSD length, determined by EM analysis in PW3 WT (r2 = 0.62; P < 0.001), EphB2 F620D/F620D (r2 = 0.42; P < 0.001), and Lnx1−/−; EphB2 F620D/F620D (r2 = 0.40; P < 0.001) mice but not in Lnx1−/− mice (left). The area of vesicles per terminal in typical synapses was significantly correlated with PSD length in PW3 WT (r2 = 0.66; P < 0.001), EphB2 F620D/F620D (r2 = 0.54; P < 0.001), and Lnx1−/−; EphB2 F620D/F620D (r2 = 0.59; P < 0.001) mice but not in Lnx1−/− mice (right). A dot presents a synapse. (E) EM analysis showed that the decreased vesicle density in terminals that formed synapses in Lnx1−/− mice was restored to normal level in Lnx1−/−; EphB2 F620D/F620D mice. (F) The increased average distance of vesicles to the presynaptic membrane in Lnx1−/− mice was restored to normal level in Lnx1−/−; EphB2 F620D/F620D mice. n = 72–165 synapses per group. n = 3–4 mice per group in D–F. (G) Representative average traces (left) and summary graph (right) showed that increased PPRs upon SPB stimulation in PW3 Lnx1−/− mice were restored to normal level in Lnx1−/−; EphB2 F620D/F620D mice. Bar, 50 ms. n = 22–27 neurons from three to four mice per group. (H) Proposed model for postsynaptic Lnx1–EphB complex–mediated retrograde regulation of MF axon terminal maturation. Lnx1 binds and stabilizes postsynaptic EphB receptors in hippocampal CA3 pyramidal neurons to sculpt postsynaptic structure, which helps to guide MF axon targeting and promote terminal maturation through a trans-synaptic regulation. Means ± SEM; one-way ANOVA with Tukey’s post hoc test (B and E–G); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Taken together, our data suggest a model in which Lnx1 serves as a specific protein stabilizer for postsynaptic EphB receptor kinases to form a protein complex on the membrane of hippocampal CA3 pyramidal neurons to sculpt postsynaptic structure, which helps to guide MF axon targeting and promote terminal maturation through trans-synaptic regulation in a retrograde, non–cell-autonomous way (Fig. 8 H).
Discussion
In this study, we reveal a postsynaptically driven mechanism for the formation of functional synapses via PDZ scaffold protein Lnx1, which controls the axon targeting and maturation of MF terminals in the developing hippocampus. In contrast with the previous research on Lnx1, which functions as an intrinsic determinant of cell fate by its interaction with the protein Numb during development (Cayouette and Raff, 2002) or as an E3 ubiquitin ligase to cause proteasome-dependent degradation for Notch signaling (Nie et al., 2002), the hippocampal-specific Lnx1 serves as a membrane stabilizer to sustain receptor proteins at postsynaptic compartments in brain to refine the hippocampal presynaptic structure in a non–cell-autonomous manner. Two variants of Lnx1 have been found in vivo, P80-Lnx1 and P70-Lnx1, to share an identical PDZ domain (Dho et al., 1998). In view of the specific expression of P70-Lnx1 isoform in the brain as shown previously (Dho et al., 1998; Xie et al., 2001) and its function on stabilizing EphB receptors, the abnormalities in the Lnx1-null mice observed in this study are attributable to the ablation of P70-Lnx1 protein that does not contain a ring-finger domain for E3 ligase activity.
Through a genetic targeting approach, we showed that ablation of postsynaptic expressed protein Lnx1 led to untrimmed presynaptic MF axon terminals during hippocampal development, which resulted in abnormalities of axon targeting and terminal maturation during dynamic coordination of post- and presynaptic compartments. Through live imaging of cocultured neurons, we studied the relevance of pre- and postsynaptic dynamics. Although a high probability of the postsynaptic altered type was observed, which has been revealed by numerous studies focused on how presynaptic inputs and signaling regulate postsynaptic structure and function (Scheiffele, 2003; Zuo et al., 2005; Alvarez and Sabatini, 2007; Südhof, 2008; Kwon and Sabatini, 2011; Li et al., 2017), we also saw a proportion of presynaptic dynamics. We found that deletion of Lnx1 decreased the rate of presynaptic altered type, including the newborn boutons and refined boutons, but did not affect the rate of postsynaptic altered type. Furthermore, we observed more abnormal MF giant boutons and thorny excrescences, with fewer release sites and docked vesicles as well as fewer mature axon terminals in typical synapses at the CA3 area formed in juvenile Lnx1−/− mice. These abnormities in synapse formation might not be limited to the MF boutons as CA3 neurons also receive axon projections from other brain regions (Witter, 2007) where EphBs are expressed (Liebl et al., 2003; Migani et al., 2007). These results suggest that Lnx1 is essential for maturation and stabilization of presynaptic terminals for precise synaptic connection. This study thus uncovers a retrograde modulation of presynaptic structure during synapse formation.
As novel interacting partners, EphB1 and EphB2 receptors were identified to bind with different PDZ domains of Lnx1, which serves as a protein stabilizer to prevent degradation in proteasome, through their PDZ-binding motif. Interestingly, we found that EphB1 and EphB2 play distinct roles in MF axon pruning and targeting, and the extracellular segment of EphB1 and intracellular domains of EphB2 are independently required. This raises a presumption that EphB receptors may be integrated differently in a heterogeneous molecular complex upon MF-CA3 neuron contact. We further revealed that EphB2 kinase dead mutant EphB2K661R/K661R showed defective MF axon pruning, while the EphB2F620D/F620D mice with constitutive kinase activity showed normal axon morphology. As the EphB2 protein was not expressed in MF axons (Xu and Henkemeyer, 2009), the defective MF axon targeting observed in EphB2K661R/K661R mice suggests a non–cell-autonomous regulation. Furthermore, the abnormal presynaptic targeting and terminal maturation observed in Lnx1-null mice were restored in the Lnx1−/−; EphB2F620D/F620D compound mutant. This indicates that not only is the EphB2 kinase activity required for normal MF axon pruning during development, but it is also sufficient to rescue the defective axon terminal morphogenesis in the absence of Lnx1, which may be attributed to the resistance of the active EphB2 to the degradation. As the kinase activation of membrane EphB receptors within CA3 pyramidal neurons promotes remodeling of postsynaptic structure (Henkemeyer et al., 2003), this may help to form a precise anchor to receive connection and control refinement of the projected axon terminals in a non–cell-autonomous manner.
Mechanistically, this study is distinct from previous studies on regulation of presynaptic function through trans-synaptic molecular signaling upon high neuronal activity (Chavis and Westbrook, 2001; Contractor et al., 2002; Jüngling et al., 2006; Regalado et al., 2006; Futai et al., 2007; Orr et al., 2017). In the specific DG-CA3 synapse, the CA3-expressed EphBs themselves can initiate pruning of MF axons in a retrograde manner through ephrin-B3 reverse signaling (Xu and Henkemeyer, 2009), which is further confirmed in this study. During postnatal development, EphB receptor signaling can be induced upon binding with secreted glycoprotein Reelin via their extracellular domain to form a massive protein complex, and this works together with ApoER2 and VLDL receptor cascade, two members of the LDL receptor family, to regulate neuron cytoskeleton in the CA3 cell layer (Bouché et al., 2013). The stabilization of the postsynaptic multiprotein complex would be critical for CA3-MF trans-synaptic regulation during the long-term developmental process of synaptogenesis.
EphB and ephrin-B receptors have been studied in sensory integration and cognitive function through mediating trans-synaptic bidirectional signals (Pasquale, 2008; Sheffler-Collins and Dalva, 2012; Sloniowski and Ethell, 2012). The integration of pre- and postsynaptic remodeling occurs either in an internucleus manner, as shown in our previous research (Zhu et al., 2016a,b), or in an inner-nucleus regulation, as presented in this study. Dysfunction of these early circuits may lead to neurodevelopmental disorders, as supported by accumulating research about the critical role of EphB receptor in brain development and function (Sheffler-Collins and Dalva, 2012; Klein and Kania, 2014; Kania and Klein, 2016). Therefore, our analysis clarifies the mechanisms underlying functional DG-CA3 circuit assembly, leading to a greater understanding of the molecular basis for brain wiring and cognitive functions.
Materials and methods
Mice and sample preparation
EphB1−/− (Williams et al., 2003), EphB1LacZ (Chenaux and Henkemeyer, 2011), EphB2−/− (Henkemeyer et al., 1996), EphB2LacZ (Henkemeyer et al., 1996), EphB2K661R (Genander et al., 2009), EphB2ΔVEV (Genander et al., 2009), EphB2F620D (Holmberg et al., 2006), Efnb3−/− (Xu et al., 2011), TdT (Ai9; Madisen et al., 2010), and Thy1-GFP-M (Feng et al., 2000) knockout and knock-in mice and genotyping methods have been described previously. TdT (Ai9) mice were crossed with a ubiquitous Cre transgene mice to allow TdT expression in brain. The Cre-activated TdT+ mice have been crossed for multiple generations and were used for primary neuronal culture. Mice were anesthetized (chloral hydrate, 350 mg/kg) and perfused with 0.1 M PBS followed by 4% PFA in phosphate buffer. The brains were then removed, postfixed, and sectioned at 30 µm using a vibratome. All experiments involving mice were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Animals under an Institutional Animal Care and Use Committee–approved protocol at an Association for Assessment and Accreditation of Laboratory Animal Care–approved facility at the Shanghai Jiao Tong University School of Medicine. Parents and pups (10–11 pups per litter) were raised in animal facilities with a constant temperature (22°C) and on a 12-h light-dark cycle. Access to food and water was unlimited. The day of birth was defined as postnatal day 0 (P0). All efforts were made to minimize the number of animals used and their suffering.
Generation of Lnx1 mutant mice
To construct the Lnx1 targeting vector, a 1.8-kb fragment of cloned mouse genomic DNA upstream of exon 3 (ATG P80-Lnx1) was used for the 5′ arm, and a 4.4-kb fragment downstream of exon4 (ATG P70-Lnx1) was used for the 3′ arm. The two arms were cloned into pPNT vector to provide the neo and TK cassettes used for positive and negative selections. To generate a LacZ marker for the normal expression of Lnx1, the targeting vector was modified to include a tau-β-gal reporter gene. Targeting vectors were electroporated into the ES cell line RI; colonies were isolated following selection in G418 and ganciclovir and expanded; and genomic DNA was screened by Southern blotting. The frequency of homologous recombination was 2 of 623 cell lines screened. Germline transmission was obtained by generating aggregation chimeras with targeted ES cells. The animals used in this study have been backcrossed to CD1 mice for multiple generations. Animals were genotyped by PCR with forward primer 5′-CCAGTAGACAGGCCCCAAGTGATTATTT-3′ and reverse primer 5′-TCGATCCACAGGGCAGAAGTCC-3′ for WT (565 bp), and forward primer 5′-CCAGTAGACAGGCCCCAAGTGATTATTT-3′ and reverse primer 5′-ACTCTTTCAGGCCGGGGTCCAT-3′ for mutant (421 bp).
Immunofluorescence
For immunofluorescence, vibratome sections were blocked with permeable buffer (0.3% Triton X-100 in PBS) containing 10% donkey serum for half an hour at room temperature and incubated with primary antibodies in permeable buffer containing 2% donkey serum overnight at 4°C. The slices were then washed three times with PBS-T (0.1% Tween-20 in PBS) for 10 min each and incubated with Alexa Fluor secondary antibodies (1:200; Molecular Probes) and NeuroTrace 633 (1:500; Molecular Probes) in the PBS buffer for 2 h at room temperature. Slices were washed in PBS-T three times, mounted on glass slides using Aqua poly/mount (Polysciences), and photographed using a confocal microscope (Leica Application Suite X) or STED imaging (Leica TCS SP8). Fluorescence microscopic images obtained were imported into ImageJ (NIH) for analysis, and all the parameters used were kept consistent during capture. For primary antibodies, we used mouse anti-calbindin 28K (1:1,000; 300; Swant), rabbit anti–β-gal (1:200; MP Biomedicals), mouse anti-lnx1 (1:200; ab22157; Abcam), goat anti-EphB2 (1:500; P54763; R&D), mouse anti–zinc-transporter-3 (ZnT3; 1:500; 197011; Synaptic Systems, rabbit anti-NeuN (1:1,000; D3S3I; Cell Signaling Technology), mouse anti-Flag (1:1,000; F3165; Sigma-Aldrich), rabbit anti-synapsin1 (1:1,000, gift from Ilya Bezprozvanny, University of Texas Southwestern Medical Center, Dallas, TX), and rabbit anti-PSD95 (1:1,000; 3450; Cell Signaling Technology).
To accurately analyze the terminals on spines (terminal-spine) and dendritic shaft (terminal-shaft), the fluorescent images were deconvolved according to the instructions of Leica TCS SP8, and 3D rendering was achieved in ImageJ using these deconvolved images. The antibody signal threshold was defined as three times brightness to the background, and brightness/contrast adjustment within linear ranges was made using ImageJ when necessary. To quantify the shape of spine, a procedure was adapted from our previous study (Xu et al., 2011). The shape of neuronal spines in slices was classified by NeuronStudio software package and an algorithm from Rodriguez et al. (2008) with the following cutoff values: AR_thin(crit) = 2.5, head-to-neck ratio (HNR(crit)) = 1.3, and head diameter (HD(crit)) = 0.3 µm. The type of these spines was determined based on the following criteria: (a) spines with HNR greater than HNR(crit) are considered to have a neck and could be either thin or mushroom types; (b) spines with HD greater than HD(crit) are classified as mushroom, otherwise thin; (c) spines lacking significant necks and less than AR_thin(crit) are considered as stubby, otherwise thin. Protrusions with length 0.2–3.0 µm and maximum width 3 µm were counted. Spine density was calculated by dividing the total spine number by the dendritic branch length. The localizations of terminals and spines were carefully identified in the 3D rendering views. The ZnT3+ synapses were defined as the overlapped connections of spines and ZnT3+ terminals with any identical red/green pixels (as shown in Fig. 2 B), otherwise ZnT3− (separated from terminals). Control and experiment conditions were adjusted with the same parameters. Acquisition of the images as well as morphometric quantification was performed under blinded conditions.
For quantification of CA3 cell number, serial coronal sections (30 µm) containing hippocampus (from bregma, −1.06 to −2.30 mm) were collected using a vibratome. NeuN immunostaining was performed on every sixth section encompassing the anterior to posterior of the CA3 area. NeuN-positive cells of CA3 area in each section of different mice were counted under blinded conditions.
For X-gal staining of embryos, E13–E15 embryos were washed with cold PBS and fixed in cold PBS + 4% PFA for 20 min with gentle agitation. After three 5-min washes with gentle agitation in cold PBS, embryos were transferred to histochemical staining solution (5 mM K4Fe(CN)6, 5 mM K3Fe(CN)6, 2 mM MgCl2, 0.02% [vol/vol] NP-40, 0.01% [wt/vol] sodium deoxycholate, 1 mg/ml 5-bromo-3-indolyl-β-d-galactopyranoside [X-Gal; Amresco], and 20 mM Tris-HCl in PBS, pH 7.3) in a 24-well plate and incubated overnight at 30°C with gentle agitation. Whole mounts were washed with PBS + 0.1% Tween-20 at room temperature for several hours before image capture.
Transmission EM
Mice were perfused with 2% PFA/2.5% glutaraldehyde in phosphate buffer, pH 7.2, for 30 min, and dissected brains were then postfixed in the same buffer overnight at 4°C. After PBS buffer rinse, samples were postfixed in 1% osmium tetroxide buffer (2 h) on ice in the dark. After a double-distilled water rinse, tissue was stained with 3% aqueous uranyl acetate (0.22-µm filtered; 1 h in the dark), dehydrated in a graded series of ethanol and propylene oxide, and embedded in Epoxy 618 resin. Samples were polymerized at 60°C for 48 h. Thin sections (60–90 nm) were cut with a diamond knife on the LKB V ultramicrotome and picked up with formvar-coated copper slot grids. Grids were stained with lead citrate and observed with transmission microscopy (PHILIP CM-120). Images from the SPB or IPB regions were captured, and the PSDs, terminals, vesicle numbers, and distance of vesicles to presynaptic membrane were counted or analyzed by ImageJ under blinded conditions (n = 3–4 animals per genotype).
Western blotting, immunoprecipitation, and isolation of synaptosome and cell-surface protein
Western blotting was performed as in a previous study (Sun et al., 2014). Briefly, hippocampal regions from WT, knockout, and knock-in mice at different ages were dissected, homogenized, and solubilized at 4°C for 1 h in lysis buffer (1% CHAPS, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.2, 5 mM EDTA, 5 mM EGTA, 1 mM PMSF, 50 mM NaF, 1 mM Na3VO4, and protease inhibitors). Primary hippocampal neurons were collected, homogenized, and solubilized in the same buffer on DIV14. For immunoprecipitation, hippocampal tissues were lysed at 4°C for 1 h in lysis buffer (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM MgCl2, 1% NP-40, 10% glycerol, 1 mM DTT, 1 mM PMSF, 50 mM NaF, 1 mM Na3VO4, and protease inhibitors) and then immunoprecipitated with indicated antibodies for 2 h and incubated with protein G beads overnight at 4°C. Bound proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with indicated antibodies. The subcellular fractions of the hippocampus from WT, knockout, and knock-in mice were purified as described previously (Pacchioni et al., 2009). Cell-surface protein of the cultured primary hippocampal neuron samples was isolated using a Pierce cell surface protein isolation kit (Thermo Fisher Scientific) per the manufacturer’s protocol. Analysis of the data was performed using ImageJ, and the mean density of each band was normalized to β-actin or GAPDH signals in the same sample and averaged. For primary antibodies, we used mouse anti-Lnx1 (1:1,000; ab22157; Abcam), rabbit anti–β-gal (1:1,000; MP Biomedicals), rabbit anti-PSD95 (1:1,000; 3450; Cell Signaling Technology), mouse antisynaptophysin (1:3,000; ab8049; Abcam), mouse anti-GAPDH (1:3,000; G8795; Sigma-Aldrich), mouse anti–β-actin (1:3,000; MA5-15739; Thermo Fisher Scientific), goat anti-EphB1 (1:200; M-19; Santa Cruz Biotechnology), goat anti-EphB2 (1:1,000; P54763; R&D), rabbit anti–N-cadherin (1:1,000; ab12221; Abcam), rabbit anti–connexin 43 (1:1,000; ab11370; Abcam), rabbit anti-claudin1 (1:1,000; ab15098; Abcam), mouse anti-striatin (1:1,000; 610838; BD), mouse anti-Flag (F3165; Sigma-Aldrich), anti–Flag-HRP and anti–HA-HRP (1:5,000; Sigma-Aldrich), rabbit anti–ephrin-B3 (1:500; 34-3600; Invitrogen), and mouse anti–phosphotyrosine 4G10 (1:500; 05-321; EMD Millipore).
DNA constructs
P70-Lnx1 and P80-Lnx1 genes were amplified from hippocampal and renal cDNA, respectively, by PCR and ligated in the EcoRI and XbaI sites of p3XFLAG-CMV-10. All the PDZ mutants of P70-Lnx1 were generated from full-length P70-Lnx1 by PCR and ligated to p3XFLAG-CMV-10.
Primary cell culture, coculture, and biochemistry
Primary cell culture of hippocampal neurons was performed as described (Xu and Henkemeyer, 2009). Briefly, hippocampal neurons were dissociated from P0 pups. The triturated cells (1 × 105 cells per well) were grown on either six-well dishes or glass coverslips coated with 10 µM polylysine overnight in 24-well dishes. Then the culture was grown in a medium of Neurobasal A media (Gibco) supplemented with B27 and 2 mM glutamine for the indicated number of days. For coculture assays, primary hippocampal neurons from Lnx1+/+ and Lnx1−/− pups were precultured for 12–14 d (transfected with GFP plasmid at DIV7 using the calcium phosphate method), and TdT+ primary hippocampal neurons from P1 TdT+ knock-in pups were dissociated as described (Baranes et al., 1996) and plated on the culture by either direct addition into the dishes or loading with coverslips for another 6 d. During the 6 d, neurons were imaged every day to measure the TdT+ axon length. On the sixth day, neurons were fixed (4% PFA and 4% sucrose in PBS) and imaged to observe the spines and boutons. For the pharmacological treatment assay, primary hippocampal neurons from WT or Lnx1−/− pups were dissociated and cultured for 14 d, and neurons or MEF cells were incubated for 12 h in the presence or absence of proteasome inhibitor (10 µM MG132; Gene Operation) or lysosomal inhibitor (100 µg/ml leupeptin and 50 mM NH4Cl; Sigma-Aldrich). After treatment, cells were lysed and subjected to Western blot analysis.
Live imaging in dual color–labeled coculture system
Live-imaging experiments were performed on a Nikon A1R confocal microscope. Primary hippocampal neurons from Lnx1+/+ and Lnx1−/− pups were precultured in 35-mm glass-bottom dishes for 12–14 d (transfected with GFP plasmid at DIV7 using the calcium phosphate method), and TdT+ primary hippocampal neurons were plated by direct addition into the dishes for another 6–7 d. Neurons were maintained at room temperature, and images were acquired every 10 min. For the presynaptic or postsynaptic alteration, we calculated the AI: |A60 − A0|/(A60 + A0), where A is the pre- or postsynaptic area and 60 or 0 indicates the time in minutes. When AI ≥ 0.2, we indicate the pre- or postsynaptic alteration as *; otherwise as ns.
Quantitative real-time PCR
Total RNA was prepared from hippocampus tissue of PW3 Lnx1 mutant and WT littermates using TRI Reagent (Sigma-Aldrich). RNAs were reverse transcribed with high-capacity cDNA reverse transcription kits (Applied Biosystems) according to the manufacturer’s instructions. The PCR mixture contained 1 µl diluted cDNA, 5 µl 2× SYBR Green PCR Master Mix (Applied Biosystems), and 200 nM of each gene-specific primer in a final volume of 10 µl. The real-time PCRs were performed using a Fast 96-Well System (Applied Biosystems). Three biological replicates for each sample were used for real-time PCR analysis, and three technical replicates were analyzed for each biological replicate. The relative copy number of β-actin RNA was quantified and used for normalization. The primer sequences are given in Table S1.
Electrophysiology
Brain coronal slices were prepared from 3-wk-old naive Lnx1 +/+ and Lnx1−/− mice. Brains were dissected quickly and chilled in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 12.5 glucose. Coronal brain slices (300 µm thick) were prepared with a vibratome and recovered in ACSF bubbled with 95% O2 and 5% CO2 at 31°C for 1 h and then maintained at room temperature (22–25°C). For EPSC recording, borosilicate glass pipettes (3–5 MΩ) were filled with an internal solution containing (in mM) 115 CsMeSO3, 10 Hepes, 2.5 MgCl2 ⋅ 6H2O, 20 CsCl2, 0.6 EGTA, 10 Na2 phosphocreatine, 0.4 Na-GTP, and 4 Mg ATP. EPSCs were recorded at −70 mV in the presence of 100 µM picrotoxin. Slices were stimulated using a bipolar concentric electrode (FHC) that was placed in the MF and connected with a stimulator (AMPI) to evoke EPSCs in CA3 pyramidal neurons. PPRs were calculated as a ratio of EPSC2 to EPSC1, separated by interstimulus intervals of 25, 50, 100, 200, and 400 ms. Data were analyzed in pClamp 10.6 (Molecular Devices), and recordings were made from an average of three cells per slice and two to three slices per mouse.
Statistical analysis
The results are presented as mean ± SEM. Statistical differences were determined by Student’s t test for two-group comparisons or ANOVA followed by Tukey test for multiple comparisons among more than two groups.
Online supplemental material
Fig. S1 shows mRNA localization of Lnx1 in brain from the Allen Brain Atlas and biochemical characterization of Lnx1 mutant. Fig. S2 shows expression and subcellular localization of Lnx1 in hippocampus. Fig. S3 shows Lnx1 effects on axon growth and pruning when cocultured with coverslips to prevent axon–cell contact. Fig. S4 shows Lnx1 effects on morphology of calbindin-positive/-negative neurons in coculture assay, spine density and mushroom ratio of CA3 neurons, and PPRs with IPB stimulation. Fig. S5 shows interactions between Lnx1 and EphB1/2 and effects of Lnx1 on membrane level of EphB1/2, mRNA level of receptors, and protein level of EphB2/EphB1-β-gal/ephrin-B3. Table S1 shows primer sequences for quantitative real-time PCR used in this study. Videos 1, 2, 3, 4, and 5 show different morphological change types including presynaptic alteration (Videos 1 and 2), postsynaptic alteration (Video 3), both alteration (Video 4), and no alteration (Video 5) in a dual color–labeled coculture system.
Supplementary Material
Acknowledgments
We thank Drs. Xiang Yu, Zhiping Pan, and Lan Bao for helpful comments on the manuscript and Liang Zhu, Guang-Ni Xu, Zi-Jun Deng, and Si Chen for laboratory technique support.
This research was supported by National Basic Research Program of China (973 Program; 2014CB965002) to N.-J. Xu, the National Natural Science Foundation of China (91232704 and 31671062) to N.-J. Xu, grants from the Shanghai Brain-Intelligence Project from the Shanghai Science and Technology Committee (16JC1420500), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (2013-25) to N.-J. Xu, the Shanghai Science and Technology Committee (11DZ2260200) to N.-J. Xu, and the NIH (MH066332) to M. Henkemeyer.
The authors declare no competing financial interests.
Author contributions: X.-D. Liu performed the experiments of morphology and histology. X.-N. Zhu and T.-L. Xu assisted with electrophysiology, M.M. Halford and M. Henkemeyer initiated the Lnx1 project and generated the mutant mouse, and M. Henkemeyer provided the various EphB1 and EphB2 mutants used in the study. X.-D. Liu and N.-J. Xu designed experiments and wrote the manuscript.
References
- Ackley, B.D., and Jin Y.. 2004. Genetic analysis of synaptic target recognition and assembly. Trends Neurosci. 27:540–547. 10.1016/j.tins.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Alvarez, V.A., and Sabatini B.L.. 2007. Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30:79–97. 10.1146/annurev.neuro.30.051606.094222 [DOI] [PubMed] [Google Scholar]
- Amaral, D.G., and Dent J.A.. 1981. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J. Comp. Neurol. 195:51–86. 10.1002/cne.901950106 [DOI] [PubMed] [Google Scholar]
- Bagri, A., Cheng H.J., Yaron A., Pleasure S.J., and Tessier-Lavigne M.. 2003. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 113:285–299. 10.1016/S0092-8674(03)00267-8 [DOI] [PubMed] [Google Scholar]
- Baranes, D., López-García J.C., Chen M., Bailey C.H., and Kandel E.R.. 1996. Reconstitution of the hippocampal mossy fiber and associational-commissural pathways in a novel dissociated cell culture system. Proc. Natl. Acad. Sci. USA. 93:4706–4711. 10.1073/pnas.93.10.4706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.R., and Lagopoulos J.. 2014. Stress and trauma: BDNF control of dendritic-spine formation and regression. Prog. Neurobiol. 112:80–99. 10.1016/j.pneurobio.2013.10.005 [DOI] [PubMed] [Google Scholar]
- Benoist, M., Gaillard S., and Castets F.. 2006. The striatin family: a new signaling platform in dendritic spines. J. Physiol. Paris. 99:146–153. 10.1016/j.jphysparis.2005.12.006 [DOI] [PubMed] [Google Scholar]
- Bhatt, D.H., Zhang S., and Gan W.B.. 2009. Dendritic spine dynamics. Annu. Rev. Physiol. 71:261–282. 10.1146/annurev.physiol.010908.163140 [DOI] [PubMed] [Google Scholar]
- Bouché, E., Romero-Ortega M.I., Henkemeyer M., Catchpole T., Leemhuis J., Frotscher M., May P., Herz J., and Bock H.H.. 2013. Reelin induces EphB activation. Cell Res. 23:473–490. 10.1038/cr.2013.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayouette, M., and Raff M.. 2002. Asymmetric segregation of Numb: a mechanism for neural specification from Drosophila to mammals. Nat. Neurosci. 5:1265–1269. 10.1038/nn1202-1265 [DOI] [PubMed] [Google Scholar]
- Chavis, P., and Westbrook G.. 2001. Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature. 411:317–321. 10.1038/35077101 [DOI] [PubMed] [Google Scholar]
- Chen, Y., Fu A.K., and Ip N.Y.. 2008. Bidirectional signaling of ErbB and Eph receptors at synapses. Neuron Glia Biol. 4:211–221. 10.1017/S1740925X09990287 [DOI] [PubMed] [Google Scholar]
- Chenaux, G., and Henkemeyer M.. 2011. Forward signaling by EphB1/EphB2 interacting with ephrin-B ligands at the optic chiasm is required to form the ipsilateral projection. Eur. J. Neurosci. 34:1620–1633. 10.1111/j.1460-9568.2011.07845.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicurel, M.E., and Harris K.M.. 1992. Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J. Comp. Neurol. 325:169–182. 10.1002/cne.903250204 [DOI] [PubMed] [Google Scholar]
- Cohen-Cory, S. 2002. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 298:770–776. 10.1126/science.1075510 [DOI] [PubMed] [Google Scholar]
- Contractor, A., Rogers C., Maron C., Henkemeyer M., Swanson G.T., and Heinemann S.F.. 2002. Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science. 296:1864–1869. 10.1126/science.1069081 [DOI] [PubMed] [Google Scholar]
- Dho, S.E., Jacob S., Wolting C.D., French M.B., Rohrschneider L.R., and McGlade C.J.. 1998. The mammalian numb phosphotyrosine-binding domain. Characterization of binding specificity and identification of a novel PDZ domain-containing numb binding protein, LNX. J. Biol. Chem. 273:9179–9187. 10.1074/jbc.273.15.9179 [DOI] [PubMed] [Google Scholar]
- El-Husseini, A.E., Schnell E., Chetkovich D.M., Nicoll R.A., and Bredt D.S.. 2000. PSD-95 involvement in maturation of excitatory synapses. Science. 290:1364–1368. [PubMed] [Google Scholar]
- Feng, W., and Zhang M.. 2009. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 10:87–99. 10.1038/nrn2540 [DOI] [PubMed] [Google Scholar]
- Feng, G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., and Sanes J.R.. 2000. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 28:41–51. 10.1016/S0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Futai, K., Kim M.J., Hashikawa T., Scheiffele P., Sheng M., and Hayashi Y.. 2007. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat. Neurosci. 10:186–195. 10.1038/nn1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner, C.C., Nash J., and Huganir R.L.. 2000. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 10:274–280. 10.1016/S0962-8924(00)01783-9 [DOI] [PubMed] [Google Scholar]
- Garner, C.C., Zhai R.G., Gundelfinger E.D., and Ziv N.E.. 2002. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 25:243–251. 10.1016/S0166-2236(02)02152-5 [DOI] [PubMed] [Google Scholar]
- Geiger, J.C., Lipka J., Segura I., Hoyer S., Schlager M.A., Wulf P.S., Weinges S., Demmers J., Hoogenraad C.C., and Acker-Palmer A.. 2014. The GRIP1/14-3-3 pathway coordinates cargo trafficking and dendrite development. Dev. Cell. 28:381–393. 10.1016/j.devcel.2014.01.018 [DOI] [PubMed] [Google Scholar]
- Genander, M., Halford M.M., Xu N.J., Eriksson M., Yu Z., Qiu Z., Martling A., Greicius G., Thakar S., Catchpole T., et al. . 2009. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 139:679–692. 10.1016/j.cell.2009.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giagtzoglou, N., Ly C.V., and Bellen H.J.. 2009. Cell adhesion, the backbone of the synapse: “vertebrate” and “invertebrate” perspectives. Cold Spring Harb. Perspect. Biol. 1:a003079. 10.1101/cshperspect.a003079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Song E., Ma S., Wang X., Gao S., Shao C., Hu S., Jia L., Tian R., Xu T., and Gao Y.. 2012. Proteomics strategy to identify substrates of LNX, a PDZ domain-containing E3 ubiquitin ligase. J. Proteome Res. 11:4847–4862. 10.1021/pr300674c [DOI] [PubMed] [Google Scholar]
- Heisler, F.F., Lee H.K., Gromova K.V., Pechmann Y., Schurek B., Ruschkies L., Schroeder M., Schweizer M., and Kneussel M.. 2014. GRIP1 interlinks N-cadherin and AMPA receptors at vesicles to promote combined cargo transport into dendrites. Proc. Natl. Acad. Sci. USA. 111:5030–5035. 10.1073/pnas.1304301111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkemeyer, M., Orioli D., Henderson J.T., Saxton T.M., Roder J., Pawson T., and Klein R.. 1996. Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell. 86:35–46. 10.1016/S0092-8674(00)80075-6 [DOI] [PubMed] [Google Scholar]
- Henkemeyer, M., Itkis O.S., Ngo M., Hickmott P.W., and Ethell I.M.. 2003. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J. Cell Biol. 163:1313–1326. 10.1083/jcb.200306033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch, T.K. 2005. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6:877–888. 10.1038/nrn1787 [DOI] [PubMed] [Google Scholar]
- Holmberg, J., Genander M., Halford M.M., Annerén C., Sondell M., Chumley M.J., Silvany R.E., Henkemeyer M., and Frisén J.. 2006. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 125:1151–1163. 10.1016/j.cell.2006.04.030 [DOI] [PubMed] [Google Scholar]
- Holtmaat, A., and Svoboda K.. 2009. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10:647–658. 10.1038/nrn2699 [DOI] [PubMed] [Google Scholar]
- Hoogenraad, C.C., Milstein A.D., Ethell I.M., Henkemeyer M., and Sheng M.. 2005. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat. Neurosci. 8:906–915. 10.1038/nn1487 [DOI] [PubMed] [Google Scholar]
- Hu, Z., Hom S., Kudze T., Tong X.J., Choi S., Aramuni G., Zhang W., and Kaplan J.M.. 2012. Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science. 337:980–984. 10.1126/science.1224896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüngling, K., Eulenburg V., Moore R., Kemler R., Lessmann V., and Gottmann K.. 2006. N-cadherin transsynaptically regulates short-term plasticity at glutamatergic synapses in embryonic stem cell-derived neurons. J. Neurosci. 26:6968–6978. 10.1523/JNEUROSCI.1013-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüttner, R., and Rathjen F.G.. 2005. Molecular analysis of axonal target specificity and synapse formation. Cell. Mol. Life Sci. 62:2811–2827. 10.1007/s00018-005-5299-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania, A., and Klein R.. 2016. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 17:240–256. 10.1038/nrm.2015.16 [DOI] [PubMed] [Google Scholar]
- Kerschensteiner, D., Morgan J.L., Parker E.D., Lewis R.M., and Wong R.O.. 2009. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature. 460:1016–1020. 10.1038/nature08236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E., and Sheng M.. 2004. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5:771–781. 10.1038/nrn1517 [DOI] [PubMed] [Google Scholar]
- Klein, R., and Kania A.. 2014. Ephrin signalling in the developing nervous system. Curr. Opin. Neurobiol. 27:16–24. 10.1016/j.conb.2014.02.006 [DOI] [PubMed] [Google Scholar]
- Kolodkin, A.L., and Tessier-Lavigne M.. 2011. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb. Perspect. Biol. 3:a001727. 10.1101/cshperspect.a001727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy, Y., Saunders A., Johnson C.A., Lowell B.B., and Sabatini B.L.. 2012. Recurrent network activity drives striatal synaptogenesis. Nature. 485:646–650. 10.1038/nature11052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, H.B., and Sabatini B.L.. 2011. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 474:100–104. 10.1038/nature09986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenihan, J.A., Saha O., Mansfield L.M., and Young P.W.. 2014. Tight, cell type-specific control of LNX expression in the nervous system, at the level of transcription, translation and protein stability. Gene. 552:39–50. 10.1016/j.gene.2014.09.011 [DOI] [PubMed] [Google Scholar]
- Li, M.Y., Miao W.Y., Wu Q.Z., He S.J., Yan G., Yang Y., Liu J.J., Taketo M.M., and Yu X.. 2017. A critical role of presynaptic cadherin/catenin/p140Cap complexes in stabilizing spines and functional synapses in the neocortex. Neuron. 94:1155–1172. [DOI] [PubMed] [Google Scholar]
- Liebl, D.J., Morris C.J., Henkemeyer M., and Parada L.F.. 2003. mRNA expression of ephrins and Eph receptor tyrosine kinases in the neonatal and adult mouse central nervous system. J. Neurosci. Res. 71:7–22. 10.1002/jnr.10457 [DOI] [PubMed] [Google Scholar]
- Low, L.K., and Cheng H.J.. 2006. Axon pruning: an essential step underlying the developmental plasticity of neuronal connections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1531–1544. 10.1098/rstb.2006.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen, L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. . 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister, A.K. 2007. Dynamic aspects of CNS synapse formation. Annu. Rev. Neurosci. 30:425–450. 10.1146/annurev.neuro.29.051605.112830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migani, P., Bartlett C., Dunlop S., Beazley L., and Rodger J.. 2007. Ephrin-B2 immunoreactivity distribution in adult mouse brain. Brain Res. 1182:60–72. 10.1016/j.brainres.2007.08.065 [DOI] [PubMed] [Google Scholar]
- Nakamura, Y., Wood C.L., Patton A.P., Jaafari N., Henley J.M., Mellor J.R., and Hanley J.G.. 2011. PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 30:719–730. 10.1038/emboj.2010.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll, R.A., and Schmitz D.. 2005. Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 6:863–876. 10.1038/nrn1786 [DOI] [PubMed] [Google Scholar]
- Nie, J., McGill M.A., Dermer M., Dho S.E., Wolting C.D., and McGlade C.J.. 2002. LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J. 21:93–102. 10.1093/emboj/21.1.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, B.O., Fetter R.D., and Davis G.W.. 2017. Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature. 550:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacchioni, A.M., Vallone J., Worley P.F., and Kalivas P.W.. 2009. Neuronal pentraxins modulate cocaine-induced neuroadaptations. J. Pharmacol. Exp. Ther. 328:183–192. 10.1124/jpet.108.143115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale, E.B. 2008. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 133:38–52. 10.1016/j.cell.2008.03.011 [DOI] [PubMed] [Google Scholar]
- Penzes, P., Johnson R.C., Sattler R., Zhang X., Huganir R.L., Kambampati V., Mains R.E., and Eipper B.A.. 2001. The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron. 29:229–242. 10.1016/S0896-6273(01)00193-3 [DOI] [PubMed] [Google Scholar]
- Regalado, M.P., Terry-Lorenzo R.T., Waites C.L., Garner C.C., and Malenka R.C.. 2006. Transsynaptic signaling by postsynaptic synapse-associated protein 97. J. Neurosci. 26:2343–2357. 10.1523/JNEUROSCI.5247-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno, M.M., Hurtado A., Wang H., Macopson J.G., Griner E.M., Betz A., Brose N., Kazanietz M.G., and Kolodkin A.L.. 2012. The RacGAP β2-Chimaerin selectively mediates axonal pruning in the hippocampus. Cell. 149:1594–1606. 10.1016/j.cell.2012.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, D.S., Northcutt G.M., and Kurschner C.. 2001. The Lnx family proteins function as molecular scaffolds for Numb family proteins. Mol. Cell. Neurosci. 18:525–540. 10.1006/mcne.2001.1024 [DOI] [PubMed] [Google Scholar]
- Rodriguez, A., Ehlenberger D.B., Dickstein D.L., Hof P.R., and Wearne S.L.. 2008. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 3:e1997. 10.1371/journal.pone.0001997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen, A., Sätzler K., Rodríguez E.P., Jonas P., Frotscher M., and Lübke J.H.. 2007. Structural determinants of transmission at large hippocampal mossy fiber synapses. J. Neurosci. 27:10434–10444. 10.1523/JNEUROSCI.1946-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala, C., and Segal M.. 2014. Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev. 94:141–188. 10.1152/physrev.00012.2013 [DOI] [PubMed] [Google Scholar]
- Scheiffele, P. 2003. Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 26:485–508. 10.1146/annurev.neuro.26.043002.094940 [DOI] [PubMed] [Google Scholar]
- Sheffler-Collins, S.I., and Dalva M.B.. 2012. EphBs: an integral link between synaptic function and synaptopathies. Trends Neurosci. 35:293–304. 10.1016/j.tins.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, K., and Cowan C.W.. 2010. Guidance molecules in synapse formation and plasticity. Cold Spring Harb. Perspect. Biol. 2:a001842. 10.1101/cshperspect.a001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, K., and Scheiffele P.. 2010. Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 33:473–507. 10.1146/annurev.neuro.051508.135302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, M., and Kim E.. 2011. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3:a005678. 10.1101/cshperspect.a005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, M., and Sala C.. 2001. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24:1–29. 10.1146/annurev.neuro.24.1.1 [DOI] [PubMed] [Google Scholar]
- Siddiqui, T.J., and Craig A.M.. 2011. Synaptic organizing complexes. Curr. Opin. Neurobiol. 21:132–143. 10.1016/j.conb.2010.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloniowski, S., and Ethell I.M.. 2012. Looking forward to EphB signaling in synapses. Semin. Cell Dev. Biol. 23:75–82. 10.1016/j.semcdb.2011.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl, G., Maxeiner S., and Willecke K.. 2005. Expression and functions of neuronal gap junctions. Nat. Rev. Neurosci. 6:191–200. 10.1038/nrn1627 [DOI] [PubMed] [Google Scholar]
- Südhof, T.C. 2008. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 455:903–911. 10.1038/nature07456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof, T.C. 2012. The presynaptic active zone. Neuron. 75:11–25. 10.1016/j.neuron.2012.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S., Zhang H., Liu J., Popugaeva E., Xu N.J., Feske S., White C.L. III, and Bezprozvanny I.. 2014. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 82:79–93. 10.1016/j.neuron.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano, G.G., and Nelson S.B.. 2004. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5:97–107. 10.1038/nrn1327 [DOI] [PubMed] [Google Scholar]
- Waites, C.L., Craig A.M., and Garner C.C.. 2005. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 28:251–274. 10.1146/annurev.neuro.27.070203.144336 [DOI] [PubMed] [Google Scholar]
- Williams, S.E., Mann F., Erskine L., Sakurai T., Wei S., Rossi D.J., Gale N.W., Holt C.E., Mason C.A., and Henkemeyer M.. 2003. Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron. 39:919–935. 10.1016/j.neuron.2003.08.017 [DOI] [PubMed] [Google Scholar]
- Witter, M.P. 2007. Intrinsic and extrinsic wiring of CA3: indications for connectional heterogeneity. Learn. Mem. 14:705–713. 10.1101/lm.725207 [DOI] [PubMed] [Google Scholar]
- Wolting, C.D., Griffiths E.K., Sarao R., Prevost B.C., Wybenga-Groot L.E., and McGlade C.J.. 2011. Biochemical and computational analysis of LNX1 interacting proteins. PLoS One. 6:e26248. 10.1371/journal.pone.0026248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Y., Zhao W., Wang W., Zhao S., Tang R., Ying K., Zhou Z., and Mao Y.. 2001. Identification of a human LNX protein containing multiple PDZ domains. Biochem. Genet. 39:117–126. 10.1023/A:1010269908398 [DOI] [PubMed] [Google Scholar]
- Xu, N.-J., and Henkemeyer M.. 2009. Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat. Neurosci. 12:268–276. 10.1038/nn.2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, N.J., Sun S., Gibson J.R., and Henkemeyer M.. 2011. A dual shaping mechanism for postsynaptic ephrin-B3 as a receptor that sculpts dendrites and synapses. Nat. Neurosci. 14:1421–1429. 10.1038/nn.2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste, R., and Bonhoeffer T.. 2004. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5:24–34. 10.1038/nrn1300 [DOI] [PubMed] [Google Scholar]
- Zhu, X.N., Liu X.D., Sun S., Zhuang H., Yang J.Y., Henkemeyer M., and Xu N.J.. 2016a. Ephrin-B3 coordinates timed axon targeting and amygdala spinogenesis for innate fear behaviour. Nat. Commun. 7:11096. 10.1038/ncomms11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X.N., Liu X.D., Zhuang H., Henkemeyer M., Yang J.Y., and Xu N.J.. 2016b. Amygdala EphB2 Signaling Regulates Glutamatergic Neuron Maturation and Innate Fear. J. Neurosci. 36:10151–10162. 10.1523/JNEUROSCI.0845-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker, R.S. 1999. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 9:305–313. 10.1016/S0959-4388(99)80045-2 [DOI] [PubMed] [Google Scholar]
- Zuo, Y., Yang G., Kwon E., and Gan W.B.. 2005. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 436:261–265. 10.1038/nature03715 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.