

Abstract
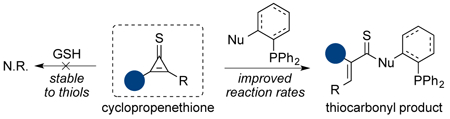
Cyclopropenethiones are reported as new bioorthogonal reagents. These motifs react readily with substituted phosphines to provide thiocarbonyl adducts. In some cases, the ligations are >300-fold faster than analogous reactions with bioorthogonal cyclopropenones. Dialkyl cyclopropenethiones are also stable in aqueous buffers and can be used for biomolecule labeling in vitro and in cell lysate. The rapid reactivity and biocompatibility of cyclopropenethiones suggest that they will be useful probes for cellular studies.
Graphical Abstract
Bioorthogonal chemistries have enabled a broad range of applications in living organisms,1,2 including biomolecule imaging,3 metabolic profiling,4–6 and targeted drug delivery.7,8 Despite their ubiquity in numerous fields, these reactions are not without limitation. Only a handful of bioorthogonal reagents are reliable in the most demanding environments, including inside cells. Additionally, many bioorthogonal probes cross-react with one another, precluding dual imaging and other multi-component studies.9 These and other applications demand new reagents and new reactions.
Our laboratory has focused on expanding the scope of bioorthogonal chemistries by developing probes that are small, stable, and tunable.10 We recently reported one class of such reagents—cyclopropenones.11,12 These motifs are stable in biological media and react robustly with ortho−substituted phosphines. The ligation involves initial formation of a reactive ketene-ylide, followed by intramolecular trapping (Figure 1A). The resulting products are stable in cellular environments. The unique mechanism of the cyclopropenone-phosphine ligation renders it compatible with many classic bioorthogonal reagents.12 Cyclopropenones are also small and innocuous to cellular enzymes and metabolic pathways.12 Dialkyl-substituted scaffolds, in particular, are well suited for time intensive studies. We used these probes in long-term cultures for site-specific protein modification.12 The most stable cycloprope nones, though, required lengthy reaction times with bioorthogonal phosphines. Faster rates could be achieved with mono-substituted scaffolds, but these probes were more susceptible to side reactions with biological nucleophiles.
Figure 1.

Bioorthogonal ligations of cyclopropenones and cyclopropenethiones. A) Cyclopropenones (CpO, X=O) react with phosphines to form ketene-ylides. These intermediates can be trapped with pendant nucleophiles to afford stable adducts. Cyclopropenethiones (CpS, X=S) were hypothesized to react similarly with phosphine probes. B) CpS scaffolds harbor lower LUMO energies than analogous CpO probes. Density functional theory (DFT) calculations were performed with Spartan, using the B3LYP level of theory and basis set 6–31G*.
We hypothesized that cyclopropenone heteroanalogs could strike the right balance between kinetic stability and rapid reactivity. We were particularly drawn to cyclopropenethione (CpS) scaffolds. CpS differs from CpO by only a single atom, and would thus likely be small enough to avoid perturbing target biomolecules or pathways. CpS is also known to participate in a variety of transformations, including photocleavage reactions13,14 and ring-opening cycloadditions.15,16 Previous work by Yoneda and Matsumura further revealed that triaryl phosphines can react with diphenyl CpS via nucleophilic addition to the alkene.17,18 Based on this precedent, we surmised that CpS derivatives could react with substituted phosphines (similar to CpO probes), ultimately providing thiocarbonyl products (Figure 1A). Thiocarbonyls are widely used in fluorescence quenching and other biophysical experiments with proteins,19,20 suggesting that they too are stable and biocompatible.21,22 Thioamides, in particular, are known to exhibit improved proteolytic stability and can increase peptide half-lives in biological media.23
To evaluate the proposed phosphine ligation, we first examined the frontier molecular orbitals of CpS derivatives. Density functional theory (DFT) calculations revealed that the LUMO values for CpS scaffolds were consistently lower than those for analogous CpO molecules (Figure 1B). The reduced LUMO values suggested the potential for increased reactivity with phosphines. Whether or not CpS molecules would also be susceptible to rapid reactions with thiols or other biological nucleophiles remained to be determined.
We synthesized a panel of CpS compounds to examine their suitability as bioorthogonal reagents. A symmetric dialkyl CpS (3a) was included for ease of product characterization. We also prepared scaffolds bearing aryl substituents (3b–c) and water-solubilizing groups (3d). All of the desired cyclopropenethiones were prepared via the corresponding cyclopropenones (2a–d, Scheme 1). Alkynes 1a–d were first treated with difluorocarbene to generate 3,3-difluorocyclopropenes.24 These intermediates were then hydrolyzed to provide 2a–d. The desired cyclopropenethiones were ultimately obtained by treating 2a–d with Lawesson’s reagent. We also attempted to prepare mono-substituted CpS probes. However, these less substituted scaffolds were not stable to concentration (data not shown), and were likely subject to intermolecular attack.
Scheme 1.

Synthesis of model cyclopropenethiones.
With the CpS scaffolds in hand, we first evaluated their stabilities in aqueous solution. Compounds 3b–d were incubated in d−PBS (pH 7.4) and monitored by 1H-NMR spectroscopy (Figures S1–S3). Probes 3b and 3d were stable for >1 week at 37 °C. The diaryl scaffold 3c degraded, resulting in an insoluble and unidentified precipitate. CpS 3c may be susceptible to dimerization or intermolecular reactivity over prolonged time periods.13,25 The stabilities of 3b and 3d toward biological nucleophiles were further examined. The molecules were incubated with L-glutathione (GSH) in d−PBS (pH 7.4) and monitored via 1H-NMR spectroscopy (Figures 2 and S4–S5). No reactivity was observed over 24 h at 37 °C, indicating that both scaffolds were stable to thiols.
Figure 2.

Cyclopropenethiones are stable to L-glutathione (GSH) at physiological pH. Cyclopropenethione 3d (5 mM) was incubated with GSH (5 mM) in d−PBS (pH 7.4) and monitored by 1H-NMR spectroscopy.
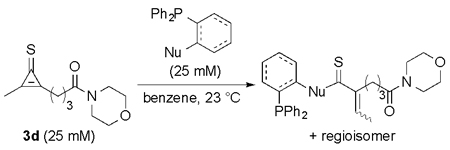
Encouraged by the biocompatibility of dialkyl CpS scaffolds, we investigated their reactivities with a panel of phosphines. Symmetric analog 3a was initially used to determine the structure of the ligated adduct. When 3a was incubated with phosphine 4, thionoester S5 was formed in high yield (Scheme S1). The expected thionoester was also observed upon treating 3d with phosphine 4. Importantly, this latter reaction proceeded ~300-fold faster than the analogous CpO-phosphine ligation (Scheme 2 and Figure S6). Such rapid reactions are desirable for live cell and tissue studies, where only small amounts of reagents are typically tolerated. By contrast, CpS 3d reacted sluggishly with thiol-substituted phosphine 5. The reduced rate may be attributed to increased steric congestion at the reactive center or decreased phosphine nucleophilicity.12,26 Dithioester products were also initially detected in the reaction of 3d with 5 (via NMR spectroscopy), but complex mixtures ultimately formed. It is likely that the dithioesters reacted further with CpS itself.
Scheme 2.

Cyclopropenethiones exhibit faster ligation rates than analogous cyclopropenones.
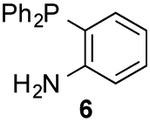
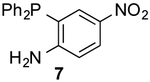
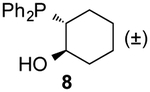
To form more stable thioamides, we evaluated CpS reactivity with amine-substituted phosphine 6. This probe did not react with 3d, though, even at elevated concentrations. Similarly poor reactivity was observed with the analogous CpO probe.12 Rapid CpO-phosphine ligations (in organic solvents) require hydrogen-bond activation (Figure S8). Thus, we hypothesized that stronger hydrogen-bond donors could boost reactivity with CpS probes. Indeed, when phosphine 7 (bearing a nitro group para to the amine) was incubated with 3d, ligation products were observed within a few hours. The lowered pKa of the pendant amine promotes reactivity, despite deactivating the phosphine via inductive effects. More nucleophilic alkyl phosphine probes also enabled efficient production of thioamides and thionoesters (Table 1 and Figure S9). Surprisingly, 3d reacted slower with phosphine 8 than phosphine 4. The opposite trend was observed with CpO analogs.12 It is possible that the CpS ligation is more dependent on initial hydrogen-bond activation. These data further imply that CpO and CpS—despite their structural similarity—might be useful probes for orthogonal labeling applications.
Table 1.
Cyclopropenethione reactivity with phosphine probes.
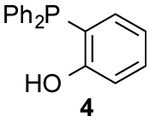
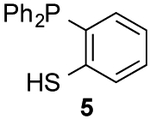
Products were not stable to isolation conditions.
Multiple addition products were observed (see Figure S7)
The reactivity and stability profiles of CpS probes suggested that dialkyl variants would be suitable for biomolecule labeling. To assess the efficiency of the CpS-phosphine ligation in this context, we performed reactions on a model protein. Hen egg-white lysozyme was non-specifically functionalized with dialkyl CpS motifs. Mass spectrometry revealed 1–5 CpS units were appended to the protein surface (Figures 3 and S10). The derivatized protein (Lys-CpS) was then treated with phosphine-biotin probe 9. This reagent was previously used to tag CpO-protein conjugates.12 Based on reactivity data from above (Figure S9), we anticipated that 9 could also efficiently ligate CpS-modified proteins to provide stable thioamide products. Indeed, rapid and quantitative conversion to the expected adducts was observed (Figures 3B and S11). The resulting conjugates were stable for >1 week at 37 °C (Figure S12). Additionally, minimal phosphine oxidation was observed upon product formation, consistent with previous reports on CpO reactivity.11–12
Figure 3.

Cyclopropenethione conjugates are readily ligated in vitro and in cell lysate. A) Lys-CpS samples were treated with a phosphine-biotin probe. B) Lys-CpS (20 μM, 1–5 modifications) was incubated with phosphine-biotin probe 9 (500 μM), and the reaction was monitored by ESI mass spectrometry. Deconvoluted mass spectra are shown. Full conversion to the expected products (Lys-Phos, 1–5 modifications) was observed. C) Lys-CpS and Lys-CpO (20 μM) were reacted with phosphine 9 (50 μM) for 0–60 min at 37 °C. Covalent adducts were detected via Western blot with streptavidin staining (top). Coomassie staining was used to assess protein loading (bottom). D) Lys-CpS (20 μM) was incubated with 9 (1 mM) in bacterial lysate for 1 h at 37 °C. Covalent adducts were detected via Western blot with streptavidin staining (top). Coomassie staining was used to assess protein loading (bottom). *Protein impurities present in commercial lysozyme stock.
Reactions with CpS scaffolds were also faster than those carried out with analogous cyclopropenones. Lysozyme samples functionalized with CpO or CpS (Lys-CpO or Lys-CpS, respectively) were prepared as above. Mass spectrometry confirmed that both conjugates were modified to a similar extent (Figure S10). The proteins were then treated with phosphine 9 and analyzed via Western blot. As shown in Figure 3C, shorter reaction times and lower phosphine concentrations were required to achieve robust labeling with Lys-CpS. Adducts were observed within minutes and reactions were complete by 30 min. By contrast, reactions with Lys-CpO required ~1 h to observe initial covalent adducts.
We further examined the CpS-phosphine ligation in a more complex setting. Probes compatible with cells and intracellular environments are often in high demand.1,27 To mimic the cellular milieu, Lys-CpS was incubated with freshly prepared cell lysate (Figure 3D). The samples were then treated with phosphine 9 and analyzed via Western blot as described above. Rapid and selective labeling was observed after just 1 h. Larger concentrations of 9 were required to achieve robust ligation, likely due to competing phosphine oxidation pathways. The lack of background reactivity observed, though, demonstrates that CpS-phosphine ligations proceed with high selectivity even in complex lysate.
In summary, we found that substituted cyclopropenethiones are viable bioorthogonal reagents. These scaffolds are stable in aqueous buffers and in the presence of biological nucleophiles. Cyclopropenethiones differ from cyclopropenones by a single atom, but react ~300-fold faster with some phosphines. Biocompatible probes with improved reaction rates are welcome additions to the bioorthogonal toolkit. CpS-phosphine ligations also proceed readily in biological buffers and cell lysate. Future experiments will examine compatibilities with live cells. Cyclopropenethiones are subject to react with strong electrophiles, and may require further tuning for successful application in some cellular environments. Cyclopropenethiones also exhibit distinct reactivity profiles compared to other bioorthogonal reagents—including cyclopropenones—suggesting additional opportunities for multi-component studies. An expanded scope of bioorthogonal reactivity will enable new pursuits in a variety of biological systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health (R01 GM126226 to J.A.P.) and the Alfred P. Sloan Foundation (J.A.P.). R.D.R. was supported by an NSF Graduate Research Fellowship. We thank the Heyduk, Nowick, and Chamberlin laboratories for providing reagents and equipment. We also acknowledge Philip Dennison (UCI) for assistance with NMR experiments, Benjamin Katz (UCI) and Felix Grün (UCI) for assistance with mass spectrometry experiments, and members of the Prescher laboratory for manuscript edits and helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details, spectroscopic data for new compounds, and additional images.
REFERENCES
- (1).Patterson DM; Nazarova LA; Prescher JA ACS Chem. Biol 2014, 9, 592. [DOI] [PubMed] [Google Scholar]
- (2).Lang K; Chin JW Chem. Rev 2014, 114, 4764. [DOI] [PubMed] [Google Scholar]
- (3).Peng T; Hang HC J. Am. Chem. Soc 2016, 138, 14423–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Prescher JA; Dube DH; Bertozzi CR Nature 2004, 430, 873. [DOI] [PubMed] [Google Scholar]
- (5).Alvarez-Castelao B; Schanzenbächer CT; Hanus C; Glock C; Dieck ST; Dörrbaum AR; Bartnik I; Nassim-Assir B; Ciirdaeva E; Mueller A; Dieterich DC; Tirrell DA; Langer JD; Schuman EM Nat. Biotechnol 2017, 35, 1196. [DOI] [PubMed] [Google Scholar]
- (6).Elliott TS; Townsley FM; Bianco A; Ernst RJ; Sachdeva A; Elsässer SJEA; Davis L; Lang K; Pisa R; Greiss S; Lilley KS; Chin JW Nat. Biotechnol 2014, 32, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Versteegen RM; Rossin R; ten Hoeve W; Janssen HM; Robillard MS Angew. Chem. Int. Ed 2013, 52, 14112. [DOI] [PubMed] [Google Scholar]
- (8).Oneto JMM; Khan I; Seebald L; Royzen M ACS Cent. Sci 2016, 2, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Patterson DM; Prescher JA Curr. Opin. Chem. Biol 2015, 28, 141. [DOI] [PubMed] [Google Scholar]
- (10).Row RD; Prescher JA Acc. Chem. Res 2018, 51, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shih H-W; Prescher JA J. Am. Chem. Soc 2015, 137, 10036. [DOI] [PubMed] [Google Scholar]
- (12).Row RD; Shih H-W; Alexander AT; Mehl RA; Prescher JA J. Am. Chem. Soc 2017, 139, 7370. [DOI] [PubMed] [Google Scholar]
- (13).Shingh S; Bhadbhade MM; Venkatesan K; Ramamurthy VJ Org. Chem 1982, 47, 3550. [Google Scholar]
- (14).Shingh S; Ramamurthy VJ Org. Chem 1985, 50, 3732. [Google Scholar]
- (15).Niu B; Jiang B; Yu L-Z; Shi M Org. Chem. Front 2018, 5, 1267. [Google Scholar]
- (16).Potts KT; Baum J Chem. Commun 1973, 0, 833. [Google Scholar]
- (17).Yoneda S; Ozaki K; Inoue T; Sugimoto A; Yanagi K; Minobe MJ Am. Chem. Soc 1985, 107, 5801. [Google Scholar]
- (18).Matsumura N; Tanaka H; Yagyu Y; Mizuno K; Inoue H; Takada K; Yasui M; Iwasaki FJ Org. Chem 1998, 63, 163. [DOI] [PubMed] [Google Scholar]
- (19).Walters CR; Szantai-Kis DM; Zhang Y; Reinert ZE; Horne WS; Chenowetha DM; Petersson EJ Chem. Sci 2017, 8, 2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Choudhary A; Raines RT ChemBioChem 2011, 12, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Batjargal S; Wang YJ; Goldberg JM; Wissner RF; Petersson EJ J. Am. Chem. Soc 2012, 134, 9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wissner RF; Batjargal S; Fadzen CM; Petersson EJ J. Am. Chem. Soc 2013, 135, 6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen X; Mietlicki-Baase EG; Barrett TM; McGrath LE; Koch-Laskowski K; Ferrie JJ; Hayes MR; Petersson EJ J. Am. Chem. Soc 2017, 139, 16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang F; Luo T; Hu J; Wang Y; Krishnan HS; Jog PV; Ganesh SK; Prakash GKS; Olah GA Angew. Chem. Int. Ed 2011, 50, 7153. [DOI] [PubMed] [Google Scholar]
- (25).Schönberg A; Mamluk M Tetrahedron Lett. 1971, 52, 4993. [Google Scholar]
- (26).Soellner MB; Nilsson BL; Raines RT J. Am. Chem. Soc 2006, 128, 8820. [DOI] [PubMed] [Google Scholar]
- (27).Shih H-W; Kamber DN; Prescher JA Curr. Opin. Chem. Biol 2014, 21, 103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.