

Abstract
Innate immune mechanisms play an important role in inflammatory chronic liver diseases. Here, we investigated the role of type I natural killer T (or iNKT) cell subsets in the progression of nonalcoholic steatohepatitis (NASH). We used α-galactosylceramide (αGalCer)/CD1d tetramers and clonotypic mAb together with intracytoplasmic cytokine staining to analyze iNKT cells in CDAA-induced murine NASH model and in human PBMCs, respectively. Cytokine secretion of hepatic iNKT cells in CDAA-fed B6 mice altered from predominantly IL-17+ to IFNγ+ and IL-4+ during NASH progression along with the down-modulation of TCR and NK1.1 expression. Importantly, steatosis, steatohepatitis and fibrosis were dependent upon the presence of iNKT cells. Hepatic stellate cell activation, infiltration of neutrophils, Kupffer cells and CD8+ T cells as well as expression of key pro-inflammatory and fibrogenic genes were significantly blunted in Jα18−/− mice and in B6 mice treated with an iNKT-inhibitory RAR-γ agonist. Gut microbial diversity was significantly impacted in Jα18−/− and in CDAA-diet fed mice. An increased frequency of CXCR3+IFNγ+T-bet+ and IL-17A+ iNKT cells was found in PBMC from NASH patients in comparison to NAFL patients or healthy controls. Consistent with their in vivo activation, iNKT cells from NASH patients remained hypo-responsive to ex-vivo stimulation with αGalCer. Accumulation of plasmacytoid DC in both mice and NASH patients suggest their role in activation of iNKT cells. In summary, our findings indicate that the differential activation of iNKT cells play a key role in mediating diet-induced hepatic steatosis and fibrosis in mice and its potential involvement in NASH progression in humans.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most frequent chronic liver disease that affects 10–20% of the population in developed countries and is increasing in prevalence with the rise of diabetes and obesity (1–3). NAFLD is characterized by abnormal accumulation of fat within liver or steatosis (NAFL) that can progress to severe inflammatory cell infiltration or nonalcoholic steatohepatitis (NASH) accompanied by necrosis and fibrosis that can lead to liver cirrhosis and hepatocellular carcinoma (4). A detailed understanding of the cellular and molecular mechanisms involved in progression from steatosis to fibrosis in NAFLD is lacking and they are crucial in the development of effective therapeutic approaches to halt this disease.
Different unconventional innate-like T cells, including γδT cells, mucosal-associated invariant T (MAIT) cells and natural killer T (NKT) cells are enriched in liver, where they can play a key role in immune homeostasis. NKT cells are the major innate-like T cells in the liver of both mice and humans. NKT cells recognize both self- and microbial lipids presented by the MHC class I-like molecule CD1d (5). CD1d-restricted NKT cells have been shown to play an important role in inflammatory conditions, including autoimmune and infectious diseases (6–9).
NKT cells can be divided in two main subsets, type I or invariant NKT (iNKT) and type II, based upon differences in TCR characteristics and some of the antigens they recognize. While both NKT cell subsets are predominantly NK1.1+ in mouse or CD161+/CD56+ in human, they also share common features in their TCR repertoire (10, 11). Murine iNKT cells express an invariant germ line encoded Vα14/Jα18 TCRα chain paired with a more diverse non-germ line TCRβ chains, including Vβ8.2, Vβ7 and Vβ2, and human iNKT cells express an invariant Vα24/Jα18 paired predominantly with Vβ11. In contrast, type II NKT cells express a relatively more diverse TCR repertoire and appear to be regulatory in nature. While the semi-invariant TCR in iNKT cells binds to CD1d in a parallel configuration that mainly involves the α chain, a type II NKT cell TCR contacts its ligand primarily via its β chain rather than α chain, suggesting that the TCR Vβ chain contributes significantly to Ag fine specificity of type II NKT cells (12). Thus, type II NKT cells use features of TCR binding shared by both iNKT cells and conventional T cells (12, 13). Therefore, both NKT cell subsets display distinct modes of Ag recognition.
One major caveat of the study of NKT cells in inflammatory liver diseases relates to the lack of specific reagents to distinguish between the two major NKT cell subsets and between NKT cells and CD3+CD56+/CD161+ conventional T cells. The most widely-used approach involves using the ability of the iNKT cells to recognize the marine sponge-derived glycolipid, alpha-galactosylceramide (αGalCer) that forms stable CD1d tetramers, to identify this subset. For type II NKT cells, there is no universal antigen marker, but we have identified a major subset of type II NKT cells that recognizes a self-glycolipid, sulfatide (13, 14). Thus, quantification of iNKT cells using α-GalCer/CD1d tetramers or type II NKT cells using sulfatide or lysophosphatidylcholine/CD1d tetramers, respectively, is crucial to identify each subset and to distinguish them from conventional T cells. For human iNKT cells, a clonotypic Ab (clone 6B11) directed against the human iNKT cell TCR can be used to differentiate them from other T cells (15). We have demonstrated that iNKT cells play a pathogenic role in Ischemia reperfusion injury, Con A-induced hepatitis, and Lieber-DeCarli liquid alcohol diet-induced liver injury, whereas the activation of type II NKT cells following administration with sulfatide or lysophosphatidylcholine induces a dominant anti-inflammatory pathway that prevents liver disease (16–19).
It has been shown that chronic feeding of choline-deficient L-amino acid-defined (CDAA) diet results in steatosis and peri-sinusoidal/peri-cellular fibrosis resembling NASH in humans (20–22). Interestingly, most of the US population does not meet the recommended intake of choline and its deficiency is known to exacerbate NAFL and NASH (23–25). In addition, humans with inadequate choline intake have oxidative damage caused by mitochondrial dysfunction and endoplasmic reticulum stress (23). Therefore, it remains unknown whether and/or how different NKT cell subsets can influence the progression from steatosis to fibrosis in a murine model of NASH and in humans. Although recent studies have suggested a potential pathogenic role for NKT cells in diet-induced liver steatosis and fibrosis (26–29), a detailed study of the role of different cytokine-secreting NKT cell subsets and related immune mechanism is lacking.
In this study, we sought to investigate the role of iNKT cell subsets in the development of NASH in a murine model following CDAA diet as well as in PBMCs from NAFLD patients. Our studies revealed the involvement of a key immune mechanism centered on different subsets of iNKT cells mediating progression from steatosis to fibrosis in experimental murine model as well as its potential role in human NAFLD. Activated IL-17- and IL-22-secreting iNKT cells were dominant in liver during the beginning of the steatosis phase, whereas IFNγ/IL-4/IL-13-secreting iNKT cells were prevalent later in the disease. In addition, activation of iNKT cells is associated with neutrophils, Kupffer cells (KCs) and CD8+ T cells infiltration and up-regulation of several key genes related to inflammation, steatosis and fibrosis. We also found a similar profile of cytokine secretion by iNKT cells in PBMC from NASH patients and, more importantly, a high frequency of CXCR3+/IFNγ+ iNKT cells in NASH patients. In addition, we found accumulation of plasmacytoid DC (pDC) into liver following CDAA diet and similar increase of pDC frequency in peripheral blood of NASH patients. Genetic depletion of pDC in BDCA2-DTR mice results in inhibition of iNKT activation, suggesting their role in activation of iNKT cells. In addition, the significant increase in the frequency of CXCR3+/IFNγ+ iNKT cells as well as pDCs in PBMC from NASH patients compared to healthy controls suggest that they could become potential novel biomarkers in the progression of NASH in humans.
Materials and Methods
Ethics statement
Animal studies were carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California San Diego. Human studies were approved by the UCSD Institutional Review Board. Informed consent was obtained from all study subjects before blood collection.
Mice
Seven to ten-week old male C57BL/6 (B6) and C57BL/6-Tg(CLEC4C-HBEGF)956Cln/J (BDCA2-DTR) (JAX stock #014176) mice were purchased from The Jackson Laboratory. CD1d−/− and Jα18−/− mice were originally generated by Van Kaer and Taniguchi (30, 31), respectively, and were kindly provided initially by Dr. Mitch Kronenberg (La Jolla Institute for Allergy and Immunology, La Jolla, CA). All mice were maintained in specific pathogen-free conditions.
Patients and healthy individuals
The study included 23 diagnosed patients with NAFLD: 5 patients with NAFL and 18 patients with NASH. In parallel, 19 healthy volunteers with no history of liver pathology were recruited. Diagnosis of NAFLD was based on the hepatic biopsies and the severity of tissue damage was evaluated on the basis of NAFLD activity score and fibrosis based on the NAFLD fibrosis risk score at the UCSD NAFLD Translational Research Unit. All patients with NASH had significantly higher NAFLD activity scores and NAFLD fibrosis scores compared with NAFL patients.
Murine NASH model
B6 and Jα18−/− mice were maintained on either a choline-deficient and L-amino-acid-defined diet (CDAA; 20% protein, 35% carbohydrate, and 45% fat, without choline; Dyets Inc., Bethlehem, PA) or a normal diet (ND; 29% protein, 58% carbohydrate, and 13% fat; LabDiet, St. Louis, MO) for 20 wk. For time-course studies, B6 mice were fed CDAA diet for 1 wk, and 1, 2, 3 or 4 months. CDAA diet has been extensively shown to induce steatohepatitis and fibrosis, which pathologically replicate the histological features of steatohepatitis and fibrosis observed in human NASH.
Isolation of liver mononuclear cells (MNCs)
To obtain MNCs, livers were harvested and pressed through a Falcon® 70μm cell strainer (Corning, NY), and suspended in HyClone™ High Glucose DMEM Medium (Thermo Fisher Scientific, Whaltham, MA). Liver MNCs were washed and the cell pellet was resuspended in 35% Percoll solution (GE Healthcare Life Sciences, Marlborough, MA) containing 100 U/ml of heparin and centrifuged at 2000 rpm at room temperature for 15 min. The supernatant was carefully removed and erythrocytes were disrupted by Red Blood Cell Lysis Buffer (Millipore Sigma, Burlington, MA). The cells were washed with DMEM supplemented with 2% of FBS and isolated liver MNCs cells were counted with a hemocytometer using trypan blue and resuspended in FACS buffer (0.02% sodium azide, 2% FBS in PBS).
Isolation of PBMC
Blood samples were taken by venipuncture into sterile, EDTA tubes. PBMCs were isolated by density gradient centrifugation on Ficoll-Hypaque (Millipore Sigma) at 2000 rpm at room temperature for 30 min. Interphase cells were removed and washed twice in PBS. The cells were counted with a hemocytometer using Türk’s solution (Millipore Sigma) and resuspended in FACS buffer (0.1% sodium azide, 1% human AB serum, 1% FBS in PBS).
Flow cytometry analysis and intracellular staining
Data were collected on a FACSCanto flow cytometer (BD) at the Flow Cytometry Research Core Facility (VA San Diego Healthcare System, San Diego) and analyzed with FlowJo v10 software (TreeStar, Ashland, OR). Mouse antibodies (clones) were as follows: anti-TCRβ (H57–597), anti-CD3 (145–2C11), anti-CD4 (GK1.5), anti-NK1.1 (PK136), anti-F4/80 (BM8), anti-CD45 (30-F11), anti-CD11c (N418), anti-CD45R/B220 (RA3–6B2), anti-CD19 (6D5), anti-I-A/I-E (M5/114.15.2), anti-Siglec H (551), anti-CD317 (mPDCA-1) (927), anti-CD103 (2E7) (from BioLegend, San Diego, CA), anti-CD11b (M1/70) and anti-Ly-6C (AL-21) (from BD Biosciences (BD), San Diego, CA). PE-conjugated αGalCer/mCD1d tetramers were generated in our laboratory, as previously described (17). To minimize nonspecific antibody binding, cells were previously incubated with anti-CD16/CD32 (Fc Block™, 2.4G2) (BD). For intracellular cytokine staining, liver MNCs were stimulated with PMA (10 ng/ml) and ionomycin (500 ng/ml) (Millipore Sigma) for 6h with addition of BD GolgiPlug containing brefeldin A at the start of culture. Cells were first stained for cell surface markers, then fixed and permeabilized using Cytofix/Cytoperm buffer and stained with the following fluorochrome-labeled mAbs: anti-IFNγ (XMG1.2), anti-IL-4 (BVD6–24G2), anti-IL-10 (JES5–16E3), anti-IL-13 (eBio13A), anti-IL-17 (eBio17B7) and anti-IL-22 (IL22JOP) (from eBioscience, San Diego, CA). Human antibodies were as follow: anti-CD3 (UCHT1), anti-CD19 (SJ25-C1), anti-CD4 (RPA-T4), anti-CD123 (CD1d42), anti-CD45 (HI30) from BD; anti-Vα24-Jα18 TCR (6B11) from eBioscience; anti-CXCR3 (49801) from R&D Systems; anti-CD14 (HCD14), anti-CD56 (5.1H11), anti-CD11c (Bu15), anti-CD303 (201A), anti-CD141 (M80), anti-CD1c (ICRF44) from BioLegend and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit from Thermo Fisher Scientific. For constitutive detection of cytokines and transcription factors in human iNKT cells, no in vitro stimulation was used, instead PBMC were stained directly for cell surface markers and, then, fixed and permeabilized as before and incubated with the following mAbs: anti-IFNγ (4S.B3), anti-IL-17A (eBio64DEC17), anti-IL-22 (22URTI), anti-IL-10 (JES3–9D7), anti-T-bet (4B10) from eBioscience, and anti-IL-4 (MP4–25D2) and anti-TNFα (MAb11) from BD.
Tazarotene treatment
Tazarotene (Tocris Biosciences, Ellisville, MO) was injected i.p. at a dose of 300 μg/mouse twice per week over the same period of CDAA feeding. Control mice were injected with DMSO/PBS. Tazarotene was initially diluted in 10% DMSO at 100 mg/ml and further diluted with PBS prior injection.
In vivo depletion of pDC
BDCA2-DTR mice and littermates were depleted of pDC by injecting 200 ng of diphtheria toxin (DT) (Millipore Sigma) on day −2 and −1 of CDAA diet. Similar DT treatment was made on day +3 and +6 post starting CDAA diet. Mice were sacrificed on day 8 and pDC depletion was verified by flow cytometry.
In vitro PBMC stimulation
PBMCs (2×106/well, duplicate) were cultured into 96-well plate in 250 μL of RPMI 1640 medium supplemented with 10% FBS, 1% L-glutamine, 1% Pen/Strep and 40 U/ml of human rIL-2 (PeproTech, Rocky Hill, NJ) with 100 ng/ml of αGalCer or without stimulation. The cells were cultured for 10 days at 37°C in an atmosphere containing 5% CO2. Supernatants of cell cultures were collected on day 4 and stored at −80oC until cytokine analysis. Cells in culture were harvested on day 10 after stimulation and stained for analysis of iNKT cells by FACS as above. Fold expansion of iNKT cells was calculated by dividing frequencies of iNKT cells in PBMC cultures containing αGalCer during 10-day expansion by the frequencies on day 0 without stimulation. Secretion of cytokines in the supernatants was determined using the BD CBA Human Th1/Th2/Th17 Cytokine Kit (BD).
Histology
Liver tissues were fixed in 10% formaldehyde solution and kept at room temperature until use. H&E staining of tissue sections (5μm) was performed at the UCSD Neuroscience Core for microscopy. Steatosis and liver fibrosis were assessed with Sirius Red/Fast Green staining. Myeloperoxidase staining was performed using anti-Myeloperoxidase Ab-1 rabbit polyclonal Ab (Thermo Fisher Scientific). All pictures were taken using the NanoZoomer 2.0-HT Slide Scanning System (Hamamatsu, Japan) and quantitated on Image J software (NIH).
Microbiome analysis
Stool collection:
Fresh fecal pellets (3–4) were collected individually from mice (each mouse was placed in a confined place to avoid cross-contamination) using sterile cotton swabs into individually labeled sterile 0.6 ml Eppendorf tube. Samples were collected during two days from colonies of mice of mixed age and sex that were maintained in our animal facility for a minimum of 2–3 weeks to 8 months. Following collection, tubes containing stool samples were stored at −80oC until analysis. For CDAA diet-fed groups, mice were age and sex matched and fecal pellets were collected similarly but at the end of the experiment.
16S rRNA gene sequencing:
DNA was extracted using the MO BIO PowerSoil DNA extraction kit according to Earth Microbiome Project (EMP) standard protocols (32) (http://www.earthmicrobiome.org/emp-standard-protocols/). PCR targeting the V4 region of the 16S rRNA bacterial gene was performed with the 515F/806R primers, utilizing the protocol described in Walters et al. (33). Amplicons were barcoded and pooled in equal concentrations for sequencing. The amplicon pool was purified with the MO BIO UltraClean PCR Clean-up kit and 2 × 150 bp sequencing was performed on the MiSeq sequencing platform at the Institute for Genomic Medicine at UCSD.
16 rRNA sequencing data processing and analysis:
Raw sequencing data was deposited in Qiita and processed via QIIME v1.9.1 (34) in Qiita using parameters for demultiplexing and quality control recommended by Bokulich et al. (35). Forward reads were trimmed to 150 bp and then processed with deblur (36), a de novo sub-Operational Taxonomic Unit picking method. The resulting sOTUs were inserted into the Greengenes tree (37) and assigned taxonomy via SEPP (SATé-Enabled Phylogenetic Placement using q2-fragment-insertion in QIIME2 (34, 38), https://qiime2.org). sOTUs were directly visualized in QIIME2 via Faith’s Phylogenetic Diversity (39) and Unweighted and Weighted UniFrac distances (40–42) were computed for each experimental group and then results were visualized in EMPeror (43, 44). All raw sequencing data is publically available in Qiita study 10781 (https://qiita.ucsd.edu/study/description/10781).
RNA, DNA, RT-PCR and Real-time PCR analysis
Total RNA was isolated from liver tissues using a PureLink RNA Mini Kit (Ambion, Thermo Fisher Scientific). RNA was quantified using NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific) and reverse transcribed using RevertAid First Strand cDNA Synthesis Kit (Ambion). To detect mRNA of Vα14Jα281 gene, 5 μg of total RNA were reverse transcribed and subjected to a nested PCR as previously described (45) using the following primer pairs (IDT, IA): For first-round PCR, Vα14 Leader, 5′-ATGAAAAAGCGCCTGAGTGCC-3′, C-rev1 reverse, 5′-CAGGAGGATTCGGAGTCCCA-3′; for nested PCR, Vα14/Jα281, forward, 5-TAAGCACAGCACGTGCACAT-3′, Vα14/Jα281, reverse, 5′-CAATCAGCTGAGTCCCAGCT-3′. Real-time PCR was performed using KAPA SYBR Fast Master Mix (Thermo Fisher Scientific) on a Step One Plus Real-Time PCR System (Applied Biosystems) with specific PCR primer pairs. Data were analyzed using the ΔΔ cycle threshold (ΔΔCT) method against ND-fed WT mice. Genomic DNA was extracted from the tail of BDCA2-DTR littermates’ mice using DNeasy Blood & Tissue Kit (Qiagen) and subjected to PCR following The Jackson Laboratory protocol (Tg(CLEC4C-HBEGF).
Western blot
Whole cell lysates of liver (50 μg) were separated on a gradient (4–20%) polyacrylamide NuPAGE gel (Thermo Fisher Scientific) and transferred onto nitrocellulose membranes and blotted with anti-α-SMA mAb (GeneTex Inc., Irvine, CA) or anti-β-actin (Santa Cruz, Dallas, TX). Proteins were detected with peroxidase-conjugated secondary Ab. Protein bands were visualized using an enhanced chemiluminescence reagent and digitized using ChemiDoc® Imagen System (Bio-Rad). Expression intensity was quantified by Image Lab.
Statistical analyses
Data were analyzed using GraphPad Prism v7 software (GraphPad Software). For all analyses, data were presented as mean ± SEM. For comparison between two groups, unpaired t-test for mouse data or two-tailed Mann Whitney test for human data was applied. For comparison between three groups (i.e., frequency of iNKT as well as cytokine secretion or transcription factor expression in human PBMCs), one-way ANOVA with Tukey’s correction was applied. . For all analyses, a probability values were indicated as follow: *p≤ 0.05, **p≤0.01, ***p≤ 0.001, ****p≤ 0.0001
Results
Activation and differential cytokine secretion profiles of iNKT cells in a diet-induced NASH model
Since chronic feeding of CDAA diet results in liver damage resembling NASH in humans (20–22), we investigated whether progression of diet-induced steatosis to fibrosis is associated with the alterations in the cytokine secretion profile of iNKT cells in liver. Liver MNCs derived from B6 mice maintained on standard ND or CDAA diet for 1 wk, 1, 2, 3 or 4 months were analyzed using αGalCer/CD1d-tetramers and intracellular cytokine-based flow cytometry approach. The results showed that IL-17+- and IL-22+-iNKT cells (NKT17) as well as IL-10+-iNKT cells (NKT10) (TCRβ+NK1.1+αGalCer/CD1d-tet+) increased significantly during progression of steatosis reaching a peak at 3 months of CDAA diet as compared to ND-fed B6 mice (Fig. 1A and Suppl. Fig. 1). However, there was no significant increase in IL-17+- and IL-22+-conventional T cells (TCRβ+NK1.1+αGalCer/CD1d-tet−) at these time points (Suppl. Fig. 2). Interestingly, IFNγ+-, IL-4+- and IL-13+-iNKT cells (NKT1/NKT2) increased significantly much later during steatosis/fibrosis compared with mice fed ND (Fig. 1A and Suppl. Fig. 1). At this later stage following CDAA diet, even conventional T cells significantly produced IFNγ but not IL-4 (Suppl. Fig. 2). In summary, the cytokine secretion profile of iNKT cells skewed from predominantly NKT17- to NKT1/NKT2-like and it correlates with the progression from steatosis to fibrosis following CDAA diet.
Figure 1. Differential activation and cytokine secretion profiles of hepatic iNKT cells in a diet-induced NASH model.
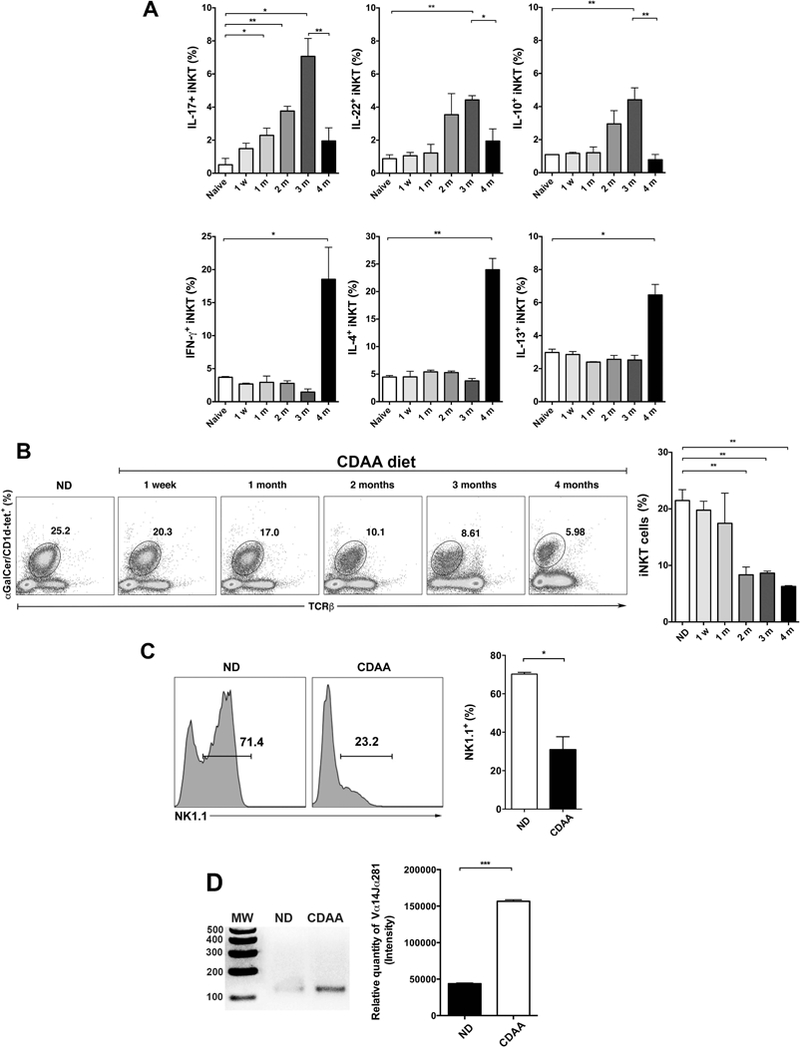
(A) Wild type C57BL/6 (B6) mice were maintained on standard ND diet or were fed CDAA diet for 1 wk, 1, 2, 3 or 4 months. Liver MNCs from each experimental group were stimulated in vitro with PMA and ionomycin for 6 h in the presence of GolgiPlug. Cells were stained with anti-TCRβ, anti-NK1.1 and αGalCer/CD1d tetramer before they were fixed, permeabilized, and stained with anti-IFNγ, anti-IL-17, anti-IL-22, anti-IL-10, anti-IL-4 and anti-IL-13 mAbs. iNKT cells were identified as double TCRβ- and αGalCer/mCD1d tetramer-positive cells (TCRβ+/αGalCer/mCD1d tetramer+) in the NK1.1+ gate. Cytokine-secreting cells were gated on iNKT cells. Bar graphs show the percentage of iNKT cells secreting IFNγ, IL-17, IL-4, IL-10, IL-13, and IL-22 in liver of ND- or CDAA diet-fed B6 mice for the indicated time points. Each group included 3 mice. (B) Representative dot plots of iNKT cells in liver of mice as indicated in (A). Numbers on dot plots indicate the percentage of αGalCer/CD1d tetramer+TCRβ+ cells within the drawn gate. Bar graph shows the percentage of tetramer+ cells in liver of mice as indicated in (A). (C) Histograms show representative NK1.1 expression on gated tetramer+ cells as indicated in (A) in liver of B6 mice fed ND or CDAA diet. Bar graph shows the percentage of NK1.1+ cells gated on αGalCer/CD1d tetramer+TCRβ+ cells. (D) Representative gel of Vα14Jα18 mRNA expression in liver of B6 mice fed ND or CDAA diet for 3 months as determined by nested PCR. Jα18−/− mice were used as negative control. Bar graph show the relative quantity of PCR product liver of B6 mice fed ND diet (n=2) or CDAA diet for 3 months (n=3) All data are presented as mean ± SEM. Data representative of three independent experiments. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001 by unpaired two-sample t-test; ns, not significant.
Since iNKT cells can down-regulate their TCR following chronic stimulation, we compared hepatic iNKT cells between B6 mice fed CDAA diet vs. ND diet using αGalCer/CD1d-tetramer staining. The results revealed a significant decrease in the tetramer staining of iNKT cells in mice fed CDAA diet compared with ND-fed mice (Fig. 1B). We next investigated whether chronic activation of iNKT cells also leads to down-regulation of cell surface expression of NK1.1 in addition to TCR. As shown in Fig. 1C, NK1.1 expression was also significantly down-regulated in iNKT cells after CDAA diet in comparison to the ND. To further examine that the reduced tetramer staining results from TCR down-modulation but not from deletion of iNKT cells following chronic activation, we used a semi-quantitative RT-PCR approach to analyze the Vα14-Jα18 TCR transcript expression. As expected, the relative quantity of hepatic Vα14-Jα18 PCR product was not decreased but significantly increased in mice fed CDAA diet compared with ND-fed mice (Fig. 1D). Collectively, our data indicate that chronic activation of hepatic iNKT cells is associated with cytokine profile changes and down-regulation of both cell surface NK1.1 and invariant TCR during CDAA diet-induced hepatic steatosis and fibrosis.
iNKT cell-deficient Jα18−/− mice are protected from diet-induced hepatic steatosis and fibrosis
To further investigate the role of iNKT cells in the pathogenesis of NASH, iNKT cell-deficient Jα18−/− and B6 mice were fed ND or CDAA diet. As shown in Figure 2A, CDAA diet significantly increased body and liver weight in both B6 and Jα18−/− mice compared with ND. Liver weight was significantly reduced in Jα18−/− mice compared with B6 mice. But, epididymal adipose tissue (EPI) weight did not differ significantly between B6 and Jα18−/− mice following CDAA diet. However, ALT levels and triglycerides were significantly reduced in Jα18−/− liver compared to B6 mice (Fig. 2B). More importantly, CDAA diet resulted in significant liver damage as indicated by marked steatosis, hepatocyte ballooning and collagen deposition compared with ND-fed B6 mice (Fig. 2C), but Jα18−/− mice had significantly less liver damage than B6 mice. These results indicate that mice lacking iNKT cells (Jα18−/−) were significantly protected from steatosis and fibrosis following CDAA diet, suggesting a pathogenic role of iNKT cells in NASH progression in mice.
Figure 2. iNKT cell-deficient Jα18−/− mice are protected from diet-induced hepatic steatosis and fibrosis.
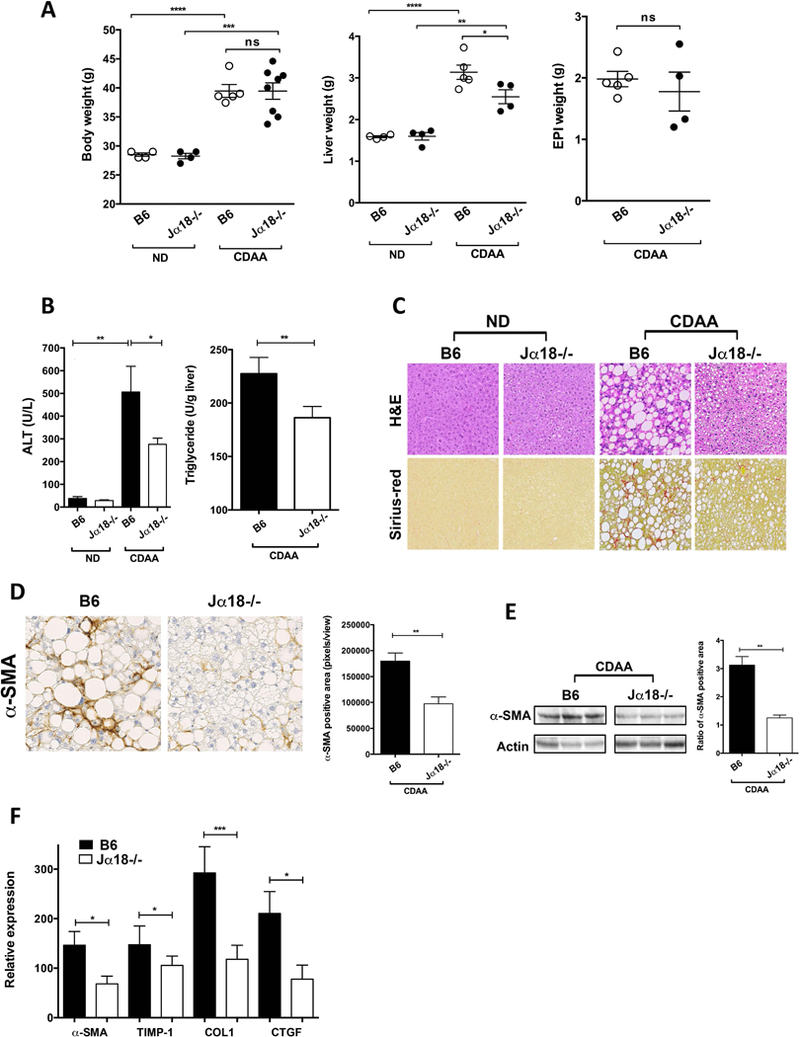
B6 and Jα18−/− mice were feed ND or CDAA diet for 20 wk. (A) Body, liver and EPI weight of B6 and Jα18−/− mice following ND or CDAA diet are shown. Each point represents an individual mouse of at least four mice in each group. (B) Bar graphs show the alanine aminotransferase (ALT) levels in the serum of B6 and Jα18−/− mice following ND or CDAA diet (left) and triglycerides levels in liver of CDAA-fed B6 and Jα18−/− mice. Each group included five mice. (C) Representative H&E and Sirius-red staining of liver sections from B6 and Jα18−/− mice fed ND or CDAA diet. Photomicrographs were prepared at 100x magnification. (D) Representative immunohistochemistry staining for alpha-smooth muscle actin (α-SMA) in liver sections from B6 and Jα18−/− mice following CDAA diet. Photomicrographs were prepared at 100x magnification. Bar graph shows the quantification of α-SMA positive areas in the indicated groups. Each group included five mice. (E) Representative western blot of α-SMA protein expression in liver homogenates from B6 and Jα18−/− mice fed CDAA diet. Bar graph shows the quantification of the ratio of α-SMA positive area in the indicated groups. Each group included five mice. (F) Bar graph show the relative expression of selected fibrogenic genes in liver tissue from B6 and Jα18−/− mice following CDAA diet as determined by quantitative RT-PCR. The expression of each gene was normalized using a reference control gene and its relative expression calculated considering the expression levels in ND-fed B6 mice. Each group included four to seven mice. All data are presented as mean ± SEM. Data representative of three independent experiments. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 by unpaired two-sample t-test; ns, not significant.
To determine whether hepatic stellate cell (HSC) activation is also influenced by iNKT cells following CDAA diet, we assessed alpha-smooth muscle actin (α-SMA) expression by immunohistochemistry in liver sections from CDAA-fed B6 and Jα18−/− mice. Our results show that α-SMA positive areas were significantly reduced in Jα18−/− liver compared with B6 (Fig. 2D). Consistent with this, α-SMA expression levels, measured via α-SMA western blot, were also reduced in Jα18−/− liver compared with B6 following CDAA diet (Fig. 2E). Accordingly, quantitative real-time RT-PCR analyses confirmed that α-SMA transcripts were significantly reduced in Jα18−/− liver compared with B6 mice (Fig. 2F). Furthermore, other key fibrogenic genes like TIMP Metallopeptidase Inhibitor 1 (TIMP-1), collagen (COL1) and Connective Tissue Growth Factor (CTGF) showed significantly reduced expression in CDAA-fed Jα18−/− mice in comparison to B6 mice (Fig. 2F). These results indicate a key role for iNKT cells in diet-induced HSC activation, steatosis, and fibrosis in liver.
Reduced liver inflammation in Jα18−/− mice following CDAA diet
Next, we examined whether hepatic inflammatory pathways are involved in protecting Jα18−/− mice from diet-induced steatosis and fibrosis. Indeed, CDAA feeding of Jα18−/− mice resulted in a significant reduction of hepatic neutrophil infiltration, analyzed by myeloperoxidase (MPO) staining, compared with CDAA-fed B6 mice (Fig. 3A). Moreover, the expression level of several key inflammatory genes, including TNFα, IL-6, KC, and MIP-2, as well as genes related to inflammasome activation, like NLRP3 (Nucleotide-binding domain, leucine rich pyrin containing 3) and IL-1β, was significantly reduced in Jα18−/− liver compared to B6 following CDAA diet (Fig. 3B). However, genes involved in gluconeogenesis or lipid metabolism did not differ between Jα18−/− and B6 mice following CDAA diet, except for down-regulation of PEPCK (phosphoenolpyruvate carboxykinase) in Jα18−/− mice (Fig. 3B). These results suggest that diet-induced liver inflammation and steatohepatitis are reduced in the absence of iNKT cells confirming their critical role in mediating diet-induced NASH.
Figure 3. Reduced liver inflammation in iNKT cell-deficient Jα18−/− mice following CDAA diet.
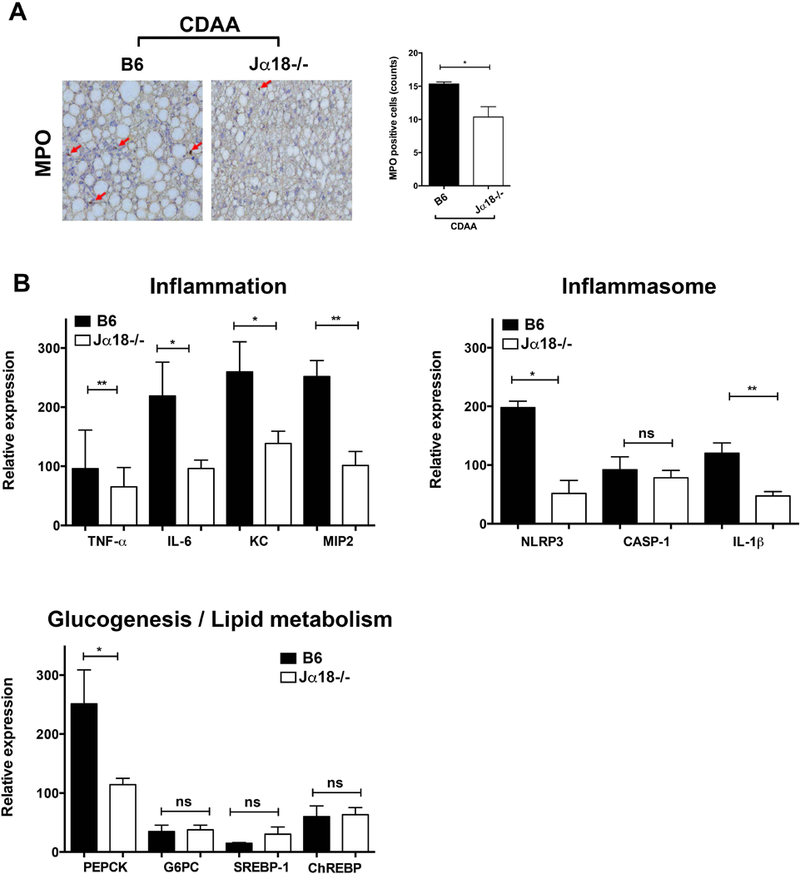
B6 and Jα18−/− mice were fed ND or CDAA diet for 20 wk. (A) Representative immunohistochemistry staining for myeloperoxidase (MPO) in liver sections of B6 and Jα18−/− mice following CDAA diet. Photomicrographs were prepared at 100x magnification. Bar graph shows the quantification of MPO positive cells in the indicated groups. Each group included four mice. (B) Bar graphs show the relative expression of selected genes in liver tissues from B6 and Jα18−/− mice following CDAA diet as determined by quantitative RT-PCR. The expression of each gene was normalized using a reference control gene and its relative expression calculated considering the expression levels in ND-fed B6 mice. Each group included five mice. All data are presented as mean ± SEM. Data representative of three independent experiments. *p < 0.05, **p ≤ 0.01 by unpaired two-sample t-test.
A RAR-γ agonist treatment inhibits effector function of iNKT cells and protects mice from CDAA-induced steatosis and fibrosis
We have recently shown that an agonist of the RAR-γ, Tazarotene, specifically inhibits iNKT cells but not type II NKT cells or conventional T cells and significantly protects mice from alcoholic liver disease and carbon tetrachloride (CCL4)-induced fibrosis (18). To determine whether functional inhibition of iNKT cells protects mice from diet-induced liver injury, CDAA-fed B6 mice were treated twice a week i.p. with Tazarotene (300 μg/mouse) or DMSO/PBS. As shown in Figure 4A, Tazarotene treatment significantly blunted CDAA-induced increase in body, liver and EPI weight as compared with untreated mice. More importantly, Tazarotene treatment significantly reduced steatosis, collagen deposition and HSCs activation following CDAA diet (Fig. 4B). Next, we investigated whether suppression of liver disease results from the inhibition of iNKT cells in Tazarotene-treated animals. We determined the function of iNKT cells in both control and treated groups of mice by measuring proliferative response and cytokine secretion after in vitro stimulation with αGalCer. As shown in Figure 4C, both proliferation and cytokine secretion by iNKT cells were significantly inhibited following Tazarotene treatment. Additionally, we found that functional inhibition of iNKT cells was not consequence of reduced numbers of iNKT cells in Tazarotene-treated mice as shown by similar numbers of αGalCer/CD1d-tetramer+ cells and Vα14-Jα18 PCR products in livers of both groups of animals (Figure 4D). Overall, these data are consistent with the data shown in CDAA-fed Jα18−/− mice. Thus, both genetic deficiency as well as functional inhibition of iNKT cells leads to the inhibition of diet-induced hepatic steatosis and fibrosis.
Figure 4. Treatment of B6 mice with a RAR-γ agonist inhibits effector function of iNKT cells and protects mice from CDAA-induced steatosis and fibrosis.
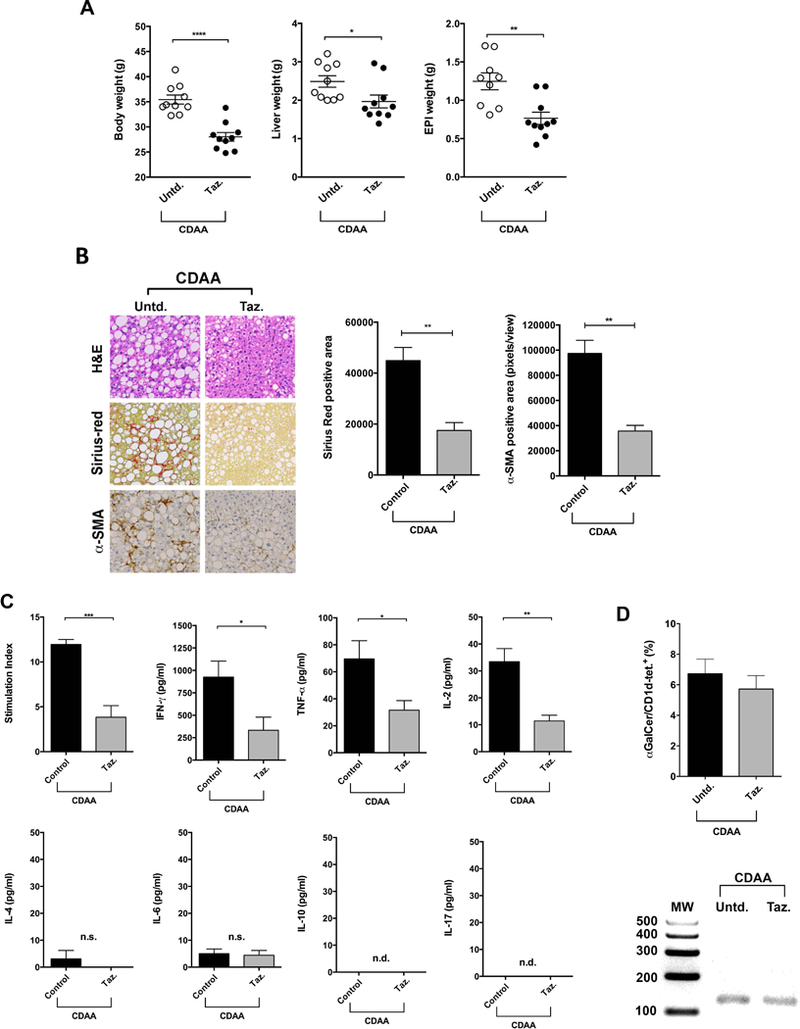
B6 mice maintained on CDAA diet for 20 wk were subjected to i.p. injection of Tazarotene (300 μg/mouse) twice per week or left untreated. (A) Body, liver and EPI weight in B6 mice untreated or treated with Tazarotene following CDAA diet are shown. Each point represents an individual mouse. Each group included ten mice. (B) Representative H&E, Sirius-red and α-SMA staining of liver sections from CDAA-fed B6 mice untreated or treated with Tazarotene. Photomicrographs were prepared at 100x magnification. Bar graphs show the quantification of Sirius-red positive area (left) and α-SMA positive areas (right) in the indicated groups. (C) Bar graphs show proliferative response (Stimulation Index) and cytokine secretion (IFNγ, TNFα, IL-2, IL-4, IL-6, IL-10 and IL-17) in splenocytes from untreated (Control) and Tazarotene-treated mice (n= 5 mice/group) in response to an in vitro challenge with αGalCer at an optimum concentration (1 ng/ml). (D) Bar graph shows the percentage of αGalCer/CD1d tetramer+TCRβ+ cells in liver of CDAA-fed B6 mice untreated or treated with Tazarotene (top panel). Representative gel of Vα14Jα18 mRNA expression in liver from untreated and Tazarotene-treated mice (bottom panel). All data are presented as mean ± SEM. Data representative of three independent experiments. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 by unpaired t-test; n.s., not significant; n.d., not detected; Untd, untreated; Taz., Tazarotene.
Hepatic infiltration by CD8+ T cells and macrophages following CDAA diet is dependent on iNKT cells
Recent data has suggested an important role of hepatic CD8+ T cells and KCs in murine models of steatosis and fibrosis (28, 46, 47). Next, we determined the role of iNKT cells in hepatic infiltration of CD8+ T cells in diet-induced liver disease. As expected, CDAA diet significantly increased the frequency of CD8+ T cells in liver MNCs compared with ND (Fig. 5A). We found that the hepatic infiltration of CD8+ T cells in CDAA-induced liver damage was significantly decreased in Jα18−/− mice as compared with B6 mice (Fig. 5A).
Figure 5. Hepatic infiltration by CD8+ T cells and macrophages following CDAA diet is dependent on iNKT cells.
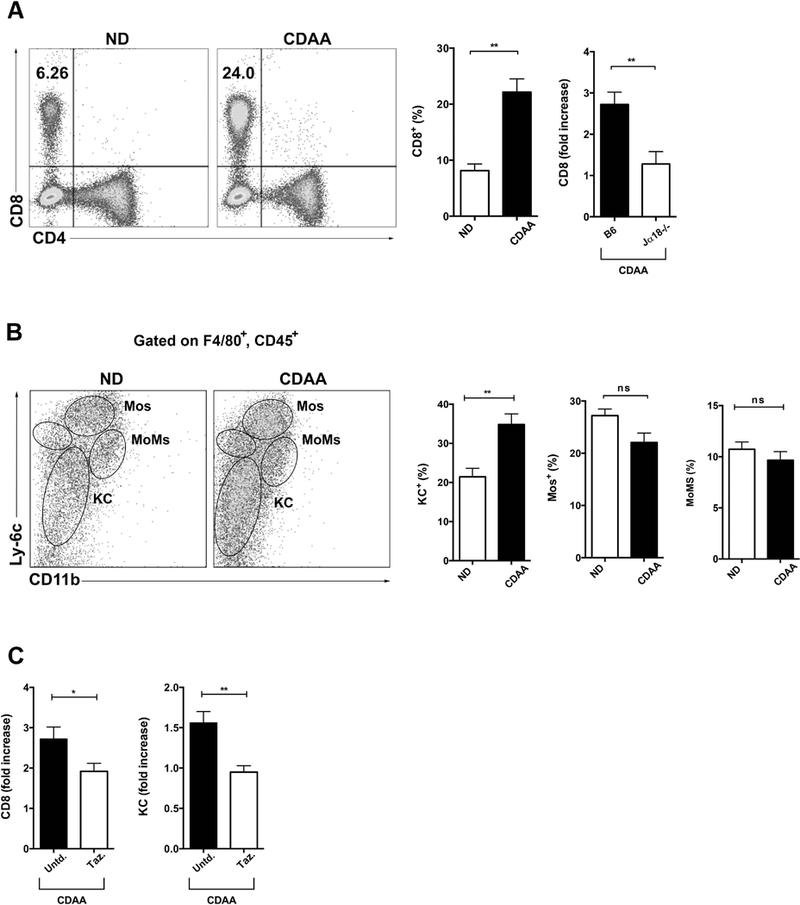
B6 and Jα18−/− mice were feed ND or CDAA diet for 20 wk. (A) Representative dot plot of CD8+ T cells in liver of B6 mice fed ND or CDAA diet. Numbers on dot plots indicate percentage of CD8+ T cells on the lymphocyte gate. Bar graphs show the percentage of CD8+ T cells (left) in liver of B6 mice after 20 wk of ND or CDAA diet and fold increase of CD8+ T cells (right) in liver of B6 and Jα18−/− mice fed CDAA diet. Fold increase was obtaining by dividing the percentages of CD8+ T cells in liver of B6 or Jα18−/− mice fed CDAA diet by that in liver of ND-fed B6 mice. At least four mice per group. (B) Representative dot plots of liver macrophages cells in B6 mice fed ND or CDAA diet. Gated on CD45+F4/80+ cells, Kupffer cells (KCs) were defined as Ly-6CloCD11blo, inflammatory monocytes (Mos) as Ly-6ChiCD11bint and monocyte-derived macrophages (MoMs) as Ly-6CintCD11bhi as described before (32). Each subset is indicated on the dot plots. Bar graphs show the percentage of KCs, Mos and MoMs in liver of B6 mice fed ND (n=4) or CDAA (n=7–9) diet. (C) Bar graphs show the fold increase of CD8+ T cells (left) and KCs (right) in liver of CDAA-fed B6 mice untreated or treated with Tazarotene. Fold increase was obtaining by dividing the percentage of CD8+ T cells or KCs in liver of untreated or Tazarotene treated CDAA-fed B6 mice by that in liver of ND-fed B6 mice. B6 mice maintained on CDAA diet for 20 wk were subjected to i.p. injection of Tazarotene (300 μg/mouse) twice per week or left untreated. All data are presented as mean ± SEM. Data representative of three independent experiments. *p < 0.05, **p ≤ 0.01 by unpaired two-sample t-test; ns, not significant; Untd, untreated; Taz, Tazarotene.
Since inflammatory macrophages also play a significant role in liver injury, we also investigated whether CDAA diet promoted hepatic macrophage infiltration. We followed three distinct subsets of pro-inflammatory macrophages within liver: Kupffer cells (KC), monocytes (Mos) and monocyte-derived macrophages (MoMs) gated on CD45+F4/80+ cells as described before (48). Indeed, the frequency of hepatic KC was significantly increased in CDAA-fed B6 mice compared with ND-fed B6 mice (Fig. 5B), however, the frequency of monocytes (Mos) and monocyte-derived macrophages (MoMs) did not differ between B6 mice feeding CDAA diet or ND (Fig. 5B).
To further confirm that the effects on CD8+ T cells and KC mediated by CDAA diet were dependent on functional iNKT cells, B6 mice maintained on CDAA diet for 20 wk were treated with Tazarotene (300 μg/mouse) as before. Tazarotene treatment significantly reduced the frequency of both CD8+ T cells and KC in liver of CDAA-fed B6 mice as compared with untreated mice (Fig. 5C). These results confirm the key role of iNKT cells in mediating liver infiltration by CD8+ T cells and KC consistent with our results in CDAA-fed Jα18−/− mice.
iNKT cells from NASH patients are activated and secrete pro-inflammatory cytokines
We next sought to determine whether there was evidence of a similar change in the frequency and cytokine profile of circulating iNKT cells in patients with NAFLD as we observed in the murine model of NASH. We first compared the frequency of iNKT cells in peripheral blood between patients with NAFLD (5 patients with NAFL and 18 patients with NASH) and 19 healthy controls using a mAb that recognize the invariant CDR3 loop of the human canonical Vα24Jα18 TCR α chain (clone 6B11) (15) in a multi-parameter flow cytometry approach to specifically identify human iNKT cells (Fig. 6A). The selective labeling of iNKT cells was validated by αGalCer/human CD1d tetramer staining that show equivalent frequencies (data not shown). The frequency of circulating iNKT cells was not significantly different between groups. However, NASH patients exhibited high frequency of circulating iNKT cells (0.34% ± 0.18 vs. 0.1% ± 0.03 in healthy controls, and vs. 0.08% ± 0.05 in NAFL patients) (Fig. 6B). We next examined the functionality of iNKT cells in NAFLD patients and healthy controls by measuring the spontaneous production of IFNγ, IL-4, IL-17A, IL-22, IL-10 and TNFα as well as the expression of CXCR3 and T-bet. Compared with both NAFL patients and healthy controls, iNKT cells from NASH patients secreted significantly more IFNγ, as measured by geometric mean fluorescence intensity (MFI: 679.4 ± 130.1 vs. 183.2 ± 30.1 in healthy controls and vs. 171.3 ± 69.3 in NAFL patients), than NAFL patients and healthy controls (Fig. 6C, first panel). This was consistent with the analysis of the transcription factor T-bet, which was also highly expressed by iNKT cells from NASH patients compared to healthy control (MFI: 9763 ± 2017 vs. 2458 ± 330.3 in healthy controls) (Fig. 6C, second panel). Consistent with a pro-inflammatory profile, iNKT cells from NASH patients expressed significantly more CXCR3 compared with iNKT cells from both NAFL patients and healthy controls (MFI: 1422 ± 194.4 vs. 559.4 ± 74.6 in healthy controls and vs. 550.8 ± 59.1 in NAFL patients) (Fig. 6C, third panel). IL-4 secretion was not significantly different in iNKT cells from NASH or NAFL patients and healthy controls (Fig. 6C, last panel). Furthermore, 55% of NASH patients (10 out of 18) had a population of iNKT cells that produce more IFNγ than IL-4 (p<0.001, Fisher test), whereas 87.5% of healthy controls (14 out of 16) had a population of iNKT cells that constitutively secrete more IL-4 than IFNγ (p=0.0083, Fisher test) (data not shown).
Figure 6. iNKT cells from NASH patients are activated and secrete pro-inflammatory cytokines.
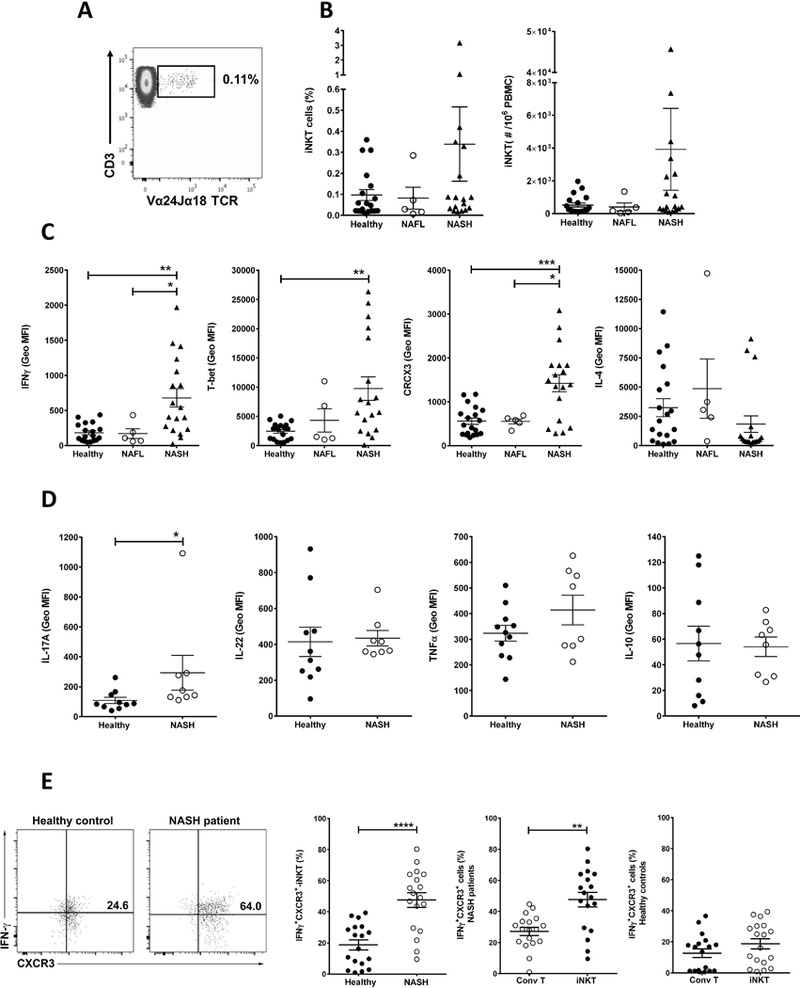
PBMC were analyzed by multi-parameter flow cytometry. iNKT cells were identified using the following gate strategy: first, lymphocytes were gated based on FSC and SSC and singlet live cells were selected. CD19+ cells were excluded and CD3+ T cells were gated in the CD19− gate. iNKT cells were defined as double CD3- and Vα24-Jα18 TCR-positive cells (CD3+Vα24Jα18TCR+) in the CD3+ gate and expressed as percentage of CD3+ T cells. Conventional T cells were identified as CD3-positive and simultaneously Vα24-Jα18 TCR-negative cells (CD3+/Vα24-Jα18 TCR−) in the live/CD19− gate. (A) Representative dot plot of iNKT cells in PBMC from healthy control. Number on dot plot indicate the percentage of iNKT cells within the drawn gate. (B) Scatter graphs show the cumulative results of the percentage (left) and numbers (right) of iNKT cells in PBMC from healthy controls (n=19) and patients with NAFL (n=5) or NASH (n=18). (C) Scatter graphs show the geometric mean fluorescent intensity of IFNγ, T-bet, CXCR3 and IL-4 staining of unstimulated iNKT cells in PBMC from healthy controls and patients with NAFL or NASH. (D) Scatter graphs show the geometric mean fluorescent intensity of IL-17A, IL-22, TNFα and IL-10 staining of unstimulated iNKT cells in PBMC of NASH patients (n=8) and healthy controls (n=10–11). (E) Representative dot plots of IFNγ+CXCR3+ double-positive cells in the iNKT gate in PBMC of healthy controls and NASH patients. Scatter graphs show the percentage of IFNγ+CXCR3+ iNKT cells in PBMC of healthy controls (n=18) and NASH patients (n=18) (left) and the percentage of IFNγ+CXCR3+ double-positive cells in the conventional CD3+ T cell gate (Conv T) and iNKT gate in PBMC of NASH patients (center) and healthy control (right). Conv T were identified as CD3+Vα24Jα18TCR− cells in the CD3+ gate. Each point represents an individual. All data are presented as mean ± SEM. *p≤ 0.05, **p≤ 0.01, ***p≤ 0.001 and ****p≤ 0.0001 by one way Anova or Mann–Whitney U test.
Additionally, iNKT cells from NASH patients significantly secreted more IL-17A compared to healthy controls (MFI 293.6 ± 116.6 vs. 108.1 ± 20.9) (Fig. 6D). However, when the level of liver fibrosis was considered, patients with advance fibrosis (Fibrosis score 2–4) had lower levels of IL-17A and higher levels of IFNγ compared to patients with Fibrosis score ≤ 1 (data not shown). Moreover, iNKT cells from NASH patients secreted similar amounts of IL-22, TNFα and IL-10 compared with iNKT cells from healthy controls (Fig. 6D). These data suggest that iNKT cells from NASH patients are more activated and pro-inflammatory as defined by higher expression of CXCR3, T-bet, IL-17A and IFNγ.
It is interesting that conventional CD3+CD4+ T cells were significantly increased in peripheral blood of NASH patients compared to healthy controls (67.6% ± 2.6 vs. 59.6% ± 2.2), and, consequently, the CD4+/CD8+ ratio was also significantly increased in NASH patients, however, the frequency of CD8+ T cells was not significant different between NASH patients and healthy controls. NAFL patients showed similar trend as NASH patients but the difference did not reach statistical significance (data not shown).
Interestingly, we detected a subset of iNKT cells that co-express IFNγ and CXCR3 (IFNγ+CXCR3+ iNKT cells) significantly increased in NASH patients compared to healthy controls (47.64% ± 4.73 vs. 18.78% ± 3.2) (Fig. 6E). Also, IFNγ+CXCR3+ iNKT cells were significantly increased in NASH patients, but not in healthy controls, compared to IFNγ+CXCR3+ conventional CD3+ T cells (CD3+Vα24Jα18TCR−) (47.64% ± 4.73 vs. 27.15% ± 2.61) (Fig. 6E). These results indicate that IFNγ+CXCR3+ iNKT cells were more prevalent in NASH patients than in healthy controls.
iNKT cells in NASH patients are chronically activated and hypo-responsive to in vitro stimulation with αGalCer
Since, iNKT cells following chronic activation in vivo can become refractory to subsequent ex vivo stimulation, we determined their ex-vivo response to αGalCer in PBMC from NASH patients and compared to those from healthy controls. As shown in Figure 7A, iNKT cells expansion was significantly decreased in NASH patients. The fold expansion of iNKT cells in the cultures was significantly reduced in NASH patients (25.6 ± 8.5) compared with healthy controls (93 ± 39.3) (Fig. 7B). Consistent with their hypo-responsiveness, the secretion of several cytokines, including IL-6, IFNγ, IL-17A and IL-4, after αGalCer stimulation was also significantly reduced in NASH patients compared with healthy controls (Fig. 7C). Collectively, these data further indicate chronic in vivo activation of iNKT cells in NASH progression.
Figure 7. iNKT cells in NASH patients are hypo-responsive to an in vitro stimulation with αGalCer.
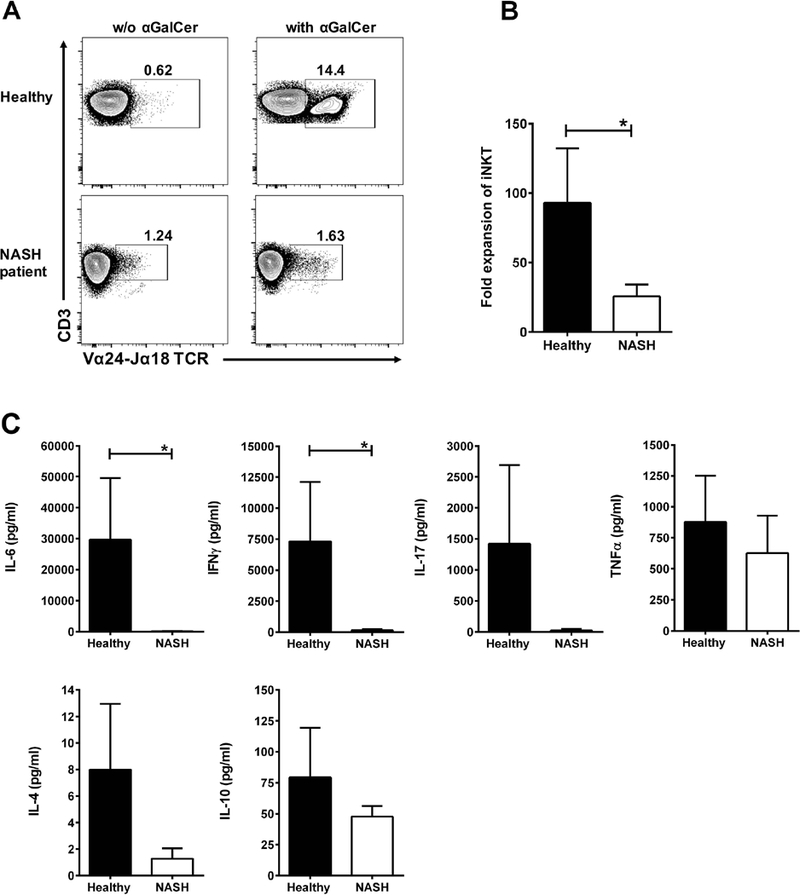
Fresh PBMCs from NASH patients (n=8) and healthy controls (n=9) were cultured (2×106/well) in duplicate for 10 days with αGalCer [100 ng/ml] in 200 μL of completed RPMI medium plus 40 U/ml of human rIL-2 in flat bottom 96-well plates at 37°C in an atmosphere containing 5% CO2. Supernatants collected after 4 days of cultured. (A) Representative contour plots of iNKT cells in PBMC of healthy controls (top) and NASH patients (bottom) after 10-day culture without or with αGalCer. Numbers on dot plots indicate the percentage of iNKT cells. (B) Bar graph show the fold expansion of iNKT cells in PBMC of healthy controls and NASH patients after 10-day culture with αGalCer. The fold expansion of iNKT cells was obtaining by dividing the percentage of iNKT cells after 10-day culture by that on day 0 (fresh PBMC). (C) Bar graphs show the quantification of IL-6, IFNγ, IL-17, TNFα, IL-4 and IL-10 (pg/ml) in culture supernatants from PBMC of healthy controls and NASH patients after 4-day culture with αGalCer. Cytokine secretion was quantified using BD™ CBA Human Th1/Th2/Th17 kit. All data are presented as mean ± SEM. *p≤ 0.05 by Mann–Whitney U test.
DC subsets are altered and involved in iNKT activation in both murine and human NASH
Since the liver is enriched in different DC subsets that can play a central role in iNKT cell activation, we determined whether they are involved in progression of NASH in a murine model and in NASH patients. We first examined the frequency of pDC (PDCA-1+SiglecH+) and two major subsets of cDCs, CD103+ cDC (CD103+CD11b−) and CD11b+ cDC (CD103−CD11b+) in liver MNCs from B6 mice fed CDAA diet. Interestingly, the pDC population was significantly increased in liver of mice fed CDAA diet compared to ND-fed B6 mice (Fig. 8A). In contrast, CD103+ cDC were significantly decreased in CDAA-fed mice compared with ND-fed mice, whereas CD11b+ cDC were not affected significantly (Fig. 8B). These changes suggest that DC subsets infiltrate into liver differentially during CDAA diet-induced liver injury. Importantly, CD1d surface expression on pDC was similar between B6 mice fed CDAA or ND diet (Fig. 8C). Next, we investigated whether pDC infiltration into liver influenced the activation of hepatic iNKT cells following CDAA diet. To examine pDC role in down-regulation of αGalCer/CD1d-tetramer+ iNKT cells, we used transgenic BDCA2-DTR mice, in which pDC cells can be specifically depleted with DT. Thus, BDCA2-DTR mice and littermates (transgene negative) were injected DT as described and liver MNCs were analyzed by FACS. We found that DT-induced pDC depletion resulted in a significant inhibition of the down-regulation of tetramer+ cells in BDCA2-DTR mice, but not in littermates, following CDAA diet (Fig. 8D). These results indicate that pDC play an important role in CDAA-induced activation and iNKT TCR down-regulation.
Figure 8. DC subsets are altered and are involved in iNKT activation in both murine and human NASH.
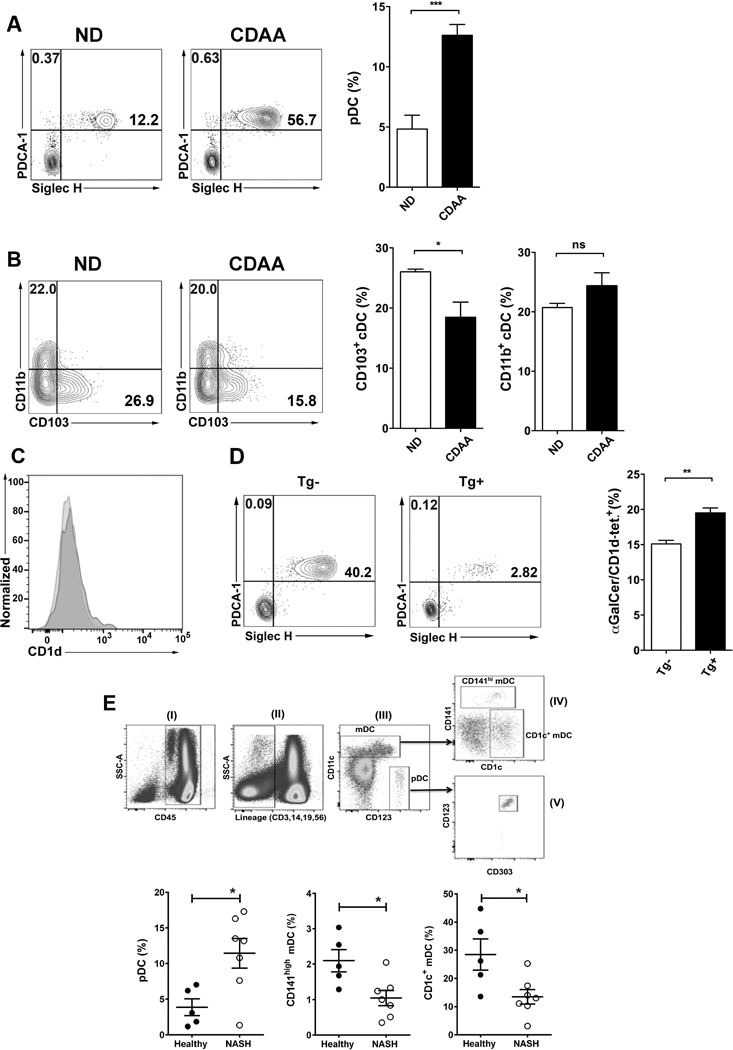
(A) Representative contour plots of pDC in liver of B6 mice fed ND or CDAA diet. pDC cells were identified as double PDCA-1- and Siglec H-positive cells (PDCA-1+Siglec H+) in the live+CD3−CD45+CD11c+CD11b−B220+ gate. Numbers on plots indicate the percentage of pDC. Bar graph shows the percentage of pDC in B6 mice fed ND (n=8) or CDAA diet (n=6). (B) Representative contour plots of the two major cDC subsets, CD103+ cDC and CD11b+ cDC, in liver of B6 mice fed ND or CDAA diet. The CD103+ cDC subset was identified as CD103-positive and simultaneously CD11b-negative cells (CD103+/CD11b−) in the live+CD3−CD19−CD45+MHC-II+CD11chi gate. The CD11b+ cDC subset was identified as CD11b-positive and simultaneously CD103-negative cells (CD11b+/CD103−) in the live+CD3−CD19−CD45+MHC-II+CD11chi gate. Numbers on plots indicate the percentage of both subsets. Bar graphs show the percentage of CD103+ cDC (left) and CD11b+ cDC (right) in the indicated groups. Each group included 3 mice. (C) Histogram shows representative CD1d expression on gated pDC cells in liver of B6 mice fed ND or CDAA diet. (D) Representative contour plots of pDC cells in liver of BDCA2-DTR transgenic mice (Tg+) and littermates (Tg−) on CDAA diet after DT administration. Bar graph shows the percentage of iNKT cells (αGalCer/CD1d tetramer+) in liver of Tg− (n=3) and Tg+ (n=3) mice after DT treatment. All data presented in (A), (B) and (D) are presented as mean ± SEM and are representative of three independent experiments. *p≤ 0.05, **p≤ 0.01, ***p≤ 0.001 by unpaired two-sample t-test; ns, not significant. (E) Representative dot plots of gating strategy to identify DC subsets in human PBMC. DC cells were gated based on FSC and SSC including both lymphocytes and monocytes. Doublets and dead cells were excluded. (I) CD45+ cells were selected on gated live singlet cells. (II) Lineage (CD3/19/56/14) negative cells were gated on CD45+ cells. (III) Based on CD11c and CD123 expression, pDC were identified as CD123+CD11c− cells while mDC were identified as CD11c+CD123− cells. (IV) Based on CD141 and CD1c expression, mDCs were divided in CD141hi cells or CD1c+ cells. (V) The pDC lineage was confirmed by expression of CD303. (F) Scatter graphs show the percentage of pDC (left), CD141hi mDC (center) and CD1c+ mDC (right) in PBMC of healthy controls (n=5) and NASH patients (n=7). Each point represents an individual. All data are presented as mean ± SEM. *p≤ 0.05 by Mann–Whitney U test.
Next, we determined whether the DCs populations are also affected in NASH patients and whether there is any similarity with the murine data. Therefore, we analyzed the frequency in peripheral blood of NASH patients and healthy controls of pDC (CD123+CD11c−) and two subsets of mDCs: CD141high mDC that seem to be the human counterpart of mouse CD103+ cDC and CD1c+ mDC that appear to be related to mouse CD11b+ cDC (Fig. 8E). Consistent with our data in murine model of NASH, the pDC population was significantly increased in peripheral blood from NASH patients compared with healthy controls (11.44% ± 2.08 vs. 3.86% ± 1.17) (Fig. 8E, bottom). In contrast, the frequency of CD141high (1.05% ± 0.22 vs. 2.1% ± 0.31%) and CD1c+ (13.46% ± 2.58 vs. 28.5% ± 5.53) mDCs was significantly reduced in NASH patients compared with healthy controls (Fig. 8E, bottom). These results indicate that these pDC and mDC subsets are altered in peripheral blood of NASH patients and may play a role in NASH.
Altered gut microbiota in Jα18−/− mice and CDAA diet-fed mice
Previous reports have suggested that gut microbiota could potentially have an impact on inflammation in NAFLD (49, 50), we next analyzed the V4 region of the 16S ribosomal RNA (rRNA) genes (32) in stool samples to assess whether the protection in iNKT cell-deficient mice (Jα18−/−) is related to the differences in microbial diversity. Since the microbiome in Jα18−/− and CDAA-treated mice was unknown, we performed a detailed characterization of the composition observed in our experiments. We found significant differences in the composition of the community between Jα18−/− and B6 mice, with greatly increased alpha diversity in Jα18−/− mice as measured by Faith’s phylogenetic diversity (PD) (Kruskal-Wallis q=9.28×10–7, Fig. 9A) (39). In addition, both strains showed significant and dramatic separation in Principal Coordinates Analysis (PCoA) space of unweighted UniFrac distances (40) (q=0.001, PERMANOVA (51), Fig. 9B). An analysis by ANCOM (52) revealed that this separation may be driven by the differential abundance of several organisms in the Jα18−/− mice (Fig. 9C & 9D). Also, alpha diversity was significantly higher in CD1d−/− mice in comparison with Jα18−/− and B6 mice, however, both CD1d−/− and Jα18−/− mice showed significant separation in PCoA analysis compared with B6 mice (Supp. Fig. 3). These results suggest that genetic deficiency of iNKT cells has a strong impact in the microbiota diversity.
Figure 9. Differential microbial diversity in mice lacking functional iNKT cells.
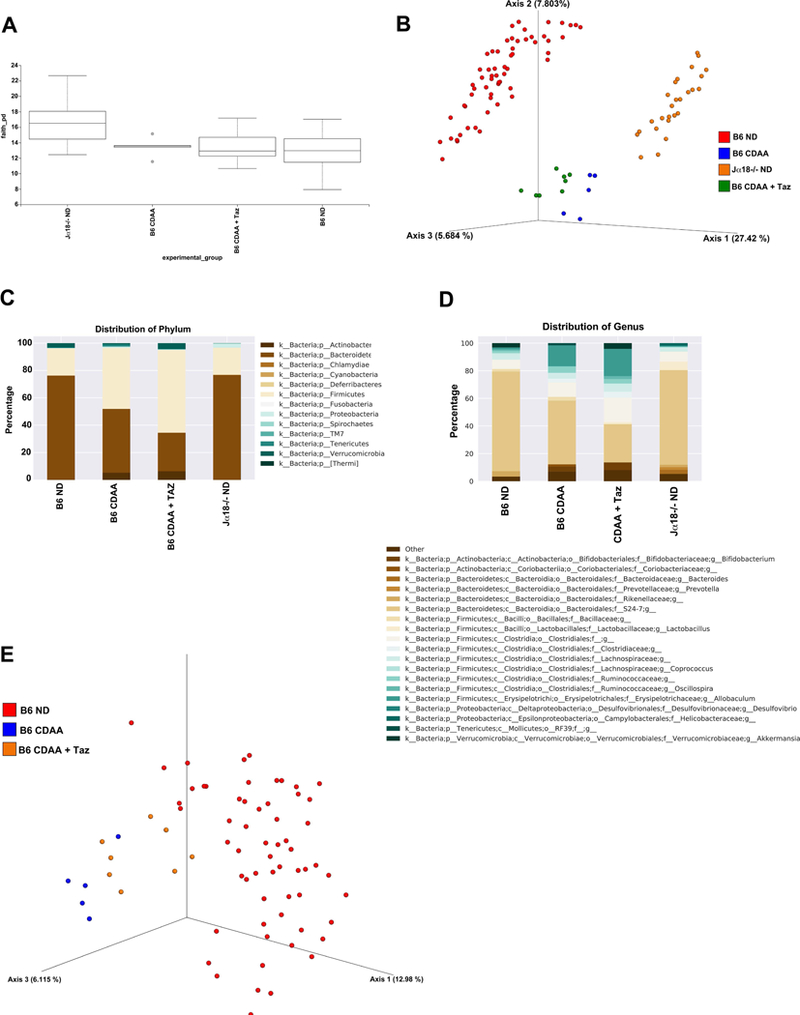
Jα18−/− (n=26) and B6 (n=60) mice were maintained on ND diet. B6 mice maintained on CDAA diet for 20 wk were subjected to i.p. injection of Tazarotene (300 μg/mouse) twice per week (n=8) or left untreated (n=5). All groups were housed in separate cages. Genomic bacterial DNA was isolated from fecal samples and the V4 region of 16S rDNA was amplified and sequenced. (A) Box-plot of Faith’s phylogenetic alpha diversity for each of the experimental groups; bars indicate the interquartile range. (B) Discrimination of mice groups based on Principal Coordinates Analysis (PCoA) of UniFrac distance. Red= B6, ND; Blue= B6, CDAA diet; Green= B6, CDAA+Taz; Orange= Jα18−/−, ND. (C) Summary of the average representation of bacterial phyla detected in these samples. (D) Summary of the average representation of bacterial genera detected in these samples. (E) Principal Coordinates Analysis of Jα18−/− vs B6 mice with and without CDAA diet and Tazarotene treatment. Red= B6, ND; Blue= B6, CDAA diet; Orange= B6, CDAA+Taz.
When comparing between groups at the phylum level, there were an increased proportion of Proteobacteria and Cyanobacteria in Jα18−/− mice as well as Deferribacteres and TM7, which were not detected in B6 mice (Fig. 9C). Similar results were previously reported in the microbiota analysis of CD1−/− mice (53). Only Verrucomicrobia was significantly decreased in Jα18−/− mice as compared to B6 mice (Fig. 9C). At the genus level, there was an increased proportion of 17 bacterial genera, including Bacteroides, Lactobacillus, Mucispirillum and Prevotella, in Jα18−/− mice (Fig. 9D). Interestingly, these four genera have been associated with CD1d−/− mice (53), suggesting that the lack of NKT cells may enable these groups organisms to occupy a greater fraction of the mouse microbiome. Beta and delta proteobacteria have also been reported to be associated with CD1d−/− mice (53), and we observed that the beta proteobacteria genus Zoogloea and delta proteobacteria genera Desulfovibrio were present at higher proportion in Jα18−/− mice than in B6 mice. Several previously unreported genera were also present at an increased proportion in Jα18−/− mice, including Helicobacteraceae family genera, Odoribacter, AF12 (family Rikenellaceae), Candidatus Arthromitus (family Clostridiaceae), Parabacteroides and F16 family genera (phylum TM7). In contrast, Bifidobacterium that was previously reported to be associated with CD1d−/− mice was present at a decreased proportion in Jα18−/− mice (53), suggesting that some of the microbiome effects may be specific to the loss of both type of NKT cells. Others like Clostridiaceae family genera (order Clostridiales), Coprococcus and Ruminococcus (order Clostridiales, family Lachnospiraceae), Allobaculum, and Akkermansia were also decreased in Jα18−/− mice (Fig. 9D).
In Tazarotene-treated B6 mice fed CDAA diet, there was no significant changes to the alpha diversity compared with B6 mice fed a ND diet or B6 mice fed CDAA diet as measured by Faith’s PD (Kruskal-Wallis q=0.88 and q=0.88 respectively, Fig. 9A). While a large separation between mice fed a ND vs CDAA diet was observed in PCoA space of unweighted UniFrac distances (q=0.1.5×10–3, PERMANOVA, Fig. 9E), there was a much smaller, though statistically significant, separation between Tazarotene-treated and untreated CDAA-fed B6 mice (q=0.004, PERMANOVA, Fig. 9E). Furthermore, our ANCOM analysis revealed that the only differences between diets at the phylum level were driven by an increased proportion of Actinobacteria and a decreased proportion of Bacteroidetes in CDAA-fed B6 mice regardless of Tazarotene treatment (Fig. 9C). At the genus level, we observed an increased proportion of Desulfovibrio and Clostridiaceae family genera previously reported in CDAA-diet fed mice (54) as well as a significant increase in Allobaculum, Erysipelotrichaceae family genera, and Coriobacteriaceae family genera, which were found only in CDAA-fed mice (Fig. 9D). There was a reduction in the proportion of the microbiome represented by Bifidobacterium S24–7 family genera, as well as Rikenellaceae family genera, which were previously reported to be decreased in children affected by NASH (55). However, a robust analysis of Tazarotene treated vs untreated CDAA-fed mice was hampered by low number of mice. Together, these results suggest that profound changes in the gut microbiome result from the CDAA diet, but that the differences in composition of the microbiome observed in Jα18−/− mice may be driven by events that occur earlier in development rather than as a result of the acute absence of iNKT cells.
Discussion
In this study, we have identified a key innate immune mechanism that is crucial for diet-induced progression from steatosis, steatohepatitis and fibrosis mediated by differential activation and cytokine secretion by hepatic iNKT cell subsets leading to HSC activation, KCs and CD8+ T cells and neutrophil infiltration into liver. Thus, in iNKT cell-deficient (Jα18−/−) mice as well as in B6 mice following functional inhibition of iNKT cells, hepatic inflammatory infiltration, steatosis and fibrosis as well as up-regulation of key inflammatory genes are significantly blunted. Interestingly, microbial dysbiosis in the absence of iNKT cells may relate to choline-metabolizing bacteria in CDAA-fed diet mice. Importantly, we also found a significant increase in the frequency of CXCR3+/IFNγ+ iNKT cells in peripheral blood of NASH patients as compared with healthy controls or NAFL patients. Additionally, rapid accumulation of pDC, but not other DC subsets into liver occurs following feeding of CDAA diet and genetic depletion of pDC resulted in significant inhibition of the activation of hepatic iNKT cells. Similarly, NASH patients had a significantly high frequency of pDC in PBMCs compared with healthy controls. Collectively our data indicate a key role for an immune mechanism mediated by the differentially activated hepatic iNKT cell subsets in progression of NASH.
NKT cells have been shown to be either pathogenic or protective in inflammatory liver diseases (9, 17, 56). Some of the discrepancy related to the role of NKT cells in NAFLD may be due to the presence of different subsets of NKT cells as well as the microbial variability in different animal facilities that has a major impact on NKT cells. Here, we have used two different approaches to demonstrate a pathogenic role of iNKT cells in not only a murine model but also importantly their involvement in NASH in humans. First, Jα18−/− mice, deficient in iNKT cells but harboring type II NKT cells, are significantly protected from CDAA-induced liver disease (Fig. 2 & 3). Secondly, functional inhibition of iNKT cells in Tazarotene-treated B6 mice also results in a significant protection from diet-induced hepatic steatosis and fibrosis (Fig. 4). Accordingly, there was similarity in the extent of protection from hepatic damage between Tazarotene-treated B6 mice and Jα18−/− mice. Although contribution from other innate-like cells, including MAIT cells, can not be completely ruled out, based upon the published data from others and our laboratory it is unlikely that they play any significant role in diet-induced liver disease: (i) CD1d−/− mice, unlike Jα18−/− mice (57) that have a full TCRα chain repertoire and increased number of MAIT cells (60), are similarly significantly protected from diet-induced hepatic steatosis and fibrosis (26, 47, 58); (ii) anti-CD1d antibody treatment also inhibits diet-induced hepatic disease (19); (iii) there is a significant increase in the number of hepatic pDC in the murine model as well as in NASH patients and their important role in activation of iNKT cells; (iv) there is a significant increase in activated pro-inflammatory iNKT cells in NASH patients in comparison to NAFL patients and healthy subjects. Collectively, our data in murine models and in NASH patients indicate that iNKT cells play an important role in inflammatory liver diseases.
Although, a reduction in iNKT cell numbers has been reported following methionine/choline-deficient (MCD) or HFD diets as well as in obese ob/ob mice and KK-A(y) diabetic mice (29, 61–65), none of these model have evaluated TCR or NK1.1 down-modulation specifically on tetramer+ cells. Interestingly, in some cases, pro-inflammatory activated KC have been shown to induce apoptosis of over-activated iNKT cells following HFD diet (66). We found that the number of αGalCer/CD1d-tetramer+ cells is progressively reduced in liver of B6 mice following CDAA diet and that the majority of tetramer+ cells had reduced surface expression of NK1.1. However, the intrahepatic mRNA level of the invariant Vα14-Jα18 TCR expressed by iNKT cells did not decrease, but rather increased following CDAA diet (Fig. 1). These results suggest that long-term feeding of CDAA diet leads to chronic iNKT cells activation and down-modulation of their surface expression of TCR and NK1.1. It is known that after stimulation with αGalCer or IL-12, iNKT cells can become undetectable due to down-modulation of surface TCR and NK1.1 expression (67–69). Furthermore, progressively increased pro-inflammatory cytokine secretion by iNKT cells following CDAA diet further indicate their chronic activation.
Another important finding in our study is that IL-17+- and IL-22+ iNKT cells (NKT17) are dominant early during liver steatosis while IFNγ+ and IL-4+ or IL-13+ (NKT1/NKT2) cells prevail later during fibrosis following CDAA diet (Fig. 1). Notably, activated IFNγ- and IL-17A-secreting iNKT cells are also present in high frequency in peripheral blood of NASH patients (Fig. 6) in comparison to those in NAFL patients or healthy controls. Additionally, NASH patients with advanced liver fibrosis had low levels of IL-17A and high levels of IFNγ compared with patients with less fibrosis (data not shown). Although an important pathogenic role for IFNγ- (Th1) and IL-17- (Th17) secreting conventional CD4+ T cells in the progression of NAFLD in both mice and humans has been suggested (70, 71), our study indicates that early cytokine secretion by iNKT cells may set the stage for changes in conventional CD4+ T cells during NASH progression. Consistent with our data, IL-17 signaling has been shown to stimulate liver inflammatory cells, like KCs and macrophages, to produce pro-inflammatory cytokines, IL-1, IL-6 and TNFα, as well as the fibrogenic cytokine, TGFβ. IL-17 also induces activation of HSC to produce collagen type I and, thus, promoting their differentiation into fibrogenic myofibroblasts via Stat3 signaling pathway (71–75). Consistently, high IL-17 levels as well as high hepatic expression of Th17 cell-related genes have been associated with hepatic steatosis in patients with NASH (76, 77). Thus, our data suggest that different iNKT cell subsets participate in the progression from steatosis to steatohepatitis and fibrosis in NASH.
Our study also suggests an important role of iNKT cells in HSC activation that mediates hepatic fibrosis following CDAA diet. Thus, CDAA feeding of B6 mice led to increased frequency of IL-13+ iNKT cells accompanied by HSC activation as shown by an increase in α-SMA positive cells. Furthermore, α-SMA staining as well as expression of genes indicative of fibrosis, including α-SMA, TIMP-1, COL1 and CTGF, was significantly reduced in liver of CDAA-fed Jα18−/− mice. Consistent with our data, MCD diet also resulted in hepatic iNKT cells stimulation of HSC activation and fibrosis through production of hedgehog (Hh) protein and osteopontin (OPN) (26). Similarly, IL-13 secretion by iNKT cells can also promote HSC activation. Accordingly, a pro-fibrotic role for TGF-β/Smad signaling and IL-13 in activation of HSC as well as in up-regulation of collagen I and fibrosis-associated genes, like α-SMA and CTGF, in HSC has been shown (78, 79). Furthermore, high serum levels of IL-13 as well as up-regulation of IL-13 and IL-13Rα2 expression have been found in activated HSC in liver biopsies from NASH patients (80).
Our data showed that CDAA diet induced a significant increase in liver infiltrating CD8+ T cells and KCs (Fig. 5) that is dependent on iNKT cells since it is reduced significantly in either Jα18−/− mice or following functional inhibition of iNKT cell in B6 mice. We propose that the activation of IFNγ-secreting iNKT cells results in cross-priming of conventional CD8+ T cells that have been shown to be pathogenic in liver disease following HFHC, HFD or CD-HFD diets (28, 47, 81, 82). Consistent with the potential pathogenic role of CD8+ T cells in liver, increased numbers of infiltrating CD8+ T cells have been found in livers from NASH patients (28, 47, 81). Our data in human PBMC indicate that CD8+ T cells are not significantly increased in peripheral blood of NASH patients compared with healthy controls and is consistent with the hypothesis that CD8+ T cells may have migrated into liver (28, 83). It has been shown that KCs also play a key pro-inflammatory role in liver damage in NASH via inflammatory cytokines, such as TNFα, IL-1β and reactive oxygen species. Thus, depletion of KCs by intravenous injection of liposomal clodronate results in improvement of hepatic steatosis, inflammatory cell infiltration and metabolic disease (84–87). Collectively, these results indicated that diet-induced HSC activation and hepatic infiltration by CD8+ T and KCs is dependent on the presence of activated iNKT cells, suggesting that iNKT cells in collaboration with CD8+ T cells and KCs are drivers in the pathogenesis of NASH.
Earlier studies related to the analysis of NKT cells in human NAFLD have caveats because of the use of non-specific markers, like CD3+CD56+, to identify iNKT cells. In our study, to differentiate iNKT cells from other T cells, including type II NKT cells, we have used a clonotypic mAb Vα24Jα18 TCR, which specifically identifies iNKT cells. Our data indicated that NASH patients have a significantly higher frequency of activated pro-inflammatory IFNγ- and IL-17-secreting iNKT cells in peripheral blood (Fig. 6) in comparison to those from NAFL patients or healthy controls, suggesting their important role in the progression from NAFL to NASH. Consistent with our findings, it has been reported that CD3+CD56+ NKT-like cells are increased in liver and peripheral blood of NAFLD patients with moderate to severe steatosis (88–90). In addition, Tajiri et al. reported that CD3+CD56+Vα24+ iNKT cells were increased in both liver and peripheral blood of NAFLD patients as the disease activity increased (91). Consistent with our data, lower frequency of peripheral CD3+CD56+Vα24+ iNKT cells was found in patients with only steatosis and no steatohepatitis (92). Additionally, our data suggest that iNKT cells are chronically activated in vivo in NASH patients as indicated by their hypo-responsiveness to ex-vivo stimulation with αGalCer (Fig. 7). Similar ex-vivo hyporesponsiveness has been shown in other clinical conditions where iNKT cells are chronically stimulated, including autoimmune disease and cancer patients (93, 94). Collectively, these data suggest a potentially important role of iNKT cells for progression from NAFL to NASH in humans. Additionally, a subset of IFNγ+CXCR3+-iNKT cells is highly represented in NASH patients, suggesting that this could constitute a novel immunological marker for NASH progression.
Our study also indicates a rapid accumulation of pDC, but not other DC subsets in liver of B6 mice following CDAA diet (Fig. 8). Furthermore, selective depletion of pDC in vivo using BDCA2-DTR mice (95) leads to a significant inhibition of hepatic iNKT TCR down-modulation, suggesting pDC involvement in iNKT cell activation. Since, complete depletion of pDC in these mice after DT administration is only sustained for short time (96), we were unable to investigate pDC-depletion effect on NASH progression. Consistently, pDCs accumulation in liver and adipose tissue has been recently shown in HFD-induced inflammation (97). We propose that damage of hepatocytes in CDAA-fed mice induces the release of mitochondrial DNA, a TLR9 ligand. Since pDC are the major early responders of TLR9, we speculate that mitochondrial DNA released from injured hepatocytes leads to accumulation of pDC in liver followed by iNKT activation in a TLR9-dependent manner (15, 98). Recently, hepatic mitochondrial damage has also been reported in NASH patients (99, 100) and increased plasma levels of mtDNA in both HFD-induced murine model as well as in NASH patients have been shown to trigger TLR9 pathway to induce steatosis (101, 102). Notably, rapid accumulation of pDC in the pancreas has been shown to initiate autoimmune diabetes in NOD mice (103). At present, we cannot rule out the importance of other cDC subsets in activation of iNKT cells in our model. It is possible that following chronic feeding of CDAA diet, altered gut microbiota could provide the necessary milieu in the gut to facilitate migration of CD103+ cDC into liver where they can cross-priming IFNγ+-iNKT cells and conventional CD8+ T cells (104, 105), Dasgupta, unpublished data). A CD103+ DC resembling lymphoid tissue resident CD8+ cDC has recently been shown to be the major APCs in iNKT cell activation (97, 106). Consistent with this hypothesis, our preliminary data using Kaede mice suggest that there is a migration of CD103+CD11b− cells but not CD11b+ DC from the major lymph nodes, draining both small and large intestines, into liver in the non-inflamed steady state (Dasgupta, unpublished data). Accordingly, following chronic feeding (4–5 months), CD11b+ cDC and CD103+ cDC are increased in liver (unpublished data). A cross-talk between pDC and cDC has been shown to be crucial in the priming of CD8+ T cells in the anti-tumor immunity (107, 108). Our results showing increased frequency of pDC in peripheral blood of NASH patients (Fig. 8) are in agreement with published data that show positive correlation between diseased livers and increased frequency of CD123+ pDC and decreased frequency of CD141+ mDC (109).
The role that the gut microbiome may play in the development of disease in the CDAA-diet fed mouse model of NASH remains unclear. It is evident that the introduction of the CDAA-diet causes a profound shift in the composition of the gut microbiome (Fig. 9) and the inhibition of iNKT cells largely does not restore the composition to reflect a normal diet nor the Jα18−/− mouse composition. As the overall diversity of the gut microbiome in B6 mice is not altered by the CDAA diet, an imbalance or dysbiotic state driven by the large changes in nutrient availability may be a driver in the development of CDAA-diet induced NASH. Intriguingly, many of the genera that appeared to be increased in CDAA-fed mice were identified to be associated with choline consumption and the increased production of trimethylamine (TMA), which is converted to the obesogenic compound trimethylamine-N-oxide (TMAO) in the liver (110). Since 16S rRNA sequencing data is compositional, it is not possible to say if the total amount of bacteria in each of these groups is increased or if others are proportionately decreased, but rather only that a shift is evident in the community as a result of an altered equilibrium (52, 111). Likewise, the lack of evidence for a given organism or group of organisms does not conclusively rule out their presence since experimental and bioinformatics techniques can provide false negatives, but does indicate an underrepresentation of the organism in the selection of sequences analyzed in this current study which may be driven by biologic effects. As this is the first report of the gut microbiome composition in Jα18−/− mice, many follow-up experiments to examine the strain-level associations and related changes to the host and gut metabolome are warranted. We are also investigating whether alterations in self- or microbial lipids are involved in activation of iNKT cells.
Collectively, our data not only show an important role of iNKT cell subsets activation in diet-induced progression from steatosis to fibrosis, but also a notable similarity with human NAFLD showing a significant increase in activation of iNKT cells in peripheral blood of NASH patients.
Supplementary Material
Acknowledgements
This work was performed with the support of the Flow Cytometry Core at the VA San Diego Health Care System, and the San Diego Veterans Medical Research Foundation. We would like to thank Gregory Humphrey, Cole Heale and Tara Schwartz for sample processing, and Gail Ackermann for assistance with metadata curation and data handling and Dr. Mitch Kronenberg, La Jolla Institute for Allergy & Immunology, for providing the MR1-tetramers.
This work was supported by National Institutes of Health Grants AA020864 (National Institutes for Alcohol Abuse and Alcoholism) and CA100660 (National Cancer Institute) to V.K. The UCSD Neuroscience Core for microscopy was supported by grant NS047101. The investigation of the mouse microbiome was supported by a Seed Grant from the UC San Diego Center for Microbiome Innovation.
Abbreviations used in this article:
- NAFLD
nonalcoholic fatty liver disease
- NAFL
nonalcoholic fatty liver
- NASH
nonalcoholic steatohepatitis
- B6
C57BL/6
- iNKT
invariant NKT
- αGalCer
α-galactosylceramide
- RAR
retinoid acid receptor
- CDAA
choline-deficient L-amino acid-defined
- pDC
plasmacytoid DC
- conv T
conventional T
References
- 1.Wree A, Broderick L, Canbay A,Hoffman HM, and Feldstein AE, 2013. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 10: 627–636. [DOI] [PubMed] [Google Scholar]
- 2.Heymann F, and Tacke F, 2016. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 13: 88–110. [DOI] [PubMed] [Google Scholar]
- 3.Greuter T,Malhi H, Gores GJ, and Shah VH, 2017. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic steatohepatitis: exploiting similarities and differences in pathogenesis. JCI Insight 2(17). doi: 10.1172/jci.insight.95354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anstee QM, and Day CP, 2013. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 10: 645–655. [DOI] [PubMed] [Google Scholar]
- 5.Brennan PJ, Brigl M, and Brenner MB, 2013. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 13: 101–117. [DOI] [PubMed] [Google Scholar]
- 6.Godfrey DI, and Kronenberg M, 2004. Going both ways: immune regulation via CD1d-dependent NKT cells. J. Clin. Invest. 114: 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godfrey DI,Uldrich AP, McCluskey J,Rossjohn J, and Moody DB, 2015. The burgeoning family of unconventional T cells. Nat. Immunol. 16: 1114–1123. [DOI] [PubMed] [Google Scholar]
- 8.Kumar V, 2013. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J. Hepatol. 59: 618–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrero I, Ware R, and Kumar V, 2015. Type II NKT Cells in Inflammation, Autoimmunity, Microbial Immunity, and Cancer. Front. Immunol. 6: 316. doi: 10.3389/fimmu.2015.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Exley M, Garcia J, Balk SP, and Porcelli S, 1997. Requirements for CD1d recognition by human invariant Valpha24+ CD4-CD8- T cells. J. Exp. Med. 186: 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Exley MA, Bigley NJ, Cheng O, Shaulov A,Tahir SM, Carter QL, Garcia J,Wang C,Patten K,Stills HF,Alt FW, Snapper SB, and Balk SP, 2003. Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology. 110: 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girardi E, Maricic I, Wang J, Mac TT,Iyer P,Kumar V, and Zajonc DM, 2012. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat. Immunol. 13: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arrenberg P, Halder R, Dai Y, Maricic I, and Kumar V, 2010. Oligoclonality and innate-like features in the TCR repertoire of type II NKT cells reactive to a beta-linked self-glycolipid. Proc. Natl. Acad. Sci. U.S.A. 107: 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, and Kumar V, 2004. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 199: 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montoya CJ, Jie HB, Al-Harthi L, Mulder C, Patino PJ, Rugeles MT, Krieg AM, Landay AL, and Wilson SB, 2006. Activation of plasmacytoid dendritic cells with TLR9 agonists initiates invariant NKT cell-mediated cross-talk with myeloid dendritic cells. J. Immunol. 177: 1028–1039. [DOI] [PubMed] [Google Scholar]
- 16.Arrenberg P, Maricic I, and Kumar V, 2011. Sulfatide-mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology. 140: 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder RC, Aguilera C, Maricic I, and Kumar V, 2007. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J. Clin. Invest. 117: 2302–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, Molle N, Mathews SA, Gao B, and Kumar V, 2015. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology. 61: 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathews S, Feng D, Maricic I, Ju C, Kumar V, and Gao B, 2016. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell. Mol. Immunol. 13: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eguchi A, De X Mollerat Du Jeu, C. D. Johnson, A Nektaria, and A. E Feldstein, 2016. Liver Bid suppression for treatment of fibrosis associated with non-alcoholic steatohepatitis. J. Hepatol. 64: 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, and Brenner DA, 2009. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 137: 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakae D, Yoshiji H, Mizumoto Y, Horiguchi K, Shiraiwa K, Tamura K, Denda A, and Konishi Y, 1992. High incidence of hepatocellular carcinomas induced by a choline deficient L-amino acid defined diet in rats. Cancer Res. 52: 5042–5045. [PubMed] [Google Scholar]
- 23.Corbin KD, and Zeisel SH, 2012. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 28: 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerrerio AL, Colvin RM,Schwartz AK, Molleston JP, Murray KF, Diehl A, Mohan P, Schwimmer JB, Lavine JE, Torbenson MS, and Scheimann AO, 2012. Choline intake in a large cohort of patients with nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 95: 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.<Zeisel SH 2010. Choline: clinical nutrigenetic/nutrigenomic approaches for identification of functions and dietary requirements. World Rev. Nutr. Diet. 101: 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.<Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H,Xie G,Philips G,Chan IS, Karaca GF, Pereira Tde A, Chen Y, Mi Z, Kuo PC, Choi SS, Guy CD, Abdelmalek MF, and Diehl AM, 2012. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 61: 1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebbard L, and George J, 2011. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 8: 35–44. [DOI] [PubMed] [Google Scholar]
- 28.Wolf MJ, Adili A,Piotrowitz K,Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, Lorentzen A, Einer C, Schulz S, Clavel T, Protzer U, Thiele C, Zischka H, Moch H, Tschop M, Tumanov AV, Haller D, Unger K, Karin M, Kopf M, Knolle P, Weber A, and Heikenwalder M, 2014. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 26: 549–564. [DOI] [PubMed] [Google Scholar]
- 29.Li Z,Soloski MJ, and Diehl AM, 2005. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 42: 880–885. [DOI] [PubMed] [Google Scholar]
- 30.Kawano T, Cui J,Koezuka Y, Toura I,Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, and Taniguchi M, 1997. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 278: 1626–1629. [DOI] [PubMed] [Google Scholar]
- 31.Mendiratta SK, Martin WD,Hong S,Boesteanu A,Joyce S, and Van Kaer L, 1997. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 6: 469–477. [DOI] [PubMed] [Google Scholar]
- 32.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vazquez-Baeza Y, Gonzalez A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, and C. Earth Microbiome Project. 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 551: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, Gilbert JA, Jansson JK, Caporaso JG, Fuhrman JA, Apprill A, and Knight R, 2016. Improved Bacterial 16S rRNA Gene (V4 and V4–5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 1. doi: 10.1128/mSystems.00009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, and Knight R, 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, and Caporaso JG, 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods. 10: 57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, and Knight R, 2017. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2. doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL, 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mirarab S, Nguyen N, and Warnow T, 2012. SEPP: SATe-enabled phylogenetic placement . Pac. Symp. Biocomput. 247–258. [DOI] [PubMed] [Google Scholar]
- 39.Faith DP 1992. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 61: 1–10. [Google Scholar]
- 40.Lozupone C, and Knight R, 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71: 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lozupone CA, Hamady M, Kelley ST, and Knight R, 2007. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73: 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen MJ, Chou LC, Hsieh TT, Lee DD, Liu KW, Yu CY, Oyang YJ, Tsai HK, and Chen CY, 2012. De novo motif discovery facilitates identification of interactions between transcription factors in Saccharomyces cerevisiae. Bioinformatics. 28: 701–708. [DOI] [PubMed] [Google Scholar]
- 43.Vazquez-Baeza Y, Pirrung M, Gonzalez A, and Knight R, 2013. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vazquez-Baeza Y, Gonzalez A, Smarr L, McDonald D, Morton JT, Navas-Molina JA, and Knight R, 2017. Bringing the Dynamic Microbiome to Life with Animations. Cell Host Microbe. 21: 7–10. [DOI] [PubMed] [Google Scholar]
- 45.Iwabuchi K, Iwabuchi C, Tone S, Itoh D, Tosa N, Negishi I, Ogasawara K, Uede T, and Onoe K, 2001. Defective development of NK1.1+ T-cell antigen receptor alphabeta+ cells in zeta-associated protein 70 null mice with an accumulation of NK1.1+ CD3- NK-like cells in the thymus. Blood. 97: 1765–1775. [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharjee J, Kumar JM, Arindkar S, Das B, Pramod U, Juyal RC, Majumdar SS, and Nagarajan P, 2014. Role of immunodeficient animal models in the development of fructose induced NAFLD. J. Nutr. Biochem. 25: 219–226. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P, and Kohli R, 2017. Hepatic Natural Killer T-cell and CD8+ T-cell Signatures in Mice with Nonalcoholic Steatohepatitis. Hepatol. Commun. 1: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, and Lecuit M, 2015. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 42: 145–158. [DOI] [PubMed] [Google Scholar]
- 49.Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK, Jones MB, Sirlin CB, Schnabl B, Brinkac L, Schork N, Chen CH, Brenner DA, Biggs W, Yooseph S, Venter JC, and Nelson KE, 2017. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 25: 1054–1062 e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, Bassirian S, Hooker J, Sy E, Richards L, Schork N, Schnabl B, Brenner DA, Sirlin CB, Chen CH, Loomba R, and Genetics of NAFLD in Twins Consortium. 2018. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology. doi: 10.1002/hep.29892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson MJ 2001. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26: 32–46. [Google Scholar]
- 52.Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, and Peddada SD, 2015. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26: 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selvanantham T, Lin Q, Guo CX, Surendra A, Fieve S, Escalante NK, Guttman DS, Streutker CJ, Robertson SJ, Philpott DJ, and Mallevaey T, 2016. NKT Cell-Deficient Mice Harbor an Altered Microbiota That Fuels Intestinal Inflammation during Chemically Induced Colitis. J. Immunol. 197: 4464–4472. [DOI] [PubMed] [Google Scholar]
- 54.Ishioka M, Miura K, Minami S, Shimura Y, and Ohnishi H, 2017. Altered Gut Microbiota Composition and Immune Response in Experimental Steatohepatitis Mouse Models. Dig. Dis. Sci. 62: 396–406. [DOI] [PubMed] [Google Scholar]
- 55.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, and Gill SR, 2013. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 57: 601–609. [DOI] [PubMed] [Google Scholar]
- 56.Bandyopadhyay K, Marrero I, and Kumar V, 2016. NKT cell subsets as key participants in liver physiology and pathology. Cell. Mol. Immunol. 13: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bedel R, Berry R, Mallevaey T, Matsuda JL, Zhang J, Godfrey DI, Rossjohn J, Kappler JW, Marrack P, and Gapin L, 2014. Effective functional maturation of invariant natural killer T cells is constrained by negative selection and T-cell antigen receptor affinity. Proc. Natl. Acad. Sci. U. S. A. 111: E119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, Nakayama T, Taniguchi M, Hirata N, Ishimori N, Tsutsui H, Onoe K, and Iwabuchi K, 2012. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One. 7: e30568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, Rautou PE, Albuquerque M, Picq O, Gupta AC, Ferrere G, Gilgenkrantz H, Kiaf B, Toubal A, Beaudoin L, Letteron P, Moreau R, Lehuen A, and Lotersztajn S, 2018. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat. Commun. 9: 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koay HF, Gherardin NA, Enders A, Loh L, Mackay LK, Almeida CF, Russ BE, Nold-Petry CA, Nold MF, Bedoui S, Chen Z, Corbett AJ, Eckle SB, Meehan B, d’Udekem Y, Konstantinov IE, Lappas M, Liu L, Goodnow CC, Fairlie DP, Rossjohn J, Chong MM, Kedzierska K, Berzins SP, Belz GT, McCluskey J, Uldrich AP, Godfrey DI, and Pellicci DG, 2016. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat. Immunol. 17: 1300–1311. [DOI] [PubMed] [Google Scholar]
- 61.Valenti L, Fracanzani AL, and Fargion S, 2009. The immunopathogenesis of alcoholic and nonalcoholic steatohepatitis: two triggers for one disease? Semin. Immunopathol. 31: 359–369. [DOI] [PubMed] [Google Scholar]
- 62.Guebre-Xabier M, Yang S, Lin HZ, Schwenk R, Krzych U, and Diehl AM, 2000. Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: potential mechanism for sensitization to liver damage. Hepatology. 31: 633–640. [DOI] [PubMed] [Google Scholar]
- 63.Locatelli I, Sutti S, Vacchiano M, Bozzola C, and Albano E, 2013. NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin. Sci. (Lond). 124: 279–287. [DOI] [PubMed] [Google Scholar]
- 64.Yamagata H, Ikejima K, Takeda K, Aoyama T, Kon K, Okumura K, and Watanabe S, 2013. Altered expression and function of hepatic natural killer T cells in obese and diabetic KK-A(y) mice. Hepatol. Res. 43: 276–288. [DOI] [PubMed] [Google Scholar]
- 65.Vasseur P, Dion S, Filliol A, Genet V, Lucas-Clerc C, Jean-Philippe G, Silvain C, Lecron JC, Piquet-Pellorce C, and Samson M, 2017. Endogenous IL-33 has no effect on the progression of fibrosis during experimental steatohepatitis. Oncotarget. 8: 48563–48574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang T, Sui Y, Lian M, Li Z, and Hua J, 2013. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS One. 8: e81949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson MT, Johansson C, Olivares-Villagomez D, Singh AK, Stanic AK, Wang CR, Joyce S, Wick MJ, and Van Kaer L, 2003. The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc. Natl. Acad. Sci. U. S. A. 100: 10913–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harada M, Seino K, Wakao H, Sakata S, Ishizuka Y, Ito T, Kojo S, Nakayama T, and Taniguchi M, 2004. Down-regulation of the invariant Valpha14 antigen receptor in NKT cells upon activation. Int. Immunol. 16: 241–247. [DOI] [PubMed] [Google Scholar]
- 69.Emoto M, Shimizu T, Koike H, Yoshizawa I, Hurwitz R, Kaufmann SH, and Emoto Y, 2010. Dissociated expression of natural killer 1.1 and T-cell receptor by invariant natural killer T cells after interleukin-12 receptor and T-cell receptor signalling. Immunology. 129: 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, Kudlich T, Hermanns HM, Bantel H, Beyersdorf N, and Geier A, 2016. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 196: 97–105. [DOI] [PubMed] [Google Scholar]
- 71.Harley IT, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, Sheridan R, Xanthakos SA, Steinbrecher KA, Sartor RB, Kohli R, Karp CL, and Divanovic S, 2014. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 59: 1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, and Diehl AM, 2010. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 51: 1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, Le Moine O, and Deviere J, 2009. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 49: 646–657. [DOI] [PubMed] [Google Scholar]
- 74.Koyama Y, and Brenner DA, 2017. Liver inflammation and fibrosis. J. Clin. Invest. 127: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, Osterreicher CH, Stickel F, Ley K, Brenner DA, and Kisseleva T, 2012. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 143: 765–776 e763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, Han X, Peng Y, Chen X, Shen L, Qiu D, Li Z, and Ma X, 2011. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 166: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, and Djouder N, 2016. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell. 30: 161–175. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, Munker S, Mullenbach R, and Weng HL, 2012. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 3: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li HY, Ju D, Zhang DW, Li H, Kong LM, Guo Y, Li C, Wang XL, Chen ZN, and Bian H, 2015. Activation of TGF-beta1-CD147 positive feedback loop in hepatic stellate cells promotes liver fibrosis. Sci. Rep. 5: 16552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, and Puri RK, 2008. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 181: 4656–4665. [DOI] [PubMed] [Google Scholar]
- 81.Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, Shen L, D’Angelo JA, Horton P, Chapman WC, Brockmeier D, Woo M, Engleman EG, Adeyi O, Hirano N, Jin T, Gehring AJ, Winer S, and Winer DA, 2017. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci. Immunol. 2. doi: 10.1126/sciimmunol.aai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Strodthoff D, Lundberg AM, Agardh HE, Ketelhuth DF, Paulsson-Berne G, Arner P, Hansson GK, and Gerdes N, 2013. Lack of invariant natural killer T cells affects lipid metabolism in adipose tissue of diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 33: 1189–1196. [DOI] [PubMed] [Google Scholar]
- 83.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, Han M, Thornton AM, Zhang H, Egger M, Luo J, Felsher DW, McVicar DW, Weber A, Heikenwalder M, and Greten TF, 2016. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 531: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, Zacks S, Fried M, Rippe RA, and Hines IN, 2010. Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. 51: 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S, and Muller M, 2010. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 51: 511–522. [DOI] [PubMed] [Google Scholar]
- 86.Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, and Brenner DA, 2009. CCR2 promotes hepatic fibrosis in mice. Hepatology. 50: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miura K, Yang L, van Rooijen N, Ohnishi H, and Seki E, 2012. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 302: G1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei Y, Zeng B, Chen J, Cui G, Lu C, Wu W, Yang J, Wei H, Xue R, Bai L, Chen Z, Li L, Iwabuchi K, Uede T, Van Kaer L, and Diao H, 2016. Enterogenous bacterial glycolipids are required for the generation of natural killer T cells mediated liver injury. Sci. Rep. 6: 36365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, and Szabo G, 2008. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 48: 1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adler M, Taylor S, Okebugwu K, Yee H, Fielding C, Fielding G, and Poles M, 2011. Intrahepatic natural killer T cell populations are increased in human hepatic steatosis. World J. Gastroenterol. 17: 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tajiri K, Shimizu Y, Tsuneyama K, and Sugiyama T, 2009. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 21: 673–680. [DOI] [PubMed] [Google Scholar]
- 92.Xu CF, Yu CH, Li YM, Xu L, Du J, and Shen Z, 2007. Association of the frequency of peripheral natural killer T cells with nonalcoholic fatty liver disease. World J. Gastroenterol. 13: 4504–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Der Vliet HJ, Nishi N, Koezuka Y, Peyrat MA, Von Blomberg BM, Van Den Eertwegh AJ, Pinedo HM, Giaccone G, and Scheper RJ, 1999. Effects of alpha-galactosylceramide (KRN7000), interleukin-12 and interleukin-7 on phenotype and cytokine profile of human Valpha24+ Vbeta11+ T cells. Immunology. 98: 557–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gutowska-Owsiak D, Birchall MA, Moots RJ, Christmas SE, and Pazmany L, 2014. Proliferatory defect of invariant population and accumulation of non-invariant CD1d-restricted natural killer T cells in the joints of RA patients. Mod. Rheumatol. 24: 434–442. [DOI] [PubMed] [Google Scholar]
- 95.Swiecki M, Gilfillan S, Vermi W, Wang Y, and Colonna M, 2010. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 33: 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mandl M, Drechsler M, Jansen Y, Neideck C, Noels H, Faussner A, Soehnlein O, Weber C, and Doring Y, 2015. Evaluation of the BDCA2-DTR Transgenic Mouse Model in Chronic and Acute Inflammation. PLoS One. 10: e0134176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, Kunnath-Velayudhan S, Carreno LJ, Venkataswamy MM, Kim J, Lazar-Molnar E, Lauvau G, Chang YT, Liu Z, Bittman R, Al-Shamkhani A, Cox LR, Jervis PJ, Veerapen N, Besra GS, and Porcelli SA, 2014. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity. 40: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marschner A, Rothenfusser S, Hornung V, Prell D, Krug A, Kerkmann M, Wellisch D, Poeck H, Greinacher A, Giese T, Endres S, and Hartmann G, 2005. CpG ODN enhance antigen-specific NKT cell activation via plasmacytoid dendritic cells. Eur. J. Immunol. 35: 2347–2357. [DOI] [PubMed] [Google Scholar]
- 99.Pirola CJ, Gianotti TF, Burgueno AL, Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castano GO, and Sookoian S, 2013. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 62: 1356–1363. [DOI] [PubMed] [Google Scholar]
- 100.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, and Roden M, 2015. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell. Metab. 21: 739–746. [DOI] [PubMed] [Google Scholar]
- 101.Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, and Mehal WZ, 2016. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Invest. 126: 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Handa P, Vemulakonda A, Kowdley KV, Uribe M, and Mendez-Sanchez N, 2016. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 22: 6965–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, and Lehuen A, 2013. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 19: 65–73. [DOI] [PubMed] [Google Scholar]
- 104.Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC, Smith AM, O’Neill CM, Lam WY, Edelson BT, Urban JF Jr., Murphy KM, and Pearce EJ, 2016. Migratory CD103+ dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J. Exp. Med. 213: 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mowat AM, and Agace WW, 2014. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 14: 667–685. [DOI] [PubMed] [Google Scholar]
- 106.Lukacs-Kornek V, and Schuppan D, 2013. Dendritic cells in liver injury and fibrosis: shortcomings and promises. J. Hepatol. 59: 1124–1126. [DOI] [PubMed] [Google Scholar]
- 107.Osmond TL, Farrand KJ, Painter GF, Ruedl C, Petersen TR, and Hermans IF, 2015. Activated NKT Cells Can Condition Different Splenic Dendritic Cell Subsets To Respond More Effectively to TLR Engagement and Enhance Cross-Priming. J. Immunol. 195: 821–831. [DOI] [PubMed] [Google Scholar]
- 108.Shimizu K, Asakura M, Shinga J, Sato Y, Kitahara S, Hoshino K, Kaisho T, Schoenberger SP, Ezaki T, and Fujii S, 2013. Invariant NKT cells induce plasmacytoid dendritic cell (DC) cross-talk with conventional DCs for efficient memory CD8+ T cell induction. J. Immunol. 190: 5609–5619. [DOI] [PubMed] [Google Scholar]
- 109.Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, Geoghegan J, Hegarty JE, Ryan EJ, and O’Farrelly C, 2014. CD141(+) myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 60: 135–142. [DOI] [PubMed] [Google Scholar]
- 110.Romano KA, Vivas EI, Amador-Noguez D, and Rey FE, 2015. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 6: e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morton JT,Sanders J,Quinn RA,McDonald D,Gonzalez A,Vazquez-Baeza Y, Navas-Molina JA, Song SJ, Metcalf JL, Hyde ER, Lladser M, Dorrestein PC, and Knight R, 2017. Balance Trees Reveal Microbial Niche Differentiation. mSystems. 2. doi: 10.1128/mSystems.00162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.