

Abstract
Reversible redox processes involving hydroquinones and quinones are ubiquitous in biological reaction networks, materials science, and catalysis. While extensively studied in intermolecular settings, less is known about intramolecular scenarios. Herein, we report hydroquinone-quinone hybrid molecules that form two-stereocenter dihydrobenzofurans via intramolecular cyclization under thermodynamic control. A π-methylhistidine peptide-catalyzed kinetic resolution allowed us to study the stereodynamic behavior of enantio- and diastereo-enriched dihydrofurans. In the course of this study, it was revealed that a reversible intramolecular redox-interconversion network connects all four possible stereoisomers via inversion of a quaternary carbon stereocenter without achiral intermediates. As a result, these findings on hydroquinone-quinone hybrid molecules provide insights into potential natural origin and synthetic access of the common dihydrobenzofuran motif.
Keywords: Stereodynamics, Chirality, Quinones, Redox Chemistry, Enantiomerization
Graphical Abstract
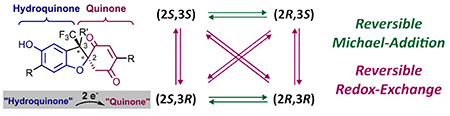
Stereodynamic dihydrobenzofurans with interconverting quaternary carbon stereocenters: Reversible intramolecular redox-interconversion in hydroquinone-quinone hybrid dihydrobenzofurans is the key to a dynamic four-state network. Peptide mediated kinetic resolution via acylation allowed studying the interconversion pathways in detail and obtaining mechanistic insights.
In biological reaction networks, electron transport often relies on reversible one- and two-electron redox steps involving hydroquinone and quinone structures (Figure 1A, left and middle).[1–2] When combined, hydroquinones and quinones form charge-transfer complexes called quinhydrones[3] that undergo reversible redox exchange[4] (Figure 1A, right), and have been studied as cofactors in artificial photosystem mimics.[5] In molecules that contain both hydroquinone and quinone groups (Figure 1B, left and middle),[6–7] this dynamic redox behavior can result in facile enantiomerization in cases where both hydroquinone and quinone are substituents at the same stereogenic center (Figure 1B, right).[8] This phenomenon of dynamic stereochemistry,[9–13] when a function of redox chemistry,[14] has also been observed with helical chirality in copper complexes upon reversible change of oxidation state.[15] The stereodynamic nature of these molecules allows for potential application as chiral redox switches[16–17] or oxidation and reduction catalysts[18–19] in stereoselective transformations.
Figure 1.
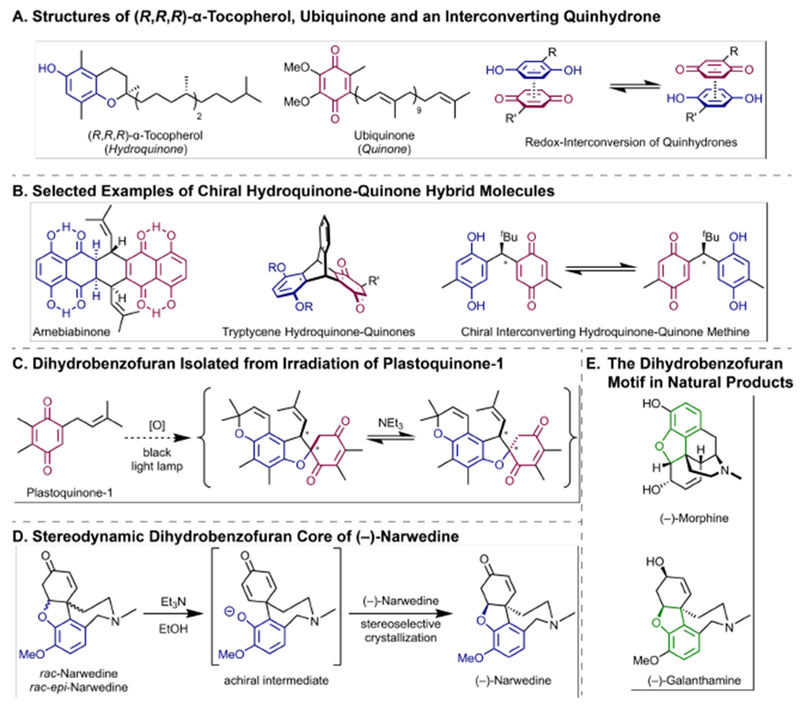
Hydroquionone-quinone hybrid compounds. A: (R,R,R)-α-Tocopherol, ubiquinone and the quinhydrone redox-equilibrium. B: Examples of “intramolecular quinhydrones”. C,D: Stereodynamic dihydrobenzofuranes. E. Dihydrobenzofuran-containing natural products.
In addition to redox processes, hydroquinones and benzoquinones are embedded in diverse complex natural products.[20–23] Hydroquinone-quinone hybrid molecules have been observed to undergo intramolecular cyclization reactions such as formation of a dihydrobenzofuran upon irradiation of plastoquinone-1 under oxidative conditions (Figure 1C).[24] A related reversible intramolecular addition is the key step in preparation of dihydrobenzofuran containing (–)-narwedine (Figure 1D) via seeding of the racemate with a sample of crystalline enantiopure (–)-narwedine (Figure 1D).[25–26] The dihydrobenzofuran core is also part of the related morphine structural family (Figure 1E). Thus, these examples demonstrate that certain dihydrobenzofurans can be seen as masked hydroquinone-quinone hybrids, however, this has not yet been investigated in a general way, especially with regard to stereodynamic aspects.
The relation between hydroquinone-quinone hybrid molecules and dihydrobenzofurans is intriguing due to possibilities for new synthetic strategies towards the latter motifs as well as the opportunity to study reversible redox chemistry as a potential intramolecular mechanism for stereo-interconversion. We hypothesized that a two-stereocenter dihydrobenzofuran (Figure 2), which, in addition, is a spiro compound,[27] could be designed that allows for the separate study of reversible Michael-type addition and intramolecular redox-exchange in a four-state stereodynamic network. Along with our aim for phenomenological and mechanistic insights, dihydrobenzofuran model compounds also provide the opportunity to demonstrate epimerization of a quaternary carbon stereocenter without achiral intermediates. This question of whether or not stereodynamic systems could be achieved with quaternary stereogenic carbon atoms has not been addressed in any system, to our knowledge, and stands in contrast to our previous study of methine-based intramolecular quinhydrones.[8] Thus, in addition to reversible bond-breaking and hindered rotation, our study was designed to highlight intramolecular redox chemistry as a fundamental interconversion pathway in organic molecules.
Figure 2.
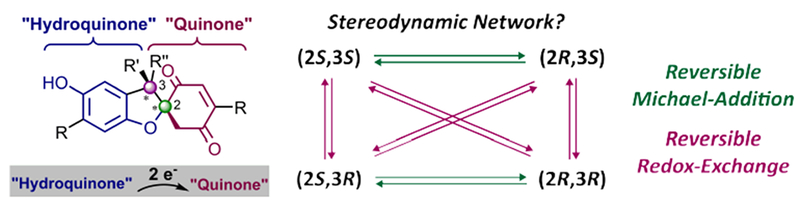
Design of a four-state double-stereodynamic dihydrobenzofuran network in which both reversible Michael-type ring-opening and -closing (green) as well as redox-interconversion (purple) are operative.
The first part of our study was dedicated to synthetic access to model dihydrobenzofuran compounds via hydroquinone-quinone hybrid molecules. Prochiral xanthenes 1 (Figure 3A, top) were selected as a starting point due to a combination of our interest in potential epimerization at quaternary carbon atoms as well as anticipated crucial implications of the Thorpe-Ingold effect[28–29] on intramolecular Michael-type addition. Oxidation resulted in formation of bis-quinone 2, but several attempts to access the bis-hydroquinone derivative only resulted in isolation of condensation product 1. This result implies that both hydroquinone moieties are in close proximity and primed for ring-closing reactions. Gratifyingly, treatment of 2 with electron-rich 2-methoxyhydroquinone cleanly formed the unsymmetrical mono-reduction product. Monitoring the reaction progress in DMSO-d6 via NMR spectroscopy revealed that no hydroquinone-quinone hybrid compound is observed during this reaction; instead, 2 is converted to hemiketal product 3 resulting from intramolecular 1,2-addition (Figure 3A, bottom). We hypothesize that under the reaction conditions, hemiketal formation stabilizes the quinone moiety and inhibits a second reduction event even in the presence of excess reductant
Figure 3.

Synthetic access to dihydrobenzofurans. A: Formation of hemiketal-stabilized hydroquinone-quinone hybrid 3. i) 6.0 equiv. (NH4)2Ce(NO3)6, CH3CN/H2O, 0 °C to r.t., 30 min, 67% (2), 44% (2a), 68% (2b). ii) NMR study: 1.0 equiv. 2 (20 mg scale), 2.0 equiv. 2-methoxyhydroquinone, DMSO-d6, r.t., overnight, quantitative conversion to 3. Preparative scale: 2.0 equiv. 2-methoxyhydroquinone, acetone, r.t., overnight, contains trace amounts of 4, 94% (3), 78% (3a), 42% (in toluene, 3b). B: Preparation of dihydrobenzofuran 4. iii) 3.0 equiv. Hünig’s base, toluene, r.t., overnight, 73% (I-4 and u-4), 81% (I-4a and u-4a), 44% (I-4b and u-4b). C,D: Thermal ellipsoid plots (50% probability level) of X-ray structures. u-4: One co-crystallized acetone is omitted for clarity.
Given the prevalence of dihydrobenzofuran core structures, we wondered whether hemiketal 3 might be formed as the kinetic product during mono-reduction, and indeed, treatment with Hünig’s Base led to full conversion of 3 to, presumably, thermodynamically favored dihydrobenzofurans u-4 and I-4 (Figure 3B), which were separated and characterized (Figure 3C and D).
Following our design principle (Figure 2), stereodynamic pathways of 4 could be initiated via reversible ring-opening to reveal transient hydroquinone-quinone hybrid compounds. Two requirements to begin mechanistic interconversion studies include: (i) enantio- and diastereo-enriched material of 4 must be accessed in order to study both potential epimerization processes taking place in concert, and (ii) enantio- and diastereo-enriched material of a derivative of 4 must be accessed, in which one of the interconversion pathways is inhibited and an isolated interconversion event can be followed. We considered proper substitution of the phenolic OH-group—so that the hydroquinone side can no longer be oxidized to the quinone—as the most straightforward way to block redox-interconversion while still allowing for reversible Michael-type ring-opening. All of the aforementioned requirements can be fulfilled using a kinetic resolution approach of diastereomerically pure u-4 via acylation of the phenolic OH-group. However, differentiation between the two enantiomers of u-4 turned out to be very difficult for well-known, venerable catalysts.[30–31] We were pleased that a brief screen of π-methylhistidine-containing peptide catalysts[32] revealed a β-turn motif—Boc-Pmh-dPro-Aib-Phe-OMe—that allowed for the isolation of starting material u-4 in 74.0:26.0 e.r. and acylation product u-5 with 36.5:63.5 e.r. (Figure 4A). Therefore, we could proceed with our stereodynamics investigations with diastereo- and enantio-enriched dihydrobenzofuran.
Figure 4.
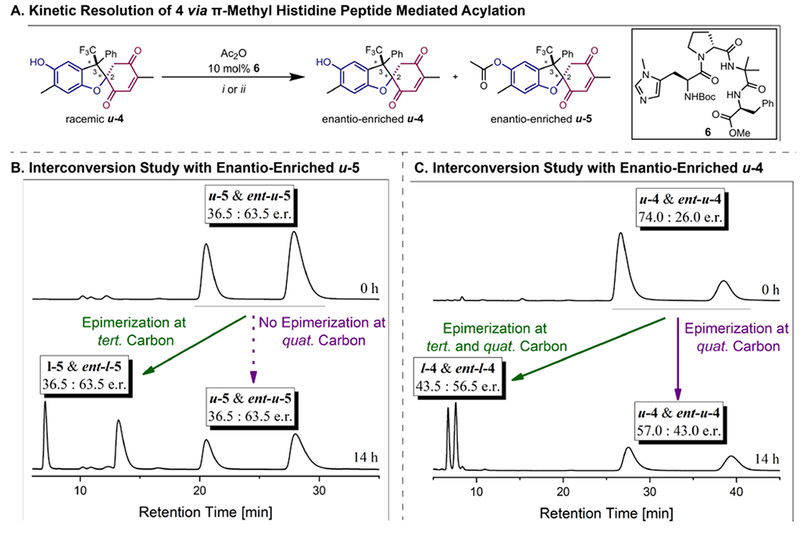
Studies of the stereodynamic dihydrobenzofuran network. A. Acylation of diastereopure u-4 with peptide catalyst 6. i) 0.6 equiv. Ac2O, CH2Cl2:toluene (1:5, v/v), −45 °C, 21 h, 19%. ii) 1.1 equiv. Ac2O, CHCl3:toluene (1:2, v/v), −18 °C, 40 h, 91%. B: Interconversion study of enantio-enriched u-5 in Hünig’s base/toluene (1:10, v/v) at RT. CHIRALPAK® IC (250 mm, i.d. 4.6 mm, particle size 5 μm), hexanes/iPrOH (90:10, v/v), 1.5 mL min−1, I-5 enantiomers: = 7.09 min and = 13.57 min, u-5 enantiomers: = 20.41 min and = 28.01 min. HPLC traces after 14 h and 34 h (see SI). C: Interconversion study of enantio-enriched u-4 in Hünig’s base/toluene (1:10, v/v) at RT. CHIRALPAK® IC (250 mm, i.d. 4.6 mm, particle size 5 μm), hexanes/iPrOH (95:5, v/v), 1.5 mL min−1, I-4 enantiomers: = 6.75 min and = 7.60 min, u-4 enantiomers: = 27.79 min and = 39.73 min. HPLC traces after 14 h and 34 h (see SI).
Studies of interconversion began with acylated, enantio-enriched u-5, and the interconversion process in toluene/Hünig’s base 10:1 (v/v) at RT was followed via CSP-HPLC (Figure 4B). Indeed, a second set of enantiomers—corresponding to I-5 and ent-I-5—was formed under these conditions (44:56 d.r. I-5: u-5 after 34 h, see SI), consistent with reversible Michael-type addition and epimerization at the tertiary 2-position. However, no interconversion at the quaternary stereocenter was observed, as both I-5 and u-5 showed unchanged enantio-enrichment after 14 h and 34 h. No side products were detected via HPLC or NMR spectroscopy after completion of the study. Thus, with a blocked phenolic OH-group, redox-interconversion was inhibited, which is in line with our initial hypothesis and allowed us to study the two processes separately.
The stereodynamic network was then probed with enantio-enriched u-4 under analogous interconversion conditions (Figure 4C) and epimerization at the tertiary carbon position proceeded as expected (37:63 d.r. I-4:u-4 after 34 h). In contrast to the previous experiment, we also observed erosion of e.r. in both diastereomers: 43.5:56.5 e.r. I-4 and 57.0:43.0 e.r. u-4, respectively, after 14 h, and 48.5:51.5 e.r. I-4 and 51.0:49.0 e.r. u-4, respectively, after 34 h (see SI). This observation represents epimerization at a quaternary carbon without breaking any constituent bond adjacent to a stereogenic tertiary carbon atom that is also interconverting.
With the observed stereodynamic properties of u-4 and u-5 in hand, we focused on identifying a plausible mechanistic scenario that applies to the dihydrobenzofuran network. The Hünig’s base-mediated deprotonation ring-opening sequence (Figure 5A)—either in the depicted direct way or via initial formation of the enolate—seems to be a general way to epimerize the dihydrobenzofuran 2-position. At least two mechanistic scenarios for epimerization of the quaternary stereocenter (3-position) were considered: i) formation of a transient mixed hydroquinone-quinone species that undergoes sequential intermolecular redox-exchanges to initially form achiral bis-quinone and bis-hydroquinone intermediates, which then ultimately lead to the hydroquinone-quinone compound of opposite absolute configuration; or ii) a base-mediated intramolecular redox-process that converts the former hydroquinone side into the quinone and vice versa (Figure 5B). Both interconversion experiments (u-4 and u-5) involve reaction solutions that appear as pale-yellow solutions throughout the entire duration of the reaction. This seems to be consistent with the second mechanistic scenario, as dark and very colorful solutions were observed in our previous study on stereodynamic hydroquinone-quinone compounds due to the presence of significant quantities of quinhydrone-like structures (Figure 1A, right).[4, 8] Additionally, neither bis-hydroquinone, xanthene 1, nor bis-quinone 2 were detected after completion of the interconversion experiment of u-4 (see SI). In order to further probe for any intermolecular redox processes, we attempted to reduce u-4 under the interconversion conditions (Figure 5C). If intermolecular redox exchange is operative, reduction of u-4 with electron-rich 2-methoxyhydroquinone to the bis-hydroquinone, or xanthene 1 after dehydration, is expected. Indeed, no reduction product was observed over the course of 34 h and 4 was recovered (see SI, section 4.5), suggesting that any ring-opened intermediate is too short-lived to engage in intermolecular redox processes, and the intramolecular mechanistic scenario (Figure 5B) is operative.
Figure 5.

Plausible mechanistic scenarios operative in the stereodynamic dihydrobenzofuran network. A. Starting from (2R,3R)-4, base-mediated reversible ring opening (green arrows) to the (R)-configured intermediate results in epimerization of the tertiary carbon stereocenter. B. A second ring-opening mechanism involves the phenolic OH-group (purple arrows) and operates via the (S)-configured intermediate and inversion of the absolute configuration at the quaternary carbon stereocenter. C. In order to probe for intermolecular redox-exchange, u-4 was treated with 2-methoxyhydroquinone under interconversion conditions, i) Hünig’s base/toluene (1:10, v/v), 34 h,
In this study, a dynamic four-stereoisomer network has been identified in 2,3,3-trisubstituted dihydrobenzofurans that involves reversible, intramolecular redox-interconversion. On a fundamental level, the dynamic stereochemistry described herein represents a unique way to invert the absolute configuration of a quaternary carbon stereocenter without breaking any constituent bond or involving any achiral intermediates, which is, also, interesting in the context of Mislow’s label paradox.[33] We anticipate the reported findings to both impact stereoselective synthetic strategies towards dihydrobenzofuran core structures, as well as highlight the implications of reversible redox-interconversion in organic molecules containing substituents in different oxidation states. Our findings of intramolecular redox chemistry might also stimulate further consideration of the involvement of unimolecular mechanisms in hydroquinone-quinone redox cofactor transformations in biological settings as well as in redox catalysts in synthetic chemistry. With deeper understanding of these fundamental processes, design and application of new redox active hydroquinone-quinone compounds in catalysis, materials science, and biochemistry is anticipated
Experimental Section
The X-ray crystallographic coordinates for 1, 2, u-4, I-4, u-4b and I-4b are deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1851146, 1851147, 1851151, 1851150, 1851149 and 1851148.
Supplementary Material
Acknowledgements
We would like to thank Prof. Gary W. Brudvig and Prof. James M. Mayer for their valuable insights throughout the project, Dr. Margaret J. Hilton for peptide catalysts, and Dr. Anthony J. Metrano and Prof. Peter Karuso for helpful discussions. This work was supported by the National Institutes of Health (NIH R01-GM096403). GS is also very grateful to the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship (STO 1175/1-1).
References
- [1].Burton GW, Ingold KU, Acc. Chem. Res 1986, 19, 194–201. [Google Scholar]
- [2].Wikström M, Sharma V, Kaila VRI, Hosler JP, Hummer G, Chem. Rev 2015, 115, 2196–2221. [DOI] [PubMed] [Google Scholar]
- [3].Wöhler F, Liebigs Ann. Chem. 1844, 51, 145–163. [Google Scholar]
- [4].Desiraju GR, Curtin DY, Paul IC, J. Org. Chem 1977, 42, 4071–4075. [Google Scholar]
- [5].Liang Z, Jianwei W, Jing Z, Cheng H, Chunying D, Angew. Chem. Int. Ed 2017, 56, 15284–15288. [Google Scholar]
- [6].Xanthopoulou NJ, Kourounakis AP, Spyroudis S, Kourounakis PN, Eur. J. Med. Chem 2003, 38, 621–626. [DOI] [PubMed] [Google Scholar]
- [7].Liu H, Jin Y-S, Song Y, Yang X-N, Yang X-W, Geng D-S, Chen H-S, J. Asian Nat. Prod. Res 2010, 12, 286–292. [DOI] [PubMed] [Google Scholar]
- [8].Kim B, Storch G, Banerjee G, Mercado BQ, Castillo-Lora J, Brudvig GW, Mayer JM, Miller SJ, J. Am. Chem. Soc 2017, 139, 15239–15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wolf C, Chem. Soc. Rev 2005, 34, 595–608. [DOI] [PubMed] [Google Scholar]
- [10].Trapp O, Schoetz G, Schurig V, Chirality 2001, 13, 403–414. [DOI] [PubMed] [Google Scholar]
- [11].Trapp O, Top. Curr. Chem 2013, 231–269. [DOI] [PubMed] [Google Scholar]
- [12].Clayden J, Moran WJ, Edwards PJ, LaPlante SR, Angew. Chem. Int. Ed 2009, 48, 6398–6401. [DOI] [PubMed] [Google Scholar]
- [13].Coates GW, Waymouth RM, Science 1995, 267, 217–219. [DOI] [PubMed] [Google Scholar]
- [14].Reichl KD, Ess DH, Radosevich AT, J. Am. Chem. Soc 2013, 135, 9354–9357. [DOI] [PubMed] [Google Scholar]
- [15].Zahn S, Canary JW, Science 2000, 288, 1404–1407. [DOI] [PubMed] [Google Scholar]
- [16].Jiří M, Andreas VJ, Shin - ichiro S, Daniel E, Jiri M, Stefan M, Angew. Chem. Int. Ed 2010, 49, 7680–7683. [Google Scholar]
- [17].Schweinfurth D, Zalibera M, Kathan M, Shen C, Mazzolini M, Trapp N, Crassous J, Gescheidt G, Diederich F, J. Am. Chem. Soc 2014, 136, 13045–13052. [DOI] [PubMed] [Google Scholar]
- [18].Wendlandt AE, Stahl SS, Angew. Chem. Int. Ed 2015, 54, 14638–14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huynh MT, Anson CW, Cavell AC, Stahl SS, Hammes-Schiffer S, J. Am. Chem. Soc 2016, 138, 15903–15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aldemir H, Richarz R, Gulder TAM, Angew. Chem. Int. Ed 2014, 53, 8286–8293. [DOI] [PubMed] [Google Scholar]
- [21].Miller LL, Stewart RF, Gillespie JP, Ramachandran V, So YH, Stermitz FR, J. Org. Chem 1978, 43, 1580–1586. [Google Scholar]
- [22].Kirby GW, Science 1967, 155, 170–173. [DOI] [PubMed] [Google Scholar]
- [23].Davin LB, Lewis NG, Curr. Opin. Biotechnol 2005, 16, 407–415. [DOI] [PubMed] [Google Scholar]
- [24].Werbin H, Creed D, Strom ET, J. Am. Chem. Soc 1971, 93, 502–511. [DOI] [PubMed] [Google Scholar]
- [25].Czollner L, Frantsits W, Küenburg B, Hedenig U, Fröhlich J, Jordis U, Tetrahedron Lett 1998, 39, 2087–2088. [Google Scholar]
- [26].Shieh W-C, Carlson JA, J. Org. Chem 1994, 59, 5463–5465. [Google Scholar]
- [27].Trapp O, Schurig V, Chem. Eur. J 2001, 7, 1495–1502. [DOI] [PubMed] [Google Scholar]
- [28].Jung ME, Piizzi G, Chem. Rev 2005, 105, 1735–1766. [DOI] [PubMed] [Google Scholar]
- [29].Beesley RM, Ingold CK, Thorpe JF, J. Chem. Soc., Trans 1915, 107, 1080–1106. [Google Scholar]
- [30].Li X, Jiang H, Uffman EW, Guo L, Zhang Y, Yang X, Birman VB, J. Org. Chem 2012, 77, 1722–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ruble JC, Latham HA, Fu GC, J. Am. Chem. Soc 1997, 119, 1492–1493. [Google Scholar]
- [32].Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ, J. Am. Chem. Soc 2008, 130, 16358–16365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mislow K, Bolstad R, J. Am. Chem. Soc 1955, 77, 6712–6713. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.