

Abstract
Helminths stimulate the secretion of T helper 2 (Th2) cytokines, like interleukin-4 (IL4) and suppress lethal graft-versus-host disease (GVHD) after bone marrow transplantation (BMT). This suppression depends on the production of immune-modulatory TGFβ and is associated with TGFβ-dependent in vivo expansion of Foxp3+ regulatory T cells (Treg). In vivo expansion of Tregs is under investigation for its potential as a therapy for GVHD. Nonetheless, the mechanism of induced and TGFβ-dependent, in vivo expansion of Tregs - in a Th2 polarized environment after helminth infection - is unknown. Here we show that helminth-induced IL4 production by host cells is critical to the induction and maintenance of TGFβ secretion, TGFβ-dependent expansion of Foxp3+ Tregs, and the suppression of GVHD. In mice with GVHD, the expanding donor Tregs express the Th2-driving transcription factor, GATA3, which is required for helminth-induced production IL4 and TGFβ. On the other hand, TGFβ is not necessary for GATA3 expression by Foxp3+ Tregs or by Foxp3− CD4 T cells. Various cell types of innate or adaptive immune compartments produce high quantities of IL4 after helminth infection. As a result, IL4-mediated suppression of GVHD does not require invariant NKT (iNKT) cells of the host - a cell type known to produce IL4 and suppress GVHD in other models. Thus, TGFβ generation – in a manner dependent on IL4 secretion by host cells and GATA3 expression - constitutes a critical effector arm of helminthic immune modulation that promotes the in vivo expansion of Tregs and suppresses GVHD.
Introduction
Allogeneic bone marrow transplantation (BMT) and hematopoietic stem cell transplantation (HSCT) are curative approaches for the treatment of both malignant and lethal nonmalignant disorders. The beneficial outcome of transplantation is curtailed by donor immune cell-mediated alloreactivity against host tissues, causing lethal and devastating graft-versus-host disease (GVHD)(1–3). Treatment options for GVHD are limited to immune-suppressive medications (i.e., steroids) that provide limited short- and no long-term benefits and cause severe toxicity. An alternative approach to the management of lethal GVHD is the administration of donor Foxp3-positive regulatory T cells (Tregs). Administration of Tregs at adequate numbers suppresses donor cell alloreactivity, and thus GVHD, yet preserves the beneficial donor cell-mediated anti-tumor (graft-versus-tumor, GVT) immunity(4, 5). However, the addition of sufficient numbers of donor Tregs is a challenging and costly goal in clinical practice(6, 7), necessitating the discovery of in vivo methods to trigger immune regulatory pathways, expand functional donor Tregs and suppress GVHD in BMT/HSCT patients.
Intestinal helminths have immune regulatory properties affecting the innate as well as adaptive immune pathways and they promote the expansion of Tregs (8, 9). Helminths or helminth products can directly stimulate immune regulatory pathways of the host; for example they can induce the expansion of Tregs (10). Several clinical trials have explored the use of helminths to suppress aberrant immunity in patients with allergic, autoimmune or immunological disorders (11, 12). Helminths can also modulate intestinal and systemic immunity through altering the composition of commensal bacteria in mammalian gut, called microbiota (13, 14). GVHD is associated with major shifts in composition of microbiota where lack of specific bacterial strains is found to predispose to more severe GVHD (15, 16). Add-back of these bacterial strains suppresses intestinal inflammation and improves the outcome of BMT in mice (16). Therefore, therapeutic manipulation of the composition of intestinal microbiota - by means of fecal microbiota transplantation, synthetic stool substitutes, add-back of bacterial strains or bacterial products - is an attractive area of basic and clinical research (12, 15, 17).
The mechanism of helminth- or microbiota-mediated immune modulation is not characterized in detail, although TGFβ appears to be a central player in helminth-induced immune suppression(18). We showed previously that TGFβ is critical to helminth-induced in vivo expansion of Tregs and helminth-induced suppression of GVHD, in a major MHC mismatch (H2b→H2d) mouse model of BMT after myeloablative conditioning regimen, total body irradiation (TBI)(19). In this model, helminth infection promoted the survival of host T cells, like interleukin 4 (IL4) producing T helper 2 (Th2) lymphocytes, TGFβ-generating Foxp3− CD4 T cells or Foxp3+ CD4 Tregs. Elements of the Th2 pathway of the host mitigate GVHD(20–22). These include invariant NKT (iNKT) cells, a group of T lymphocytes whose antigen recognition is restricted to lipid antigens. Stimulation of host iNKT cells by cell-specific ligands or an immune regulatory conditioning regimen - called total lymphoid irradiation (TLI) - promotes the expansion of Tregs and suppress GVHD, in a manner dependent on IL4 production by host iNKT cells(21–23). Generation of IL4 and other Th2 cytokines is driven by the transcription factor, GATA3(24). GATA3 is also expressed by Foxp3+ Tregs, contributing to in vivo maintenance and function of regulatory T cells (25, 26). The link between IL4/Th2 pathway and Treg expansion – the latter being dependent on TGFβ in helminth infection(19) - is controversial: IL4 can stimulate or inhibit Tregs(21, 27–30). Moreover, Th2 and TGFβ pathways can inhibit each other(31, 32) and how both pathways remain active after helminth infection is unknown.
Here, we report on the role of host cell Th2 cytokine IL4 production in helminth-induced TGFβ generation and suppression of GVHD. In a model of BMT – where we demonstrated previously that helminth-induced expansion of Tregs and suppression of GVHD depends on TGFβ (19) - we show now that helminth-induced generation of TGFβ, TGFβ-dependent expansion of Tregs and suppression of GVHD requires the production of IL4 by host cells. Furthermore, helminth-induced production of IL4 and TGFβ requires GATA3. With various types of immune cells stimulated to produce IL4 after helminth infection (33–36), host iNKT lymphocytes are not required as a necessary source of this cytokine. Taken together, our results demonstrate a novel link between Th2 pathway and Treg expansion (21, 22, 37, 38), where helminth-induced TGFβ secretion – critical to expansion of Tregs and suppression of GVHD(19) - is driven by Th2 (IL4/GATA3) pathway.
Materials and Methods
Mice and Heligmosomoides polygyrus bakeri (Hpb) administration.
Wild type (WT) C57BL/6 (H2b), WT BALB/c (H2d), IL4−/− (H2d) mice, mice that express the Cre endonuclease driven by a CD4 promoter (H2b), mice conditionally deficient for GATA3 (GATA3 flox/flox) (H2b) and mice with a T cell-specific defect in TGFβ signaling due to overexpression of a truncated TGFβ receptor II (Cd4-TGFBR2; also called TGFβRII dominant negative) (H2b) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice deficient in iNKT cells (Jα18−/−; H2d) were defined before (39) and were bred at the University of Iowa. For helminth-induced immune conditioning, 5–6-week-old male WT BALB/c, Jα18−/− or IL4−/− mice were inoculated with 150 Heligmosomoides polygyrus bakeri (Hpb) third stage larvae (L3) by oral gavage. We used a modified Baermann method (40) to obtain and enrich for infective Hpb L3 (original specimens archived at the U.S. National Helminthological Collection, no. 81930) from stool of helminth (Hpb)-infected mice. Infective larvae were stored at 4°C until used. Mice were maintained and used in accordance with the University of Iowa Animal Care and Use Committee Guidelines.
Cell purification for GVHD induction.
Donor bone marrow (BM) cells were obtained from the femurs and tibias of uninfected, 5–8-week-old male WT C57BL/6 mice. Samples were depleted of T-cells (T cell-depleted; TCD) using mouse panT cell beads (Dynal Biotech) according to the manufacturer’s instructions. Samples of spleen cells from uninfected, 5–8-week-old C57BL/6 mice were magnetically enriched for donor T lymphocytes (CD3+), using a T cell isolation kit (Miltenyi Biotech).
Total body irradiation (TBI) and GVHD induction.
Our studies utilized an acute lethal GVHD model with MHC I/II mismatch (19, 41). Three-week Hbp-infected and uninfected male wild-type BALB/c, Jα18−/− or IL4−/− recipients (H2d) were subjected to total body irradiation (TBI) using a Cs137 source (a total of 850 cGy in two doses given four hours apart) and were administered 10×106 T cell-depleted bone marrow (TCD-BM) cells and 1.5×106 splenic T lymphocytes from uninfected C57BL/6 WT (H2b) donors. Mice were monitored daily for survival for up to 100 days. Disease severity was scored based on animal weight, posture, activity, fur texture and skin integrity(42–44). In parallel experiments, uninfected and Hpb-infected mice were sacrificed 6 days after BMT and subjected to analysis of cell composition by flow cytometry, grading of inflammation by histopathology and quantitation of serum or donor T cell-produced cytokines.
Cell purification for in vitro cultures.
To assay TGFβ cytokine secretion, CD4+ T cells were purified from splenic and mesenteric lymph nodes (MLN) of Hpb-infected and uninfected male WT BALB/c, Jα18−/− or IL4−/− mice, using a CD4 T cell isolation kit (Miltenyi Biotech); this resulted in >98% enrichment for CD4 T cells (data not shown). To assay Hpb-mediated suppression of cytokine production by WT (C57BL/6; H2b) donor T cells during GVHD, donor CD3+ T cells from uninfected and Hpb-infected WT BALB/c, Jα18−/− or IL4−/− BMT recipients (hosts) (all H2d) were sorted from total splenocytes based on staining with anti-CD3 FITC and anti-H2b PE. Sorting was performed 6 days after GVHD induction, using a FACS Vantage SE DiVa cell sorter (Becton Dickinson).
Flow cytometry.
Six days after BMT, uninfected and Hpb-infected mice were sacrificed. The spleen and MLN were isolated for the analysis of cell composition. For surface staining, cells were suspended at 2×107 cells/ml in PBS with 2% FCS, and Fc receptors were blocked with a 2.4G2 mAb (Clone: 93, BioLegend). Antibodies for surface staining were: anti-CD3 FITC, anti-CD3 PE-Cy7 (Clone: 145–2C11), anti-CD4 PE-Cy7 (Clone: GK1.5; eBioscience), anti-H2b PE, anti-H2d PE, and anti-H2b APC (Clones: SF1–1.1, SF1–1.1.1, AF6.88.5; BD Biosciences). For intracellular Foxp3 staining, the Foxp3 staining buffer and anti-Foxp3 PE, Foxp3 PE-Cy7 or Foxp3 APC antibodies (Clone: FJK-16S; eBioscience) were used in accordance with the manufacturer’s instructions. For intracellular GATA3 staining, anti-GATA3 PE (Clone: L50–823; BD Biosciences) or isotype control IgG1, kappa was used.
In vitro cell culture, cytokine ELISA and intracellular staining for IL4.
For TGFβ ELISA, MLN cells from uninfected or Hpb-infected male WT BALB/c, Jα18−/− or IL4−/− mice that did not undergo BMT were stimulated with anti-CD3 (Clone: 145–2C11, eBioscience) and anti-CD28 (Clone: 37.51; eBioscience) (each at 1 μg/ml) for 48 hours, in cell culture medium with 1% FCS and 1 mg/ml BSA(45, 46). In some experiments, purified MLN CD4 T cells were stimulated with plate-bound anti-CD3 and soluble anti-CD28 (each at 1 μg/ml) in the same medium. TGFβ cytokine concentration in acidified and re-alkalinized supernatants was determined using antibody pairs from R&D Systems, according to manufacturer’s instructions. Results were displayed after subtracting the TGFβ concentration in culture supernatants from TGFβ concentrations in the culture media. IFNγ, IL4 and IL10 concentrations in supernatants from parallel cultures with medium containing 10% FCS (45, 46) were analyzed using again antibody pairs from R&D Systems, according to manufacturer’s instructions. To determine IL4-mediated maintenance of TGFβ secretion, anti-IL4 blocking (Clone: 11B11) or isotype control (rat IgG1 kappa) antibody (eBioscience) were added to cultures, each at 5 μg/ml end-concentration. To determine the frequency of IL4 producing cells, splenic or MLN cell cultures were stimulated with anti-CD3/28, and brefeldin A (Thermo Fisher) was added at the last 12 hours to the cultures and at 1:1,000 dilution. Cells were stained for anti-CD4, Foxp3 and IL4 APC (Clone: 11B11; Thermo Fisher). In BMT recipients, IFNγ and TNFα secretion was determined by ELISA from the serum or from sorted donor splenic T cell (CD3+ and H2b+) of uninfected and Hpb-infected mice 6 days after BMT, as described above. Cells were stimulated with plate-bound anti-CD3 and soluble anti-CD28 (each at 1 μg/ml) for 48 hours in lymphocyte growth medium containing 10% FCS(47). Supernatants or sera were analyzed for IFNγ and TNFα using antibody pairs from R&D Systems.
Histopathology.
Six days after BMT, colons and lungs from uninfected or Hpb-infected mice were fixed in 4% neutral buffered formalin and processed, and 6 μm sections were stained with hematoxylin and eosin. Tissues were analyzed for GVHD-related inflammation, and the severity of inflammation was scored in blinded fashion by DEE. GVHD-related colitis was graded based on the degree of inflammation and the frequency of crypt apoptosis: inflammation was graded as previously described (48) and as none (score: 0), mild (1), moderate (2), severe without ulcer (3) and severe with ulcer (4); crypt apoptosis was graded as none (score: 0), less than 2 per 10 crypts (1), 2–5 per 10 crypts (2), majority (>5) of crypts containing apoptotic bodies (3), majority of crypts containing more than one apoptotic body (4). The minimal score in this grading system for colonic disease was 0 and the maximum score was 8. GVHD-related lung inflammation was graded based on the presence of perivascular cuffing, vasculitis, peribronchiolar cuffing and alveolar hemorrhage (49). The minimal score in this grading system for lung inflammation was 0 and the maximum was 4.
Statistical analysis.
Differences in survival between groups were determined by Kaplan Meier’s log rank test. Differences in cell number and composition, serum IFNγ and TNFα content, IFNγ and TNFα generation by splenic donor T cells, TGFβ, IFNγ, IL4, IL10 cytokine output of in vitro stimulated cell cultures and histopathological GVHD scores between groups were determined using unpaired Welch’s t-test using the software GraphPad Prism.
Results
Helminthic conditioning of mucosal T cell responses is independent of host iNKT cells.
Helminths that promote the expansion of Tregs and suppress GVHD stimulate the survival of Th2 polarized and IL4 producing host cells after conditioning with total body irradiation (TBI) (19). Activated Th2 pathway can coincide with a mixed chimeric environment in states of immune tolerance after transplantation (50). Indeed, host iNKT cells also survive after conditioning with total lymphoid irradiation (TLI), secrete IL4 and alleviate GVHD by promoting the expansion of Foxp3+ Tregs (21, 23). Therefore, we investigated the role of host iNKT cells in helminth-induced suppression of graft-versus-host disease. As TGFβ is required for helminthic suppression of GVHD and helminth-induced expansion of Tregs (19), we first investigated whether host iNKT cells are required for helminth-induced TGFβ production. We performed our analysis in Hpb-infected and uninfected iNKT−/− (Jα18−/−) mice prior BMT. T cells are the major source of TGFβ after helminth infection (19, 45, 46). Following anti-CD3/28 stimulation, mesenteric lymph node (MLN) cells from Jα18−/− mice colonized with the mouse nematode, Heligmosomoides polygyrus bakeri (Hpb) showed increased TGFβ secretion relative to uninfected mice (Figure 1A). In addition, the TGFβ secretion observed in cells from Jα18−/− mice was similar to levels reported in WT strains (19, 46). Hpb infection suppressed IFNγ, induced IL4 as well as IL10 secretion in MLN cells from iNKT-deficient (Jα18−/−) and WT BALB/c mice (Figure 1 B-D). These data are consistent with a requirement for TGFβ in helminthic conditioning of T cell responses (46) and demonstrate that this can occur in the absence of iNKT cells.
Figure 1. Helminth-induced T cell-stimulated TGFβ production and modulation of cytokine production does not require host iNKT cells.
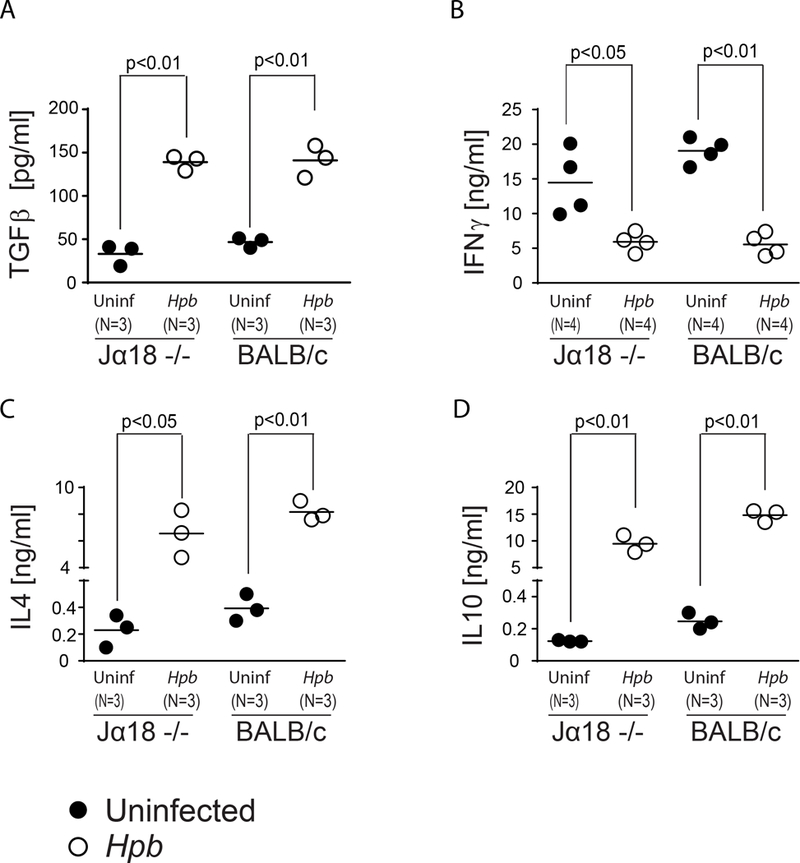
TGFβ (A), IFNγ (B), IL4 (C) and IL10 (D) concentrations in supernatants from 48-hour cultures of MLN cells from Hpb-infected and uninfected 8–9 week old male Jα18−/− and WT BALB/c mice, as measured by ELISA. Cells were cultured in vitro with anti-CD3 and anti-CD28. Data show mean (bar) from multiple independent experiments (scatter plots) where each dot (N) represents mean value of a single independent experiment calculated from multiple (≥3) repeats (p values <0.05 between uninfected and Hpb-infected as indicated for each panel; differences between groups determined by unpaired Welch’s t-test).
Hpb-induced suppression of inflammatory cytokine generation does not require host iNKT cells.
GVHD is caused by donor T cells. Next, we tested whether Hpb infection suppressed WT donor T cell inflammatory response in Jα18−/− hosts. Inflammatory responses by WT C57BL/6 donor T cells were analyzed by in vitro IFNγ and TNFα secretion of purified donor T cells 6 days after BMT. Supernatants were collected from purified splenic donor T cell cultures, stimulated in vitro with plate-bound anti-CD3 and soluble anti-CD28, and assessed for IFNγ as well as TNFα content. Hpb infection suppressed IFNγ and TNFα secretion by WT donor T cells in Jα18−/− BMT mice (Figure 2A and B). Similarly, serum inflammatory cytokine (IFNγ and TNFα) content was also suppressed in Hpb-infected Jα18−/− BMT recipients (Figure 2C and D).
Figure 2. Helminth-induced suppression of inflammatory cytokine generation in BMT mice does not require host iNKT cells.
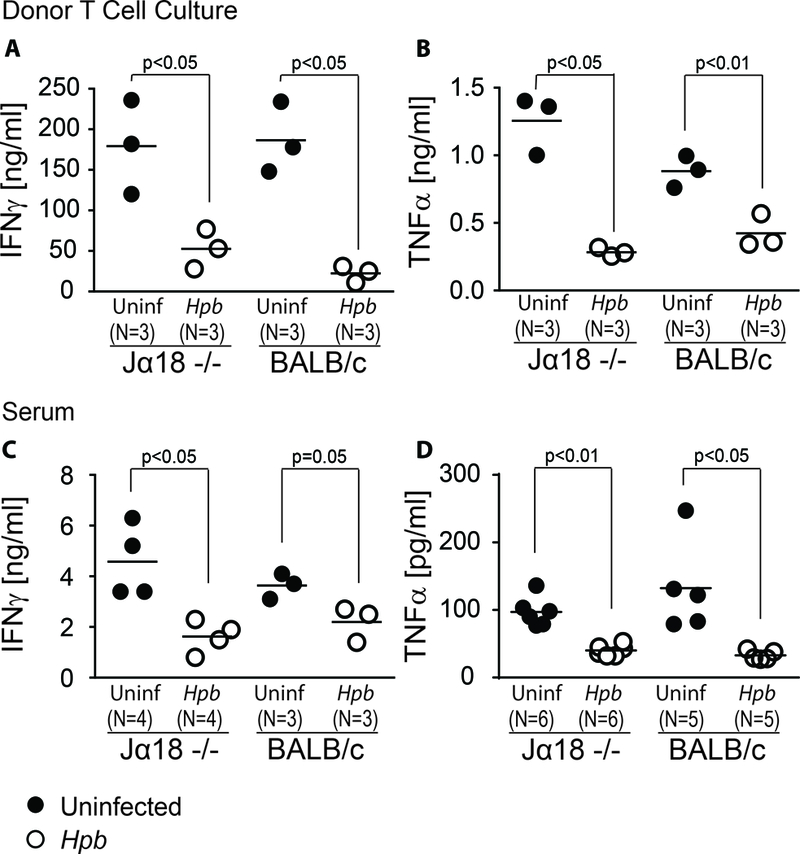
Concentrations of IFNγ (A) and TNFα (B) in supernatants of splenic donor T cell cultures isolated from uninfected or Hpb-infected iNKT-deficient, Jα18−/− or WT BALB/c BMT mice were assessed by ELISA. WT C57BL/6 donor T cells were FACS-sorted as described in Materials and Methods and seeded in triplicate at 105 cells per well. Wells were coated in anti-CD3 and cells were cultured with additional soluble anti-CD28. Serum concentrations of IFNγ (C) and TNFα (D) in uninfected and Hpb-infected animals six days after BMT were assessed by ELISA. Each symbol (dot) represents an independent experiment (N) and is calculated as the average of ≥3 wells from an individual mouse; bars represent the mean from multiple samples; p values <0.05 between uninfected and Hpb-infected as indicated for each panel; differences between groups determined by unpaired Welch’s t-test.
Helminth-induced expansion of Tregs and protection from lethal GVHD does not host require iNKT cells.
Foxp3+ Tregs of recipient (host) or donor origin (4, 51, 52) suppress GVHD and helminths promote the expansion of Tregs. Therefore, we investigated whether host iNKT cells are necessary for helminth-induced expansion of Tregs. Hpb infection resulted in an increase in Treg expansion in Jα18−/− BMT hosts of WT donor cells (Table I). Our findings suggest that helminths employ other host immune suppressive pathways, besides iNKT cells to induce the expansion of Tregs. When we analyzed GVHD-related inflammation in lungs and the colons of uninfected and Hpb-infected Jα18−/− BMT mice, we observed significant suppression of inflammation by helminths in these target organs (Table II), and Hpb infection promoted survival in Jα18−/− BMT mice (Figure 3A). Again, similar to WT BMT recipients (19), helminth infection did not alter the weight loss associated with GVHD in Jα18−/− BMT hosts (Figure 3B). Compared to WT BALB/c BMT mice, where helminths decreased sharply GVHD-related disease score (19), helminth-induced improvement in GVHD disease score in Jα18−/− BMT mice remained modest (Figure 3C). Helminth-stimulated expansion of WT C57BL/6 donor Tregs in Jα18−/− BMT mice (Table I) was dependent on TGFβ, because helminths did not stimulate the expansion of donor Tregs from TGFβ receptor II dominant negative (TGFβ RII DN) mice, whose T cells do not sense TGFβ due to over expression of a truncated TGFβ receptor (Figure 3D)(53). Based on the above parameters analyzed, our results showed that helminthic suppression of GVHD does not require host iNKT cells.
Table I.
The percentage of donor and host MLN Tregs increase in Hpb-infected Jα18−/− BMT mice.
| T cell | Foxp3+ CD4 Treg (%, Mean±SEM)* | Number of Foxp3+ CD4 Treg (Mean±SEM) | ||||
|---|---|---|---|---|---|---|
| Uninfected | Hpb | p value | Uninfected | Hpb | p value | |
| Donor (N=6)** | 1.42±0.11 | 1.99±0.14 | p<0.05 | 2.29±0.90×103 | 11.90±2.93×103 | p<0.01 |
| Host (N=6)** | 8.64±3.04 | 8.77±0.97 | NS | 2.71±0.71×103 | 46.92±10.31×103 | p<0.01 |
p value as indicated between uninfected and Hpb-infected; NS: Not significant.
The percentage of donor Foxp3+ CD4 Tregs among all CD3+ donor T cells or the percentage of host Foxp3+ CD4 Tregs among all CD3+ host T cells are displayed.
The number of Foxp3+ MLN donor or recipient Tregs/mouse was calculated using the total number of mice used in each experiment, the total number of cells isolated from MLN cells and the percentage of Foxp3+ CD4+ cells gated on CD3+ lymphocytes.
The number of independent experiments.
Table II.
Hpb colonization suppresses GVHD-related inflammation in Jα18−/− and WT BALB/c BMT mice.
| Organ | Histology Score (Mean±SD) | |||||
|---|---|---|---|---|---|---|
| Jα18−/− (Mean±SD) | BALB/c WT (Mean±SD) | |||||
| Uninfected | Hpb | p value | Uninfected | Hpb | p value | |
| Lung (N=6)* | 3.3±0.8 | 1.8±0.8 | p<0.01 | 3.7±0.5 | 1.7±0.5 | p<0.001 |
| Colon (N=6)* | 6.7±0.5 | 4.5±1.0 | p<0.01 | 7.2±0.8 | 3.2±1.5 | p<0.001 |
p value as indicated between uninfected and Hpb-infected.
Number of independent samples from each group - uninfected or Hpb-infected.
Figure 3. Helminth-induced suppression of lethal GVHD and promotion of survival does not require host iNKT cells.
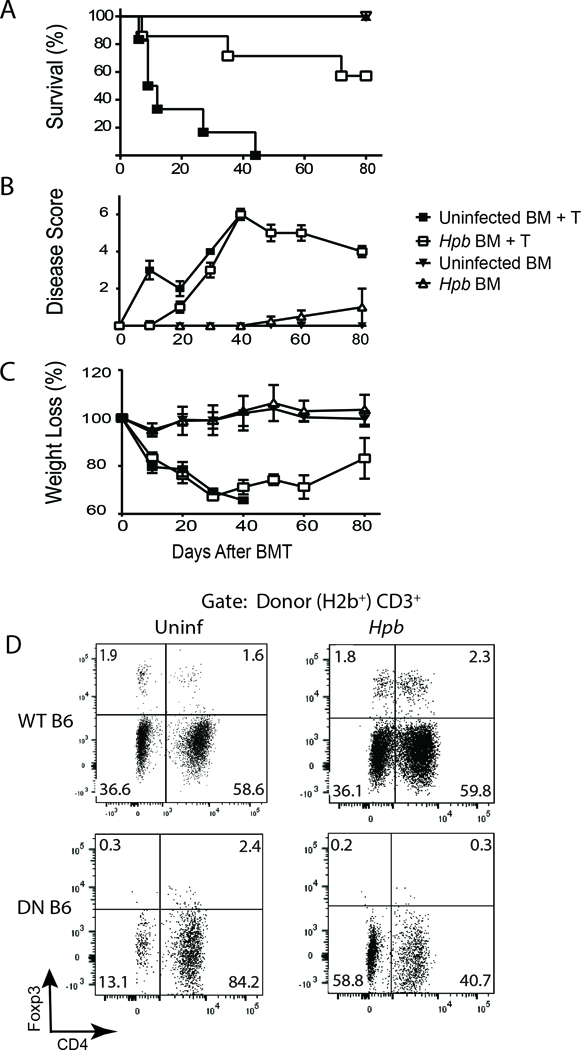
(A) Kaplan-Meier survival curves for Hpb-infected or uninfected iNKT-deficient (Jα18−/−) male BMT recipients that received T cell-depleted bone marrow (TCD-BM) cells (TCD BM) only, or TCD-BM plus total splenic T (TCD-BM + T) cells, from 5–6 week old male WT C57BL/6 donor mice. Cumulative data from two independent experiments. Uninfected TCD-BM only: N = 5; Hpb-infected TCD-BM only: N = 5; uninfected TCD-BM + T: N = 6; Hpb-infected TCD-BM + T: N = 7; p<0.001 between uninfected TCD-BM + T and Hpb-infected TCD-BM + T. (B) GVHD disease score and (C) weight change of the same group of mice. Weight loss for each group of mice is displayed as percent weight change at different time points compared to initial weight. (D) Representative dot plots from MLN cells isolated 6 days after BMT, from uninfected (Uninf) or Hpb-infected Jα18−/− BMT recipients of WT C57BL/6 (WT B6) or TGFβRII DN (DN B6) splenic T cell donors. For BMT, splenic donor T cells were obtained from uninfected mice and all groups also received donor TCD-BM (T cell-depleted BM) cells from uninfected C57BL/6 mice. MLN cells were stained for CD3, CD4, H2b, H2d and Foxp3. Cells were gated on donor (H2b+) CD3+ T cells. Numbers represent the percentage of events in each quadrant and the percentage of Foxp3+ CD4 Tregs in the right upper quadrant. Representative example from 3 parallel independent experiments.
Helminthic suppression of acute GVHD is dependent on IL4 production by BMT recipients.
As we show above, helminths do not require host iNKT cells to stimulate IL4, TGFβ secretion and to induce TGFβ-dependent Treg expansion. Helminths stimulate IL4 production by various cells (33–36). IL4 production by host iNKT cells was found critical in expansion of Tregs and mitigation of GVHD (21). Although our results did not attest to a critical role of host iNKT cells in suppressing GVHD, we investigated the role of host cell IL4 production – in general - in Hpb-mediated suppression of graft-versus-host disease. We explored the production of inflammatory cytokines from WT (C57BL/6) donor T cells 6 days after bone marrow transfer into Hpb-infected or uninfected IL4−/− or WT BALB/c BMT hosts. Supernatants from splenic WT donor T cells stimulated in vitro with plate-bound anti-CD3 and soluble anti-CD28 were assayed for IFNγ and TNFα content. WT donor T cell inflammatory cytokine secretion (IFNγ and TNFα) isolated from IL4−/− BMT hosts were not subject to Hpb-mediated suppression of secretion of inflammatory cytokines, although those isolated from WT BALB/c mice were (Figure 4A and B), as previously reported (19). TNFα secretion by WT donor T cells isolated from uninfected and helminth-infected IL4−/− BMT mice was low compared to TNFα secretion by WT donor T cells isolated from uninfected and helminth-infected WT BALB/c BMT mice (Figure 4A and B). Inflammatory cytokine content was also analyzed in the sera of Hpb-infected and uninfected IL4−/− or BALB/c WT mice 6 days after BMT (Figure 4C and D). As in the case of cytokine secretion by donor T cells, the absence of IL4 production by host cells had no effect, regardless of Hpb infection (Figure 4C and D). In contrast, levels of serum T helper 1 (Th1) inflammatory cytokines were significantly reduced in Hpb-infected WT BALB/c BMT recipients (Figure 4C and D). These data demonstrate that the suppression of inflammatory cytokine output in BMT mice by helminths is dependent on IL4 production by the host, suggesting that this cytokine is critical to helminthic conditioning of the host prior to BMT.
Figure 4. Helminth-induced suppression of inflammatory cytokine generation in BMT mice requires host cell IL4 production.
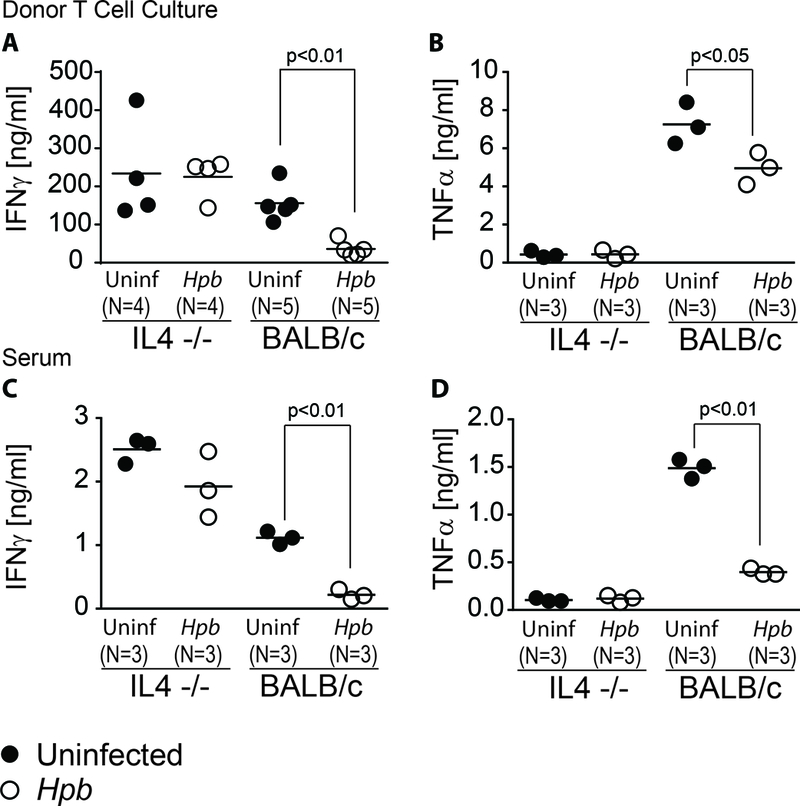
Concentrations of IFNγ (A) and TNFα (B) in supernatants of splenic donor T cell cultures isolated from uninfected or Hpb-infected IL4−/− or WT BALB/c BMT mice were assessed by ELISA. WT donor T cells were FACS-sorted as described in Materials and Methods and seeded in triplicate at 105 cells per well. Wells were coated in anti-CD3 and cells were cultured with additional soluble anti-CD28. Serum concentrations of IFNγ (C) and TNFα (D) in uninfected and Hpb-infected animals six days after BMT were assessed by ELISA. Each symbol (dot) represents an independent experiment (N) and is calculated as the average of ≥3 wells from an individual mouse; bars represent the mean from multiple samples; p values <0.05 between uninfected and Hpb-infected as indicated for each panel; differences between groups determined by unpaired Welch’s t-test.
In parallel we harvested colons and lungs of Hpb-infected and uninfected IL4−/− and WT BALB/c mice 6 days after BMT. Abundant mononuclear cell infiltrates and apoptotic bodies were present in the colons of Hpb-infected and uninfected IL4−/− mice as well as uninfected WT BALB/c mice (Figure 5). Similarly, dense infiltrates were evident in the lungs in the same groups. Unlike in WT BALB/c(19) (Figure 5) or Jα18−/− (Table II) BMT recipients, Hpb colonization did not reduce the histopathological GVHD disease score in IL4−/− BMT mice (Figure 5).
Figure 5. Helminths do not suppress GVHD-related end-organ damage in lung and the colon in IL4−/− BMT mice.
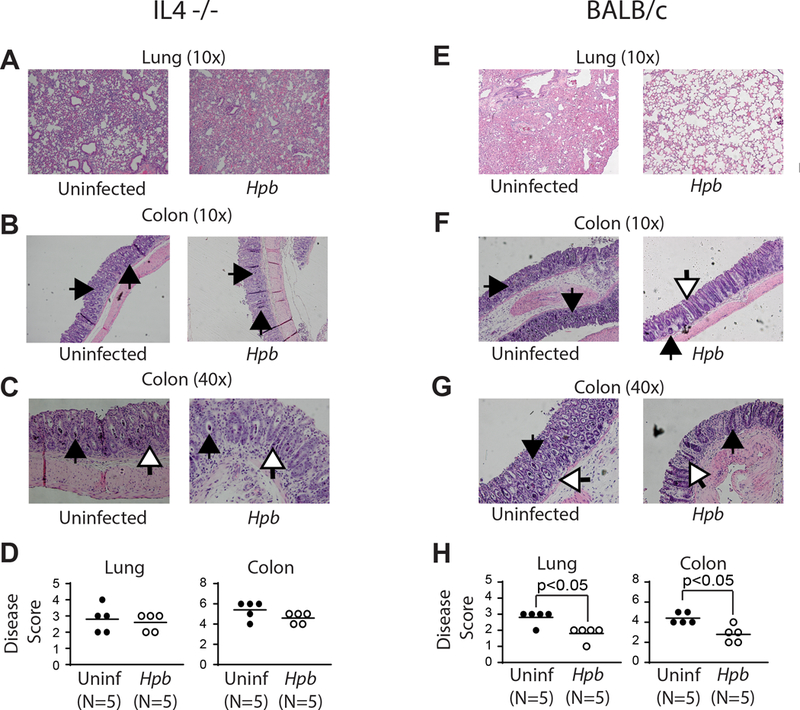
Histopathological analysis of lung (10x magnification) (A, E) and the colon (10x magnification)(B, F), (40x magnification)(C, G) from uninfected and Hpb-infected IL4−/− (A-D) or WT BALB/c (E-H) BMT mice. Organs were harvested 6 days after BMT, tissue preparation and scoring between groups (D, H) was performed as detailed in Methods. Inflammation in the colon was characterized by mononuclear cell infiltrates, apoptotic cells filling crypts (black arrows) and apoptotic bodies (white arrows). Each symbol (dot) is an independent experiment (N) and represents the histopathology score from an individual mouse; bars represent the mean from multiple samples; p values <0.05 between uninfected and Hpb-infected as indicated for each panel; differences between groups determined by unpaired Welch’s t-test.
Next we investigated whether helminth-induced suppression of GVHD is dependent on IL4. Hpb-infected and uninfected IL4−/− mice that received only TCD-BM cells showed minimal signs of disease throughout the 100-day follow-up period and survived for the duration of the experiment (Figure 6). By contrast, Hpb-infected and uninfected IL4−/− mice that received splenic T cells in addition to TCD-BM cells had increased disease scores, a reduction in body weight and, ultimately, all succumbed to disease 40–50 days after BMT (Figure 6). When we repeated the BMT survival experiments in Jα18−/− and IL4−/− hosts in parallel with WT BALB/c mice, we observed that helminthic protection from lethal GVHD was similar between Jα18−/− and WT BALB/c strains (Figure 7A), whereas helminth-infected IL4−/− BMT mice – unlike their WT BALB/c counterparts - were not protected from lethal GVHD (Figure 7B). These results solidify the role of host cell IL4 production – generated by various innate and adaptive immune cells after helminth infection - in Hpb-induced control of GVHD.
Figure 6. Helminth-induced suppression of GVHD and promotion of survival are dependent on host cell IL4 production. (Upper panel).
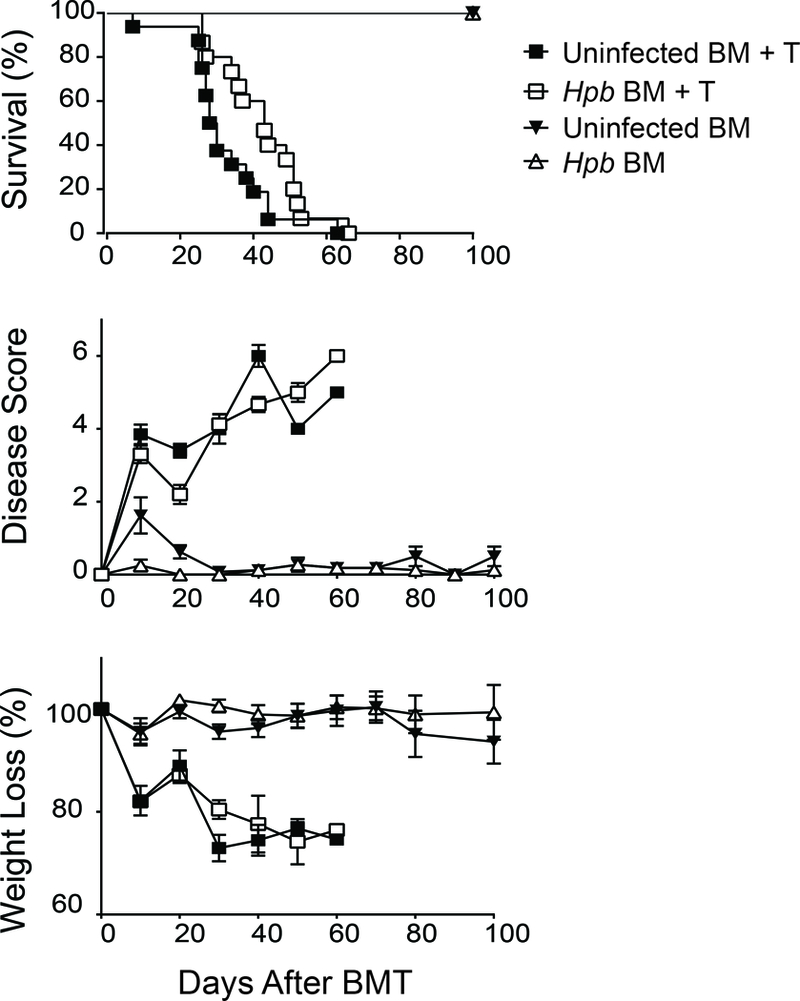
Kaplan-Meier survival curves of Hpb-infected or uninfected IL4−/− male BMT recipients that received T cell-depleted (TCD-BM) cells (TCD BM) or TCD-BM plus total splenic T cells (TCD-BM + T) from 5–6 week old male WT C57BL/6 donor mice. Cumulative data from three independent experiments. N: cumulative number of BMT mice in each group; N = 10: uninfected TCD-BM; N = 10: Hpb-infected TCD-BM; N = 15: uninfected TCD-BM + T; N = 15: Hpb-infected TCD-BM + T. (Middle panel) GVHD disease score and (Lower panel) weight change of the same group of mice. Weight loss for each group of mice is displayed as percent weight change at different time points compared to initial weight.
Figure 7. Helminth-induced suppression of lethal GVHD and promotion of survival does not require host iNKT cells but requires IL4 generation by host cells.
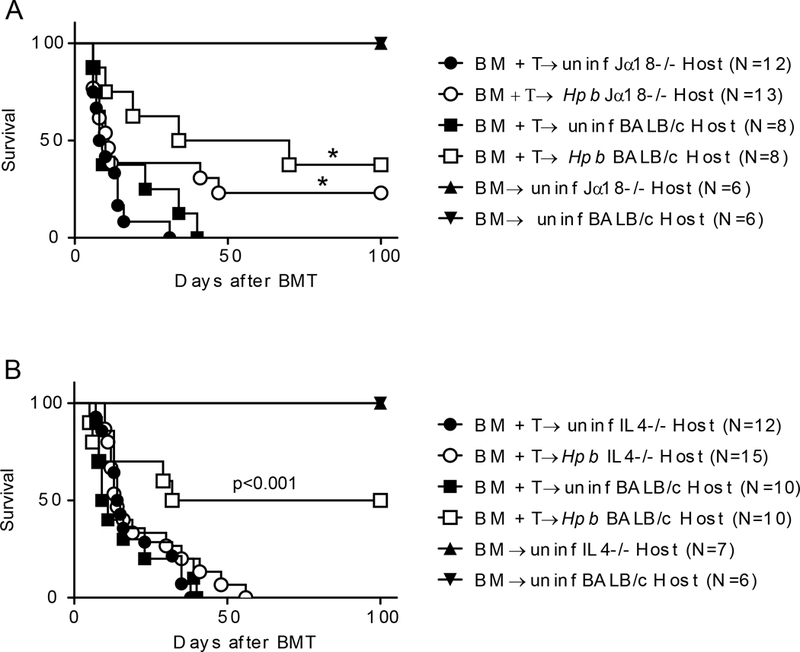
(A) Kaplan-Meier survival curves for Hpb-infected or uninfected iNKT-deficient (Jα18−/−) and WT BALB/c male BMT recipients that received TCD-BM cells (TCD BM) only, or TCD-BM plus total splenic T (TCD-BM + T) cells, from 5–6 week old male WT C57BL/6 donor mice. Cumulative data from multiple independent experiments that involved Jα18−/− and BALB/c WT hosts (N: cumulative number of BMT mice from multiple experiments); *p: NS between WT C57BL/6 TCD-BM + T donor cells into Hpb-infected Jα18−/− and WT C57BL/6 TCD-BM + T donor cells into Hpb-infected BALB/c WT hosts. (B) Kaplan-Meier survival curves for Hpb-infected or uninfected IL4−/− and WT BALB/c male BMT recipients of C57BL/6 WT donors; p<0.001 between TCD-BM + T donor cells into Hpb-infected IL4−/− and TCD-BM + T donor cells into Hpb-infected BALB/c WT hosts.
Helminth-induced expansion of GVHD-suppressing donor Tregs is dependent on IL4 production by BMT recipients.
Next, we tested whether helminth-induced expansion of donor Tregs is IL4 dependent. Relative to uninfected WT BMT recipient mice, counterparts infected with Hpb showed an increase in the percentage and total number of donor as well as host Tregs, in both the spleen and MLN, 6 days after BMT (Figures 8 and 9), similar to our previous results in WT BMT hosts (19). However, in uninfected IL4−/− BMT recipients, analysis of the spleen and MLN at the 6-day time point revealed a sharp decrease in the percentage and number of Tregs of both origins donor and host Tregs (Figures 8 and 9). Moreover, Hpb infection had no effect on the percentage or total number of host and donor Tregs in this context (Figures 8 and 9). These observations revealed that host IL4 plays a broad role in the expansion of donor Tregs, and emphasize the importance of Th2-mediated suppression of GVHD. Thus, infection with an intestinal helminth stimulated Th2 immunity and immune regulatory pathways.
Figure 8. Helminth-induced increase in Foxp3+ Treg percentage requires host cell IL4 production.
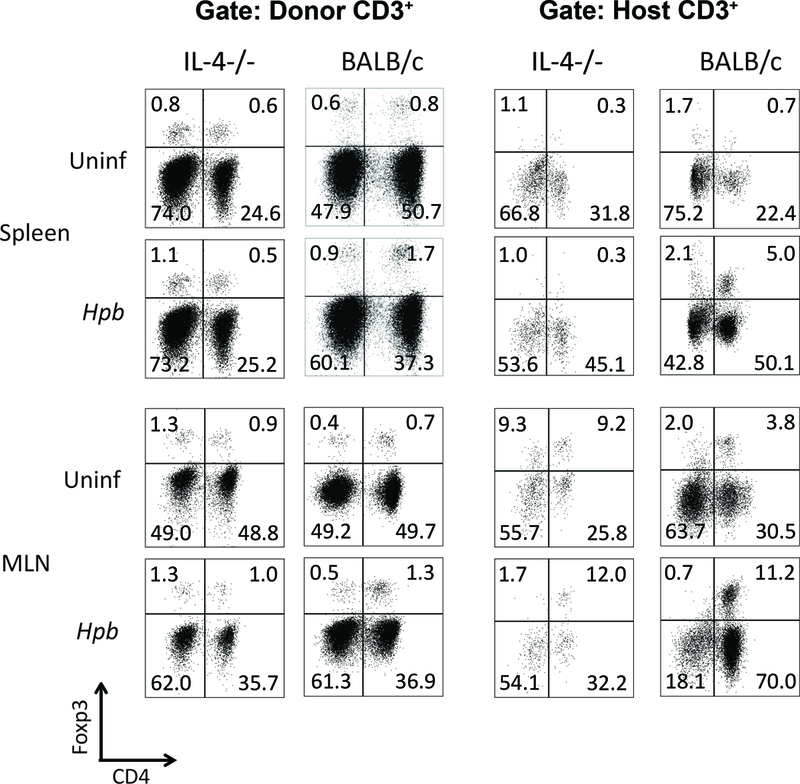
Representative dot plots from spleen (upper rows) and MLN (lower rows) cells isolated from uninfected and Hpb-infected IL4−/− or WT (BALB/c) mice 6 days after BMT. Spleen and MLN cells were stained for CD3, CD4, H2b, H2d and Foxp3. Cells were gated on donor or host CD3+ T cells. Numbers represent the percentage of events in each quadrant and the percentage of Foxp3+ CD4 Tregs in the right upper quadrant.
Figure 9. Helminth-induced increase in Foxp3+ Treg percentage and number requires host cell IL4 production.
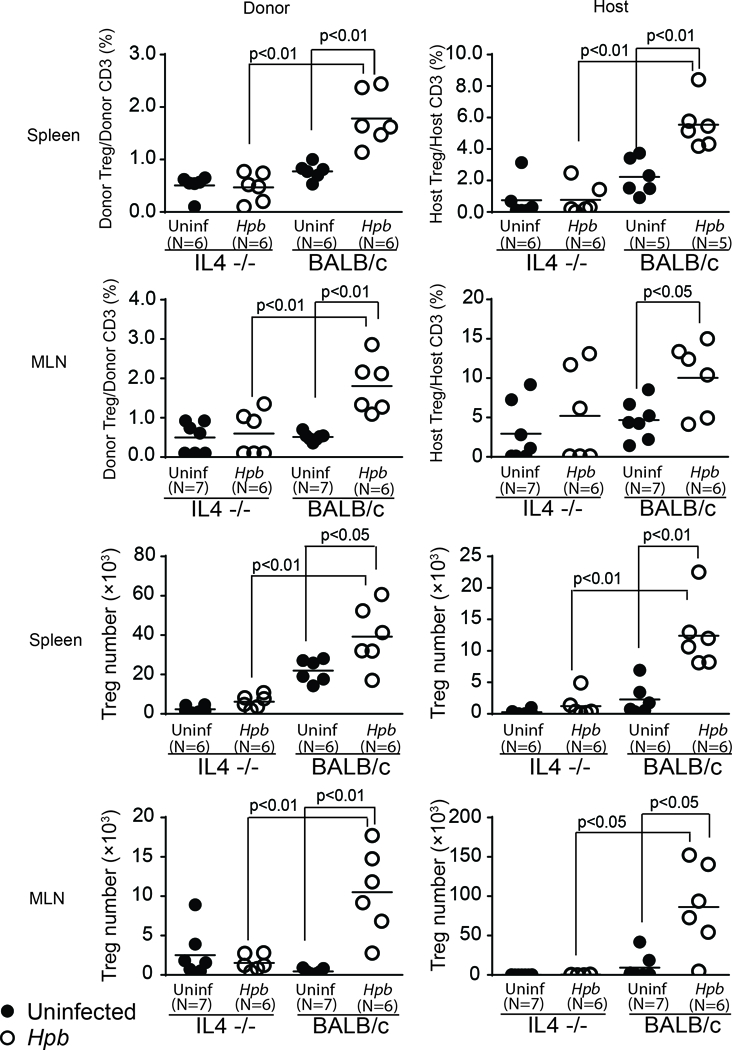
Data from multiple samples (N) were analyzed as detailed in Figure 7 and displayed as dot-plot distribution with means (bar); significant p values (<0.05) as shown in each graph between groups; differences between groups were determined by unpaired Welch’s t-test.
Helminth-induced TGFβ production is IL4-dependent.
Helminth infection induces the production of TGFβ and IL4 (46, 47). We have demonstrated that helminthic suppression of GVHD and induction of Tregs is dependent on these two cytokines (Figure 3–9, (19)). As TGFβ is the cytokine critical for helminth-induced expansion of Tregs and the suppression of GVHD, we investigated the role of IL4 in helminthic induction of TGFβ. WT and IL4−/− mice were colonized with Hpb, and TGFβ secretion by MLN cells following stimulation with anti-CD3/28 was assessed. In MLN cells obtained from Hpb-infected WT mice, the increase in TGFβ secretion was significantly higher than in uninfected WT cells (Figure 10A). In contrast, TGFβ cytokine secretion was not induced in MLN cell cultures from IL4−/− Hpb-infected mice, suggesting that IL4 is necessary for Hpb-induced TGFβ output (Figure 10A). Hpb infection also stimulates IL4 and TGFβ secretion from purified T cells (19, 47). Therefore, we investigated a possible direct effect of IL4 on TGFβ secretion in MLN T cells from Hpb-infected WT mice. Treatment of WT Hpb-infected MLN T cells with anti-IL4 antibodies reduced TGFβ secretion by ~50% relative to cells that were treated with an isotype control antibody or cells that were left untreated. (Figure 10B). These data suggest that IL4 is necessary not only for the induction of TGFβ, but also for maintaining its secretion. In the absence of IL4 and TGFβ in Hpb-infected IL4−/− mice, helminthic suppression of Th1 and helminthic induction of IL10 were impaired (Supplemental Figure 1). When we explored the origin of IL4 among CD4 T cell subsets in helminth-infected WT BALB/c and Jα18−/− mice, we observed that almost all IL4-producing cells were Foxp3− CD4 T cells (Th2 lymphocytes) and IL4 production from Foxp3+ CD4 Tregs was undetectable after primary infection with Hpb (Figure 10C). We previously reported TGFβ production is evident from Foxp3+ CD4 Tregs and Foxp3− CD4 T cells ((19). By contrast, we show here that IL4 production was only evident in Foxp3- CD4 T cells (Figure 10C). Thus, IL4 generated by Foxp3− Th2 cells appears to be critical to the induction and maintenance of TGFβ production by Foxp3− CD4 T cells and Foxp3+ CD4 Tregs.
Figure 10. Helminthic induction and maintenance of TGFβ production requires IL4 production by Foxp3− CD4 T cells.
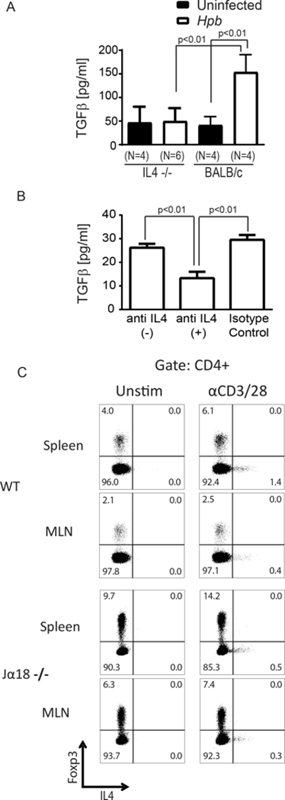
(A) TGFβ concentration in supernatants of anti-CD3/28-stimulated MLN cultures from Hpb-infected and uninfected 8–9 week old male IL4−/− or WT BALB/c mice, as measured by ELISA. Data show mean±SD from ≥3 independent experiments, with each experiment containing multiple determinations (N indicates the number of independent determinations). p value as indicated on the figure between Hpb-infected vs. uninfected groups; differences between groups determined by unpaired Welch’s t-test. (B) Anti-CD3/28 stimulated MLN T cells from Hpb-infected WT BALB/c mice, as described in Methods were cultured with anti-IL4 blocking (anti-IL4 (+)) isotype control antibodies (Isotype Control), or no antibody added (anti-IL4(–)), as indicated. Supernatants were analyzed for TGFβ content by ELISA. Data show mean±SD from a representative experiment of 5 independent experiments, with each experiment containing multiple determinations; p values as shown between groups; differences between groups determined by unpaired Welch’s t-test. (C) Representative dot plots of anti-CD3/28-stimulated splenocyte and MLN cultures from Hpb-infected 8–9 week old male WT BALB/c or Jα18−/− mice, with Brefeldin A added to cultures for the last 12 hours. Cells were stained for CD4, Foxp3 and IL4 using Foxp3 staining protocol. Data is representative example of 3 independent experiments for each group.
GATA3 drives IL4 and TGFβ production by T cells after helminth infection.
GATA3 is an essential transcription factor in Th2 development and it plays an important role in IL4 production (24). GATA3 is also expressed in Foxp3+ Tregs and contributes to the function of these cells (25, 26). Furthermore, GATA3−/− Tregs fail to expand in the intestine after colonization with Hpb (25). Because expansion of Tregs in BMT mice after helminth infection is dependent on TGFβ, we analyzed the relationship between GATA3 expression, IL4 and TGFβ production. WT donor Foxp3+ CD4 Tregs and Foxp3- CD4 T cells expressed GATA3 in BMT mice with or without helminth infection (Figure 11A). In helminth-infected mice without BMT, IL4 and TGFβ production was only evident in cultures of GATA3 sufficient – and not in cultures of GATA3 deficient - T cells (Figure 11B). Furthermore, when we analyzed GATA protein expression in MLN T cells from uninfected and helminth-infected TGFβ RII DN mice, whose T cells do not sense TGFβ, and their C57BL/6 counterparts, we observed that GATA3 expression did not require TGFβ (Figure 11C). Together, these results attest to a novel role of Th2 pathway (GATA3/IL4) in TGFβ generation and helminthic suppression of GVHD. Hence, GATA3-driven and IL4-mediated stimulation of TGFβ production fills a gap in knowledge of the events in GVHD that occur between triggering of the Th2 pathway and the induction of Tregs.
Figure 11. GATA3 is expressed on Foxp3− CD4 T cells and Foxp3+ CD4 Tregs during GVHD and is critical to IL4 and TGFβ production by T cells.
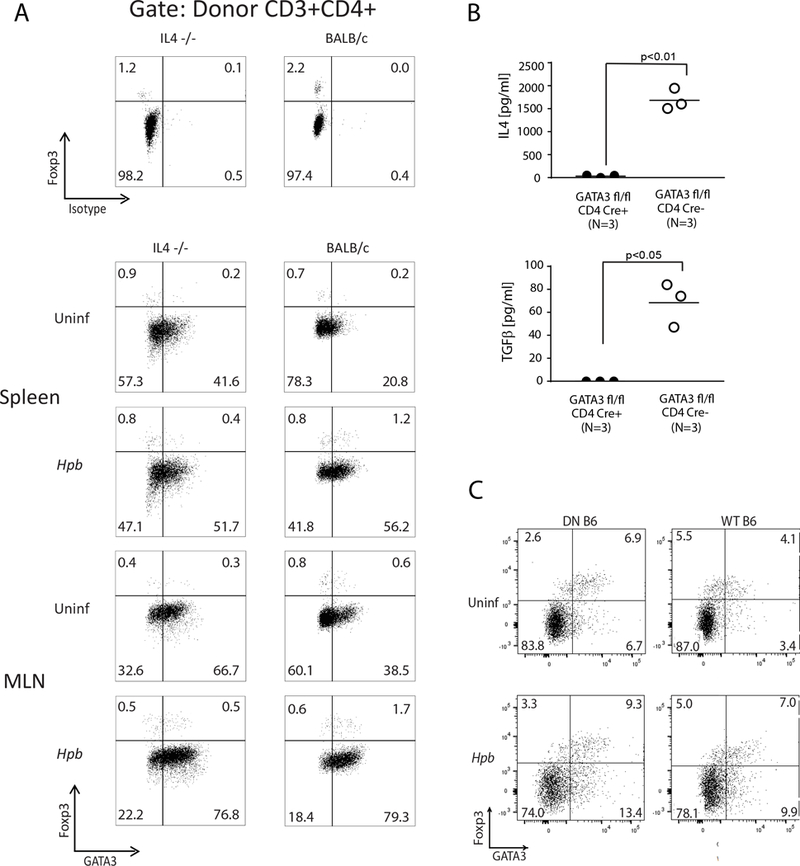
(A) Representative dot plots from spleen and MLN cells isolated from uninfected and Hpb-infected IL4−/− (left) or WT (BALB/c) (right) BMT recipients of WT (C57BL/6) donors, 6 days after BMT. Spleen and MLN cells were stained for CD3, CD4, H2b, Foxp3 and GATA3. Cells were gated on WT C57BL/6 (H2b+) donor CD3+ CD4+ T cells. Parallel splenocyte and MLN cell isolates were stained for CD3, CD4, H2b, Foxp3 and isotype antibody (instead of GATA3) (upper panels). Numbers represent the percentage of events in each quadrant and GATA3− and GATA3+ CD4 Tregs in left upper and the right upper quadrants, respectively. Representative example from 3 independent experiments. (B) Purified CD4 T cells from helminth-infected mice with T cell specific deficiency for GATA3 (GATA3 fl/fl x CD4 Cre+) and from helminth-infected control GATA3 sufficient mice (GATA3 fl/fl x CD4 Cre−) were stimulated plate-bound anti-CD3 and soluble anti-CD28 for 48 hours. Culture supernatants were analyzed by ELISA. Data show mean (bar) from multiple independent experiments (scatter plots) where each dot (N) represents mean value of a single independent experiment calculated from multiple (≥3) repeats (p values between GATA3 deficient and GATA3 sufficient groups as indicated in each panel; differences between groups determined by unpaired Welch’s t-test). (C) Representative dot plots of splenocytes from uninfected (Uninf) and Hpb-infected TGFβ RII DN (DN B6) or C57BL/6 WT (WT B6) mice. Cells were stained for CD3, CD4, Foxp3 and GATA3. Cells were gated on CD3+ CD4+ T cells. Representative example from 3 independent experiments.
Discussion
Modulation of intestinal immune pathways is critical to suppression of devastating GVHD and aberrant immune reactivity in various disorders. Intestinal colonization by helminthic parasites suppresses aberrant immunity in mice and accumulated evidence has linked the helminth-induced Th2 pathway to suppression of inflammation(8). Nonetheless, helminth-induced suppression of aberrant immunity also requires immune regulatory cytokine TGFβ, which inhibits Th2 signaling in some in vitro or in vivo conditions(31). Moreover, TGFβ is critical for the expansion of GVHD-suppressing Tregs in helminth-colonized BMT mice (19). Although the Th2 cytokine IL4 was shown to drive the expansion of Tregs in other models of BMT (21, 22), the link between Th2 signaling, the TGFβ pathway and the activation of Tregs after helminth infection remains obscure.
In this manuscript we provide compelling evidence that helminth-induced activation of the Th2 cytokine IL4 drives TGFβ generation and TGFβ-dependent immune suppression. We demonstrated that IL4 production after helminth infection is driven by the Th2 transcription factor, GATA3, which is critical to the induction and maintenance of TGFβ in Hpb-infected mice. Furthermore, colonization of IL4−/− BMT mice with Hbp fails to promote the expansion of wild-type donor Tregs, resulting in an inability to suppress GVHD. In contrast, Hpb infection of WT BALB/c or Jα18−/− BMT recipients triggers TGFβ-dependent expansion of wild-type donor Foxp3+ Tregs, which dampen lethal alloreactive responses.
Helminth infection is associated with the survival of Th2-polarized host T cells and Foxp3+ Tregs following conditioning with total body irradiation (TBI). To understand the mechanism that links helminth-induced Th2 polarization of host cells to the TGFβ-dependent expansion of donor Tregs, we utilized BMT models that employ myeloablative radiation (TBI) as conditioning, because Hpb infection in this model preserves host lymphoid cells and generates a Th2-polarizing environment – similar to BMT models after conditioning by total lymphoid irradiation (TLI) (21–23). BMT experiments after TLI have demonstrated that the production of IL4 by host iNKT lymphocytes is critical for the suppression of acute GVHD(21). However, when we investigated the role of these cells in suppressing acute GVHD in Hpb-infected mice following conditioning with TBI, we observed helminth-induced suppression of GVHD in iNKT−/− (Jα18−/−) BMT recipients. These results support the notion that helminths influence immune cell subsets of the host other than iNKT in the gut, and that those immune cell subsets are able to generate IL4 and suppress acute GVHD after TBI. Hence, stimulation of host iNKT cells by TLI (21, 23) or glycolipid ligands (22) are no longer needed for helminth-induced IL4 secretion by host cells. Our results are consistent with previous studies that showed enhanced IL4 production by various cell types after helminth infection, where each cell type – rather than being unique - contributed separately to optimal IL4 production by the host (33–36). These studies suggest that helminth-induced type 2 immunity and IL4 production requires coordinated action of various cell types (54, 55).
IL4 binds to the IL4 receptor, whose transduction activates the transcription factor STAT6. STAT6, in turn, stimulates the expression of master regulator of Th2 pathway, GATA3 (56). Although activation of STAT6 or GATA3 was shown to inhibit Tregs (57, 58), Tregs in Hpb-infected and Th2-polarized mice ubiquitously express GATA3(25). Hpb infection stimulates the expansion of Tregs in inflammatory conditions, like GVHD (19) and GATA3 expression by Tregs is critical to the maintenance of suppressive function of these cells in inflammation (25). Similarly, the STAT6-dependent Th2 pathway, which is essential to suppressing lethal inflammation in GVHD (22, 59), has been proposed to constitute a nonredundant signal - second to TcR - in stimulating the expansion and maintenance of peripheral Tregs(60). We propose that it is the IL4/Th2-dependent TGFβ generation – rather than a direct effect of IL4 or Th2 pathway on T lymphocytes - that triggers the expansion of Tregs for the following reasons: First, Th2 pathway is intact in T cells with TGFβ signaling defects (61). Second, helminths stimulate Th2 pathway in T cells that are deficient in TGFβ signaling (46) and do not affect GATA3 protein expression by Foxp3+ Tregs (Figure 11C). Third, helminths fail to promote the expansion of Tregs that do not sense TGFβ (19), although helminths promote the conversion of Foxp3+ Tregs to Foxp3− CD4 Th2 cells (62) after adoptive transfer and this appears to be a direct effect of IL4 on Tregs. We also propose that helminth-stimulated Th2 pathway suppresses inflammatory Th1 cells through TGFβ-dependent circuitries because helminth-triggered Th2 cells do not suppress inflammatory bowel disease or acute GVHD, if TGFβ signaling to T cells is abrogated (19, 46).
The expression of Th2 transcription factor GATA3 on Tregs is known to contribute to the function of Tregs through an unknown mechanism (25, 26). We show that GATA3 is required for helminth-induced production of TGFβ by T cells and TGFβ generated by Tregs can be essential for intestinal immune regulation(63). Although Tregs from TGFβ RII DN mice retain their ability to suppress inflammation(64), these cells do not generate TGFβ(65) but express GATA3, as we show here. With our data that GATA3 is critical to TGFβ generation, cellular mechanisms that lead to GATA3-dependent immune regulation by Tregs and the role of TGFβ in these immune suppressive pathways remain to be established.
In our BMT experiments, host cell IL4 also appears to be necessary for WT donor T cell TNFα secretion. Besides being a well-known inflammatory mediator, TNFα activates and stimulates TGFβ generation by Tregs(66, 67). It will be interesting to know whether TNFα plays a role in IL4-mediated TGFβ secretion after helminth infection.
After primary infection with Hpb, cultures of splenocytes and MLN cells from WT BALB/c and Jα18−/− mice showed IL4 production from Foxp3− CD4 T cells but not from Foxp3+ Tregs. Although Foxp3+ Tregs can be induced to generate IL4 after secondary infection(62), our results raise the possibility that IL4 production by Foxp3− cells drive the TGFβ production by Foxp3+ CD4 Tregs after primary helminth infection. How IL4 signaling to Tregs contributes to GATA3 expression, TGFβ generation and immune regulation – besides conversion to Th2 cells after adoptive transfer (62) – remains to be established.
In the current study, we focused on immune conditioning of host cells by helminths. Besides cells of the host, donor T cells of helminth-infected BMT mice increase their IL4 production(19). Several previous reports have indicated that donor Th2 cells can also alter the course of GVHD(68–71). It will be important to investigate the role of the donor Th2 pathway in helminth-induced suppression of GVHD.
Although TGFβ is critical for the expansion of Tregs during the first days following BMT, alternative TGFβ-independent mechanisms of Treg expansion in a Th2 polarized environment have been reported (20). Another example implicated IL2 signaling as a determinant of the expansion and function of Tregs (72). Experimental evidence, such as the requirement of STAT5 activation by IL2 for early IL4 production (73), indicates that these pathways can coordinate Th2 development, TGFβ secretion and Treg expansion - instead of working independently from each other. Further research on these pathways can also help understand why IL4 inhibits Treg expansion or development in some (28, 32) and activates Tregs in other experimental settings (21, 29, 30). Similarly, in-depth exploration of these pathways (20) may explain why helminthic improvement of clinical GVHD disease score in Jα18−/− BMT recipients is modest, although helminths stimulate TGFβ-dependent regulatory pathways, suppress markedly inflammation in lung as well as the colon and promote survival in Jα18−/− BMT mice.
Collectively, our results address two important questions: First, how do Th2 and TGFβ pathways – that can inhibit each other - co-exist after helminth infection? We answer this by showing that enhanced TGFβ generation is Th2 (GATA3/IL4) dependent. Second, how can the Th2 pathway constitute the second signal in peripheral development and maintenance of Tregs (60)? We provide experimental evidence of a link between GATA3/IL4 - thus Th2 pathway - and TGFβ-dependent Treg expansion in vivo. Our results deserve further attention in clinical and translational research in BMT. The FDA has not approved any medication for use in GVHD, and immune suppressive drugs administered to BMT recipients with GVHD do not provide clear benefit, rather cause severe toxicity. Although the delivery of Tregs has been shown to prevent GVHD in both animal models and BMT patients, in vitro propagation or fresh isolation of enormous numbers of Tregs (6, 7) are required, at a cost that is prohibitive in clinical practice. An alternative to these obstacles is the induction of Treg expansion in vivo. A more detailed understanding of mechanisms that contribute to Treg expansion in vivo, including the link between the Th2 signaling pathway and Treg expansion in helminth infection, is expected to facilitate the development of novel therapeutics for this deadly and devastating disease.
Supplementary Material
Acknowledgments
This study was supported by research funds from the National Institute of Health R56 AI 116715 (MNI); R01 HL56067, AI34495, HL11879 (BRB), DK443119 (RSB) and Department of Veterans Affairs BX002906 (MNI), BX002715 (DEE).
Footnotes
Conflict of Interest Disclosure
The authors declare no conflict of financial interest
References
- 1.Zeiser R, Socie G, and Blazar BR, 2016. Pathogenesis of acute graft-versus-host disease: from intestinal microbiota alterations to donor T cell activation. British journal of haematology 175: 191–207. [DOI] [PubMed] [Google Scholar]
- 2.Teshima T, Reddy P, and Zeiser R, 2016. Acute Graft-versus-Host Disease: Novel Biological Insights. Biol Blood Marrow Transplant 22: 11–16. [DOI] [PubMed] [Google Scholar]
- 3.Sung AD, and Chao NJ, 2013. Concise review: acute graft-versus-host disease: immunobiology, prevention, and treatment. Stem Cells Transl Med 2: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohrt HE, Pillai AB, Lowsky R, and Strober S, 2010. NKT cells, Treg, and their interactions in bone marrow transplantation. European journal of immunology 40: 1862–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beres AJ, and Drobyski WR, 2013. The role of regulatory T cells in the biology of graft versus host disease. Front Immunol 4: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, Bushell A, Cools N, Geissler EK, Gregori S, Marieke van Ham S, Hilkens C, Hutchinson JA, Lombardi G, Madrigal JA, Marek-Trzonkowska N, Martinez-Caceres EM, Roncarolo MG, Sanchez-Ramon S, Saudemont A, and Sawitzki B, 2015. Hurdles in therapy with regulatory T cells. Sci Transl Med 7: 304ps318. [DOI] [PubMed] [Google Scholar]
- 7.Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, Curtsinger J, Verneris MR, MacMillan ML, Levine BL, Riley JL, June CH, Le C, Weisdorf DJ, McGlave PB, Blazar BR, and Wagner JE, 2016. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 127: 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinstock JV, and Elliott DE, 2014. Helminth Infections Decrease Host Susceptibility to Immune-Mediated Diseases. Journal of immunology 193: 3239–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gause WC, and Maizels RM, 2016. Macrobiota - helminths as active participants and partners of the microbiota in host intestinal homeostasis. Curr Opin Microbiol 32: 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, Finney CA, Greenwood EJ, Knox DP, Wilson MS, Belkaid Y, Rudensky AY, and Maizels RM, 2010. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J.Exp.Med. 207: 2331–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleming JO, and Weinstock JV, 2015. Clinical trials of helminth therapy in autoimmune diseases: rationale and findings. Parasite Immunol 37: 277–292. [DOI] [PubMed] [Google Scholar]
- 12.Ince MN, Blazar BR, Edmond MB, Tricot G, and Wannemuehler MJ, 2016. Understanding Luminal Microorganisms and Their Potential Effectiveness in Treating Intestinal Inflammation. Inflamm Bowel Dis 22: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walk ST, Blum AM, Ewing SA, Weinstock JV, and Young VB, 2010. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm.Bowel.Dis. 16: 1841–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, Honda K, Gause WC, Blaser MJ, Bonneau RA, Lim YA, Loke P, and Cadwell K, 2016. Helminth infection promotes colonization resistance via type 2 immunity. Science 352: 608–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peled JU, Jenq RR, Holler E, and van den Brink MR, 2016. Role of gut flora after bone marrow transplantation. Nat Microbiol 1: 16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, Liu C, West ML, Singer NV, Equinda MJ, Gobourne A, Lipuma L, Young LF, Smith OM, Ghosh A, Hanash AM, Goldberg JD, Aoyama K, Blazar BR, Pamer EG, and van den Brink MR, 2012. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J.Exp.Med. 209: 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claes IJ, Vargas Garcia CE, and Lebeer S, 2015. Novel opportunities for the exploitation of host-microbiome interactions in the intestine. Curr Opin Biotechnol 32: 28–34. [DOI] [PubMed] [Google Scholar]
- 18.Grencis RK 2015. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu Rev Immunol 33: 201–225. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Chen HL, Bannick N, Henry M, Holm AN, Metwali A, Urban JF Jr., Rothman PB, Weiner GJ, Blazar BR, Elliott DE, and Ince MN, 2015. Intestinal helminths regulate lethal acute graft-versus-host disease and preserve the graft-versus-tumor effect in mice. Journal of immunology 194: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Merwe M, Abdelsamed HA, Seth A, Ong T, Vogel P, and Pillai AB, 2013. Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplantation. Journal of immunology 191: 5764–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillai AB, George TI, Dutt S, and Strober S, 2009. Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood 113: 4458–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hashimoto D, Asakura S, Miyake S, Yamamura T, Van Kaer L, Liu C, Tanimoto M, and Teshima T, 2005. Stimulation of host NKT cells by synthetic glycolipid regulates acute graft-versus-host disease by inducing Th2 polarization of donor T cells. J Immunol 174: 551–556. [DOI] [PubMed] [Google Scholar]
- 23.Pillai AB, George TI, Dutt S, Teo P, and Strober S, 2007. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J.Immunol. 178: 6242–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, Tumes DJ, and Okamoto Y, 2017. Th2 Cells in Health and Disease. Annu Rev Immunol 35: 53–84. [DOI] [PubMed] [Google Scholar]
- 25.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, Paul WE, Bosselut R, Wei G, Zhao K, Oukka M, Zhu J, and Belkaid Y, 2011. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest 121: 4503–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu F, Sharma S, Edwards J, Feigenbaum L, and Zhu J, 2015. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol 16: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapoval S, Dasgupta P, Dorsey NJ, and Keegan AD, 2010. Regulation of the T helper cell type 2 (Th2)/T regulatory cell (Treg) balance by IL-4 and STAT6. J Leukoc Biol 87: 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, and Qin FX, 2007. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A 104: 18169–18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace L, Pioli C, and Doria G, 2005. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. Journal of immunology 174: 7645–7653. [DOI] [PubMed] [Google Scholar]
- 30.Maerten P, Shen C, Bullens DM, Van Assche G, Van Gool S, Geboes K, Rutgeerts P, and Ceuppens JL, 2005. Effects of interleukin 4 on CD25+CD4+ regulatory T cell function. J Autoimmun 25: 112–120. [DOI] [PubMed] [Google Scholar]
- 31.Li MO, and Flavell RA, 2008. TGF-beta: a master of all T cell trades. Cell 134: 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, and Kuchroo VK, 2008. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol 9: 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urban JF Jr., Katona IM, Paul WE, and Finkelman FD, 1991. Interleukin 4 is important in protective immunity to a gastrointestinal nematode infection in mice. Proc.Natl.Acad.Sci.U.S.A 88: 5513–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu P, Zhou X, Chen SJ, Moorman M, Morris SC, Finkelman FD, Linsley P, Urban JF, and Gause WC, 1994. CTLA-4 ligands are required to induce an in vivo interleukin 4 response to a gastrointestinal nematode parasite. J Exp Med 180: 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Ruckerl D, Seddon B, MacDonald AS, McKenzie A, and Wilson MS, 2016. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol 9: 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith KA, Harcus Y, Garbi N, Hammerling GJ, MacDonald AS, and Maizels RM, 2012. Type 2 innate immunity in helminth infection is induced redundantly and acts autonomously following CD11c(+) cell depletion. Infection and immunity 80: 3481–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, and Blazar BR, 2010. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood 116: 5738–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagaraj S, Youn JI, and Gabrilovich DI, 2013. Reciprocal relationship between myeloid-derived suppressor cells and T cells. Journal of immunology 191: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, and Taniguchi M, 1997. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 278: 1623–1626. [DOI] [PubMed] [Google Scholar]
- 40.Persson L 1974. A modified baermann apparatus for the recovery of infective nematode larvae from herbage and manure. Zentralblatt fur Veterinarmedizin. Reihe B. Journal of veterinary medicine. Series B 21: 483–488. [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann P, Ermann J, Edinger M, Fathman CG, and Strober S, 2002. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J.Exp.Med. 196: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, and Ferrara JL, 1996. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 88: 3230–3239. [PubMed] [Google Scholar]
- 43.Tran IT, Sandy AR, Carulli AJ, Ebens C, Chung J, Shan GT, Radojcic V, Friedman A, Gridley T, Shelton A, Reddy P, Samuelson LC, Yan M, Siebel CW, and Maillard I, 2013. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J.Clin.Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, Chao NJ, and Yang Y, 2012. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 120: 2899–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ince MN, Elliott DE, Setiawan T, Blum A, Metwali A, Wang Y, Urban JF Jr., and Weinstock JV, 2006. Heligmosomoides polygyrus induces TLR4 on murine mucosal T cells that produce TGFbeta after lipopolysaccharide stimulation. J.Immunol. 176: 726–729. [DOI] [PubMed] [Google Scholar]
- 46.Ince MN, Elliott DE, Setiawan T, Metwali A, Blum A, Chen HL, Urban JF, Flavell RA, and Weinstock JV, 2009. Role of T cell TGF-beta signaling in intestinal cytokine responses and helminthic immune modulation. Eur.J.Immunol. 39: 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Setiawan T, Metwali A, Blum AM, Ince MN, Urban JF Jr., Elliott DE, and Weinstock JV, 2007. Heligmosomoides polygyrus promotes regulatory T-cell cytokine production in the murine normal distal intestine. Infect.Immun. 75: 4655–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan DH, Anderson BE, McNiff JM, Jain D, Shlomchik MJ, and Shlomchik WD, 2004. Target antigens determine graft-versus-host disease phenotype. J.Immunol. 173: 5467–5475. [DOI] [PubMed] [Google Scholar]
- 49.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, and Serody JS, 2009. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood 113: 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sachs DH, Kawai T, and Sykes M, 2014. Induction of tolerance through mixed chimerism. Cold Spring Harb Perspect Med 4: a015529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayer AL, Jones M, Chirinos J, de Armas L, Schreiber TH, Malek TR, and Levy RB, 2009. Host CD4+CD25+ T cells can expand and comprise a major component of the Treg compartment after experimental HCT. Blood 113: 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoue T, Ikegame K, Kaida K, Okada M, Yoshihara S, Tamaki H, Fujimori Y, Soma T, and Ogawa H, 2016. Host Foxp3+CD4+ Regulatory T Cells Act as a Negative Regulator of Dendritic Cells in the Peritransplantation Period. Journal of immunology 196: 469–483. [DOI] [PubMed] [Google Scholar]
- 53.Gorelik L, and Flavell RA, 2000. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 12: 171–181. [DOI] [PubMed] [Google Scholar]
- 54.Mohrs K, Wakil AE, Killeen N, Locksley RM, and Mohrs M, 2005. A two-step process for cytokine production revealed by IL-4 dual-reporter mice. Immunity 23: 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, Honjo T, Mohrs K, Mohrs M, Randall T, and Lund FE, 2009. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity 30: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paul WE 2010. What determines Th2 differentiation, in vitro and in vivo? Immunol Cell Biol 88: 236–239. [DOI] [PubMed] [Google Scholar]
- 57.Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, Kato S, Yoshimura A, and Kobayashi T, 2008. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem 283: 14955–14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, Akdis CA, Blaser K, and Schmidt-Weber CB, 2007. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol 5: e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reddy P, Teshima T, Hildebrandt G, Williams DL, Liu C, Cooke KR, and Ferrara JL, 2003. Pretreatment of donors with interleukin-18 attenuates acute graft-versus-host disease via STAT6 and preserves graft-versus-leukemia effects. Blood 101: 2877–2885. [DOI] [PubMed] [Google Scholar]
- 60.Sanchez-Guajardo V, Tanchot C, O’Malley JT, Kaplan MH, Garcia S, and Freitas AA, 2007. Agonist-driven development of CD4+CD25+Foxp3+ regulatory T cells requires a second signal mediated by Stat6. Journal of immunology 178: 7550–7556. [DOI] [PubMed] [Google Scholar]
- 61.Li MO, Wan YY, Sanjabi S, Robertson AK, and Flavell RA, 2006. Transforming growth factor-beta regulation of immune responses. Annu.Rev.Immunol. 24: 99–146. [DOI] [PubMed] [Google Scholar]
- 62.Pelly VS, Coomes SM, Kannan Y, Gialitakis M, Entwistle LJ, Perez-Lloret J, Czieso S, Okoye IS, Ruckerl D, Allen JE, Brombacher F, and Wilson MS, 2017. Interleukin 4 promotes the development of ex-Foxp3 Th2 cells during immunity to intestinal helminths. J Exp Med 214: 1809–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li MO, Wan YY, and Flavell RA, 2007. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26: 579–591. [DOI] [PubMed] [Google Scholar]
- 64.Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, and Powrie F, 2005. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med 201: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ince MN, Elliott DE, Setiawan T, Metwali A, Blum A, Chen HL, Urban JF, Flavell RA, and Weinstock JV, 2009. Role of T cell TGF-beta signaling in intestinal cytokine responses and helminthic immune modulation. Eur J Immunol 39: 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X, Wu X, Zhou Q, Howard OM, Netea MG, and Oppenheim JJ, 2013. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. Journal of immunology 190: 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, Pan Y, Schneidawind D, Meyer E, and Negrin RS, 2016. TNF-alpha priming enhances CD4+FoxP3+ regulatory T-cell suppressive function in murine GVHD prevention and treatment. Blood 128: 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nikolic B, Lee S, Bronson RT, Grusby MJ, and Sykes M, 2000. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest 105: 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy WJ, Welniak LA, Taub DD, Wiltrout RH, Taylor PA, Vallera DA, Kopf M, Young H, Longo DL, and Blazar BR, 1998. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Clin Invest 102: 1742–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneidawind D, Pierini A, Alvarez M, Pan Y, Baker J, Buechele C, Luong RH, Meyer EH, and Negrin RS, 2014. CD4+ invariant natural killer T cells protect from murine GVHD lethality through expansion of donor CD4+CD25+FoxP3+ regulatory T cells. Blood 124: 3320–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coghill JM, Sarantopoulos S, Moran TP, Murphy WJ, Blazar BR, and Serody JS, 2011. Effector CD4+ T cells, the cytokines they generate, and GVHD: something old and something new. Blood 117: 3268–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, and Soiffer RJ, 2011. Interleukin-2 and regulatory T cells in graft-versus-host disease. N.Engl.J.Med. 365: 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamane H, Zhu J, and Paul WE, 2005. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med 202: 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.