

Abstract
Centromeric chromatin defines the site of kinetochore formation and ensures faithful chromosome segregation. Centromeric identity is epigenetically specified by the incorporation of CENP-A nucleosomes. DNA replication presents a challenge for inheritance of centromeric identity because nucleosomes are removed to allow for replication fork progression. Despite this challenge, CENP-A nucleosomes are stably retained through S phase. We used BioID to identify proteins transiently associated with CENP-A during DNA replication. We found that during S phase, HJURP transiently associates with centromeres and binds to pre-existing CENP-A, suggesting a distinct role for HJURP in CENP-A retention. We demonstrate that HJURP is required for centromeric nucleosome inheritance during S phase. HJURP co-purifies with the MCM2–7 helicase complex and together with the MCM2 subunit binds CENP-A simultaneously. Therefore, pre-existing CENP-A nucleosomes require an S phase function of the HJURP chaperone and interaction with MCM2 to ensure faithful inheritance of centromere identity through DNA replication.
eTOC
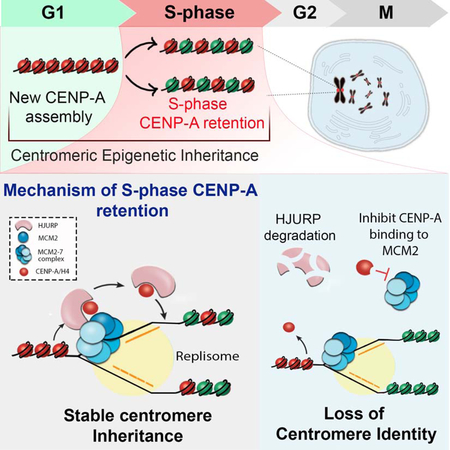
Inheritance of centromere identity requires the transmission of CENP-A across DNA replication, when nucleosomes are disassembled ahead of the replication fork. Zasadzinska et al. demonstrate that CENP-A requires the dedicated CENP-A chaperone HJURP and interaction with the replicative helicase complex to retain and redeposit CENP-A following DNA replication.
GRAPHICAL ABSTRACT
Introduction
Centromeres are unique chromatin domains present on each chromosome that facilitate recruitment of the constitutive centromere-associated network (CCAN) and kinetochore, which work together to ensure equal chromosome segregation during mitosis (Amano et al., 2009; Cheeseman and Desai, 2008; Earnshaw et al., 1986; Foltz et al., 2006; Izuta et al., 2006; McKinley and Cheeseman, 2016; Nishihashi et al., 2002; Okada et al., 2006; Saitoh et al., 1992; Sugata et al., 1999). In most eukaryotes, deposition of centromere specific nucleosomes containing the histone H3 variant CENP-A serves as an epigenetic mark critical for centromere specification and inheritance, independent of the underlying DNA sequence (Allshire and Karpen, 2008; Cleveland et al., 2003).
In contrast to the canonical H3.1 histone variant, new CENP-A incorporation in human cells is uncoupled from DNA replication and occurs during early G1 (Jansen et al., 2007). The vertebrate Holliday Junction Recognition Protein (HJURP), the yeast homolog Scm3, and functional homolog CAL1 in Drosophila, specifically recognize prenucleosomal CENP-A and facilitate its deposition at the centromere (Barnhart et al., 2011; Bernad et al., 2011; Camahort et al., 2007; Chen et al., 2014; Dechassa et al., 2011; Dunleavy et al., 2009; Erhardt et al., 2008; Foltz et al., 2009; Goshima et al., 2007; Mizuguchi et al., 2007; Pidoux et al., 2009; Shuaib et al., 2010; Stoler et al., 2007; Williams et al., 2009). HJURP is known for its role in early G1 deposition of new CENP-A nucleosomes (Dunleavy et al., 2009; Foltz et al., 2009). Centromeric recruitment of HJURP or budding yeast Scm3 in complex with CENP-A/H4 requires the Mis18 complex (Barnhart et al., 2011; Camahort et al., 2007; Fujita et al., 2007; Hayashi et al., 2004; Mizuguchi et al., 2007; Moree et al., 2011; Pidoux et al., 2009; Stoler et al., 2007; Williams et al., 2009). In humans, the Mis18 complex is comprised of Mis18α, Mis18β and Mis18BP1 subunits (Fujita et al., 2007; Maddox et al., 2007). The Mis18 complex directs HJURP to centromeres through a physical interaction between HJURP centromere targeting domain and the Mis18α-β C-terminal coiled-coil domains (Nardi et al., 2016; Wang et al., 2014). The recruitment of CENP-A deposition pathway proteins to centromeres is cell cycle regulated and both HJURP and the Mis18 complex are controlled by Cdk and Plk1 mediated phosphorylation (McKinley and Cheeseman, 2016; Muller et al., 2014; Pan et al., 2017; Silva et al., 2012; Spiller et al., 2017; Stankovic et al., 2017).
DNA replication requires the disassembly of existing chromatin ahead of the replication fork, and reassembly of evicted histones once DNA synthesis is completed (Annunziato et al., 1981; McKnight and Miller, 1977; Sogo et al., 1986). A proportion of parental H3.1 nucleosomes are inherited across DNA replication to preserve chromatin modifications and epigenetic states. The MCM2–7 helicase complex facilitates DNA unwinding at replication origins during replication initiation, and travels with the replication forks to unwind DNA ahead of the progressing replisome (Bochman and Schwacha, 2009; Boos et al., 2012). Although a mechanism of how parental nucleosomes are retained is not fully understood, experiments in yeast and human cells suggested that the PCNA in complex with CAF-1, as well as the MCM2 helicase in complex with Asf1 or FACT are important players in recycling parental nucleosomes at the replication fork (Alabert and Groth, 2012; Burgess and Zhang, 2013; Foltman et al., 2013; Gerard et al., 2006; Huang et al., 2015; Ransom et al., 2010; Shibahara and Stillman, 1999). An analogous role for the interaction between MCMs and the CENP-A chaperone HJURP in nucleosome retention has also been proposed (Huang et al., 2015). MCM2 co-purifies with nucleosomal H3-H4, and directly binds all histone H3 variants, including CENP-A, through its N-terminal histone biding motif (HBD) (Foltman et al., 2013; Groth et al., 2007; Huang et al., 2015; Jasencakova et al., 2010; Richet et al., 2015).
Centromeric nucleosomes are stably inherited across multiple generations suggesting that CENP-A nucleosomes are faithfully retained through DNA replication (Bodor et al., 2013; Falk et al., 2015; Guo et al., 2017; Jansen et al., 2007; Mellone et al., 2011). CENP-A nucleosomes are dispersed into daughter strands, and the gaps resulting from CENP-A dilution are occupied by histone H3.1 and H3.3 nucleosomes (Bodor et al., 2013; Dunleavy et al., 2011; Falk et al., 2015; Jansen et al., 2007; Ross et al., 2016). This suggests that existing CENP-A is specifically reassembled onto centromeric DNA following DNA synthesis; however, the mechanism that regulates CENP-A maintenance is unknown. We posit that CENP-A retention at centromeres during S phase is facilitated by a unique mechanism that specifically distinguishes CENP-A from bulk histones. Likewise, distinct mechanisms may also be required to ensure the stable inheritance of PTM based epigenetic signals on canonical histones during DNA replication.
Here we employed BioID in an unbiased experimental approach to identify proteins associated with CENP-A during DNA replication. We demonstrated that HJURP is associated with existing CENP-A during DNA replication. We identify a distinct role for HJURP chaperone outside of G1 phase and show that HJURP plays a role in maintaining centromere identity during DNA replication. To delineate a specific role for the protein in S phase, we used Auxin-inducible degron strategy for HJURP depletion and show that loss of HJURP during DNA replication leads to loss of CENP-A retention. We demonstrate that CENP-A also requires interaction with the MCM2 protein for stable inheritance. CENP-ARK->AA mutants that are deficient in MCM2 binding in vitro fail to be efficiently retained at the centromere through DNA replication. Furthermore, HJURP and MCM2 can bind CENP-A simultaneously. These data demonstrate that the mechanism of S phase retention of the CENP-A nucleosomes requires CENP-A specific deposition machinery including HJURP together with the activity of MCM2.
Results
Identification of proteins associated with CENP-A during DNA replication
CENP-A nucleosomes are dispersed between daughter DNA strands during replication; however, the mechanism that facilitates CENP-A inheritance is largely unknown. We hypothesize that interactions involved in CENP-A retention during S phase occur transiently; therefore, in order to identify the proteins involved in this process we used the BioID proximity based in vivo labelling assay coupled with mass spectrometry (MS)(Roux et al., 2012). In this strategy, the BirA* enzyme mediates a covalent biotin attachment to lysine residues of stable and transiently associated proteins. Biotinylated proteins are then purified under denaturing conditions using streptavidin-beads and analyzed by mass spectrometry (Fig. 1A). CENP-A or histone H3.1 were fused to the BirA* biotin ligase and stable cell lines were generated expressing the fusion proteins (Fig. 1B). Biotin addition to the culture medium was used to induce CENP-A or H3.1 mediated labeling. Biotinylated proteins were visualized by Cy3-conjugated streptavidin and analyzed by immunoblot (Fig. 1C,D). The CENP-A–BirA*-HA cellular localization as well as its biotinylation profile colocalize with centromere marker CENP-T, indicating that we can specifically biotinylate proteins associated with centromeric chromatin. The H3.1–BirA*-HA localizes to the bulk chromatin and mediates biotinylation of protein factors associated with general chromatin (Fig.1 B,C). We isolated proteins biotinylated by either CENP-A or H3.1 from randomly cycling cells using streptavidin purification. By immunoblot, we identified factors known to be closely associated with CENP-A and H3.1 histones including HJURP or Asf1α, respectively (Fig. 1D). We also detected histone H2B in the pull-down fractions, suggesting that we can induce biotinylation mediated by nucleosomal CENP-A–BirA*HA and H3.1–BirA*-HA (Fig. 1D).
Figure 1. Labeling of proteins transiently associated with CENP-A and H3.1 nucleosomes.
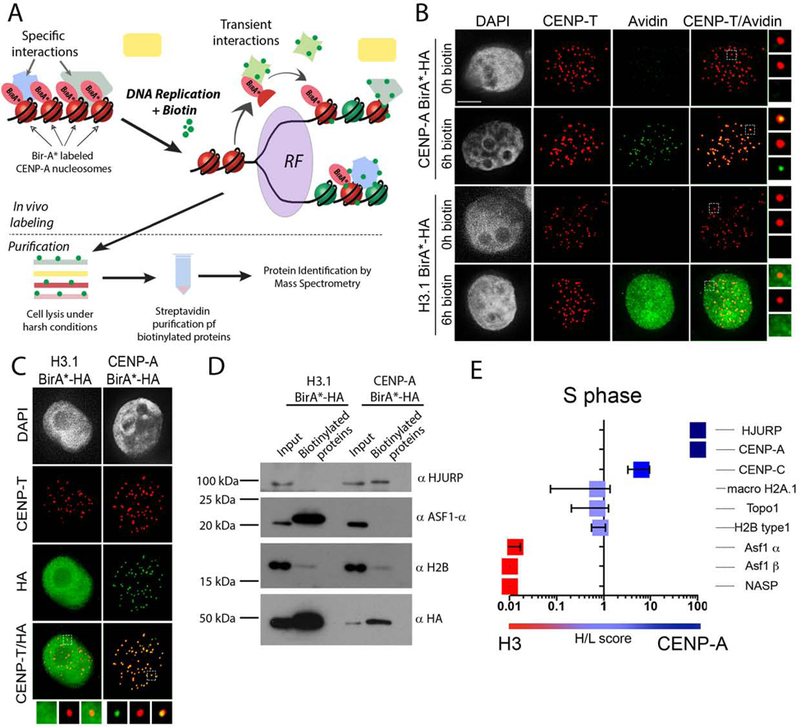
(A) Schematic representation of the experimental approach. (B) (C) Representative images of cells stably expressing indicated proteins fused to the BirA* ligase and HA tag, and incubated with medium supplemented with or without biotin for 5 hours. DNA is visualized by DAPI staining, immunofluorescence for CENP-T is shown in red, biotinylated proteins (B) or BirA*-HA fusion proteins (C) are shown in green. Scale bar is 5μm. (D) Streptavidin purification of biotinylated proteins from indicated cells lines analyzed by immunoblot. Cell media was supplemented with or without biotin for 24 hours. (E) Graph showing SILAC comparison between CENP-A-BirA*-HA biotinylation profile (heavy) versus biotinylation profile of H3.1-BirA-HA* (light) in cells undergoing S phase. Corresponding H/L scores for selected proteins are displayed. The graph represents an average of two independently performed experiments +/− SEM.
We used the BioID approach to biotinylate and purify proteins specifically and transiently associated with CENP-A during DNA replication, and identified proteins by mass spectrometry. Cell lines expressing BirA*-fusion proteins were synchronized by double thymidine block and release and the biotinylation profiles mediated by CENP-A–BirA* versus histone H3.1-BirA* during DNA replication were quantitatively compared using SILAC (Fig. 1E, S1, Table1). Heavy/Light ratios above 1 indicate an enrichment of the protein in the CENP-A–BirA* condition. This allowed us to identify proteins specifically associated with CENP-A during DNA replication relative to general chromatin. Using this strategy, we observed strong enrichment for the known S phase histone chaperone protein Asf1α, Asf1β, and NASP with histone H3.1, thus validating our approach (Fig. 1E). In addition, we observed no enrichment for Macro H2A.1, Topo1 or histone H2B, which are common to both CENP-A and histone H3.1 containing chromatin. Surprisingly, our analysis shows that the CENP-A specific chaperone HJURP is associated with CENP-A during DNA replication. Comparisons of CENP-A–BirA* labeled proteins in S phase arrested cells and randomly cycling cells shows no enrichment, suggesting that CENP-A associates with HJURP in S phase as well as in G1 (Fig. S1D, Table1).
HJURP is associated with chromatin assembled CENP-A during S phase
The association of HJURP with CENP-A during DNA replication suggests that HJURP may be involved in facilitating the retention of pre-existing CENP-A as centromeric nucleosomes are disrupted during replication. In order to demonstrate that pre-existing CENP-A already present within chromatin associates with HJURP during DNA replication we generated stable cell lines expressing CENP-A–BirA*-HA or H3.1–BirA*-HA under a Dox-inducible promoter. Cells were treated with Dox for 96 hours and then Dox was washed out to shut down CENP-A expression and ensure the sole source of CENP–BirA* was from already incorporated CENP-A. Biotinylation was induced in thymidine-synchronized cells undergoing S phase, arrested in early S phase or asynchronous cell populations by providing biotin for 6 hours (Fig. 2A). We observed that both HJURP and Mis18BP1 were biotinylated by nucleosomal CENP-A in asynchronous cells (Fig. 2B right panel, Fig S2). Consistent with our MS purification above, we detected an interaction of nucleosomal CENP-A with HJURP and Mis18BP1 during early S phase, and the degree of biotinylation increased in cells that underwent DNA replication (Fig. 2B left panel, Fig S2).
Figure 2. CENP-A deposition proteins are associated with centromeres during DNA replication.
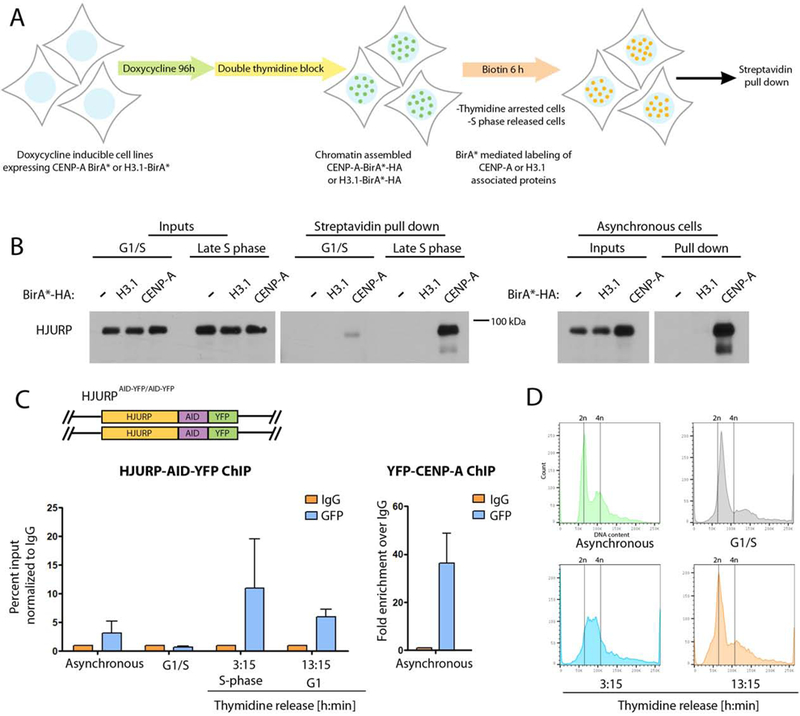
(A) Schematic representation of the experimental approach used in B. Cells were blocked at the G1/S boundary or early S phase by addition of thymidine, and allowed to under S phase following thymidine removal. (B) Cells expressing BirA*-fused proteins under doxycycline inducible promoter were treated as shown in A. Biotinylated proteins were isolated by streptavidin purification following by immunoblot analysis with an HJURP antibody. The experiment was conducted twice, each experiment was conducted using 0.9*107 cells. Input and pull down fractions in the experiment were run on independent gels. (C) Schematic representation of the DLD1-Tir1 cell line where HJURP was endogenously tagged at both alleles with AID-YFP. ChiP from HJURP-AID-YFP cells (left) or YFP-CENP-A cells (right) at indicated time points. ChIP was performed using anti-GFP antibody or normal rabbit IgG. RT-PCR was performed using primers specific for α-satellite DNA of chromosome 7. The graph represents an average of two independent experiments, +/− SEM. (D) FACS profiles of cells used as an input for ChIP.
To further support the association of HJURP with centromeric chromatin during S phase we performed ChIP for chromatin associated HJURP. Using CRISPR-Cas9 mediated gene editing we tagged endogenous HJURP with an auxin-inducible degron (AID) and YFP tag (HJURP-AID-YFP) in a DLD1 cell line stably expressing the E3 ubiquitin ligase Tir1 (Fig. 2C top panel) (see below). ChIP for endogenously-tagged HJURP-AID-YFP was performed using anti-GFP antibody from asynchronous population, cells blocked in early S phase by thymidine arrest, and those progressing through S phase or undergoing G1 following release from thymidine for the indicated times (Fig 2C). The FACS profiles performed on indicated cell populations confirm the desired cell populations (Fig. 2D). qPCR analysis was conducted using primers amplifying α-satellite DNA from chromosome 7 (Tsuda et al., 1997). The amount of endogenous HJURP at the centromere increased as cells progressed through S phase compared to S phase blocked cells or asynchronously dividing population (Fig. 2C). The amount of HJURP detected in S phase was comparable to what we observed in the G1 population, when new CENP-A is being deposited. A similar enrichment of HJURP and Mis18BP1 was observed at chromatin during S phase in cells stably expressing GFP-HJURP and GFP-Mis18BP1 created by lentiviral transduction (Fig. S3A).
The accumulation of HJURP at centromeres has been observed during early G1 using immunofluorescence (Dunleavy et al., 2009; Foltz et al., 2009); however, HJURP has not been previously observed at centromeres during DNA replication (Bui et al., 2012). We reasoned that HJURP may interact with the centromere in a dynamic nature during DNA replication and may be difficult to detect using standard immunofluorescence. Therefore, we generated HJURP–BirA*-HA and Mis18BP1–BirA*-HA constructs which can be used to create a biotin trace of transient occupancy of the protein at the centromere during S phase. Biotinylation mediated by HJURP–BirA* or Mis18BP1–BirA* was induced in cells arrested in early S phase or cells undergoing DNA replication (Fig. S3B,C). Biotinylation of centromere associated proteins was apparent in HJURP–BirA* and Mis18BP1–BirA* expressing cells in blocked cells as detected by fluorophore conjugated streptavidin and in cells undergoing DNA replication, indicating HJURP and Mis18BP1 are present at the centromere while cells are undergoing S phase. Consistent with S phase association, all three Mis18 complex subunits (Mis18α, Mis18β and Mis18BP1) were previously found to be associated with nascent DNA in human cells (Alabert et al., 2014).
We hypothesized that protein-turnover may contribute to the transient association of HJURP with the centromere during S phase, as other proteins are actively degraded during DNA replication to control their function (Roseaulin et al., 2013a; Roseaulin et al., 2013b). Therefore, we assessed HJURP-GFP, Mis18α-GFP and Mis18BP1-GFP recruitment in stably expressing cells during S phase in the presence of the proteasome inhibitor MG132. Cells were released into S phase and 3 hours post release treated with MG132 for 2 hours. GFP-tagged HJURP, Mis18α and Mis18BP1 all accumulated specifically at centromeres in response to the addition of MG132 (Fig. 3A,B). Although GFP-tagged proteins levels increased at centromeres, this was not due to a direct increase in stability of either HJURP, Mis18α or Mis18BP in response to MG132 treatment (Fig. 3C). Live cell imaging showed that the accumulation of GFP-HJURP occurred within 20 minutes of MG132 treatment (Fig. 3D, S4), and was unique to cells undergoing S phase, and did not occur at high frequency in randomly cycling cells (Fig. 3E).
Figure 3. CENP-A deposition proteins accumulate at centromeres during DNA replication in response to MG132 treatment.
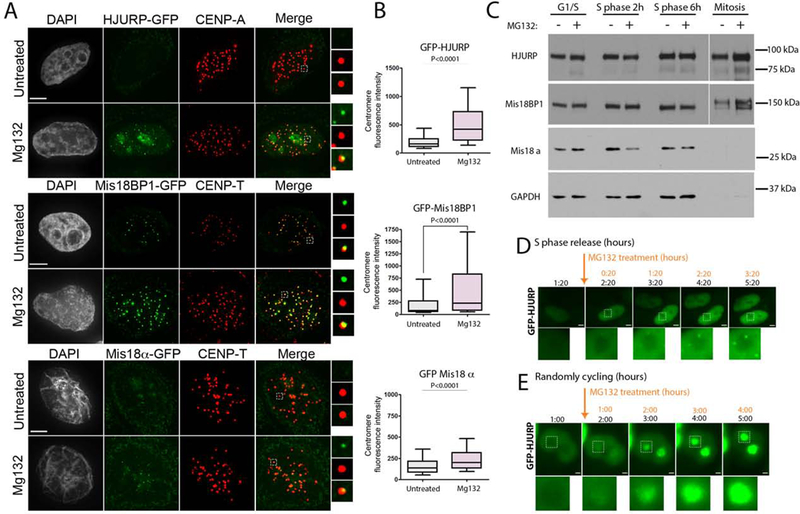
(A) Representative images of cells expressing GFP-fused HJURP, Mis18BP1 or Mis18α during S phase with or without MG132 treatment. Scale bar is 5μm. (B) Quantification of the GFP fluorescence intensity in A. Data was plotted using box-and-whisker plot: 10–90 percentile. The statistical significance was calculated using unpaired t-test and the p values are indicated, n > 434 (C) Immunoblot analysis of HJURP, Mis18BP1 and Mis18α protein levels in response to MG132 treatment at indicated cell cycle stages. The separated section of the blot corresponds to a longer exposure required due to unequal loading (see GAPDH). (D)(E) Live-cell images of cells expressing GFP-HJURP undergoing S phase. (D) or in asynchronous (E) cell populations. Cells were treated with MG132 during imaging starting from the time points indicated by the arrow. Scale bar is 5μm and 2μm, respectively.
HJURP is required for CENP-A inheritance during DNA replication
The association of HJURP with CENP-A during S phase suggests that, in addition to its role in G1-coupled new CENP-A deposition, the HJURP chaperone may also contribute to CENP-A inheritance during DNA replication. In order to specifically test if HJURP is required for CENP-A retention during S phase, we employed the auxin-inducible degron (AID) system for the rapid depletion of proteins upon treatment with IAA (Nishimura et al., 2009). IAA treatment of an endogenously-tagged homozygous HJURP-AID-YFP cell line (Fig. 2C) for 24 hours resulted in reduction of HJURP protein levels below the limit of detection (Fig. 4B). Consistent with the essential role of HJURP, cell survival was drastically reduced in response to the IAA treatment in HJURP-AID-YFP cells, whereas the parental DLD1-Tir1 cells were unaffected (Fig. 4C).
Figure 4. HJURP is required for CENPA retention across S phase.
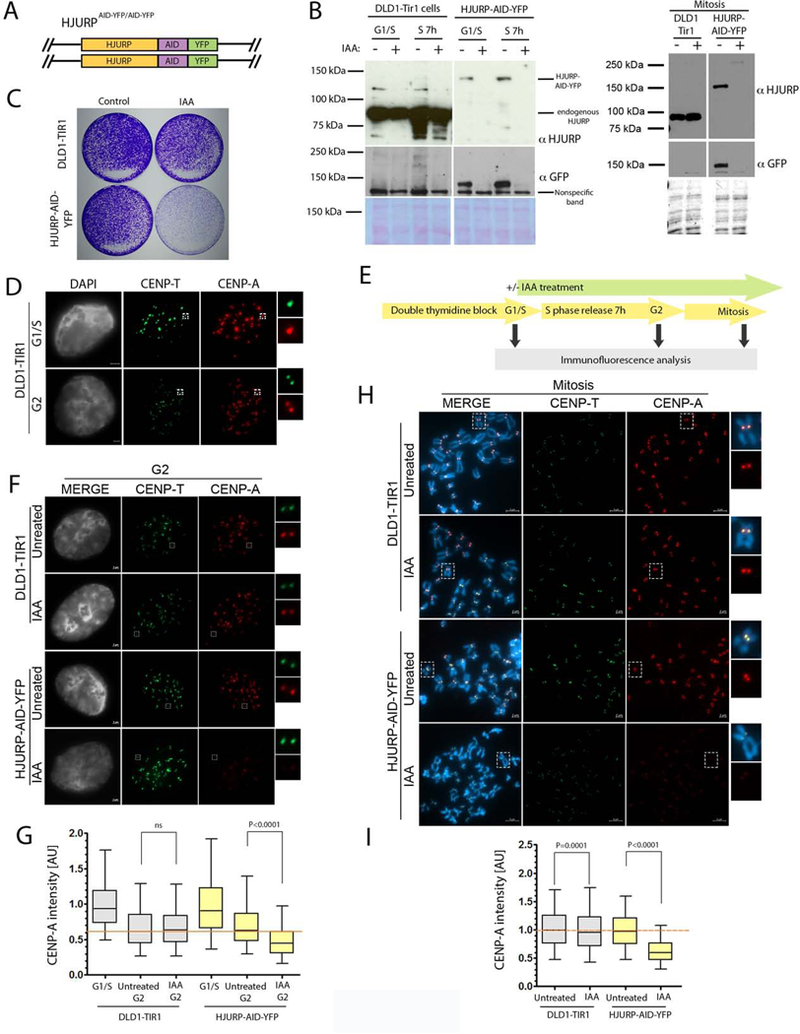
(A) Schematic representation DLD1-Tir1 cell line where HJURP was endogenously tagged with AID-YFP at both alleles. (B) Immunoblot analysis showing the efficiency of IAA dependent HJURP degradation. Ponceau was used as a loading control. The blots in B were separated into two sections due to removal of extraneous samples present on the same blot. (C) Clonogenic assay using the parental DLD1-Tir1 and HJURP-AID-YFP cells plus or minus IAA treatment for 10 days. (D) Representative images of DLD1-Tir1 cells in G1/S arrest and G2 phase. Insets are showing single centromeres and sister centromeres in G1/S and G2 phase cells, respectively. DNA was visualized by DAPI, immunofluorescence for CENP-T is shown in green and CENP-A is shown in red. Scale bar is 2μm. (E) Schematic representation of the experiment in F and H. (F)(H) Representative images of cells in G2 phase and mitosis, respectively, and treated as shown in E. DNA was visualized by DAPI, immunofluorescence for CENP-T is shown in green and CENP-A is shown in red. Scale bar is indicated. (G)(I) Quantification of F and H, respectively. The data normalized to G1/S phase condition (G) and untreated condition (I) within cell lines. Normalized data from four (G) or three (I) independent experiments was plotted using box-and-whisker plot: 5–95 percentile, n > 2946 (G) and 8414 centromeres (I). The statistical significance was calculated using unpaired t-test and the p values are indicated. For reference, red line indicates level of CENP-A retention in (G) untreated parental control in G2 phase or (I) untreated parental control in mitosis. The percent loss of CENP-A was calculated to be 31.53% and 36.68% in G and I, respectively.
Prior work has demonstrated that existing CENP-A nucleosomes show no turnover across the cell cycle (Bodor et al., 2013; Jansen et al., 2007). Consistent with these reports, we observed a similar amount of CENP-A at centromeres in cells blocked in early S phase compared with centromeres that had progressed 7-hours through DNA replication (Fig. S5C,E). Therefore, CENP-A nucleosome are quantitatively retained during DNA replication. To directly test the role of HJURP in CENP-A inheritance, we examined the stability of endogenous CENP-A at the centromere through S–phase when endogenous HJURP-AID-YFP was degraded by addition of IAA. Parental DLD1-Tir1 or HJURP-AID-YFP cell lines were synchronized and treated with or without IAA for 90 mins prior to S phase release in order to degrade existing HJURP in these cells (Fig. 4E). DNA replication occurs asynchronously between centromeres of different chromosomes during mid to late S phase (O’Keefe et al., 1992; Ten Hagen et al., 1990). Therefore, the retention of endogenous CENP-A was analyzed in G2-phase cells. Fully replicated late G2 centromeres were easily identified as separated sisters. These cells had not yet entered mitosis based on the decondensed state of the chromatin (Fig. 4D,F).
Under control conditions CENP-A levels decreased by approximately 50% at G2 centromeres compared with centromeres in early S phase (Fig. 4D,F,G), which is consistent with redistribution of centromeric nucleosomes into daughter strands as cells progress through DNA replication followed by separation of sister centromeres. The parental DLD1-Tir1 cell line showed no change of CENP-A levels in G2 cells upon IAA treatment. However, IAA treatment of HJURP-AID-YFP cells resulted in a 31% decrease of centromeric CENP-A in G2 cells (Fig. 4F,G). Similarly, analysis of mitotic cell populations revealed that CENP-A inheritance was reduced by 37% in cells where HJURP was degraded, compared to controls (Fig. 4H,I). Consistent with these results, the siRNA mediated depletion of HJURP prior to S phase entry also resulted in significant reduction of endogenous CENP-A at the centromeres (49%). (Fig. S5A-C, S5E-G). These data suggest that CENP-A requires HJURP activity to faithfully transit the replication fork.
In order to specifically examine the retention of pre-existing CENP-A nucleosomes, we generated a stable cell line in which endogenous CENP-A was tagged with the SNAP-tag (Jansen et al., 2007) and endogenous HJURP was tagged with the AID-YFP (Fig. 5A). This dual tagged-protein approach allowed us to label existing CENP-A nucleosomes and determine their fate across the cell cycle when HJURP is degraded. HJURP-AID-YFP localizes to centromeres in telophase cells and is efficiently depleted upon IAA treatment (Fig. 5B,C). In synchronized cells, pre-existing CENP-A was labeled with TMR* in the thymidine arrested population, cells were then released into S phase with or without IAA treatment and assayed at the following G1/S boundary 20 hours later. In agreement with previous studies, we observed approximately 50% reduction of pre-existing CENP-A levels following 1 cell cycle in control cells when compared to the initial G1/S TMR* signal (Jansen et al., 2007). This reduction is attributed to CENP-A dilution into sister chromatids during DNA replication (Fig. 5D,E). HJURP-AID degradation resulted in significant loss of pre-existing CENP-A from the chromatin, and the CENP-A levels in these cells were reduced by 38% (Fig. 5D,E). Previous work suggested that HJURP may be involved in the DNA damage response (Kato et al., 2007). In order to preclude a potential role for DNA damage response induced by thymidine arrest, pre-existing CENP-A was labeled with TMR* in asynchronous cell population (CENP-A initial signal), and CENP-A-TMR* levels were analyzed 24 (or 27) hours later in cells subjected to IAA or control treatments (Fig. 5F, G). The degradation of HJURP-AID led to a 28% loss of pre-existing CENP-A nucleosomes, consistent with what was observed in synchronized cells. We therefore conclude that HJURP is required for the inheritance of pre-existing CENP-A nucleosomes across DNA replication independent of its role in new CENP-A deposition.
Figure 5. HJURP is required for CENPA inheritance of existing CENP-A nucleosomes.
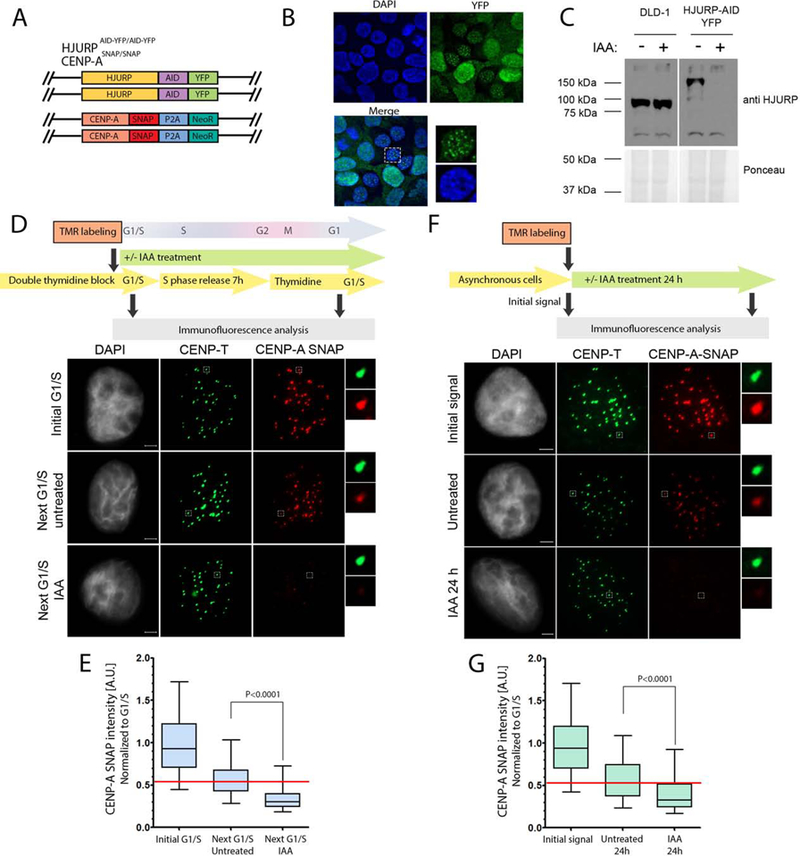
(A) Schematic representation of the DLD1-Tir1 cell line where HJURP was endogenously tagged with AID-YFP and CENP-A was endogenously tagged with the SNAP tag. (B) The immunofluorescence images of the localization profile of HJURP-AID-YFP in cell line shown in A. (C) Immunoblot analysis of the efficiency of IAA dependent HJURP degradation during mitosis demonstrated by staining with HJURP antibody. Ponceau staining was used as a loading control. The blots were separated in two sections due to removal of empty lanes from the original blot. (D) (F) Schematic representation of the experiment (top). Representative images of cells at indicated time points and treated as shown in the top panel. DNA was visualized by DAPI, immunofluorescence for CENP-T is shown in green and CENP-A is shown in red (bottom). Scale bar is 2μm. (E)(G) Quantification of D and F, respectively. The data was normalized to initial G1/S condition (E) or initial signal (G) within each individual experiment. Normalized data from two (E) or three (G) independent experiments was plotted using box-and-whisker plot: 5–95 percentile, n at least 4674 (E) and 2295 (G). The statistical significance was calculated using unpaired t-test and the p values are indicated. For reference, red line indicates the median level of CENP-A retention in untreated control. The percent loss of CENP-A was calculated to be 38.41% and 27.91% in E and G, respectively.
Since Mis18BP1 and Mis18α were associated with centromeres during S phase, and Mis18BP1 interacts with existing CENP-A during DNA replication (Fig. S2B, S3), we tested if these components contribute to CENP-A S phase stability. We depleted Mis18α and Mis18β, using siRNA treatment prior to DNA replication, and measured centromeric CENP-A levels in thymidine arrested cells (G1/S), or cells that had been released into S phase for 7 hours (Fig S5). The siRNA treatment resulted in efficient depletion of both Mis18α and Mis18β subunits (Fig S5B). We detected a very modest 5% decrease in CENP-A levels in response to the Mis18β siRNA treatment. The Mis18α depletion resulted in 19% decrease of CENP-A in these cells (Fig S5B, D-E). The role of Mis18BP1 in CENP-A retention during S phase was tested using endogenously AID-tagged Mis18BP1 (Fig. S6). The steady-state levels of endogenous CENP-A at the centromere through S–phase were analyzed when endogenous Mis18BP1-AID was degraded by addition of IAA. Analogous to experiments performed for HJURP-AID-YFP, the Mis18BP1-AID cell line was synchronized and treated with or without IAA for 90 mins prior to S phase release in order to degrade existing Mis18BP1. Cells were subsequently released into S phase with or without IAA for 8 hours and the nocodazole treatment was included in order to arrest cells in mitosis (Fig S6A,E). The IAA treatment resulted in Mis18BP1-AID degradation below the limit of detection by immunoblot (Fig S6C). The analysis of mitotic spreads derived form Mis18BP1-AID cells revealed that CENP-A inheritance was unaffected when Mis18BP1 was degraded in response to IAA treatment (Fig. S6B,D). These data suggest that, while Mis18α/may contribute to CENP-A stability, Mis18BP1 is not required for the ability of CENP-A to faithfully transit the replication fork.
CENP-A interaction with MCM-2 is required for retention during DNA replication
The MCM2 protein of the replicative MCM2–7 helicase complex directly binds to histones and is proposed to contribute to the retention of parental H3 nucleosomes during DNA replication (Huang et al., 2015; Richet et al., 2015). MCM2 has been shown to bind both histone H3.1 and CENP-A, and amino acids R63 and K64 within H3.1 contribute to the interaction with MCM2 (Huang et al., 2015). The R63 and K64 residues are conserved among all human histone H3 variants (Fig. S5A). Therefore, we introduced alanine at positions R63 and K64 of CENP-A (CENP-ARK->AA) and determined whether these mutations affect the interaction with MCM2 (Fig. 6A, S7A). We generated recombinant CENP-ARK->AA or wild type CENP-A complexed with histone H4, and the MCM2-HBD fragment (a.a. 43–160) previously reported to be sufficient for interacting with the histone H3 and H4 complex. We tested the efficiency of interaction of MCM2-HBD with histone variants by in vitro pull down and SPR (Fig. 6 B,C). Consistent with previous studies, we detected a physical interaction of MCM2-HBD with both histone H3/H4 and WT CENP-A/H4 complexes; however, histone H3/H4 showed higher affinity for MCM2 binding when compared to CENP-A/H4. The CENP-A/H4 complex containing the CENP-ARK->AA mutant failed to bind MCM2-HBD as efficiently as WT CENP-A (Fig. 6 B,C).
Figure 6. MCM2 binds CENP-A and is involved in its maintenance during DNA replication.
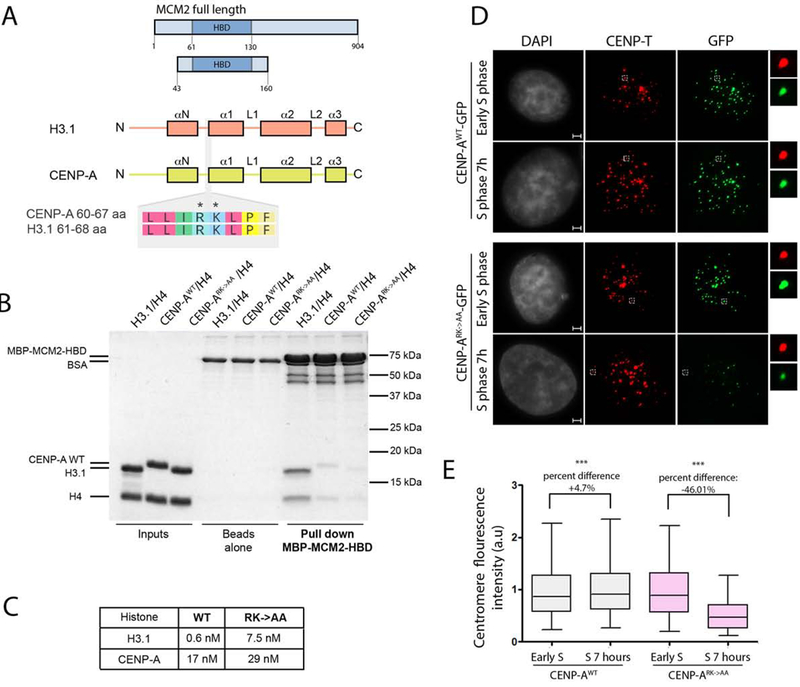
(A) Schematic representation of constructs used in B and C. The CENP-A and H3.1 domain structure is shown. The alignment of an 8 amino acid stretch corresponding to both histones demonstrates the conservation of Arginine 63 and Lysine 64 between the variants. (B) MBP-MCM2-HBD in vitro pull down demonstrating the interaction with indicated histone variants in the wild type and mutant form. (C) Table indicating Kd values measured SPR to assess the strength of interaction between MBP-MCM2-HBD and indicated histone variants in the wild type and mutant form. (D) Representative images of HeLa cells expressing either CENP-AWT-GFP or CENP-ARK->AA-GFP mutant at indicated cell cycle stages. DNA was visualized by DAPI, immunofluorescence for CENP-T is shown in red, expressed CENP-A was detected by GFP signal. Scale bar is 2μm. (E) Quantification of D. The data vas plotted using box-and-whisker plot: 5–95 percentile, n > 5660. The percent change of the levels of centromeric CENP-AWT-GFP and CENP-ARK->AA-GFP forms between experimental time points is indicated, (***) indicates p value <0.0001, statistical significance was calculated using unpaired t-test.
In order to test whether the interaction of MCM2 with CENP-A is important for retention of centromeric nucleosomes during DNA replication, we tested the efficiency of inheritance across DNA replication of nucleosomes containing the CENP-ARK->AA mutant. Cell lines stably expressing either CENP-AWT-GFP or CENP-ARK->AA-GFP were synchronized using double thymidine block and release. Both the CENP-A wild type and CENP-ARK->AA mutant were efficiently incorporated into centromeric chromatin, indicating that the CENP-A mutation did not alter de novo CENP-A deposition in G1 (Fig. 6D). Fluorescence intensity of GFP-tagged CENP-A at the centromere was compared between thymidine arrested cells and cells that had been released into S phase for 7 hours. While the level of CENP-AWT at the centromere was nearly identical before and after S phase, levels of the CENP-ARK->AA mutant were significant reduced following S phase, indicating that the CENP-ARK->AA mutant failed to be efficiently retained through DNA replication. Although reduced, the CENP-ARK->AA mutant was not completely lost from the centromeres when cells underwent DNA replication, which is likely due to the fact that MCM2 also makes contacts with the histone H4 and is consistent with our in vitro experiments as well as others (Huang et al., 2015; Richet et al., 2015).
Asf1 together with MCM2 was proposed to facilitate recycling of canonical histones during DNA replication (Huang et al., 2015; Richet et al., 2015) ). We therefore hypothesized that perhaps HJURP, analogous to Asf1, interacts with MCM2 to facilitate recycling of CENP-A containing nucleosomes specifically at centromeres. To determine whether MCM2 and HJURP interact in vivo, endogenous HJURP-AID-YFP was immunoprecipitated using anti GFP antibody. Endogenous MCM2 was co-immunoprecipitated with HJURP (Fig. 7A), consistent with previous experiments (Huang et al., 2015). Likewise, HEK293 cells transiently transfected with GFP-HJURP showed a similar interaction with endogenous MCM2 (Fig. 7B). To determine if the HJURP associates with the chromatin bound intact MCM complex, the endogenous MCM complex was immunoprecipitated from MNase digested cell lysates using an anti-MCM6 antibody. In addition to the other MCM components, the MCM6 co-immunoprecipitated HJURP, and we confirmed that this interaction is DNA independent by treating immunopreciptates with DNAse prior to elution (Fig. 7C, Fig. S7B,C).
Figure 7. HJURP copurifies with the MCM2–7 helicase complex and simultaneously interact with MCM2-CENP-A/H4 proteins.
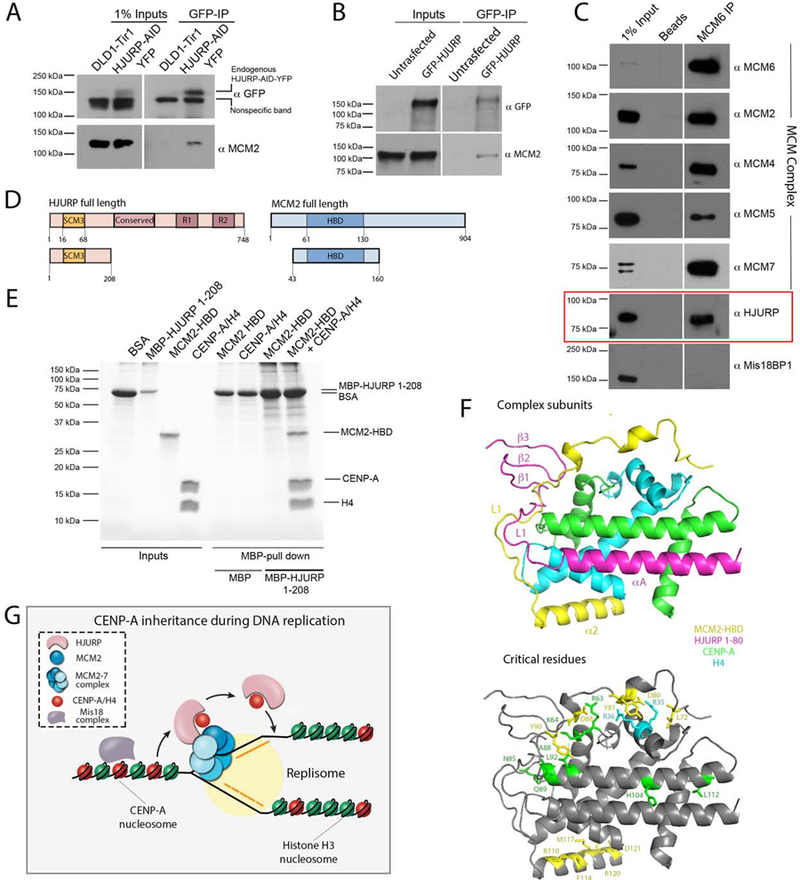
(A) Immunoblot analysis of GFP immunoprecipitation (IP) demonstrating the interaction of endogenous HJURP with endogenous MCM2. HJURP-AID-YFP DLD-1 cells were used as an input for the IP and samples were analyzed with indicated antibodies. (B) Immunoblot analysis of GFP IP performed from HEK293 cells overexpressing HJURP-GFP. Samples were analyzed with indicated antibodies. The blots in (A) and (B) were separated in two sections as the input and IP fractions correspond to different exposure times. (C) Immunoblot analysis of MCM6 IP performed from HEK293-derived cell lysates treated with Micrococcal nuclease. Samples were analyzed with indicated antibodies. (D) Schematic representation of constructs used in E. (E) Coomassie gel of MBP-HJURP1−208 in vitro pull down demonstrating the interaction with MCM2-HBD only in the presence of CENP-A/H4 heterodimer. (F) The superimposed model of 3R45 and 5BNX crystal structures where H3.3 and CENP-A were used as a reference. MCM2-HBD is shown in yellow, HJURP1−80, CENP-A and H4 are shown in pink, green, and aqua, respectively. Residues critical for facilitating the interaction of MCM2 with histones and mediating CENP-A recognition by HJURP are depicted in bottom panel. (G) The model of inheritance of CENP-A nucleosomes across DNA replication. MCM2–7 helicase complex is involved in unwinding chromatin ahead of the replication fork. HJURP is associated with MCM2–7 complex, and both MCM2 and HJURP can bind CENP-A nucleosomes simultaneously. The ability of CENP-A to be recognized by HJURP through the CATD domain and MCM2 through the R63-K64 motif are both essential for facilitating CENP-A retention across S phase and maintaining centromere identity.
HJURP and MCM2 were both shown previously to directly bind the CENP-A/histone H4 heterodimer, (Cho and Harrison, 2011; Dunleavy et al., 2009; Foltz et al., 2009; Hu et al., 2011; Huang et al., 2015) and we confirm those observations here (Fig. 6 and 7). In order to determine whether MCM2 and HJURP are able to simultaneously bind CENP-A and histone H4 we tested if we could assemble the HJURP-MCM2-CENP-A/H4 complex in vitro. Recombinant MCM2-HBD and MBP-HJURP1−208 fragments, sufficient for CENP-A binding, were purified from bacteria (Fig. 7D). MBP pull down assays were performed in the presence and absence of recombinant CENP-A/H4. MCM2-HBD co-purified with MBP-HJURP1−208 fragment only in the presence of CENP-A and histone H4, indicating that HJURP and MCM2 can bind CENP-A simultaneously (Fig. 7E, S7D).
In order to understand the interaction between HJURP-CENP-A-H4-MCM2 complex we superimposed the crystal structures of CENP-A/H4/HJURP1−80 (3R45) (Hu et al., 2011) and MCM261−130/H3.3/H4 (5BNX) (Huang et al., 2015) using H3 histone variants as a reference (Fig. 7F). This model structure demonstrates that the critical binding interfaces conferring MCM2 interactions (MCM2 residues L72, D80, D88, Y81, Y90, R110, E114, M117, R120, D121) with histone H3 and H4 heterodimer (CENP-A R63-K64; H4 R35-R36) are compatible with HJURP binding. However, the MCM2 L1 loop and HJURP β-sheet domain and the L1 loop—which blocks the DNA binding interface of CENP-A/H4—appear to occupy similar regions in the model. This perhaps implies that HJURP adopts a different conformation when bound to CENP-A/H4 in the presence of MCM2, or vice versa. Furthermore, the Q89, H104 and L112 residues of CENP-A were previously demonstrated to be sufficient to confer HJURP recognition, and in our structural model this interface is accessible for HJURP binding in the presence of MCM2 (Fig 7F bottom panel) (Bassett et al., 2012). The N85, H104, L112 residues of CENP-A were also shown to be sufficient for HJURP recognition; however, the N85 residue seems to be inaccessible in our model and may suggest that this mode of binding is not utilized when CENP-A is bound to MCM2 (Bassett et al., 2012). Collectively our data demonstrate that MCM2 is required for inheritance of CENP-A nucleosomes, and MCM2 together with HJURP chaperone bind CENP-A to facilitate its retention during DNA replication.
Discussion
The complete and stable inheritance of centromere identity requires both the assembly of new CENP-A nucleosomes in G1, and their retention during DNA replication. Nucleosomes are disassembled ahead of the DNA replication machinery in order to allow for DNA synthesis, and this process presents a challenge for the stability of CENP-A nucleosomes. Existing CENP-A is stably retained at the centromere for multiple cell divisions, suggesting that existing CENP-A is stably propagated during DNA synthesis (Bodor et al., 2013; Falk et al., 2015; Jansen et al., 2007). In this study, we identified a mechanism that facilitates inheritance of existing CENP-A nucleosomes during S phase. We found a distinct function for HJURP outside of its known G1 phase role and demonstrated the association of HJURP with centromeres and parental CENP-A during DNA replication. Our work revealed that HJURP collaborates with the MCM2–7 helicase complex to facilitate retention of evicted CENP-A (Fig. 7G).
The S phase role of HJURP in CENP-A retention that we describe here is consistent with the early observation that the degree of CENP-A loss in randomly cycling cells treated with HJURP siRNA for 48 hours is greater than what is expected if HJURP only affected new CENP-A loading (Dunleavy et al., 2009). Our experiments demonstrate the transient association of existing CENP-A with HJURP during DNA replication (Fig. 2A-C, S2 A,C). Prior work showed that the CENP-A centromere targeting domain (CATD), which binds HJURP, is essential to confer CENP-A nucleosome stability, and is consistent with the role we propose for HJURP in retaining CENP-A during DNA replication (Bodor et al., 2013). We observed that while depletion of AID-tagged HJURP reduced CENP-A retention, it did not completely eliminate CENP-A stability during S phase. This could be due to incomplete degradation of HJURP-AID, or it may suggest that MCM2 or other factors are able to retain CENP-A, albeit less efficiently. It will be interesting to determine whether under conditions of reduced HJURP, canonical histone chaperones such as ASF1 are involved in CENP-A retention.
Although our experiments demonstrate that Mis18BP1 and Mis18α are associated with centromeric chromatin during DNA, we did not observe an effect of Mis18BP1 depletion on CENP-A stability across S phase, suggesting that the presence of these proteins is linked to other functions. Depletion of Mis18α and Mis18β by siRNA showed a small reduction of CENP- A stability, suggesting a potential role for the Mis18 α and β subunits, independent of Mis18BP1. The Mis18 complex was proposed to be involved in regulating centromeric chromatin states, including histone modification and DNA methylation (Hayashi et al., 2004; Kim et al., 2012; Ohzeki et al., 2016). The presence of the Mis18 complex in S phase, that we observed in our studies, may facilitate the maintenance of epigenetic marks of centromeric chromatin specifically during DNA replication. The relatively mild effect of Mis18α/β depletion leaves open the possibility that additional factors are required for HJURP-dependent redeposition of CENP-A specially at centromeres in S phase.
Previous immunofluorescence based studies did not detect the presence of HJURP at centromeres in S phase cells (Bui et al., 2012; Dunleavy et al., 2009; Foltz et al., 2009). However, our more sensitive approaches using BioID labeling and ChIP identified an association of HJURP with the replicating centromere, suggesting a transient association of HJURP at S phase centromeric chromatin. Consistent with the dynamic process of DNA replication, ChIP experiments detected more abundant association of endogenous HJURP with centromeric DNA of chromosome 7 in cells released from thymidine block for over 3h when compared to thymidine arrested cells. This profile is consistent with reports showing that human centromeres are replicated from mid to late S phase (O’Keefe et al., 1992; Ten Hagen et al., 1990).
CENP-A mRNA transcripts rise specifically in G2, and peak in mitosis. However, prior experiments using CENP-A driven by a constitutive promoter, and thus providing new CENP-A throughout the cell cycle, observed no loading during S phase (Shelby et al., 1997). Since we observe an interaction with HJURP and CENP-A in S phase, this suggests that HJURP preferentially interacts with existing CENP-A rather than new CENP-A during S phase. Consistent with this idea, the expression of HJURP Ser412, Ser448, and Ser473 mutants that prematurely localize to centromeres in S and G2 phase assembled new CENP-A only in G2 but not in S phase (Muller et al., 2014; Stankovic et al., 2017). Ser412, Ser448, and Ser473 are key residues of HJURP that undergo a decrease in phosphorylation at the M/G1 transition. How HJURP may delineate new and old CENP-A is not yet known.
Parental H3-H4 heterotetramers are recycled during DNA replication and old histones are not mixed with newly synthesized dimers during nucleosome re-formation following replication (Leffak, 1984; Leffak et al., 1977; Yamasu and Senshu, 1990). Regardless of this pattern of inheritance, existing H3 nucleosomes require the Asf1 chaperone, which disrupts the H3-H4 heterotetramer, and binds the H3-H4 heterodimer (English et al., 2005; Groth et al., 2007). CAF1 interacts with PCNA and was shown to play a role in retention of parental nucleosomes (Shibahara and Stillman, 1999). CAF-1 binds two H3-H4 dimers and promotes formation of a H3-H4 heterotetramer (Winkler et al., 2012). Likewise, HJURP binds to the CENP-A/H4 heterodimer (Cho and Harrison, 2011; Zhou et al., 2011), and therefore CENP-A may also undergo a CENP-A/H4 heterodimer intermediate during DNA replication, similar to canonical nucleosomes. Previously, we showed that HJURP dimerization is required for assembly of new CENP-A nucleosomes (Zasadzinska et al., 2013). The ability of HJURP to dimerize might also be required to facilitate the inheritance of pre-existing CENP-A/H4 heterotetramers.
We and others have demonstrated that CENP-A directly binds the MCM2 chaperone (Huang et al., 2015)(Fig. 6C). We also show that MCM2 and HJURP can simultaneously interact with CENP-A/H4 in vitro (Fig. 7). Furthermore, disrupting the MCM2 binding interface within CENP-A by two amino acid substitutions (R63A and K64A) impairs the stable inheritance of the CENP-A nucleosome. We propose a model whereby CENP-A nucleosomes evicted ahead of the replication fork are recycled through the collaboration of HJURP and MCM2 chaperones (Fig. 7G). Although we did not identify the MCM2–7 helicase components in our BioID screen, MCM4 was previously co-purify with CENP-A from human cells (Huttlin et al., 2017), suggesting that CENP-A is associated with MCM2 within the context of the intact MCM2–7 complex. In vivo, HJURP co-immunoprecipitates with the MCM2–7 complex, and additional direct contacts between HJURP and other MCM2–7 helicase subunits may underlie this interaction. Alternatively, the interaction may be mediated by the common binding of MCM2 and HJURP to the CENP-A/h4 heterodimer, similar to the interaction of ASF1 with the MCM helicase complex (Groth et al., 2007). MCM2 also collaborates with the FACT complex to recycle parental canonical histones that have been evicted from chromatin during DNA replication and transcription in yeast (Foltman et al., 2013) and it will be interesting in determine how additional canonical chaperones contribute to CENP-A inheritance in S phase.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DLD-1 cell lines (male) expressing Tir1 were obtained from A. Holland (Johns Hopkins University). HEK293 (female), HeLa (female), DLD1-Tir1 and lines derived from these parental cell lines were cultured at 37 °C in 5% CO2. All cells lines were maintained in DMEM supplemented with 10% FBS (OPTIMA) and 1% Pen/Strep.
METHOD DETAILS
BioID and Mass Spectrometry.
Affinity purification of biotinylated proteins was performed as previously described in (Roux et al., 2012). In brief, cells were incubated in DMEM 10% FBS media supplemented with 50 μM biotin for 6 hours (25× stock solution of biotin was prepared in DMEM at 1.25 mM concentration). Cells were washed three times with PBS and harvested. Cell pellets were lysed at 25°C in 1 ml lysis buffer (50 mM Tris, pH 7.4, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM DTT, and Complete protease inhibitor [Roche]). Cell lysates were sonicated and then supplemented with Triton X-100 to 2% final concentration, and subjected to another round of sonication. Subsequently, cell lysates were diluted with an equal volume of cold (4°C) 50 mM Tris (pH 7.4) and subjected to additional sonication. Cell lysates were then centrifuged at 10,000 relative centrifugal force for 5 mins at 4°C. Supernatants were collected and protein concentration was measured using the BCA assay. For mass spectrometry, heavy and light components were mixed at 1:1 ratio. Supernatants were incubated with Streptavidin Magnetic Beads (BioLabs) for overnight. Beads were then collected and washed twice with 1 ml buffer containing 2% SDS. The beads were then washed once with buffer containing 0.1% deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, 50 mM Hepes, pH 7.5, and once with buffer containing 250 mM LiCl, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA, and 10 mM Tris, pH 8.1. Beads were then washed twice with buffer containing 50 mM Tris, pH 7.4, and 50 mM NaCl. Biotinylated proteins were eluted from the beads with 100 µl of 2x Laemmli SDS-sample buffer (4% SDS, 20% glycerol, 0.004% bromophenol blue, 0.125M Tris-Cl, pH 6.8, 10% 2-mercaptoethanol (added immediately before use)) saturated with biotin at 98°C. For the small-scale experiments, 7*10^6 cells were used as input, 15% of the pull down fractions were analyzed by immunoblot. For dox inducible cell lines expressing BirA* fusion proteins, 1.8*10^7 cells were used as an input, 50% of pull down samples were subjected to immunoblot. For the mass spectrometry analysis, proteins were eluted with 2× Laemmli SDS-sample buffer, without the bromophenol blue and glycerol, saturated with biotin at 98°C, and the protein concentration was measured using the BCA assay.
Eluted samples were diluted wit to 1ml with water, supplemented with 250 ul 100% TCA (final concentration of TCA 20%) and incubated overnight at 4°C. Samples were centrifuged at 16,000 rpm for 30 mins and the protein pellet was washed 5 times with 1 ml of ice cold acetone. Protein pellets were dried in speed vac, and then resuspended in buffer containing 100 mM ammonium bicarbonate (pH 8), 0.1% Rapigest and 10% ACN. Samples were supplemented with DTT at 5mM final concentration, and incubated at room temp for 1 hour. Iodacetamide was added at 12.5 mM final concentration, and samples were incubated in the dark for 1 hour. Proteins were digested with mass spec grade Trypsin (Trysin Gold from Promega). Trypsin was added at 1/20 ratio based on the amount of proteins measured in elutes. Samples were incubated for 15 hours at 37°C with shaking. The digestion was quenched with mass spec grade formic acid at 1% final concentration.
Sample mixtures were digested in 9ul volumes and injected directly onto an Easy Spray nano HPLC column ES801 (Thermo Scientific), packed with PepMap RSLC C18 media (2um, 100A, 50 um × 15 cm). An Easy nano LC (Thermo Scientific) delivered mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) as a gradient of 2 – 25% B over 60 min and 25 −50% B in 30 min at a flow rate of 250 nL / min. Mas spectra were collected using a Q Exactive Plus Orbitrap (Thermo Scientific) mass spectrometer at a resolution setting of 70,000 (FWHM @ 200 m/z) in full MS mode scanning from 300 – 2000 m/z and performing data dependent MS/MS acquisition (top 10) with a resolution setting of 17,500 (FWHM @ 200 m/z). LC-MS data were analyzed using Proteome Discoverer software, version 1.4 (Thermo Scientific). MS and MS/MS spectra were searched using the Sequest HT algorithm. Trypsin-generated peptides were identified using a FASTA database of human protein sequences (Unirpot, October 2015) as well as a decoy database with scrambled sequences. False positives were filtered using a false discovery rate of 1%. All peptides were quantified in a label-free manner using the MS1 extracted ion chromatogram (XIC) peak area with a tolerance of 2 ppm. Ratios of [heavy: light] peptides were calculated and averaged for each identified protein in order to perform SILAC relative quantitation of proteins.
Chromatin immunoprecipitation.
ChIP was performed as previously described in (Mayo et al., 2003). In brief, cells were synchronized using double thymidine block and released into S phase for 3 hours. Cells were grown on 15cm tissue culture dishes and 90% confluent at the day of fixation. Cells were then cross-linked on the plate by adding formaldehyde at a final concentration of 1% for 10 mins at 37°C. Glycine was added at a final concentration of 0.125 M to stop the cross-linking reaction. Cells were then washed twice with PBS, harvested and stored in −80 °C. Cell pellets were thawed on ice and lysed in 1ml of Farnham Lysis Buffer (5mM PIPES (KOH) pH 8.0, 85 mM KCl, 0.5 % NP40, and protease inhibitors [Roche]). Cell lysates were incubated for 10 mins on ice with shaking. Nuclei were pelleted at 800g for 2 minutes and resuspended in 250 ul of Lysis Buffer (1% SDS, 10mM EDTA, 50 mM Tris-HCl pH 8.0, protease inhibitors [Roche]). Lysates were incubated on ice for 30 minutes with shaking, and subsequently sonicated. Lysates were centrifuged at 13000 rpm for 10 minutes; supernatants were collected and measured for the protein concentration. An equal amount of protein per sample was then diluted 10 times with the Dilution Buffer (1.1% TritonX100, 1.2mM EDTA, 16.7mM Tris-HCl pH8.0, 167 mM NaCl, protease inhibitors [Roche]). Lysates were pre cleared with Protein A agarose beads and IgG for 30 minutes at 4°C. Agarose beads were then centrifuged at 1300 rpm for 2 minutes and the supernatants were supplemented with GFP antibody (custom polyclonal antibody, Cocalico) or rabbit IgG and incubated for 17 hours at 4°C. Protein A agarose beads were added and samples were incubated for 1 hour at 4°C. Agarose beads were then pelleted by spinning down at 1300 rpm for 1 minute at 4°C, and subsequently washed twice with Low sat Wash Buffer (0.1% SDS, 1% Triton X100, 2mM EDTA, 20mMTris HCl pH8.0, protease inhibitors [Roche]). Beads were then washed once with High Salt Wash Buffer (0.1% SDS, 1% Triton X100, 2mMEDTA, 20mM Tris-HCl pH 8.0,500 mM NaCl, protease inhibitors [Roche]), twice with LiCl Wash Buffer (0.25M LiCl, 1% NP40, 1% deoxycholate, 1mM EDTA, 10 mM Tris-HCl pH 8.0), and twice with TE buffer. Each wash was performed for 5 minutes at RT. Beads were incubated with 75 ul of Elution Buffer (0.1M NaHCO3, 1% SDS) at RT for 15 minutes. Samples were then centrifuged at 2000 rpm for 2 minutes and eluates were collected. The elution step was repeated; the elution fractions were combined and supplemented with NaCl to a final concentration of 0.3 M following by 17 hours incubation at 65 °C. DNA was purified with PCR purification kit (Qiagen) and stored at −20°C. Following ChIP, DNA was quantified by qPCR using standard procedures on a StepOne™ Real-Time PCR System. Primers for qPCR were used as previously described in Ohzeki et al., 2012: (Forward: GGCATATGTGCAAGTGGATATAC; Reverse: TATCCACTTGCAGAC), and the value of enrichment was derived by relative amount to input and ratio to IgG. In brief, signals obtained from the ChIP for each IgG IP and GFP IP sample derived from each time point tested were divided by signals obtained from an input corresponding to each time point tested in the experiment. A 1% of starting chromatin was used as an input and adjusted for the dilution factor. 15% of eluted DNA corresponding for each sample was subjected to qPCR analysis. The signal over background was then calculated for each sample, where the ChIP signals were divided by the corresponding IgG signals for each time point tested. The data was then plotted as the fold enrichment in signal relative to the background signal, and SEM are displayed.
Cell Culture and Transfection.
HEK293, HeLa, DLD1-Tir1 and lines derived from these parental cell lines were cultured in a 37 °C incubator in 5% CO2 in DMEM supplemented with 10% FBS (OPTIMA) and 1% Pen/Strep. Cells for the DNA or siRNA transfection were seeded onto six-well plate prior to transfection and transfected when they reached 60% of confluency (HeLa, HEK293, DLD1-Tir1 cells). DNA transfection was conducted with the Lipofectamine 2000 reagent using standard protocol (Thermo Fisher Scientific) with 2ug of plasmid DNA per well. siRNA transfection was performed using the RNAiMAX transfection reagent (Invitrogen) using standard protocol. Cells were treated with either 25nM of HJURP siRNA (Dharmacon),45 nM Mis18a siRNA (Ambion), 45nM Mis18b siRNA (Ambion) or GAPD control siRNA (Invitrogen) for 38–40 hours.
Cell synchronization.
Cells were synchronized with double thymidine block and release. Thymidine was added to culture medium at 20mM for 18 hours. Following the first thymidine treatment cells were washed twice with PBS and released into S phase for 8 hours, and subsequently treated with second thymidine block. Cells were released form the second thymidine arrest as indicated in the text.
DNA content analysis.
Cells were synchronized and subsequently harvested using PBS supplemented with 3 mM EDTA. Cells were then washed with PBS and centrifuged at 1000 rpm for 5 min. Cell pellets were resuspended in 200ul PBS, fixed with 5mls of 70% Ethanol and stored at 4°C. Fixed cells were centrifuged at 1600 rpm for 5 min and washed with PBS + 1% FBS. Cell pellets were then resuspended in fresh PI/RNaseA solution (10ug/ml propidium iodide, 250ug/ml RNase A in PBS + 1% FBS) and incubated at 37°C for 30 minutes. Samples were analyzed for their DNA content using flow cytometry.
SNAP labelling.
DLD1-Tir1 cells expressing endogenously tagged CENP-A-SNAP were plated on poly-lysine-coated glass coverslips. Asynchronous population or thymidine arrested cells was incubated in DMEM 10% FBS and labelled with 2 uM TMR-Star (Covalys) in complete growth medium for 20 min at 37°C. Cells were subsequently washed twice with each PBS, and DMEM and incubated for 30 min. Following incubation cells were washed twice with each PBS and DMEM. Asynchronous population was then incubated from 24 to 27 hours with or without IAA. Thymidine arrested population was treated +/−IAA for 60–90 minutes and subsequently released into S phase in the presence or absence of IAA. Cells were then pre-extracted with 0.1% Triton-X in PBS (3 minutes), fixed with 4% formaldehyde (10 minutes) and quenched with 100mM Tris, pH 7.5 (5 minutes), stained and analyzed by immunofluorescence microscopy.
Indirect immunofluorescence.
Cells were pre-extracted with 0.1% Triton-X in PBS for 3 minutes, fixed with 4% formaldehyde for 10 minutes and subsequently quenched with 100mM Tris, pH 7.5 for 5 minutes. Fixed cells were incubated in blocking solution (0.1% Triton-X in PBS, 0.2% BSA, 2% FBS) for 1.5 h at RT, and incubated with indicated primary antibodies for 1.5 h. anti-CENP-T (custom polyclonal antibody, Cocalico), anti-CENP-A (MA1–20832, ThermoFisher) and anti-HA antibodies (mAb HA.11) were used at 1:6000, 1:1000, and 1:1000 dilution, respectively and detected using fluorophore conjugated secondary antibodies (Cy3, Cy5 or FITC, Jackson Immuno Inc.). Cy3-conjigatesd streptavidin (Jackson Immuno Inc.) was used at 1:1000 dilution. DNA was visualized with 0.2mg/ml DAPI in PBS and coverslips were mounted in Prolong Gold (Invitrogen).
Immunoprecipitation and immunoblots.
Cells were harvested stored at −80C. In case of overexpression experiments, cells were harvested 24 h post transfection and stored at −80C. Cell pellets were thawed on ice and resuspended in RIPA lysis buffer (150mM NaCl, 1% NP-40, 0.3% deoxycholate, 0.15% SDS, 50mM Tris HCl pH 7.5, 1mM EDTA, 10% glycerol, protease inhibitors, 0.1mM PMSF, 5mM NaF, 10mM β-Glycerophosphate, 0.2mM NaV). For MCM6 IP lysis buffer was supplemented with 1mM ATP. Cell lysates were incubated for 15 minutes with rigorous vortexing periodically. In case pf MCM6 IP experiments cell lysates were diluted with an equal volume of dilution buffer (50 mM Tris HCl pH 7.5, 1mM ATP, protease inhibitors, 0.1mM PMSF, 5mM NaF, 10mM β-Glycerophosphate, 0.2mM NaV, 5mM CaCl2) and subjected to MNase digestion for 4 minutes. Mnase treated lysates were quenched with 6mM EGTA. Lysates were centrifuged at 10000 rpm for 5 min at 4°C, and pre-cleared with Protein A agarose (Biorad) for 1 h at 4°C. Precleared extracts were then supplemented with anti-GFP antibody (1:1000, Cell Signaling) and incubated for 17 hours at 4°C. Extracts were subsequently incubated with Protein A Dynabeads (Invitrogen) on ice for 45 min, and washed once with RIPA buffer, followed by three washes in PBST (PBS + 0.1% Tween). For MCM6 IP the beads were washed 3 times with RIPA buffer supplemented with 1mM ATP and 3 times with was buffer 2 (PBS supplemented with: 0.1% Tween, 1mM ATP, protease inhibitors, 0.1mM PMSF, 5mM NaF, 10mM β-Glycerophosphate, 0.2mM NaV). Purified proteins were eluted by boiling in SDS sample buffer for 10 minutes. Immunoblotting of precipitated proteins and cell lines was conducted using the following primary antibodies; anti-MCM2 (Bethyl Laboratories, A300–122A, used at 1:1000 dilution), anti-MCM4 (Santa Cruz, sc-28317, used at 1:1000 dilution), anti-MCM5 (Santa Cruz, sc-165994, used at 1:1000 dilution), anti-MCM6, (Abcam, ab201683, used at 1:500 dilution), anti-MCM7 (Santa Cruz, sc-9966, used at 1:1000 dilution), anti-HJURP (Foltz et al., 2009), (used at 1:1000 dilution), anti-histone H2B (07–370, Millipore, used at 1:2000 dilution), Mis18BP1 (Silva et al., 2012) (used at 1:1000 dilution), anti-Asf1α (gift from A. Dutta, used at 1:1000 dilution), Mis18α (gift of A. Losada, used at 1:1000 dilution), and anti-CENP-A (ThermoFisher, MA1–20832, used at 1:1000 dilution).
Stable isotope labeling.
SILAC labeling with light and heavy analogs of Lysine and Arginine was performed in DMEM Media for SILAC (Thermo scientific) supplemented with either Arginine- HCl and Lysine- 2HCl or 13C6-Arginine HCl and 13C6-Lysine HCl (13C Molecular), respectively. The medium was supplemented with 10% Dialyzed Fetal Bovine Serum (JR Scientific). Cells were adopted for heavy and light medium for 20 cell divisions.
Surface Plasmon Resonance.
SPR was performed on a Reichert4SPR instrument (Reichert Technologies). Biotinylated MCM243−160 was immobilized on a NeutrAvidin SPR sensor chip. Sufficient immobilization was obtained by flowing 100 μL 1 mg/ml protein through channel in SPR buffer: 20 mM HEPES pH 7.5, 500 mM NaCl, 1 mM DTT, 10 mM MgCl2, 5 % glycerol, 0.1 % Triton X-100 and 0.1 mg/ml BSA. Serial dilutions of histone proteins (H3/H4 and CENP-A/H4) in SPR buffer were injected over the chip in cycles, and the bindings were monitored. After each injection, the chip was washed with regeneration buffer (1M Tris HCl pH4) to dissociate all histone proteins from hMCM2. Data was processed by Reichert’s Surface Plasmon Resonance software. Kinetic constants were fitted to the binding curves, by a global fitting of all curves using 1:1 binding model.
Imaging.
Images were acquired using a 100× oil-immersion Olympus objective lens on a DeltaVision microscope or the 100× oil-immersion objective lens on a Zeiss LSM800 microscope. Collected images are presented as maximum stacked images. Images in Figures 1, 3A, 5, 6, S3 and S4 were subjected to deconvolution prior stacking. Channels were scaled identically within panels. In Figure 6D, green and red channels were scaled within cell lines.
Stable cell lines.
All cell lines expressing BiRA-HA* fusion proteins were generated using Flp-In™ T-REx™ System. The CENP-A-GFP CENP-A-R63AK64A-GFP stable cell line was generated using lentiviral transduction in HeLa cell line. HJURPAID-YFP and HJURPAID-YFP/CENP-ASNAP cell lines were generated using transient transfection of DLD1-Tir1 cells. In brief, using DLD-1 Flp-In T-Rex cells stably expressing Tir1 (Holland et al., 2012) as a starting point, CENP-A was tagged with a C-terminal SNAP tag using CRISPR-Cas9 as published (Guo et al., 2017), and a clone with CENP-A tagged on both alleles was used for experiments after verification by sequencing and CENP-A immunoblot. HJURP was tagged with a C-terminal AID-YFP tag using CRISPR-Cas-9, using the oligonucleotides (5’-CACCGAAACTAAAAGTGTGTAGCT-3’ and 5’-AAACAGCTACACACTTTTAGTTTC-3’) to target its 3’ UTR. To generate the repair template, the AID-YFP sequence was amplified from the published pcDNA5-FRT-TO-H2B-AID-YFP construct (Holland et al., 2012), and 5′ and 3′ HJURP homology arms of∼800 bp each were amplified from DLD-1 genomic DNA. All three pieces were inserted into a pUC19 backbone using HiFi DNA Assembly (NEB) and co-transfected with the HJURP gRNAs using published conditions (Guo et al., 2017). After transfection, YFP positive cells were isolated by FACS into 96-well plates, and clones were screened by microscopy for YFP signal. Cells were harvested and FACS sorted into single clones. Clones were verified using immunoblot and genotyping methodology.
Mitotic chromosome spreads.
Cells were arrested for 17 hours in 0.1 μg/ml nocodazole in DMEM Mitotic cells were harvested by mitotic shake off and centrifuged. Cell pellets were subsequently washed with 1ml of PBS and centrifuged. Cells were resuspended in hypotonic solution (20 mM HEPES, pH 7.0, 1 mM MgCl2, 0.2 mM CaCl2, 20 mM KCl, LPC, and 0.5 μg/ml Colcemid) and incubated on ice for 10 mins. Cells were then spun down onto glass slides using cytospin, washed with PBS and subsequently fixed with 4% formaldehyde for 10 mins. Cells were then quenched with 100mM Tris pH 7.5 for 5 minutes and stored at 4°C.
Recombinant protein purification.
Biotinylated human MCM2Δ1 (residue 43–160) was prepared by expressing Avitag-His6 tagged hMCM2Δ1 in BL21(DE3)pLysS cells at 18℃ for 18 hours after induced by 0.2 mM IPTG and the addition of 50 μM of D-biotin. The protein was purified by cobalt affinity chromatography and Superdex 200 size exclusion chromatography. MBP-His6-tagged hMCM2Δ1 was expressed in BL21(DE3)pLysS cells at 18°C. The fusion protein was purified by cobalt affinity chromatography. Histone H3RK->AA mutant was generated by site-directed mutagenesis. Recombinant human histone H3 and H4 were expressed, reconstituted and purified according to (Dyer et al., 2004). Human CENP-A (wild type or mutant) and histone H4 were expressed by bicistronic expression vector in Rosetta cells at 37°C for 3 hours (Tan et al., 2005). The cells were sonicated and cleared by centrifugation in 45mM sodium phosphate pH 6.8, 900mM NaCl, 1mM PMSF and 5mM BME. The CENP-A/H4 tetramer complex was then applied to hydroxyapatite resin (Bio-Rad), and eluted in buffer containing 45mM sodium phosphate pH 6.8, 3M NaCl, 1mM PMSF and 5mM BME. The eluted pool was changed to 20mM HEPES pH 7.5, 600mM NaCl and 1mM DTT by dialysis, before further purification by IEX (Source S, GE Lifesciences).
In vitro recombinant protein pull-downs
Thirty microliters of 50 μM MBP-His-MCM243−160 was incubated with 20 μl of amylose resin (New England Biolabs) in P300 buffer (NaP pH 7.0, 300mM NaCl). After washing the resin twice with 150 μl P300 buffer and equilibration with PD buffer (20 mM HEPES pH 7.5, 250mM NaCl, 5 mM BME, 10 mM MgCl2, 5 % glycerol, 0.5 % NP40 and 0.1 mg/ml BSA). 50 μl of 15 μM recombinant histone proteins were added and allowed to bind to the immobilized MBP-His-MCM243−160 protein. Unbound histone proteins were removed by washing with PD buffer. The immobilized MBP-His-MCM243−160 bound protein complexes were then eluted from the resin by adding 25 µl 2x gel sampling buffer and heating at 95°C for 5 min. Samples were fractionated on an 15% acrylamide SDS-PAGE gel and visualized by Coomassie Blue staining.
Recombinant MBP-HJURP1−208, MCM2-HBD proteins were purified by size exclusion chromatography using Superose 6 column and stored in SEC buffer (300mM Nacl, 20mM HEPES pH 7.5, 1mM DTT, 10% glycerol). MBP-HJURP1–208 and biotinylated MCM243−160 were then diluted with equal volumes of 2× pull down buffer (200 mM NaCl, 20mM HEPES ph 7.5, 1mM DTT, 20mM MgCl2, 1% NP40, 0.2 mg/ml BSA). Amylose beads (New England Biolabs) were washed 3 times with 2× pull down buffer and subsequently incubated with MBP-HJURP1−208 fragment for 1 hour at 4°C. The biotinylated MCM243−160 was then added at equimolar stoichiometry with respect to MBP-HJURP1−208 fragment. The samples were subsequently supplemented with either recombinant CENP-A/H histones at equimolar stoichiometry or dilution buffer (250 mM NaCl, 20mM HEPES pH 7.5, 1mM DTT, 10 mM MgCl2, 0.5% NP40, 0.1 mg/ml BSA). Protein complexes were incubated for 4 hours at 4°C, and subsequently washed 3 times with dilution buffer. The beads were then resuspended in 2xSB, boiled and analyzed by Coomassie stained SDS-PAGE.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis and quantification
For image analysis, the integrated intensities were derived from raw images that were subjected to ImageJ. The CRAQ plugin was employed in order to identify centotromeric signal and the centromere marker was used as a reference. All quantification data was analyzed in GraphPad Prism software, the statistical significance was assessed using unpaired t-test. The graphs were generated using GraphPad Prism software and displayed percentiles are as indicated in figure legends. Each figure legend contains a description regarding the sample size and or number of replicates corresponding to individual panels as well as type of graph displayed.
The Mass spec quantification
LC-MS data were analyzed using Proteome Discoverer software, version 1.4 (Thermo Scientific) that was used to derive the H/L scores for proteins identified in individual samples. The H/L scores were derived from 4 technical replicates and based on the “high peptide confidence” setting. The Mass Spec data shown in Fig1E and FigS1A-B was plotted in GraphPad Prism software, the SD displayed in Fig1E was calculated in GraphPad Prism software.
DNA content analysis
The DNA content analysis was conducted using the FlowJo software that was also used to derive the graphs demonstrating cell cycle profiles of analyzed samples.
Supplementary Material
Highlights.
CENP-A nucleosomes are stably inherited across S-phase
The CENP-A chaperone HJURP is associated with pre-existing CENP-A during S-phase
CENP-A inheritance requires HJURP and the MCM2 subunit of the replicative helicase.
CENP-A can bind MCM2 and HJURP simultaneously
Acknowledgements
We thank D. Burke, P.T. Stukenberg, Y. Wang and members of the D.R.F. lab for helpful comments, and A.F. Straight, P.T. Stukenberg, D.Matson, A. Holland and I. Cheeseman for reagents. We also acknowledge G. Minasov for help with the structure comparison. D.R.F. was supported by NIH R01GM111907 and by a Zell Scholar grant from the Robert L. Lurie Cancer Center. E. Z. was supported by a pre-doctoral fellowship from the American Heart Association (15PRE25700271). We also acknowledge support by NIH R01GM082989 (B.E.B.) and NIH F30CA186430 (L.Y.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The complete Mass Spec data sets used in this study are displayed in Table 1.
References
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, and Groth A (2012). Chromatin replication and epigenome maintenance. Nature reviews Molecular cell biology 13, 153–167. [DOI] [PubMed] [Google Scholar]
- Allshire RC, and Karpen GH (2008). Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9, 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Suzuki A, Hori T, Backer C, Okawa K, Cheeseman IM, and Fukagawa T (2009). The CENP-S complex is essential for the stable assembly of outer kinetochore structure. The Journal of cell biology 186, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato AT, Schindler RK, Thomas CA Jr., and Seale RL (1981). Dual nature of newly replicated chromatin. Evidence for nucleosomal and non-nucleosomal DNA at the site of native replication forks. The Journal of biological chemistry 256, 11880–11886. [PubMed] [Google Scholar]
- Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, and Foltz DR (2011). HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. The Journal of cell biology 194, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, DeNizio J, Barnhart-Dailey MC, Panchenko T, Sekulic N, Rogers DJ, Foltz DR, and Black BE (2012). HJURP uses distinct CENP-A surfaces to recognize and to stabilize CENP-A/histone H4 for centromere assembly. Dev Cell 22, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernad R, Sanchez P, Rivera T, Rodriguez-Corsino M, Boyarchuk E, Vassias I, Ray-Gallet D, Arnaoutov A, Dasso M, Almouzni G, et al. (2011). Xenopus HJURP and condensin II are required for CENP-A assembly. The Journal of cell biology 192, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman ML, and Schwacha A (2009). The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiology and molecular biology reviews : MMBR 73, 652–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Valente LP, Mata JF, Black BE, and Jansen LE (2013). Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Molecular biology of the cell 24, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos D, Frigola J, and Diffley JF (2012). Activation of the replicative DNA helicase: breaking up is hard to do. Current opinion in cell biology 24, 423–430. [DOI] [PubMed] [Google Scholar]
- Bui M, Dimitriadis EK, Hoischen C, An E, Quenet D, Giebe S, Nita-Lazar A, Diekmann S, and Dalal Y (2012). Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell 150, 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, and Zhang Z (2013). Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol 20, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camahort R, Li B, Florens L, Swanson SK, Washburn MP, and Gerton JL (2007). Scm3 is essential to recruit the histone h3 variant cse4 to centromeres and to maintain a functional kinetochore. Mol Cell 26, 853–865. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, and Desai A (2008). Molecular architecture of the kinetochore-microtubule interface. Nature reviews Molecular cell biology 9, 33–46. [DOI] [PubMed] [Google Scholar]
- Chen CC, Dechassa ML, Bettini E, Ledoux MB, Belisario C, Heun P, Luger K, and Mellone BG (2014). CAL1 is the Drosophila CENP-A assembly factor. The Journal of cell biology 204, 313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho US, and Harrison SC (2011). Recognition of the centromere-specific histone Cse4 by the chaperone Scm3. Proceedings of the National Academy of Sciences of the United States of America 108, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, and Sullivan KF (2003). Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421. [DOI] [PubMed] [Google Scholar]
- Dechassa ML, Wyns K, Li M, Hall MA, Wang MD, and Luger K (2011). Structure and Scm3-mediated assembly of budding yeast centromeric nucleosomes. Nat Commun 2, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Almouzni G, and Karpen GH (2011). H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, and Almouzni-Pettinotti G (2009). HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485–497. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, and Luger K (2004). Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods in enzymology 375, 23–44. [DOI] [PubMed] [Google Scholar]
- Earnshaw W, Bordwell B, Marino C, and Rothfield N (1986). Three human chromosomal autoantigens are recognized by sera from patients with anti-centromere antibodies. J Clin Invest 77, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English CM, Maluf NK, Tripet B, Churchill ME, and Tyler JK (2005). ASF1 binds to a heterodimer of histones H3 and H4: a two-step mechanism for the assembly of the H3-H4 heterotetramer on DNA. Biochemistry 44, 13673–13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Mellone BG, Betts CM, Zhang W, Karpen GH, and Straight AF (2008). Genome-wide analysis reveals a cell cycle-dependent mechanism controlling centromere propagation. The Journal of cell biology 183, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LE, Van Duyne GD, Vinogradov SA, et al. (2015). Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science 348, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, and Labib K (2013). Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep 3, 892–904. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, and Cleveland DW (2009). Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 137, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR 3rd, and Cleveland DW (2006). The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol 8, 458–469. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, and Yanagida M (2007). Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell 12, 17–30. [DOI] [PubMed] [Google Scholar]
- Gerard A, Koundrioukoff S, Ramillon V, Sergere JC, Mailand N, Quivy JP, and Almouzni G (2006). The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO reports 7, 817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, and Stuurman N (2007). Genes required for mitotic spindle assembly in Drosophila S2 cells. Science (New York, NY) 316, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, and Almouzni G (2007). Regulation of replication fork progression through histone supply and demand. Science 318, 1928–1931. [DOI] [PubMed] [Google Scholar]
- Guo LY, Allu PK, Zandarashvili L, McKinley KL, Sekulic N, Dawicki-McKenna JM, Fachinetti D, Logsdon GA, Jamiolkowski RM, Cleveland DW, et al. (2017). Centromeres are maintained by fastening CENP-A to DNA and directing an arginine anchor-dependent nucleosome transition. Nature communications 8, 15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, and Yanagida M (2004). Mis16 and Mis18 are required for CENP-A loading and histone deacetylation at centromeres. Cell 118, 715–729. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Han JS, and Cleveland DW (2012). Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 109, E3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Liu Y, Wang M, Fang J, Huang H, Yang N, Li Y, Wang J, Yao X, Shi Y, et al. (2011). Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev 25, 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Stromme CB, Saredi G, Hodl M, Strandsby A, Gonzalez-Aguilera C, Chen S, Groth A, and Patel DJ (2015). A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat Struct Mol Biol 22, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuta H, Ikeno M, Suzuki N, Tomonaga T, Nozaki N, Obuse C, Kisu Y, Goshima N, Nomura F, Nomura N, et al. (2006). Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP-A chromatin of human cells. Genes Cells 11, 673–684. [DOI] [PubMed] [Google Scholar]
- Jansen LE, Black BE, Foltz DR, and Cleveland DW (2007). Propagation of centromeric chromatin requires exit from mitosis. The Journal of cell biology 176, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, and Groth A (2010). Replication stress interferes with histone recycling and predeposition marking of new histones. Molecular cell 37, 736–743. [DOI] [PubMed] [Google Scholar]
- Kato T, Sato N, Hayama S, Yamabuki T, Ito T, Miyamoto M, Kondo S, Nakamura Y, and Daigo Y (2007). Activation of Holliday junction recognizing protein involved in the chromosomal stability and immortality of cancer cells. Cancer research 67, 8544–8553. [DOI] [PubMed] [Google Scholar]
- Kim IS, Lee M, Park KC, Jeon Y, Park JH, Hwang EJ, Jeon TI, Ko S, Lee H, Baek SH, et al. (2012). Roles of Mis18alpha in Epigenetic Regulation of Centromeric Chromatin and CENP-A Loading. Mol Cell [DOI] [PubMed]
- Leffak IM (1984). Conservative segregation of nucleosome core histones. Nature 307, 82–85. [DOI] [PubMed] [Google Scholar]
- Leffak IM, Grainger R, and Weintraub H (1977). Conservative assembly and segregation of nucleosomal histones. Cell 12, 837–845. [DOI] [PubMed] [Google Scholar]
- Maddox PS, Hyndman F, Monen J, Oegema K, and Desai A (2007). Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. The Journal of cell biology 176, 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, and Jones DR (2003). Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the Akt pathway. The Journal of biological chemistry 278, 18980–18989. [DOI] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2016). The molecular basis for centromere identity and function. Nature reviews Molecular cell biology 17, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL, and Miller OL Jr. (1977). Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell 12, 795–804. [DOI] [PubMed] [Google Scholar]
- Mellone BG, Grive KJ, Shteyn V, Bowers SR, Oderberg I, and Karpen GH (2011). Assembly of Drosophila centromeric chromatin proteins during mitosis. PLoS Genet 7, e1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Xiao H, Wisniewski J, Smith MM, and Wu C (2007). Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell 129, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Moree B, Meyer CB, Fuller CJ, and Straight AF (2011). CENP-C recruits M18BP1 to centromeres to promote CENP-A chromatin assembly. The Journal of cell biology 194, 855–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Montes de Oca R, Lacoste N, Dingli F, Loew D, and Almouzni G (2014). Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3(CENP-A) loading. Cell Rep 8, 190–203. [DOI] [PubMed] [Google Scholar]
- Nardi IK, Zasadzinska E, Stellfox ME, Knippler CM, and Foltz DR (2016). Licensing of Centromeric Chromatin Assembly through the Mis18alpha-Mis18beta Heterotetramer. Mol Cell 61, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihashi A, Haraguchi T, Hiraoka Y, Ikemura T, Regnier V, Dodson H, Earnshaw WC, and Fukagawa T (2002). CENP-I is essential for centromere function in vertebrate cells. Dev Cell 2, 463–476. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- O’Keefe RT, Henderson SC, and Spector DL (1992). Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 116, 1095–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Shono N, Otake K, Martins NM, Kugou K, Kimura H, Nagase T, Larionov V, Earnshaw WC, and Masumoto H (2016). KAT7/HBO1/MYST2 Regulates CENP-A Chromatin Assembly by Antagonizing Suv39h1-Mediated Centromere Inactivation. Dev Cell 37, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Cheeseman IM, Hori T, Okawa K, McLeod IX, Yates JR 3rd, Desai A, and Fukagawa T (2006). The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat Cell Biol 8, 446–457. [DOI] [PubMed] [Google Scholar]
- Pan D, Klare K, Petrovic A, Take A, Walstein K, Singh P, Rondelet A, Bird AW, and Musacchio A (2017). CDK-regulated dimerization of M18BP1 on a Mis18 hexamer is necessary for CENP-A loading. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux AL, Choi ES, Abbott JK, Liu X, Kagansky A, Castillo AG, Hamilton GL, Richardson W, Rappsilber J, He X, et al. (2009). Fission yeast Scm3: A CENP-A receptor required for integrity of subkinetochore chromatin. Mol Cell 33, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom M, Dennehey BK, and Tyler JK (2010). Chaperoning histones during DNA replication and repair. Cell 140, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet N, Liu D, Legrand P, Velours C, Corpet A, Gaubert A, Bakail M, Moal-Raisin G, Guerois R, Compper C, et al. (2015). Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res 43, 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseaulin LC, Noguchi C, Martinez E, Ziegler MA, Toda T, and Noguchi E (2013a). Coordinated degradation of replisome components ensures genome stability upon replication stress in the absence of the replication fork protection complex. PLoS genetics 9, e1003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseaulin LC, Noguchi C, and Noguchi E (2013b). Proteasome-dependent degradation of replisome components regulates faithful DNA replication. Cell cycle (Georgetown, Tex) 12, 2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JE, Woodlief KS, and Sullivan BA (2016). Inheritance of the CENP-A chromatin domain is spatially and temporally constrained at human centromeres. Epigenetics Chromatin 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. The Journal of cell biology 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Tomkiel J, Cooke CA, Ratrie H 3rd, Maurer M, Rothfield NF, and Earnshaw WC (1992). CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell 70, 115–125. [DOI] [PubMed] [Google Scholar]
- Shelby RD, Vafa O, and Sullivan KF (1997). Assembly of CENP-A into centromeric chromatin requires a cooperative array of nucleosomal DNA contact sites. J Cell Biol 136, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K, and Stillman B (1999). Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96, 575–585. [DOI] [PubMed] [Google Scholar]
- Shuaib M, Ouararhni K, Dimitrov S, and Hamiche A (2010). HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proceedings of the National Academy of Sciences of the United States of America 107, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Bodor DL, Stellfox ME, Martins NM, Hochegger H, Foltz DR, and Jansen LE (2012). Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell 22, 52–63. [DOI] [PubMed] [Google Scholar]
- Sogo JM, Stahl H, Koller T, and Knippers R (1986). Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. Journal of molecular biology 189, 189–204. [DOI] [PubMed] [Google Scholar]
- Spiller F, Medina-Pritchard B, Abad MA, Wear MA, Molina O, Earnshaw WC, and Jeyaprakash AA (2017). Molecular basis for Cdk1-regulated timing of Mis18 complex assembly and CENP-A deposition. EMBO reports 18, 894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic A, Guo LY, Mata JF, Bodor DL, Cao XJ, Bailey AO, Shabanowitz J, Hunt DF, Garcia BA, Black BE, et al. (2017). A Dual Inhibitory Mechanism Sufficient to Maintain Cell-Cycle-Restricted CENP-A Assembly. Mol Cell 65, 231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler S, Rogers K, Weitze S, Morey L, Fitzgerald-Hayes M, and Baker RE (2007). Scm3, an essential Saccharomyces cerevisiae centromere protein required for G2/M progression and Cse4 localization. Proceedings of the National Academy of Sciences of the United States of America 104, 10571–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugata N, Munekata E, and Todokoro K (1999). Characterization of a novel kinetochore protein, CENP-H. The Journal of biological chemistry 274, 27343–27346. [DOI] [PubMed] [Google Scholar]
- Tan S, Kern RC, and Selleck W (2005). The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein expression and purification 40, 385–395. [DOI] [PubMed] [Google Scholar]
- Ten Hagen K.G., Gilbert DM, Willard HF, and Cohen SN (1990). Replication timing of DNA sequences associated with human centromeres and telomeres. Molecular and cellular biology 10, 6348–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H, Takarabe T, Inazawa J, and Hirohashi S (1997). Detection of Numerical Alterations of Chromosomes 3, 7, 17 and X in Low-grade Intracystic Papillary Tumors of the Breast by Multi-color Fluorescence In Situ Hybridization. Breast cancer (Tokyo, Japan) 4, 247–252. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu X, Dou Z, Chen L, Jiang H, Fu C, Fu G, Liu D, Zhang J, Zhu T, et al. (2014). Mitotic Regulator Mis18beta Interacts with and Specifies the Centromeric Assembly of Molecular Chaperone Holliday Junction Recognition Protein (HJURP). The Journal of biological chemistry 289, 8326–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Hayashi T, Yanagida M, and Russell P (2009). Fission yeast Scm3 mediates stable assembly of Cnp1/CENP-A into centromeric chromatin. Mol Cell 33, 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, Zhou H, Dar MA, Zhang Z, and Luger K (2012). Yeast CAF-1 assembles histone (H3-H4)2 tetramers prior to DNA deposition. Nucleic acids research 40, 10139–10149. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yamasu K, and Senshu T (1990). Conservative segregation of tetrameric units of H3 and H4 histones during nucleosome replication. Journal of biochemistry 107, 15–20. [DOI] [PubMed] [Google Scholar]
- Zasadzinska E, Barnhart-Dailey MC, Kuich PH, and Foltz DR (2013). Dimerization of the CENP-A assembly factor HJURP is required for centromeric nucleosome deposition. The EMBO journal 32, 2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Feng H, Zhou BR, Ghirlando R, Hu K, Zwolak A, Miller Jenkins L.M., Xiao H, Tjandra N, Wu C, et al. (2011). Structural basis for recognition of centromere histone variant CenH3 by the chaperone Scm3. Nature 472, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.