

SUMMARY
Nutrient availability influences the production and degradation of materials that are required for cell growth and survival. Autophagy is a nutrient-regulated process that is used to degrade cytoplasmic materials, and has been associated with human diseases. Solute transporters influence nutrient availability and sensing, yet we know little about how transporters influence autophagy. Here we screen for solute transporters that are required for autophagy-dependent cell death, and identify CG11665/hermes. We show that hermes is required for both autophagy during steroid-triggered salivary gland cell death and TNF-induced non-apoptotic eye cell death. hermes encodes a proton-coupled monocarboxylate transporter that preferentially transports pyruvate over lactate. mTOR signaling is elevated in hermes mutant cells, and decreased mTOR function suppresses the hermes salivary gland cell death phenotype. Hermes is most similar to human SLC16A11, a protein that was recently implicated in type 2 diabetes, thus providing a link between pyruvate, mTOR, autophagy and possibly metabolic disorders.
Keywords: monocarboxylate transporter, autophagy, cell death, Drosophila, mTOR, pyruvate
In Brief
Velentzas et al. identify a requirement for the pyruvate transporter Hermes in autophagy that is associated with cell death in Drosophila. Hermes influences mTOR signaling and is similar to the type 2 diabetes-associated human protein SLC16A11, thus suggesting a further link between autophagy and metabolic disorders.
GRAPHICAL ABSTRACT
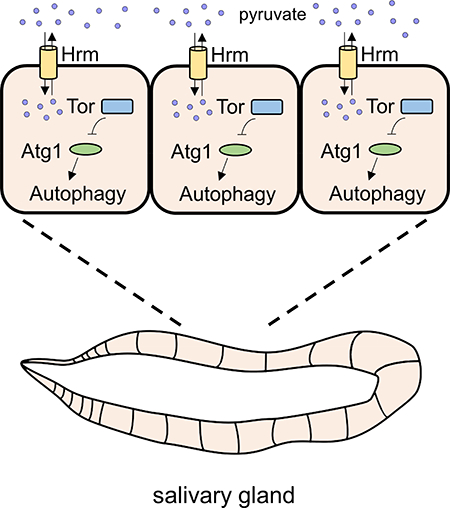
INTRODUCTION
Nutrient availability, sensing and homeostasis are paramount for cell metabolism, health and survival (Finkel, 2015). Significantly, altered cell metabolism is associated with aging and multiple human disorders. For example, changes in dopamine metabolism in Parkinson’s disease patient-derived neurons causes lysosomal dysfunction and α-synuclein accumulation (Burbulla et al., 2017), and type 2 diabetes and nonalcoholic fatty liver disease are caused by systemic changes in metabolism reviewed in (Czech, 2017; Samuel and Shulman, 2018). Cancer cells, unlike most normal tissues, prefer conversion of glucose into lactate, even in the presence of sufficient oxygen that can support mitochondrial oxidative phosphorylation (Warburg effect) (Warburg et al., 1927), and numerous studies have shown that cancer cells alter their metabolism in order to survive and proliferate (Faubert et al., 2014; Sousa et al., 2016). Metabolic reprogramming is also associated with inflammatory (Kamdar et al., 2016) and cardiovascular diseases (Franssens et al., 2016), and it is becoming clear that metabolic dysfunction is a risk factor for many age-associated diseases (Finkel, 2015).
Autophagy is a process that is used to deliver cytoplasmic materials to the lysosome for degradation (Mizushima and Komatsu, 2011). As a key catabolic process that is regulated by the nutrient sensor mechanistic target of rapamycin (mTOR) (Kamada et al., 2000; Kim et al., 2011; Scott et al., 2007), autophagy is an important node in the metabolic network of all cells (Rabinowitz and White, 2010). Like metabolism, autophagy has been implicated in multiple age-associated disorders, including neurodegenerative diseases, diabetes, cancer, inflammatory diseases and others (Levine and Kroemer, 2008; Mizushima et al., 2008), and is consider a potential therapeutic target (Rubinsztein et al., 2012). Autophagy can be induced by various stimuli, such as metabolic changes (Singh et al., 2009), hypoxia (Liu et al., 2013) and hormones (Baehrecke, 2000), and studies in animals indicate that autophagy has complex regulation and functions in different cell contexts (Zhang and Baehrecke, 2015). Therefore, a comprehensive understanding of how autophagy is regulated in different cell contexts is important to understanding organismal metabolism, development and disease.
Autophagy has been considered a cell survival process because of pioneering work that used yeast nutrient deprivation as a model (Tsukada and Ohsumi, 1993), but has also been implicated in cell death in animals (Baehrecke, 2005; Yuan and Kroemer, 2010). Autophagy that is associated with cell death has been most studied in Drosophila salivary gland cells that die during development. In salivary glands, an increase in the steroid hormone 20-hydroxyecdysone (ecdysone) triggers cell death that requires both caspases and autophagy (Berry and Baehrecke, 2007). Autophagy in dying salivary gland cells also requires mTOR-regulated cell growth arrest (Berry and Baehrecke, 2007), suggesting an important relationship between nutrient sensing and autophagy that is associated with these dying cells during development.
Nutritional signals influence mTOR activity to coordinate protein and lipid production with their catabolism by autophagy (Saxton and Sabatini, 2017). Multiple pathways influence mTOR activity, including solute transporters (Nicklin et al., 2009; Wang et al., 2015). The Solute Carrier (SLC) superfamily consists of membrane proteins that sustain homeostasis by regulating transport of many types of substances across lipid membranes.. Members of the SLC superfamily play an important role in various processes, including nutrient sensing (Rebsamen et al., 2015), metabolic homeostasis (Terker et al., 2015) and cell death (Hoffman et al., 2014). Similar to regulators of metabolism and autophagy, members of the superfamily have been implicated in various human disorders, such as Parkinson’s disease (Salazar et al., 2008), nonalcoholic fatty liver disease (DeBosch et al., 2016), type 2 diabetes (Rusu et al., 2017), and cancer (Xue et al., 2016).
Here we report the identification of members of the SLC superfamily that are required for proper salivary gland cell death, including five genes CG11665/hermes, CG5805, CG13384, CG8177 and CG10413, that prior to this study had no known function. Significantly, we discovered that hermes, a gene that encodes a proton-coupled monocarboxylate transporter that transports pyruvate, participates in TNF-induced non-apoptotic programmed cell death and is required for autophagy but not apoptotic caspase function during salivary gland degradation. Loss of hermes leads to elevated mTOR signaling, and decreased mTOR function suppresses the hermes salivary gland persistence phenotype. Interestingly the closest human homolog of hermes is SLC16A11, a gene that was recently implicated in type 2 diabetes (Rusu et al., 2017). These findings provide a unique link between pyruvate transport, mTOR and the regulation of autophagy that is associated with cell death, and have implications for metabolic diseases.
RESULTS
Identification of SLC superfamily members that function during developmentally programmed autophagic cell death
Drosophila salivary gland cell death depends on mTOR regulated cell growth arrest and autophagy (Berry and Baehrecke, 2007). Previous links between the SLC superfamily of proteins with nutrient sensing and mTOR prompted us to screen for genes that encode SLC proteins that are required for salivary gland degradation. We used the DRSC Integrative Ortholog Prediction Tool (DIOPT - http://www.flyrnai.org/diopt) (Hu et al., 2011) to identify the predicted orthologues of all human genes that encode members of the SLC superfamily from HGNC (https://www.genenames.org). Since all SLC members are transporters, we also acquired a list of all the genes in Drosophila that are predicted to have a transporter activity from Flybase (http://flybase.org), and excluded the genes that were not predicted to have a transporter activity. We then analyzed our time-series gene expression data from purified salivary glands (Lee et al., 2003) to identify SLC gene transcript levels that are induced prior to autophagic cell death. We identified 25 genes, which are predicted to be members of the SLC superfamily, and have upregulated transcripts levels in salivary glands prior to cell death (Table S1).
We decreased the function of each of the 25 genes identified in salivary glands using RNAi knockdown. Animals containing UAS-RNAi combined with fkh-GAL4 to drive RNAi expression specifically in salivary glands were compared to control animals with UAS-RNAi that is not expressed in salivary glands. These animals were fixed 24 hours after puparium formation (8 hours after this tissue is completely cleared in wild type animals), embedded in paraffin, sectioned, and stained for analyses of the persistence of salivary gland material by light microscopy. We tested two UAS-RNAi lines that target distinct sequences per gene when possible and set a cutoff of at least 50% penetrance of animals that contain a persistent salivary gland phenotype.
We identified twelve SLC genes that are required for degradation of Drosophila salivary glands, and have not been previously implicated in this process (Table S1). Five genes, CG11665, CG5805, CG13384, CG8177 and CG10413 have no known function in Drosophila and we decided to investigate them further. Downregulation of CG11665 in the salivary gland resulted in a gland degradation failure in 100% of the animals tested compared to 25% of control animals (Figure 1A and B). Knockdown of CG5805 resulted in a gland fragment phenotype in 96% of the animals compared to 25% in control animals (Figure 1C and D). Decreased CG13384 function impaired gland degradation in 95% of the animals compared to 30% in control animals (Figure 1E and F). Downregulation of CG8177 resulted in a gland degradation defect in 91% of the animals tested and none of the control animals (Figure 1G and H, Figure S1). Finally, RNAi knockdown of CG10413 resulted in a gland fragment phenotype in 67% of the tested animals compared to 20% of control animals (Figure 1I and J).
Figure 1. SLC genes that are required for salivary gland cell death.

(A) Samples from control animals (w/+; +; UAS-CG11665IR/+), n= 16 (left), and those with salivary gland-specific knockdown of CG11665 (fkh-GAL4/w; +; UAS-CG11665IR/+), n = 18 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (B) Quantification of data from (A). (C) Samples from control animals (w/+; +; UAS-CG5805IR/+), n= 20 (left), and those with salivary gland-specific knockdown of CG5805 (fkh-GAL4/w; +; UAS-CG5805IR/+), n = 24 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (D) Quantification of data from (C). (E) Samples from control animals (w/+; +; UAS-CG13384IR/+), n= 20 (left), and those with salivary gland-specific knockdown of CG13384 (fkh-GAL4/w; +; UAS-CG13384IR/+), n = 20 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (F) Quantification of data from (E). (G) Samples from control animals (w/+; UAS-CG8177IR/+; +), n= 20 (left), and those with salivary gland-specific knockdown of CG8177 (fkh-GAL4/w; UAS-CG8177IR/+; +), n = 23 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (H) Quantification of data from (G). (I) Samples from control animals (w/+; UAS-CG10413IR/+; +), n= 20 (left), and those with salivary gland-specific knockdown of CG10413 (fkh-GAL4/w; UAS-CG10413IR/+; +), n = 24 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (J) Quantification of data from (I). See also Figure S1.
Our incomplete understanding of the uncharacterized CG11665, CG5805, CG13384, CG8177 and CG10413 genes stimulated us to consider if they influence cell death in other contexts. The Drosophila eye has served as a useful model for studies of both apoptosis and TNF (Eiger in flies)-induced non-apoptotic programmed cell death (Kanda et al., 2011). Expression of the pro-apoptotic factor Hid in the developing eye leads to caspase activation and a small eye phenotype (Figure S2A) that is suppressed in all animals examined by co-expression of the caspase inhibitor p35 (Figure S2A). Knockdown of either CG11665, CG5805, CG13384, CG8177 or CG10413 failed to suppress Hid-induced cell death (Figure S2A). In contrast to Hid-induced cell death, Eiger-triggered cell death in the eye is weakly suppressed by expression of the caspase inhibitor p35 (Kanda et al., 2011), but was completely suppressed by knockdown of Bsk (Drosophila Jnk) in all animals that were analyzed (Figure S2B). Interestingly, of the five genes, only CG11665 knockdown suppressed Eiger-induced eye size reduction (Figure 2A-H). These results indicate that CG11665 plays a role in non-apoptotic programmed cell death.
Figure 2. Down regulation of CG11665 suppresses eiger-induced non-apoptotic cell death.

(A) Eye from a wild type animal. (B) Eye from a control animal that expresses Eiger specifically in the eye disc (GMR-Egr). (C) Eye from an animal that expresses Eiger and CG11665IR. (D) Eye from an animal that expresses Eiger and CG5805IR. (E) Eye from an animal that expresses Eiger and CG13384IR. (F) Eye from an animal that expresses Eiger and CG8177IR. (G) Eye from an animal that expresses Eiger and CG10413IR. (H) Quantification of data from (B-G). Percent of animals with suppression of Eiger-induced eye size reduction (n > 50 animals/genotype). Statistical significance was determined using a Chi-square test. All animals were 3–5 days old. See also Figure S2.
CG11665/hermes functions in a caspase-independent manner
CG11665 encodes a protein predicted to have a monocarboxylic acid transmembrane transporter activity. CG11665 is related to the monocarboxylate transporter 16 family (Halestrap and Wilson, 2012), which has undergone several rounds of gene duplication in humans since the divergence of humans and flies. We named CG11665 hermes/hrm after the Greek god of transportation, who also guides the souls to the underworld. In order to better understand the function of the monocarboxylate transporter encoded by hrm, we generated a loss-of-function deletion mutant Drosophila strain using CRISPR/cas9. We designed two guide (g)RNAs that target deletion of exons 3, 4, 5 and part of exon 6 in hrm, and isolated a viable Drosophila strain that had a deletion of the sequence between those two sites (Figure 3A). This resulted in a nearly complete deletion of the predicted open reading frame of the hrm (Figure 3A), suggesting this should be a strong loss-of-function mutant allele that we named hrmΔ1. hrmΔ1 mutant flies are homozygous viable, fertile and possess no obvious exterior morphological defects.
Figure 3. CG11665/hernes mutation results in persistence of salivary gland material.

(A) Schematic representation of CG11665/hermes. Arrows indicate the CRISPR targeted sites and the resulted deletion. (B) Samples from control animals (w; hrmΔ1/+; +), n=39 (left) and hrm mutants (w; hrmΔ1/Df(2R)BSC696; +), n=51 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (C) Quantification of data from (B). (D) Samples from control hrm mutants animals (w; hrmΔ1/hrmΔ1, UAS-GFP; sg-GAL4/+), n=11 (left) and hrm mutants that express a hrm transgene (w; hrmΔ1/hrmΔ1, UAS-GFP; sg-GAL4/UAS-hrm), n=20 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (E) Quantification of data from (D). See also Figure S3.
We next tested if deletion of hrm influenced salivary gland degradation. These experiments were conducted by analyses of our newly isolated hrmΔ1 mutant in combination with a deficiency for this genomic region to reduce concerns about potential secondary site mutations that could be induced by CRISPR. We found that 90% of the mutant animals possessed a salivary gland degradation defect, while only 8% of the control animals had a defect (Figure 3B and C). We next investigated if we could rescue the mutant salivary gland degradation defect by expression of a hrm cDNA transgene. For that purpose, we used sgGAL4 to drive expression of a UAS-hrm transgene specifically in the salivary glands of homozygous hrm mutant animals. We found that only 10% of the mutant animals expressing hrm in salivary glands had a defect compared to the 73% of the mutant animals without the sg-GAL4 driver (Figure 3D and E). These data indicate that hrm is required for salivary gland degradation.
Salivary gland degradation requires cell-growth arrest and an increase in the steroid hormone 20-hydroxyecdysone (Berry and Baehrecke, 2007; Jiang et al., 1997). Using the cell-growth reporter tGPH (Britton et al., 2002) we found that salivary glands from hrmΔ1 animals possess normal cell-growth arrest (Figure S3A and B). In addition, deletion of hrm did not alter the steroid response factor BR-C (Figures S3C and D). These data indicate that hrm is required for larval salivary gland clearance, but alters neither cell growth arrest nor steroid signaling.
Salivary gland cell death requires both caspases and autophagy for complete salivary gland degradation (Berry and Baehrecke, 2007), and inhibition of either of these two pathways leads to a partial gland degradation phenotype with persistent cell fragments. By contrast, when both pathways are inhibited simultaneously, the persisting tissue resembles more intact gland fragments. To determine whether hrm influences caspase-induced cell death in salivary glands, we expressed the caspase inhibitor p35 in hrm mutant animals. Expression of p35 in a hrmΔ1/+ heterozygous genotype resulted in a persistence of cell fragment phenotype in 53% of the animals, while 47% of the animals possessed a gland fragment phenotype (Figure 4A and B and Figure S4B). By contrast, expression of p35 in hrmΔ1 homozygous mutant animals resulted in a persistence of gland fragment phenotype in 88% of the animals tested, while only 12% had a cell fragment phenotype (Figure 4A and B and Figure S4C). These data indicate that hrm and caspases function in an additive manner. To further test the relationship between hrm and caspases, we stained salivary glands with antibodies against cleaved caspase 3. During normal development, cleaved caspase 3 antigen is present at 13.5h after puparium formation. When we analyzed salivary glands for cleaved caspase 3 antigen 13.5 h after puparium formation, we found that glands from both control and homozygous hrm mutant animals possessed cleaved caspase 3 (Figure 4C and D). These data indicate that hrm is not involved in the regulation of caspases during salivary gland degradation, and suggest that hrm regulates a different pathway that influences degradation of this tissue.
Figure 4. Loss of hermes does not influence caspases during salivary gland degradation.

(A) Samples from hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; fkh-GAL4/+), n=20 (left), animals with salivary gland-specific expression of p35 (w; hrmΔ1/+; fkh-GAL4/UAS-p35), n=19 (middle), and hrm mutants with salivary gland-specific expression of p35 (w; hrmΔ1/Df(2R)BSC696; fkh-GAL4/UAS-p35), n=16 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. Arrows indicate persisting salivary gland luminal structures. See also Figure S4. (B) Quantification of data from (A). Statistical significance was determined using a Chi-square test comparing the percentages of gland fragments. (C) Salivary glands dissected from control (w; hrmΔ1/+; +), n=4 (left), animals and hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; +), n=8 (right), 13.5h after puparium formation and stained with cleaved Caspase-3 antibody (green) and Hoechst (blue). Scale bars represent 20μm. (D) Quantification of data from (C). Data are represented as mean ± SEM. Statistical significance was determined using a Student’s t test.
CG11665/hermes is required for autophagy
In order to investigate whether hrm influences autophagy during salivary gland cell death, we tested if a mutation in the autophagy gene Atg13 can enhance the failure in salivary gland degradation phenotype of hrm mutant animals. All homozygous Atg13 mutant animals possessed a cell fragment phenotype (Figure 5A and B). Animals with homozygous mutations in both hrm and Atg13 did not result in an enhanced phenotype with all animals exhibiting a cell fragment phenotype (Figure 5A and B), suggesting that these two genes function in the same pathway.
Figure 5. hermes is required for autophagy during salivary gland degradation.

(A) Samples from hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; +), n=51 (left), Atg13 mutant animals (w; hrmΔ1/+; Atg13Δ74), n = 20 (middle), and mutant animals for both hrm and Atg13 (w; hrmΔ1/Df(2R)BSC696; Atg13Δ74), n=13 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (B) Quantification of data from (A). Statistical significance was determined using a Chi-square test comparing the percentages of gland fragments. (C) Samples from control hrm mutants animals (w; hrmΔ1/Df(2R)BSC696; fkh-GAL4/+), n=20 (left) and hrm mutants specifically expressing Atg1 in the salivary glands (w; hrmΔ1/Df(2R)BSC696; fkh-GAL4/UAS-Atg1GS10797), n=17 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (D) Quantification of data from (C). Statistical significance was determined using a Chi-square test comparing the percentages of gland fragments. (E) Salivary glands dissected from control animals expressing mCherry-Atg8a (red) (w; hrmΔ1, mCherry-Atg8a /+; +), n=10, (left) and hrm mutant animals (w; hrmΔ1, mCherry-Atg8a/Df(2R)BSC696; +), n=8, (right) 13.5h after puparium formation and stained with Hoechst (blue). Scale bars represent 20μm. (F) Quantification of data from (E). Data are represented as mean ± SEM. Statistical significance was determined using a Student’s t test. (G) Salivary glands dissected from control (w; hrmΔ1/+; +) animals, n=5, (left) and hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; +), n=10, (right) 13.5h after puparium formation and stained with Ref(2)p/p62 antibody (green) and Hoechst (blue). Scale bars represent 20μm. (H) Quantification of data from (G). Data are represented as mean ± SEM. Statistical significance was determined using a Student’s t test.
Expression of Atg1 is sufficient to induce autophagy that depends on core Atg gene function in salivary glands (Berry and Baehrecke, 2007). Therefore, we tested if expression of Atg1 in hrm mutant animals is sufficient to suppress the hrm mutant salivary gland persistence phenotype. Expression of Atg1 suppressed the salivary gland phenotype, with 6% of the animals possessing cell fragments compared to 100% of hrm mutant control animals that had a cell fragments phenotype (Figure 5C and D). These data indicate that hrm functions upstream of Atg1.
To further investigate the role of this monocarboxylate transporter in autophagy, we tested if loss of hrm influences markers of autophagy by assaying for Atg8a puncta formation and Ref(2)p/p62 accumulation. We found that at 13.5 h after puparium formation, salivary glands from hrm mutant animals possess significantly fewer mCherry-Atg8a puncta than salivary glands from control animals (Figure 5E and F). Consistent with these data, hrm mutant salivary glands accumulate significantly more of the autophagy cargo receptor Ref(2)p/p62 protein than salivary glands isolated from control animals at the same stage (Figure 5G and H). Combined, these data indicate that hrm is required for autophagy in dying salivary glands.
CG11665/hermes is a plasma membrane and H+-coupled monocarboxylate transporter that influences mTOR
We investigated the localization of Hrm by introducing a c-terminal sfGFP tag in the endogenous locus of hrm using CRISPR/cas9 technology (Figure S5A). We analyzed larval salivary glands from hrm-sfGFP animals and found that Hrm localizes in the cortical plasma membrane region of cells (Figure 6A). To exclude the possibility that the signal observed was due to non-specific integration of the GFP, we downregulate hrm by RNAi in animals with c-sfGFP-tagged hrm and we found that those animals did not have any GFP signal (Figure 6A). In addition, to test if the GFP tag has any impact on the function of the protein, we performed histological analyses of animals 24h APF. Only 10% of the hrm-sfGFP animals had a gland cell fragment phenotype compared to 65% in homozygous hrmΔ1 mutant animals indicating that the c-terminal GFP tag has no impact on the function of the protein (Figure S5B and C). In addition, similar results were acquired when we introduced a smaller c-terminal FLAG tag in the endogenous locus of hrm using CRISPR/cas9 technology (Figure S5D and E), further validating that Hrm localizes in the plasma membrane of the cells.
Figure 6. hermes is a plasma membrane monocarboxylate transporter that interacts with the ancillary protein Bsg.

(A) Salivary glands dissected from 3rd instar control larvae (w; +; +) (left), hrm-sfGFP larvae (w; hrm-sfGFP/+; +), (middle) and hrm-sfGFP larvae with salivary gland-specific knockdown of hrm (w; hrm-sfGFP/+; Fkh-GAL4/UAS-hrmIR) (right). Scale bars represent 20μm. (B) Fluorescence traces of control cells (gray), and cells expressing MCT-1 (black) and hrm (red) before, during and after extracellular monocarboxylate (10mM) perfusion, n = 10. Plotted data are mean + SEM (shading) for clarity. (C) Relative proton transport rate after extracellular monocarboxylate (10mM) perfusion. The data (mean ± SEM) are normalized to the MCT-1 transport rate upon removal of lactate. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparison test, * p<0.05. (D) Salivary glands dissected from hrm-sfGFP 3rd instar larvae with cell specific (red cells) knockdown of Bsg (y,w,hs>flip/w; hrm-sfGFP/UAS-BsgIR; act>CD2>GAL4, UAS-RFP/+) imaged for Hrm-sfGFP (green-left) and RFP (red-middle). (E) Samples from control animals (w/+; UAS-BsgIR/+; +), n= 20 (left), and those with salivary gland-specific knockdown of Bsg (fkh-GAL4/w; UAS-BsgIR/+; +), n = 24 (right), analyzed with histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (F) Quantification of data from (E). See also Figure S5 and S6.
Monocarboxylate transporters have been divided in two categories, category I (H+coupled monocarboxylate transporters - MCTs) and category II (hydrophobic monocarboxylate transporters) (Halestrap, 2013). Category I MCTs have three conserved charged residues (K38, D309 and R313) that appear to be crucial for the transport of the monocarboxylates (Wilson et al., 2009). Interestingly, alignment of Hrm with human category I MCTs revealed that Hrm possesses these conserved residues (K74, D503 and R507) (Figure S5F). Given the sequence similarity to mammalian monocarboxylate transporters (MCTs), we determined whether Hrm mediated proton-coupled transport of monocarboxylates. To visualize proton transport at the plasma membrane, we labeled the glycocalyces of HEK 293T cells with a pH-sensitive fluorophore (Zhang et al., 2016) and monitored the change in fluorescence (ΔF/F0) upon 10 mM monocarboxylate application (Figure 6B). Because HEK293T cells endogenously express low levels of MCTs, we compared the rates of proton-coupled pyruvate and lactate transport from cells expressing either rMCT-1 or Hrm to mock transfected (control) cells. Extracellular perfusion of 10 mM pyruvate initially resulted in a rapid loss of fluorescence (due to proton transport into the cell), which slowly recovered as transport activity slowed and protons in the media reprotonated the cell-attached pH-sensitive fluorophores (Figure 6B, left).Compared to control cells, the fluorescent signal from rMCT1 and Hrm transfected cells recovered faster and reversed direction, indicating the exogenous expression of rMCT-1 and Hrm significantly increased proton-coupled transport of pyruvate into the cell such that proton efflux became thermodynamically favored. Washout of pyruvate resulted in a rapid efflux of protons from the cell; the rate of transport for rMCT-1 and Hrm expressing cells was ~ 2-fold faster than control cells. The rate of proton-coupled transport of lactate also increased with rMCT-1 expression (Figure 6B, right); however, Hrm expression had no significant effect on lactate transport rate compared to control cells. To determine a relative transport rate, we measured the rate of proton efflux upon removal of extracellular monocarboxylate, subtracted the background rate (control cells), and normalized the transport rates to rMCT-1 transport of lactate (Figure 6C). As was previously reported (Broer et al., 1998), rMCT-1 transports lactate approximately twice as fast as pyruvate at 10 mM (Figure 6C). In contrast, only proton-coupled pyruvate transport was detectable for Hrm. Proton-coupled pyruvate transport by Hrm was inhibited by the MCT inhibitor, 2-cyano-3-(4-hydroxyphenyl)-2-propenoic acid (CHC). At 0.3 mM CHC, Hrm activity was reduced by 60 ± 3% whereas both expressed (rMCT-1) and endogenous MCT-1 were inhibited to a lesser extent: 43 ± 3% and 39 ± 3%, respectively (± SEM; n = 6 – 8 cells), indicating that the IC50 of CHC is lower for Hrm. In total, these data demonstrate that Hrm is a bona fide proton-coupled monocarboxylate transporter that transports pyruvate faster than lactate.
Hrm is similar to the human SLC16A11 protein (Table S1). Interestingly, SLC16A11 was recently implicated in type 2 diabetes (Rusu et al., 2017). SLC16A11 type 2 diabetes risk variants disrupt its interaction with the ancillary protein Basigin (Bsg), and this interaction is required for correct cell-surface localization of SLC16A11. To test if Hrm also requires Bsg for correct cell-surface localization, we downregulated bsg in a subset of salivary gland cells using RNAi and analyzed the localization of Hrm-sfGFP. We found that cells with normal bsg expression (RFP-negative) display correct plasma membrane Hrm-sfGFP localization (Figure 6D). By contrast, neighboring cells with downregulated bsg levels (RFP-positive) display decreased plasma membrane localization of Hrm-sfGFP (Figure 6D). In order to investigate if the disruption of the cell membrane localization of Hrm is sufficient for incomplete salivary gland degradation, we downregulated bsg specifically in salivary glands. All animals with decreased bsg function had a strong salivary gland persistence phenotype while only 20% of the control animals had a weak cell fragment persistence phenotype (Figure 6E and F). Importantly, similar results were obtained using a second RNAi strain that targets a different bsg sequence (Figure S6A and B).
SLCs influence nutrient sensing and metabolism (Wang et al., 2015). In addition, knockdown of SLC16A11 in primary human hepatocytes resulted in an increase in lipids (Rusu et al., 2017). In order to investigate if deletion of hrm results in accumulation of lipids, we stained salivary glands dissected from animals 13.5h after puparium formation with Nile red. We found that the number of lipid droplets in glands is unaffected by the deletion of hrm (Figure 7A and B). Interestingly, the size of the lipid droplets is significantly increased in salivary glands from homozygous hrmΔ1 mutant animals (Figure 7A and B). These data suggest that like knockdown of SLC16A11 in human hepatocytes, loss of hrm alters lipid metabolism.
Figure 7. hermes alters lipid metabolism and mTor signaling.

(A) Salivary glands dissected from control animals (w; hrmΔ1/+; +), n=13, (left) and hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; +), n=13, (right) 13.5h after puparium formation and stained with Nile Red (red) and Hoechst (blue). Scale bars represent 20μm. (B) Quantification of data from (A). Data are represented as mean ± SEM. Statistical significance was determined using a Student’s t test. (C) Western blot analyses of phospho-S6K and actin 13.5 h after puparium formation in salivary gland extracts from control (w; hrmΔ1/+; +), n=8, and hrm mutant animals (w; hrmΔ1/Df(2R)BSC696; +), n=8. (D) Quantification of data from (C). All samples are normalized to actin and plotted relative to control sample. Data are represented as mean ± SEM. (E) Samples from control hrm mutant animals (w; hrmΔ1/hrmΔ1, UAS-GFP; sg-GAL4/+), n=11 (left) and hrm mutants specifically expressing torTED in the salivary glands (w; hrmΔ1, UAStorTED/hrmΔ1, UAS-GFP; sg-GAL4/+), n=20 (right), analyzed by histology for the presence of salivary gland material (red dotted circle) 24h after puparium formation. (F) Quantification of data from (E).
SLC transporters have been implicated in cell nutrient sensing through the regulation of mTOR (Rebsamen et al., 2015; Wyant et al., 2017), a key regulator of cell metabolism. The Atg1 kinase is downstream of mTOR (Kim et al., 2011), and expression of Atg1 rescues hrm salivary gland degradation (Figure 5C and D). Therefore, we tested if mTOR signaling is altered in hrm mutant salivary glands by analyses of the phosphorylation of S6 kinase. Indeed, hrm mutant salivary glands possess elevated levels of phosphorylation of S6 kinase (Figure 7C and D). These results suggest that hrm mutant salivary glands possess sustained mTOR activity that suppresses autophagy. To test the hypothesis that mTOR is activated in hrm mutants, we investigated if expression of a dominant negative form of mTOR (torTED) suppresses the hrm mutant persistence of salivary glands. Expression of torTED in the salivary glands of homozygous hrmΔ1 animals resulted in a persistence of cell fragment phenotype in only 15% of the animals, compared to 73% in homozygous hrmΔ1 animals (Figure 7E and F). Combined, these results suggest that hrm mutant salivary glands possess sustained mTOR activity that suppresses autophagy.
DISCUSSION
Three forms of cell death have been described in developing animals (Schweichel and Merker, 1973), including apoptosis that is mediated by caspases, cell death that involves autophagy, and necrosis. Interestingly, changes in cell metabolism have been connected to all three types of cell death (Green et al., 2014). By studying developmentally programmed cell death, we identified hermes, which encodes a monocarboxylate transporter that participates in Eiger (TNF)-triggered and autophagic cell death.
SLCs are membrane proteins that transport metabolites across lipid membranes and play an important role in a variety of biological processes. The SLC superfamily consists of over 400 genes in humans, and 30% of those remain uncharacterized (Perland and Fredriksson, 2017). In this study, we have used bioinformatics and gene expression analyses to identify SLCs that are required for Drosophila salivary gland degradation during development. We identified 12 SLC genes that are required for Drosophila salivary gland degradation, thus providing a better understanding of how metabolites impact cell death. In addition to the mononcarboxylate transporter that we characterized in detail, we also identified putative transporters of amino acids (CG5805, slif, CG13384, and CG10413), nucleotide sugars (senju) and organic anions (Oatp74D and Oatp30B) among others that play a role in this process, further validating the importance of metabolism in salivary gland degradation.
hermes encodes a proton-coupled monocarboxylate transporter (MCT) that transports pyruvate and is required for autophagy during cell death. Members of the human SLC16 MCT family play a critical role in cell metabolism (Halestrap and Wilson, 2012), and have been associated with aggressive tumors (Baek et al., 2014), exercise-induced hyperinsulinemia (EIHI) (Otonkoski et al., 2007) and other disorders (Dumitrescu et al., 2004; Kloeckener-Gruissem et al., 2008). In addition, hermes is required for eiger (TNF) induced non-apoptotic cell death. Interestingly, reduced function of other members of the solute transporter family prevented TNF-induced programmed necrosis (Hitomi et al., 2008), however, the mechanisms by which these transporters contribute to diseases are not completely understood. A recent study in Drosophila indicated that genes encoding enzymes that catalyze the conversion of monocarboxylates are required for eiger-induced cell death (Kanda et al., 2011). They suggest that eiger-induced metabolic reprogramming, reduction of ATP levels and ROS production are responsible for the induction of non-apoptotic cell death. Therefore, a better understanding of how monocarboxylate transporters affect autophagy and cell death during animal development may provide valuable insight into the treatment of malignancies.
Some monocarboxylate transporters interact with ancillary proteins, such as Bsg and Emb (Poole and Halestrap, 1997), and these ancillary factors are required for correct plasma membrane localization (Wilson et al., 2002). Here we show that the Drosophila Bsg is also required for correct localization of Hermes to the plasma membrane. Interestingly, downregulation of bsg in salivary glands resulted in an enhanced defect in salivary gland clearance. This increase in the severity of the phenotype might suggest that there are other MCTs that require Bsg for correct localization, and that those MCTs are also important for salivary gland degradation. Bsg may also possess functions that are unrelated to MCTs, and play a role in salivary gland degradation. For example, Drosophila Bsg is interacts with integrin and is required for normal cell architecture (Curtin et al., 2005) and synaptic vesicle release (Besse et al., 2007). Further investigation is needed to clarify the role of bsg in cell death.
mTOR-dependent cell growth arrest is required for salivary gland cell death (Berry and Baehrecke, 2007). Interestingly, hermes did not influence salivary gland cell growth arrest, but had an impact on autophagy. We show that salivary gland cells have mTOR activity at the same time as autophagy, as evidenced by the phosphorylated S6 kinase, a well-known target of mTOR. One possible explanation for active autophagy at the same time that S6 kinase is phosphorylated and protein synthesis occurs is that there is a threshold requirement of mTOR activity for its inhibition of autophagy. Indeed, although autophagy is usually repressed when mTOR is active, it has been shown that growing tumor cells with activated Ras possess active autophagy (Guo et al., 2011). Interestingly, decreased hermes function resulted in increased levels of phosphorylated S6 kinase that were associated with the inhibition of autophagy, and these data are consistent with our results showing that mTOR is required for the hermes persistent salivary gland phenotype. It is also possible that pyruvate levels could influence AMP Kinase that in turn influences Atg1 activity and autophagy, a process that is known to clear lipids (Singh et al., 2009). Therefore, salivary gland cells possess some attributes of transformed cells that may enable the dissection of this complex mechanism in a developmental context.
SLC16A11 is the human protein similar to Hermes. Recently, SLC16A11 was linked to type 2 diabetes (Rusu et al., 2017). In that study, downregulation of SLC16A11 in human hepatocytes resulted in metabolic changes that are associated with increased risk for type 2 diabetes. Here we show that hermes induces similar metabolic changes and mTOR signaling. mTOR signaling is a key nutrient sensing pathway, and increased activation of mTOR has been linked to insulin resistance and type 2 diabetes (Polak et al., 2008; Um et al., 2004). Interestingly, autophagy has also been implicated in diabetes, and a decrease in autophagy levels increases the risk of type 2 diabetes development (Ebato et al., 2008; Lim et al., 2014). Furthermore, Hermes transports pyruvate, and pyruvate was previously shown to influence autophagy in liver cells (Holen et al., 1996). Future investigation of the mechanisms that connect the regulation of autophagy and metabolism during development could lead to new insights into treatment of diseases that are associated with altered metabolism.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Eric Baehrecke (eric.baehrecke@umassmed.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Drosophila strains
Drosophila melanogaster strains used in this study are listed in the key resources table except for stocks used for the SLC family members screen, which are located in Table S1. Fly crosses and experiments were performed at 25°C on media con sisting of 6.5g/L agar, 63g/L yeast, 60g/L cornmeal, 60mL/L molasses, 4mL/L acid mix and 0.13% Tegosept. Canton-S was used as the wild-type strain. In all the experiments a mixed population of male and female animals was used. The age and developmental stage of the animals used is indicated in the figure legends.
Human cell line
HEK293T cells (fetus hypotriploid) were cultured in high glucose DMEM medium (Gibco) with 10% fetal bovine serum (Gibco) and 100 units/mL penicillin/streptomycin (Gibco) at 37°C in 5% CO2.
METHOD DETAILS
Histology
Histology was performed as previously described (Muro et al., 2006). Briefly, flies were maintained at 25oC and aged to 24 h after puparium formation. Whole pupae were fixed in FAAG (80% ethanol, 4% Formaldehyde, 5% Acetic Acid, 1% Glutaraldehyde) overnight at 4oC, embedded in paraffin, sectioned and stained with Weigert’s Hematoxylin and Pollack Trichrome. Stained sections were examined using a Zeiss AxioImager Z1 microscope.
hermesΔ1 deletion
For the generation of hermes mutant flies we used the CRISPR/Cas9 system (Port et al., 2014). We designed gRNAs that target a sequence in the second intron of hermes (GTCAGAGCTGTCCTTTCACTGGG) and in the sixth exon of hermes (GGTCTAGAATATTATAGTACTGG) (Figure 3A), and inserted them in the pCFD3d vector. Both plasmids were injected in Drosophila embryos expressing Cas9 protein in the germline (w1118; PBac{vas-Cas9}VK00027, Bloomington Stock Center). We screened for deletions by genomic DNA amplification using primers that targeted 300bp outside the deletion gRNA sites (5’ CTGCACAAAACCGTCCCAAG 3’, 5’ TGTGGGCTTGTTAATGCACG 3’).
hermes-sfGFP and hermes-2xFLAG alleles
For the generation of hermes-sfGFP and hermes-2xFLAG flies we used the CRISPR/Cas9 system (Port et al., 2014). We designed a gRNA that targets the c-terminal of hermes (GGCTGTCGATAGCTACTAAATGG) (Figure 6A) and inserted it in the U6droBsagRNA vector (Drosophila Genomics Resource Center). For hermes-sfGFP, 1kbp regions from both sides of the targeted site were inserted into the pScarlessHD-sfGFP-DsRed vector (Drosophila Genomics Resource Center). For hermes-2xFLAG, the following small single-stranded donor DNA (ssODN) was synthesized: 5’- GCAAAGCTAATGCGGAGGAGCAACTAGATGCACAAATCGAAAAAGCGGCTGTCGATAGCT ACGGCtccGGGGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGTAAATG cTCAAACAACTCTCGTACCAAACCACGTACAATAACAATTAGATTTAAGGCTTAAAACG −3’. Both plasmids or plasmid and ssODN were injected (UMass Medical School, Model Organism CRISPR Core) in Drosophila embryos expressing Cas9 protein in the germline (w1118; PBac{vas-Cas9}VK00027, Bloomington Stock Center). We screened for flies positive for RFP signal in the eye and verified the correct insertion by direct sequencing of the target region.
Transgenic strains
To generate the UAS-hermes strain, the cDNA of hermes was amplified from the RE60337 clone (Drosophila Genomics Resource Center) and it was inserted in the pUAST-attB transformation vector. The construct was injected into y1 w67c23; P{CaryP}attP2 (Bloomington Stock Center) flies by BestGene Inc. (Chino Hills, CA).
Protein extracts and western blotting
Salivary glands were dissected from 13h after puparium formation control and hrmΔ1 animals. Salivary glands were homogenized in Laemmli buffer (Bio-Rad) and boiled for 5 min at 100oC. Salivary glands from hrm-2xFLAG animals were homogenized in 1x Laemmli buffer and incubated for 5min at 42oC. Proteins were separated on 4%–20% SDS polyacrylamide gels (Bio-Rad). Proteins were transferred to 0.45 mm nitrocellulose membrane (Bio-Rad) according to standard procedures. We used mouse anti-Broad Complex (1:500, Developmental Studies Hybridoma Bank), mouse anti-FLAG (1:1000, Sigma-Aldrich), rabbit anti-phospho-S6K (1:500, Cell Signaling Technologies) and mouse anti-actin (1:1000, Developmental Studies Hybridoma Bank) primary antibodies.
Immunolabeling
For immunostaining, salivary glands were dissected in PBS and fixed overnight in 4% formaldehyde in PBS at 4oC. They were blocked in PBS, 1% BSA, and 0.1% Tween (PBSBT) and incubated with a primary antibody (rabbit anti-cleaved caspase-3 1:400; Cell Signaling Technology, rabbit anti-Ref(2)p 1:2000, gift from G. Juhasz) overnight at 4oC. Next, the salivary glands were washed in PBSBT for 2h at room temperature, incubated with the appropriate secondary antibody for 2h at room temperature, and washed for 1h in PBSBT. Salivary glands were mounted in Vectashield (Vector Laboratories). For mCherry-Atg8a analysis, salivary glands were dissected in PBS, fixed in 4% formaldehyde in PBS for 2h and mounted in Vectashield (Vector Laboratories). Imaging was performed on a Zeiss LSM700 confocal microscope.
Cell culture transfection and pH-DIBO cell surface labeling
HEK293T cells were cultured in high glucose DMEM medium (Gibco) with 10% fetal bovine serum (Gibco) and 100 units/mL penicillin/streptomycin (Gibco) at 37°C in 5% CO2. The cells were seeded at 60–75% confluency in 35 mm dishes. After 24h, cells were transiently transfected with a 1 μg of plasmid DNA and 5 μL of Lipofectamine 2000 (Invitrogen) in OptiMEM (Gibco) for 4 hours. To visualize transfected cells, 0.25 μg of pEGFP-C3 plasmid was added to transfections that did not contain GFP-tagged transporters. After terminating the transfections, the cells were transferred to Glass bottom culture dishes (MatTek) coated by fibronectin and incubated in media containing 50 μM of Ac4ManNAz for 2 days, which was replenished after 24 h. After 2 days, transfected HEK293T cells were directly labeled with pHDIBO in Opti-MEM at 37°C in 5% CO2 for 30 min.
Extracellular pH measurement
After pH-DIBO labeling, cells were washed twice with bath solution (145mM NaCl, 5.4 mM KCl, 5 mM CaCl2 and 0.1 mM HEPES, pH 7.0). A 35-mm chamber insert (RC-33DL, Warner) was put into the glass bottom dish for decreasing volume. The dish was then mounted onto a Quick Exchange Platform QE-1 (Warner) for imaging. Monocarboxylates and α-Cyano-4hydroxycinnamic acid (Sigma) were perfused in/out by using a gravity perfusion system at about 10 mL/min. Cells were imaged at 1 Hz using a CoolLED pE-4000 light source, 63 × 1.4 N.A. oil immersion objective, and a Zyla sCMOS camera (Andor). Fluorescent images were collected and processed using open source software (micro-manager (Edelstein et al., 2010) and ImageJ). F0 is the average fluorescence intensity of first five data points.
Nile Red staining
For Nile Red staining, salivary glands were dissected in PBS and fixed in 4% formaldehyde in PBS for 90 min. Salivary glands were rinsed several times with PBS and incubated for 10 min in PBS with 1μg/mL Nile Red (Sigma-Aldrich). Subsequently, salivary glands were rinsed with PBS, mounted in Vectashield (Vector Laboratories) and imaged on a Zeiss LSM700 confocal microscope.
QUANTIFICATION AND STATISTICAL ANALYSES
For animal studies, sample sizes were determined empirically based on previous studies to ensure appropriate statistical power. No animals were excluded from statistical analyses, the experiments were not randomized, and the investigators were not blinded. p-value was calculated using a chi-square test. For puncta quantification analyze particles function of ImageJ was used. p-value was calculated using a two-tailed unpaired t test. Additional information on statistical details can be found in the figure legends.
Supplementary Material
Highlights.
Members of the SLC superfamily are required for cell death during development
Identification of hermes, a gene that is required for autophagy during cell death
Hermes is a proton-coupled pyruvate transporter
Hermes influences mTOR to regulate autophagy
ACKNOWLEDGEMENTS
We thank M. Brodsky for his help in generating the hrmΔ1 animals, the Bloomington Stock Center and the VDRC for flies, and the Baehrecke lab for constructive comments. This work was supported by NIH grant GM079431 to EHB.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baehrecke EH (2000). Steroid regulation of programmed cell death during Drosophila development. Cell Death Differ 7, 1057–1062. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH (2005). Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6, 505–510. [DOI] [PubMed] [Google Scholar]
- Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES, et al. (2014). MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep 9, 2233–2249. [DOI] [PubMed] [Google Scholar]
- Berry DL, and Baehrecke EH (2007). Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse F, Mertel S, Kittel RJ, Wichmann C, Rasse TM, Sigrist SJ, and Ephrussi A (2007). The Ig cell adhesion molecule Basigin controls compartmentalization and vesicle release at Drosophila melanogaster synapses. J Cell Biol 177, 843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, and Edgar BA (2002). Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2, 239–249. [DOI] [PubMed] [Google Scholar]
- Broer S, Schneider HP, Broer A, Rahman B, Hamprecht B, and Deitmer JW (1998). Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem J 333 (Pt 1), 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YY, and Neufeld TP (2009). An Atg1/Atg13 complex with multiple roles in TORmediated autophagy regulation. Mol Biol Cell 20, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin KD, Meinertzhagen IA, and Wyman RJ (2005). Basigin (EMMPRIN/CD147) interacts with integrin to affect cellular architecture. J Cell Sci 118, 2649–2660. [DOI] [PubMed] [Google Scholar]
- Czech MP (2017). Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, et al. (2016). Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal 9, ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Chang TK, Nicolson S, Shravage B, Simin R, Baehrecke EH, and Kumar S (2012). Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell death and differentiation 19, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, and Refetoff S (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8, 325–332. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2010). Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14, Unit14 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A 111, 2554–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T (2015). The metabolic regulation of aging. Nat Med 21, 1416–1423. [DOI] [PubMed] [Google Scholar]
- Franssens BT, Westerink J, van der Graaf Y, Nathoe HM, Visseren FLJ, and group S.s. (2016). Metabolic consequences of adipose tissue dysfunction and not adiposity per se increase the risk of cardiovascular events and mortality in patients with type 2 diabetes. Int J Cardiol 222, 72–77. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, and Kroemer G (2014). Cell biology. Metabolic control of cell death. Science 345, 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, and Steller H (1995). The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP (2013). The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med 34, 337–349. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, and Wilson MC (2012). The monocarboxylate transporter family--role and regulation. IUBMB Life 64, 109–119. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, and Rubin GM (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, and Yuan J (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, et al. (2014). SLC25A23 augments mitochondrial Ca(2)(+) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell 25, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holen I, Gordon PB, Stromhaug PE, and Seglen PO (1996). Role of cAMP in the regulation of hepatocytic autophagy. Eur J Biochem 236, 163–170. [DOI] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Baehrecke EH, and Thummel CS (1997). Steroid regulated programmed cell death during Drosophila metamorphosis. Development 124, 4673–4683. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, and Ohsumi Y (2000). Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150, 1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamdar K, Khakpour S, Chen J, Leone V, Brulc J, Mangatu T, Antonopoulos DA, Chang EB, Kahn SA, Kirschner BS, et al. (2016). Genetic and Metabolic Signals during Acute Enteric Bacterial Infection Alter the Microbiota and Drive Progression to Chronic Inflammatory Disease. Cell Host Microbe 19, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Igaki T, Okano H, and Miura M (2011). Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc Natl Acad Sci U S A 108, 18977–18982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloeckener-Gruissem B, Vandekerckhove K, Nürnberg G, Neidhardt J, Zeitz C, Nürnberg P, Schipper I, and Berger W (2008). Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet 82, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-Y, Clough EA, Yellon P, Teslovich TM, Stephan DA, and Baehrecke EH (2003). Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol 13, 350–357. [DOI] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, et al. (2014). Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 5, 4934. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shoji-Kawata S, Sumpter RMJ, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. (2013). Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 110, 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, and Komatsu M (2011). Autophagy: Renovation of Cells and Tissues Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Yan M, and Basler K (2002). Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, and Hay BA (2006). The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133, 3305–3315. [DOI] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, Schuit F, Quintens R, Sipila I, Mayatepek E, et al. (2007). Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet 81, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perland E, and Fredriksson R (2017). Classification Systems of Secondary Active Transporters. Trends Pharmacol Sci 38, 305–315. [DOI] [PubMed] [Google Scholar]
- Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, and Hall MN (2008). Adiposespecific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8, 399–410. [DOI] [PubMed] [Google Scholar]
- Poole RC, and Halestrap AP (1997). Interaction of the erythrocyte lactate transporter (monocarboxylate transporter 1) with an integral 70-kDa membrane glycoprotein of the immunoglobulin superfamily. J Biol Chem 272, 14624–14628. [DOI] [PubMed] [Google Scholar]
- Port F, Chen HM, Lee T, and Bullock SL (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci U S A 111, E2967–E2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, and White E (2010). Autophagy and metabolism. Science 330, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, et al. (2015). SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, and Levine B (2012). Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11, 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusu V, Hoch E, Mercader JM, Tenen DE, Gymrek M, Hartigan CR, DeRan M, von Grotthuss M, Fontanillas P, Spooner A, et al. (2017). Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 170, 199–212 e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, et al. (2008). Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A 105, 18578–18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2018). Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab 27, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E, and Bertozzi CR (2000). Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweichel J-U, and Merker H-J (1973). The morphology of various types of cell death in prenatal tissues. Teratology 7, 253–266. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, and Neufeld TP (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 17, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, and Neufeld TP (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 7, 167–178. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, et al. (2015). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, and Ohsumi Y (1993). Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333, 169–174. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205. [DOI] [PubMed] [Google Scholar]
- Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. (2015). Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, and Negelein E (1927). The Metabolism of Tumors in the Body. J Gen Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Meredith D, Bunnun C, Sessions RB, and Halestrap AP (2009). Studies on the DIDS-binding site of monocarboxylate transporter 1 suggest a homology model of the open conformation and a plausible translocation cycle. J Biol Chem 284, 20011–20021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Meredith D, and Halestrap AP (2002). Fluorescence resonance energy transfer studies on the interaction between the lactate transporter MCT1 and CD147 provide information on the topology and stoichiometry of the complex in situ. J Biol Chem 277, 3666–3672. [DOI] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, and Sabatini DM (2017). mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 171, 642–654 e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, Pant A, Gyorffy B, Zhan M, Carter-Su C, et al. (2016). Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab 24, 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, and Kroemer G (2010). Alternative cell death mechanisms in development and beyond. Genes Dev 24, 2592–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, and Baehrecke EH (2015). Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol 25, 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bellve K, Fogarty K, and Kobertz WR (2016). Fluorescent Visualization of Cellular Proton Fluxes. Cell Chem Biol 23, 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.