

Abstract
UV irradiation-induced oxidative stress and inflammation contribute to the development of skin diseases. Therefore, targeting oxidative stress and inflammation might contribute to reduce skin diseases. Resolvin D1 (RvD1) is a bioactive metabolite generated during inflammation to actively orchestrate the resolution of inflammation. However, the therapeutic potential of RvD1 in UVB skin inflammation remains undetermined, which was, therefore, the aim of the present study. The intraperitoneal treatment with RvD1 (3-100 ng/mouse) reduced UVB irradiation-induced skin edema, myeloperoxidase activity, matrix metalloproteinase 9 activity, and reduced glutathione depletion with consistent effects observed with the dose of 30 ng/mouse, which was selected to the following experiments. RvD1 inhibited UVB reduction of catalase activity, and hydroperoxide formation, superoxide anion production, and gp91phox mRNA expression. RvD1 also increased the Nrf2 and its downstream targets NQO1 and HO-1 mRNA expression. Regarding cytokines, RvD1 inhibited UVB-induced production of IL-1β, IL-6, IL-33, TNF-α, TGF-β, and IL-10. These immuno-biochemical alterations by RvD1 treatment had as consequence the reduction of UVB-induced epidermal thickness, sunburn and mast cell counts, and collagen degradation. Therefore, RvD1 inhibited UVB-induced skin oxidative stress and inflammation, rendering this resolving lipid mediator as a promising therapeutic agent.
Keywords: resolvin, inflammation, oxidative stress, UVB irradiation, lipid mediator
Introduction
The skin is the largest organ of the human body and the main protection barrier of the organism against chemical, physical, and biological aggressors (Afaq et al., 2005; Fonseca et al., 2011; Khavkin and Ellis, 2011). External aggressors include exposure to UV irradiation, the main cause of skin damage. Acute exposure to UV irradiation may lead to a number of effects such as edema, sunburn, erythema, inflammation, and prolonged or chronic exposure can lead, for instance, to immunosuppression, premature aging, and skin cancer (Afaq et al., 2005; Fonseca et al., 2011; Tewari et al., 2013a; Martinez et al., 2015b).
The skin damage induced by UV irradiation occurs due to overproduction of reactive oxygen species (ROS), such as the superoxide anion (Ivan et al., 2014), consequently leading to depletion of endogenous antioxidant, such as reduced glutathione (GSH) (Zaid et al., 2007; Halliwell, 2009). The imbalance between generation and removal of free radicals in the body leads to a pro-oxidant state that can lead to cell damage, including cell death (Júnior et al., 2001). The UVB irradiation spectrum is considered the most damaging and harmful UV irradiation for the human skin (Afaq et al., 2005; Hupel et al., 2011; Figueiredo et al., 2014), because its main consequence is direct DNA damage, mainly in keratinocytes. In addition, exposure to UVB irradiation activates the skin immune system components, triggering inflammatory response through the release of inflammatory mediators such as cytokines that will orchestrate the inflammatory response (Bowden, 2004; Hildesheim et al., 2004; Oliveros et al., 2009; Maverakis et al., 2010; Balogh et al., 2011). Considering the synergistic effect of the production of ROS and inflammatory mediators, the improvement of the endogenous antioxidant system and the resolution of inflammation become promising approaches to prevent and treat UVB irradiation-induced skin damage (Fonseca et al., 2010; Serhan, 2014).
Resolvin D1 (RvD1) is a bioactive metabolite generated in response to inflammation by enzymatic conversion of docosahexaenoic acid (DHA) (Seki et al., 2010). The RvD1 belongs to the group of lipid mediators that play important roles in the resolution phase of inflammation (Chan and Moore, 2010; Recchiuti and Serhan, 2012; Moro et al., 2016). These lipid mediators have been a major focus in recent years due to their anti-inflammatory and pro-resolution abilities in various disease models. Their actions include reduction of neutrophil chemotaxis, induction of neutrophil apoptosis, chemoattraction of non-phlogistic macrophages, reduction of dendritic cell migration to the lymph nodes and IL-12 production, and increasing macrophage-mediated clearance of cell debris, apoptotic cells, and invading microorganisms (Serhan et al., 2008; Serhan, 2014).
Studies have shown that RvD1 is able to inhibit the inflammatory response and promote the resolution of inflammation by reducing the production of the pro-inflammatory cytokines TNF-α and IL-1β in mouse models of acute lung injury induced by lipopolysaccharides (LPS). In addition, the ability of RvD1 to reduce oxidative stress in lung injury was demonstrated through increased transcription of the gene encoding the enzyme heme-oxygenase 1 (HO-1) (Wang et al., 2014). Other studies have demonstrated the reduction of inflammatory responses in asthma and acute lung injurie with the use of RvD1, in addition to suppression chemokine production and oxidative stress induced by cigarette smoke extract (Haworth and Levy, 2007; Rogerio et al., 2012; Hsiao et al., 2013).
These results make reasonable to envisage that RvD1 has, in principle, the potential to reduce inflammation and oxidative stress in other disease conditions. However, there is no evidence on the effect of RvD1 in UVB irradiation-induced skin inflammation and oxidative stress, which we reason to be worthy investigating in the present study.
Materials and methods
Chemicals
Chemicals were obtained from the following sources: resolvin D1 from Cayman Chemical (Ann Arbor, Michigan, USA); brilliant blue R, reduced glutathione (GSH), hexadecyltrimethylammonium bromide (HTAB), o-dianisidine dihydrochloride, 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB), nitroblue tetrazolium (NBT), and bisacrylamide from Sigma-Aldrich (St. Louis, MO, USA); tert-butyl hydroperoxide from Acros (Pittsburgh, PA, USA); tris from Amresco (Solon, OH, USA); ELISA kits for determination of cytokine from eBioscience (San Diego, CA, USA); and acrylamide, sodium dodecyl sulfate (SDS), platinum SYBRGreen, and superscript III kits from Invitrogen. All other reagents used were from pharmaceutical grade.
Animals
Sex matched hairless mice (HRS/J) weighing 20–30 g were obtained from the University Hospital of Londrina State University under the following conditions: 12 h dark/12 h light cycle and 23 ± 2°C temperature. The mice were maintained with free access to water and food throughout the experiment. The animal protocol used in this study was approved by Animal Ethics Committee (CEUA process number 1447.2015.10) of the Londrina State University.
Experimental protocol
Five mice per group were randomly assigned to six groups. The groups were: non-irradiated control treated with vehicle (saline), irradiated control treated with vehicle (saline), irradiated treated with RvD1 3 ng/mice, irradiated treated with RvD1 10 ng/mice, irradiated treated with RvD1 30 ng/mice, irradiated treated with RvD1 100 ng/mice.
Treatment doses of RvD1 were 3, 10, 30, and 100 ng/ mouse, via intraperitoneal administration, 1 h before and 7 h after the beginning of UV irradiation (Martinez et al., 2015a). Animals in the control groups received treatment with vehicle (saline) used in the dilution of the drug. The doses of RvD1 used in the treatments were selected based on the therapeutic effects of studies published in other disease models (Spite et al., 2009a; Hsiao et al., 2013; Wang et al., 2014) and on dose-response curves tested in the present study.
Based on the results obtained in the assays evaluating skin edema, GSH, MPO and MMP-9, one dose of RvD1 was selected to the following experiments quantitating oxidative stress and oxidative stress-related mRNA expression (catalase, hydropexide formation, and superoxide anion production, and qPCR to determine gp19phox, Nrf2, Nqo-1, and HO-1 mRNA expression), cytokine production (IL-1β, IL-6, IL-33, TNF-α, TGF-β, and IL-10), and skin tissue alterations (epidermal thickness, sunburn cell counts, mast cell counts, collagen degradation). The time points of sample collection after UVB irradiation and assays to be performed at each time point were determined in standardization experiments. Cytokine production, oxidative stress and antioxidant markers were evaluated at earlier time points than the tissue alterations since tissue alterations are consequences of mediator production (Campanini et al., 2013; Martinez et al., 2015b, 2017).
Irradiation
The light source used in the experiments to induce oxidative stress and acute inflammatory process in hairless mice was a fluorescent UVB lamp model PHILIPS TL/12 40W RS (MEDICAL-NETHERLANDS). The lamp emits irradiation in the range of 270 to 400 nm with a peak emission at 313 nm. The dose of irradiation used to induce inflammation and oxidative stress was 4.14 J/cm2 (Campanini et al., 2013).
Mice were kept at a distance of 20 cm from the lamp as previously described (Campanini et al., 2013) and were irradiated simultaneously. The hairless mice were terminally anesthetized with 5% isoflurane (Abbott [Abbott Park, IL, USA]) 12 h after the end of UVB irradiation and the full thickness of the dorsal skins were removed for edema, MPO activity, MMP-9 activity and GSH assays and histology. Moreover, the hairless mice were anesthetized with 5% isoflurane, following by decapitation at 2 h for catalase and NBT assays, and 4 h for evaluation of production hydroperoxides, cytokines measurement and PCR after the end of UVB exposure and the dorsal skins were removed. Each parameter was evaluated at a specific time, which was previously determined (Campanini et al., 2013; Martinez et al., 2015b). The dorsal skin samples were collected and stored at −80°C until analysis. The samples collected by verification of cutaneous edema were weighed immediately after collecting and by histology were fixed in buffered formaldehyde.
Skin edema
UV irradiation increases the permeability of the vascular endothelium causing edema (Dusting and Macdonald, 1995). In order to evaluate the skin edema associated with the inflammatory process, the dorsal skin samples were collected from the animals with the aid of a mold with a fixed area (5 mm diameter). Edema was expressed by the variation of skin weight between the different control and treated groups (Ivan et al., 2014).
Myeloperoxidase (MPO) activity
Myeloperoxidase (MPO) activity was quantitated to be used as a marker of the leukocyte infiltrate (monocytes/macrophages and neutrophils) on the skin after UVB irradiation (Katiyar and Meeran, 2007).
The skin samples were collected in 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide (HTAB), homogenized with Tissue-Tearor (Biospec 985370) and centrifuged (16,100 g for 2 min at 4°C). Briefly, 30 μL of the resulting supernatant from each sample were mixed with 50 mM phosphate buffer (pH 6.0) containing 0.167 mg/mL o-dianisidine and 0.015% hydrogen peroxide. MPO activity was determined spectrophotometrically at 450 nm (EnSpire, Perkin Elmer). The MPO activity of the samples was obtained by comparison with the MPO activity of a standard neutrophil curve. The results were expressed in neutrophil numbers per mg of skin (Casagrande et al., 2006).
Analyses of skin proteinase substrate-embedded enzymography
For the analyze of MMP-9, the polyacrylamide gel zymography technique with sodium dodecyl sulfate was applied (SDS), a method used to detect proteases. The analysis detects enzymes that degrade the gelatin present in the gel (Kim et al., 2007; Fonseca et al., 2011).
Skin samples were diluted (1: 4) and homogenized in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM calcium chloride (CaCl2) and 1% proteinase inhibitors (phenanthroline, phenylmethylsulfonyl fluoride and N—ethylmaleimide) with the aid of Tissue-Tearor (Biospec 985370). Thereafter, the homogenates were centrifuged (12,000 g, 10 min, 4°C) twice. Supernatant aliquots (25 μL) were mixed with 5 μL of 0.1 M Tris-HCl (pH 7.4) containing 20% glycerol, 4% SDS, and 0.005% xylene cyanol and applied on electrophoresis gel (13.5% acrylamide and 0.025% gelatin). After electrophoresis, the gels were washed for 1 h with 2.5% Triton X-100 under constant shaking, incubated overnight in 0.05 M Tris-HCl (pH 7.4) and 0.01M CaCl2 at 37°C. The next day, the gels were stained with brilliant blue R and destaining in 20% acetic acid. The zones of enzymatic activity were detected as regions of negative staining against a dark background. The proteolytic activity was analyzed quantitatively by comparing the results of the samples of the treated animals with the controls not treated by the Image J program (NIH, Bethesda, MD, USA) (Onoue et al., 2003; Casagrande et al., 2006).
Quantification of endogenous antioxidant reduced glutathione (GSH)
Skin samples were diluted in 0.02M EDTA and triturated using Tissue-Tearor (Biospec 985370). Whole homogenates were treated with 50% trichloroacetic acid. The mixture was then centrifuged at 2,700 g for 10 min at 4°C. The supernatant was removed and recentrifuged at 2,700 g for a further 15 min at 4°C. The final supernatant was removed for analysis. For the assay, the reaction mixture contained 50 μL of the sample supernatant, 100 μL of 0.4 M Tris buffer pH 8.9 and 5 μL of a 1.9 mg/mL solution of 5,5'-dithio-bis- (2-nitrobenzoic acid; DTNB) in methanol. The absorbance was determined in a spectrophotometer (EnSpire, Perkin Elmer) after 5 min of incubation at 405 nm. The standard curve was prepared with 0 to 150 μM GSH. The results were expressed in as μM of GSH per mg of skin (Srinivasan et al., 2007).
Levels of the of endogenous antioxidant catalase (CAT)
The method is based on the concentration decay of hydrogen peroxide (H2O2) which is directly proportional to the absorbance decrease at 240 nm. The difference in absorbance per unit time is the measure of catalase activity (Aebi, 1984).
Skin samples were homogenized in 500 μL of 0.02M EDTA using the Tissue-Tearor homogenizer (Biospec 985370). The homogenate was centrifuged at 2,700 g for 10 min at 4°C twice. The determination of CAT activity on skin was performed on microplate by addition of 10 μL sample, 160 μL 1M Tris-HCl buffer with 5 mM EDTA pH 8.0, 20 μL deionized water, and 20 μL 200 mM H2O2. A white was included for each sample prepared with 10 μL of the sample supernatant, 180 μL of 1M Tris-HCl buffer with 5 mM EDTA pH 8.0, and 20 μL of deionized water. The rate at which H2O2 is reduced by the action of CAT was evaluated by decreasing the absorbance value by the difference between the initial reading and reading 30 s after the addition of 200 mM H2O2. The reading was performed on a microplate spectrophotometer (Enspire, Perkin Elmer) at 240 nm with a temperature maintained at 25°C. The catalase values were expressed as unit of CAT/ mg skin/ minute (Aebi, 1984).
Assay for lipid peroxidation (LPO)
Lipid peroxidation is one of the most important organic expressions of oxidative stress (Yagi, 1987). Oxidation of lipids was measured by the formation of hydroperoxides, which are the primary products in lipid peroxidation (Linggnert et al., 1979).
The hydroperoxide production was evaluated by the chemiluminescence method previously described (Martinez et al., 2015b). The method is based on the determination of the chemiluminescence initiated by the tert-butyl hydroperoxide (Gonzalez Flecha et al., 1991).
The dorsal skin samples were homogenized in 800 μL of phosphate buffer (pH 7.4) using a Tissue-Tearor (BIOSPEC 985370) and centrifuged at 700 g for 2 min at 4°C. For the assay, 250 μL of the supernatant was diluted to 1,730 μL of reaction medium (120 mM KCl, 30 mM phosphate buffer pH 7.4) and 20 μL of 3 mM tert-butyl hydroperoxide. The reading was conducted in a β–counter Beckman® LS 6000SC (FULLERTON, CA, USA) in a non-coincident counting for 30 s with a response range between 300 and 620 nm. The experiment was performed in the darkin order to avoid vial phosphorescence activated by light at 30°C for 2 h. The results were measured in counts per min (cpm) per mg of skin.
Evaluation of the production of superoxide anion ()
Superoxide anion assay was performed through the nitroblue tetrazolium assay (NBT). Skin samples were homogenized with Tissue-Tearor (BIOSPEC 985370) in 0.02 M EDTA and centrifuged (2,000 g for 20 s at 4°C). For the reaction, the supernatant (50 μL) was incubated in microplates for 1 h. Then the supernatant was removed and NBT (1 mg/mL) added to the fixed cells. After 15 min, the NBT was carefully removed and 20 μL of 100% methanol was added to the precipitate to settle. The compound formed by the reduction of NBT (formazan) was solubilized with 120 μL of 2M KOH and 140 μL of dimethylsufoxide (DMSO). The reduction of NBT to formazan was measured in a microplate spectrophotometer reader (ENSPIRE, PERKIN ELMER) at 620 nm and the results are presented as optical density (OD) per 10 mg of skin (Campanini et al., 2013).
Cytokine measurement
The dorsal skin samples were homogenized in saline (500 μL) with Tissue-Tearor (Biospec 985370) and centrifuged at 2,000 g for 15 min at 4°C. The supernatants were used to quantify the cytokines IL-1, IL-6, IL-33, TNF-α, TGF-β, and IL-10 aby enzyme-linked immunosorbent assay (ELISA) according to manufacturer's instructions (eBioscience). The results were obtained by comparing the optical densities at 490 nm of the samples with the densities of the respective cytokine standard curves (Verri et al., 2008).
Quantitative polymerase chain reaction (QPCR)
Followed the method described elsewhere (Campanini et al., 2013; Martinez et al., 2016b). Briefly, samples were homogenized in trizol reagent for total RNA extraction. The purity of total RNA was measured spectrophotometrically, and the wavelength absorption ratio (260/280 nm) was between 1.8 and 2.0 for all preparations. Reverse transcription of total RNA to cDNA was carried out using a Superscript III kit (Invitrogen) and oligo (dT)12–18 primers. Real-time PCR (qPCR) was performed with Platinum SYBRGreen kits (Invitrogen) in a 50 μL reaction volume following the manufacturer's cycling conditions. Melting curve analysis was performed in order to verify that only one product was amplified. Samples with more than one peak were excluded. qPCR was performed in a LightCycler Nano Instrument (Roche). The relative gene expression was measured using the comparative 2−−(ΔΔCq) method. The expression of gyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA was used as a control for tissue integrity in all samples. The primers used were gp91phox, sense: 5′-AGCTATGAGGTGGTGATGTTAGTGG-3′, antisense: 5′- CACAATATTTGTACCAGACAGACTTGAG-3′; Nrf2, sense: 5′-TCACACGAGATGAGCTTAGGGCAA-3′, antisense: 5′-TACAGTTCTGGGCGGCGACTTTAT-3′; Nqo-1, sense: 5′-TGGCCGAACACAAGAAGCTG-3′, antisense: 5′-GCTACGAGCACTCTCTCAAACC-3′; HO-1, sense: 5′-CCCAAAACTGGCCTGTAAAA-3′; antisense: 5′-CGTGGTCAGTCAACATGGAT-3′; and Gapdh sense: 5′-ATGACATCAAGAAGGTGGTG-3, antisense: 5′-CATACCA- GGAAATGAGCTTG-3′;
Skin histologic evaluation
The dorsal skin samples were collected in formol 10%, fixed in paraformaldehyde 4%, dehydrated in ascending concentrations of ethanol, cleared in xylene, embedded in paraffin and sectioned to a thickness of 5 μm. The sections were stained with hematoxylin and eosin, toluidine blue and Masson's trichrome stain.
The sections stained with H & E were examined using light microscopy at 40x magnification for determination of epidermal thickness (Deng et al., 2015) and a 100x magnification for counting the number of sunburn cells (Schwarz et al., 1995). For mast cell count, the sections were stained with toluidine blue and analyzed under light microscopy at 40x magnification. Both analyses were done with the software Infinity Analyze (Lumenera® Software). The sections stained with masson's trichrome were examined using light microscopy at a magnification of 10x to visualize changes in collagen fibers by analyzing the intensity of the blue coloration in the dermal areas of the skin exposed to UVB with the aid of the Image J software (NIH) (Song et al., 2016).
Statistical analysis
The bars in the results indicate the mean values ± standard error of the mean (SEM) of 5 mice per group per experiment and are representative of two separate experiments. Data were statistically analyzed by ANOVA followed by Tukey's t-test. Statistical analyses were performed using GraphPad Prism 7 software (GraphPad Software Inc., San Diego, CA, USA). Results were considered significantly different when p < 0.05.
Results
Resolvin D1 (RvD1) reduces UVB irradiation-induced skin edema and MPO activity
The anti-inflammatory action of RvD1 was first assessed by the edema assay and MPO activity (neutrophil marker). Skin edema was inhibited by RvD1 treatment only by the dose of 30 ng/mice (Figure 1A). On the other hand, all four doses of RvD1 showed a similar inhibition of MPO activity (Figure 1B). Based on the result obtained in edema, the dose of 30 ng/mouse was chosen for the next assays. Receptor expression in the target cellular population affects the efficacy of pro-resolution lipids as observed for aspirin-triggered lipoxin A4 (ATLA4). ATLA4 has dose-dependent effect over leukocyte chemoattraction. Nevertheless, the genetic induction of lipoxin receptor ALXR/FPR2 in myeloid cells enhances the activity of ATLA4 in a manner that 10 ng achieves the same efficacy of 1μg of ATLA4 (Chiang et al., 2005) Despite the wide range of doses tested, the effect of RvD1 over UVB irradiation was not dose-dependent. It is likely that further investigation on delivery route, formulation and time of treatment may improve the efficacy of RvD1 by reaching the right cellular target at the best time point and dose.
Figure 1.

RvD1 reduces UVB irradiation-induced skin edema and MPO activity. The skin inflammation was determined in samples collected 12 h after the end of irradiation. (A) Skin edema and (B) MPO activity. Bars represent means ± SEM of 5 mice per group and are representative of two separate experiments. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-irradiated control group; #p < 0.05 compared to the irradiated control group (vehicle)].
RvD1 reduces UVB irradiation-induced increase of epidermal thickness and apoptosis of keratinocytes
Epidermal thickness is used as a quantitative parameter to assess inflammation (Martinez et al., 2015b). Measurement of hematoxylin and eosin stained tissue sections indicated that dorsal skin epidermal thickness was significantly increased following UVB irradiation in the irradiated control group compared to the non-irradiated control group. In contrast, epidermal hypertrophy was significantly reduced by 57.48% compared with irradiated control group when mice were treated with RvD1 (Figure 2A).
Figure 2.

RvD1 reduces UVB irradiation-induced increase of epidermal thickness and sunburn cell counts. Epidermal thickness and sunburn cell counts were evaluated using hematoxylin and eosin staining (H & E) in skin samples collected 12 h after the end of irradiation. Epidermal thickness (μm) (A) and the number of sunburn cells (B). No sunburn cells were detected in the negative control group, which was indicated as ND. The sections stained with H & E were examined using light microscopy at 40x (A) magnification and 100x (B). Bars are representative of two separate experiments and represent means ± SEM of 5 mice per group per experiment. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-radiated control group; #p < 0.05 compared to the radiated control group (vehicle)].
One of the consequences of acute exposure to UV radiation is the activation of apoptosis of epidermal keratinocytes, which are defined as shrunken cells within the epidermis that exhibit eosinophilic cytoplasm and condensed nucleus, and called sunburn cells (Bayerl et al., 1995). UV radiation induced an increase on sunburn cell counts, which was inhibited by 39.47% compared with irradiated control group by RvD1 treatment (Figure 2B).
RvD1 reduces UVB irradiation-induced increase of mast cells
UVB irradiation induces a significant increase in the number of mast cells in the skin as well as their degranulation releasing varied pro-inflammatory mediators (Hart et al., 2001). In this study, treatment with RvD1 inhibited the UVB irradiation increase of mast cells counts (Figure 3).
Figure 3.

RvD1 reduces UV irradiation-induced increase of mast cell counts. Mast cell counts were evaluated using blue toluidine in skin samples collected 12 h after the end of irradiation. The number of mast cells (A) in the sections stained with blue toluidine were examined using light microscopy at 40x magnification (B). Bars are representative of two separate experiments and represent means ± SEM of 5 mice per group per experiment. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-radiated control group; #p < 0.05 compared to the radiated control group (vehicle)].
RvD1 reduces UVB irradiation-induced skin MMP-9 activity and damage of collagen fiber
MMP-9 is a gelatinase involved in the degradation of the elastic fiber network and collagen matrix, thus, it is involved in tissue remodeling and collagen fiber density (Jenkins, 2002). MMP-9-induced damage to the collagenous matrix of the skin is one of the hallmarks of photoaging and non-melanoma skin cancer (Brennan et al., 2003; Grady et al., 2007). Treatment with RvD1 reduced the UVB irradiation-induced activity of MMP-9 in a dose-dependent manner with the peak of RvD1 activity obtained with the dose of 30 ng/mouse. The doses of 30 and 100 ng/mouse of RvD1 reduced the activity of MMP-9 (Figures 4A,B).
Figure 4.

RvD1 inhibits UVB irradiation-induced increase of MMP-9 activity and damage to skin collagen fiber of hairless mice. The MMP-9 activity and collagen fiber formation were determined in samples collected 12 h after the end of irradiation. (A) Representative image of gelatin zymography and (B) MMP-9 activity. Collagen fiber formation was evaluated using Masson's trichrome staining. Collagen fiber intensity and bundles shown in blue were analyzed by Image J Program (10x magnification) (C). Collagen intensity (D). Bars are representative of two separate experiments and represent means ± SEM of 5 mice per group per experiment. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-radiated control group; #p < 0.05 compared to the radiated control group (vehicle)].
The tissue sections were subjected to Masson's trichrome staining in order to determine changes in collagen fiber density in the dermal areas of the UVB-exposed dorsal skin (Song et al., 2016). Notably, the collagen fibers stained in blue in the group pretreated with RvD1 30 ng/mice showed lower levels of damage in collagen fiber compared with irradiated group (Figures 4C,D).
RvD1 inhibits UVB irradiation-induced cytokine production
Cytokines such as IL-1β, IL-6, IL-33, and TNF-α are involved in many cellular and tissue alterations in UVB irradiation, which include the recruitment of neutrophils that produce ROS and MMP-9, thus, with implications on inflammation and tissue remodeling (Garcia et al., 1999; Witko-Sarsat et al., 2000; Robinson et al., 2004; Verri et al., 2006; Walz and Cayabyab, 2017). Cytokines also affect vascular permeability, inducing tissue edema (Joosten et al., 2006; Zarpelon et al., 2013; Staurengo-Ferrari et al., 2018). Other cytokines such as TGF-β and IL-10 limit inflammation and orchestrate tissue repair (Verri et al., 2006; Penn et al., 2012). Therefore, cytokines were quantitated. UVB irradiation induced the production of pro-inflammatory (IL-1β, IL-6, IL-33, and TNF-α) and anti-inflammatory (TGF-β and IL-10) cytokines in hairless mice skin, which were inhibited by RvD1 treatment (Figures 5A–F). Therefore, RvD1 treatment reduced the cytokine production irrespectively whether these were pro-inflammatory or anti-inflammatory cytokines.
Figure 5.
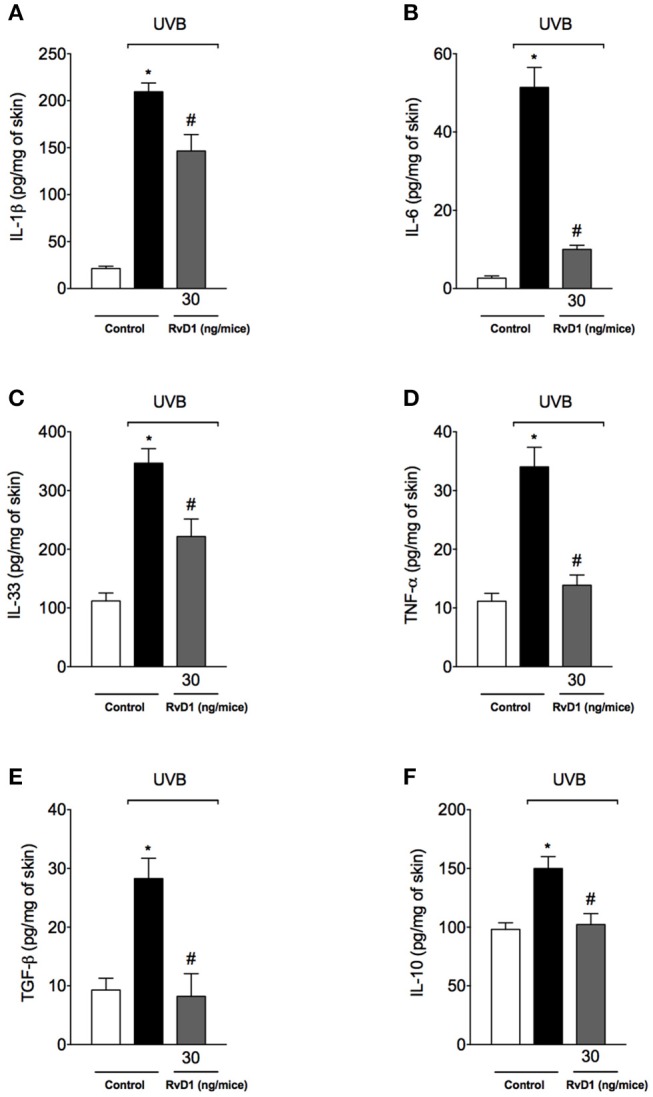
RvD1 inhibits UVB irradiation-induced cytokine production (A) IL-1β (B) IL-6 (C) IL-33 and (D) TNF-α and inhibits anti-inflammatory cytokines production (E) TGF-β and (F) IL-10. Cytokines were determined in skin samples collected 4 h after the end of irradiation. Bars represent means ± SEM of 5 mice per group and are representative of two separate experiments. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-radiated control group; #p < 0.05 compared to the radiated control group (vehicle)].
RvD1 reduces UVB irradiation-induced oxidative stress, and enhances mRNA expression of genes involved in antioxidant response and skin antioxidants
UVB irradiation increased the hydroperoxide and superoxide anion production in the skin of irradiated control group compared to non-irradiated control group. Treatment with RvD1 at the dose of 30 ng/mouse reversed this effect by reducing LOOH and superoxide anion production (Figures 6A,B). The NADPH oxidase is an important source of superoxide anion during inflammation (Anrather et al., 2006), and UVB irradiation caused an increase of the NADPH oxidase subunit gp91phox mRNA expression, which was also reduced by RvD1 (Figure 6C) corroborating the data on superoxide anion production (Figure 6B).
Figure 6.

RvD1 inhibits UVB irradiation-induced oxidative stress, mRNA expression of oxidative stress-related genes and antioxidant depletion. Lipid peroxidation (A) was determined by t-butyl lipid hydroperoxides (LOOH)-initiated chemiluminescence (QL) at 4 h, superoxide anion production (B) was determined by nitroblue tetrazolium (NBT) reduction assay at 2h; gp91phox, Nrf2 (D), Nqo-1 (E), and HO-1 (F) mRNA expression (C) were determined by quantitative polymerase chain reaction (qPCR) at 4 h after the end of irradiation; GSH levels (G) and CAT activity (H) were determined at 12 h and 2h after the end of irradiation, respectively. Bars represent means ± SEM of 5 mice per group per experiment and are representative of two separate experiments. Statistical analysis was performed by one-way ANOVA followed by Tukey's test. [*p < 0.05 compared to the non-radiated control group; #p < 0.05 compared to the radiated control group (vehicle)].
GSH, NQO-1, and HO-1 are downstream targets of Nrf2 (Loboda et al., 2016). We observed that UVB irradiation decreased Nrf2 and Nqo-1 mRNA expression and an increase on HO-1 mRNA expression in the skin at 4 h. RvD1 treatment inhibited the reduction of Nrf2 mRNA expression, not only reversed the down-modulation of Nqo-1, but increased its mRNA expression, and also further enhanced HO-1 mRNA expression in the skin (Figures 6D,E,F).
The effects of RvD1 were determined by reduced glutathione (GSH) quantitation and catalase activity. The dose of UVB irradiation used in the experiment was able to significantly reduce the endogenous antioxidant GSH and catalase in the irradiated control group compared to the non-irradiated control group. The treatment with 30 ng/mice of RvD1 inhibited the reduction of GSH levels following UVB irradiation (Figure 6G). In the GSH assays, a bell-shaped curve was observed, corroborating the adequality of the chosen dose. Catalase activity was reduced by the UVB irradiation and RvD1 inhibited this reduction (Figure 6H).
Discussion
UV irradiation causes a number of adverse biological effects in the skin, such as premature aging and skin cancer (Afaq et al., 2005; Fonseca et al., 2011; Tewari et al., 2013b; Martinez et al., 2015a). The deleterious effect of UV irradiation in the skin, in particular UVB (280–315 nm), occurs due to excessive free radical production and direct DNA damage (Fernández-García, 2014). ROS are directly involved in the induction of the inflammatory process and oxidative stress, because they stimulate the secretion of cytokines (Ivan et al., 2014) and the depletion of endogenous antioxidants (Zaid et al., 2007; Halliwell, 2009). Recently, we demonstrated for the first time that lipoxin A4, a pro-resolution lipid mediator derived from the arachidonic acid, reduces UVB irradiation-induced skin inflammation and oxidative stress (Martinez et al., 2018). However, there was no evidence on the therapeutic effect of a pro-resolving lipid mediators derived from DHA metabolism on UVB skin inflammation and oxidative stress. In the present study, we demonstrated that RvD1 reduced the skin inflammation and oxidative stress caused by UVB irradiation by reducing the production of cytokines and ROS resulting in diminished skin cellular infiltrate, keratinocyte apoptosis, and collagen degradation. Thus, RvD1 presents a pronounced inhibition of UVB irradiation detrimental effects suggesting its therapeutic potential.
RvD1 inhibited UV-induced skin edema and thickening of the epidermis. These data involve a complex range of cellular events. The acute inflammatory process triggered by exposure to UV irradiation causes specific cellular events, including increased permeability of the vascular endothelium, impairment of lymphatics function, infiltration of polymorphonuclear leukocytes, activation of inflammatory macrophages, lymphocytes, and mast cells that release pro-inflammatory molecules at the site of the lesion, which consequently lead to skin edema, and proliferation of keratinocytes and epidermal cells (Cotran and Collins, 2009). In line with the mechanisms of UVB skin edema and RvD1 effects, RvD1 attenuates pulmonary edema in a model of lipopolysaccharide (LPS)-induced acute lung injury by reducing occludin and zona occludin-1 tight junction proteins deterioration (Xie et al., 2013) and vascular injury-induced neointimal hyperplasia (Miyahara et al., 2013). Therefore, RvD1 may reduce edema by targeting vascular permeability, but also cellular proliferation and recruitment.
UV irradiation induces the increase of mast cells in the dermis (Grimbaldeston et al., 2003) and keratinocyte damages causing modifications toward sunburn cell phenotype (Bayerl et al., 1995). The presence of mast cells in the dermis correlates directly with the degree of susceptibility to systemic immunosuppression induced by long term UVB, and suppression of the immune system allows UV-induced tumors not to be destroyed (Hart et al., 1998). At early time points, mast cells contribute to UV-induced skin inflammation. (Harvima and Nilsson, 2011). Mast cells are known to release a great variety of inflammatory mediators upon degranulation including leukotrienes (LT), histamine and prostaglandin E2 (PGE2) (Krystel-Whittemore et al., 2016). RvD1 treatment decreased the UVB-induced increase of mast cell counts in the present study. However, it is possible that RvD1 also inhibits the function of the mediators released by mast cells. For instance, RvD1 can inhibit LTD4- and histamine-induced conjunctival goblet cell secretion (Dartt et al., 2011)and intracellular calcium increase(Li et al., 2013), respectively. Further, RvD1 inhibits 48/80-induced mast cell degranulation and PGE2 production (Grabauskas et al., 2018), which could possibly involve the inhibition of the release of other mast cell mediators. Evidence using the mast cell deficient W/Wv mouse shows that mast cells and their derived prostaglandins contribute to UVB-induced skin edema, however, mast cells and their prostaglandins do not contribute to sunburn cell formation (Ikai et al., 1985). Sunburn cells are also used as markers of skin damage caused by UVB. When UV irradiation exceeds the protective response threshold of keratinocytes, these cells undergo apoptosis and die. The reduction in the number of sunburn cells indicates an increase in the photoprotection of keratinocytes. Histopathological analysis, showed that RvD1 treatment decreased the number of sunburn cells compared to the irradiated control group.
Neutrophils have the potential to increase the damage caused by UV irradiation, as they are able to release a variety of substances that are harmful to cells and tissue such as ROS (Garcia et al., 1999; Robinson et al., 2004) and serine proteases that contribute to the alternative processing of TNF-α and IL-1β (Meyer-Hoffert and Wiedow, 2011). It has been reported that RvD1 reduced MPO activity in liver injury model induced by carbon tetrachloride (CCl4) (Chen et al., 2016). In addition, in the acute cigarette smoke-induced lung inflammation model, RvD1 inhibits neutrophilic inflammation and increase neutrophil efferocytosis (Hsiao et al., 2013). We found that treatment with RvD1 decreased the recruitment of neutrophils induced by UVB irradiation, determined by MPO activity. Neutrophils also secrete MMP-9 (Meyer-Hoffert and Wiedow, 2011), a proteolytic enzyme that degrades extracellular matrix collagen during pathological processes such as photoaging (Kossodo et al., 2004). RvD1 treatment inhibited the increase of MMP-9 activity upon exposure to UVB irradiation, which lined up well with the reduction of collagen fibers degradation and production of TNF-α and IL-1β.
Treatment with RvD1 inhibited the increase of pro-inflammatory cytokines IL-1β, IL-6, IL-33, and TNF-α, and anti-inflammatory TGF-β and IL-10 induced by UVB exposure. UVB induces the activation of inflammasomes NLRC4, NLRP3, and AIM2 to activate caspase-1 to cleave pro-IL-1β to its active and secreted form, IL-1β (Feldmeyer et al., 2007; Sollberger et al., 2015; Hung et al., 2017). However, despite the contribution of caspase-1 to keratinocyte apoptosis, this event is independent on inflammasomes (Hasegawa et al., 2016). In addition to NLRP3 activation, UVB-induced damaged DNA also triggers the production of IL-6 (Feldmeyer et al., 2007; Sollberger et al., 2015). Mast cells degranulate upon UVB stimulus releasing TNF-α (Walsh, 1995) and targeting the TNFR1 receptor (Zhuang et al., 1999) and TNF-α mRNA half-life with pentoxifylline (Schwarz et al., 1997) showed that TNF-α plays a role in the development of sunburn cells (Schwarz et al., 1995). This evidence suggests that the RvD1 inhibition of TNF-α production may contribute to the reduced sunburn cell counts we observed. IL-1β, IL-6, TNF-α, and IL-33 have a role in recruiting leukocytes (Verri et al., 2010). For instance, UV induces the IL-33 expression by keratinocytes and dermal fibroblasts, which recruit dermal mast cells and skin-infiltrating neutrophils (Byrne et al., 2011). IL-33, IL-1β, and TNF-α also contribute to the development of inflammatory edema (Joosten et al., 2006; Zarpelon et al., 2013; Staurengo-Ferrari et al., 2018). Therefore, these data on inhibitory effect of RvD1 over UVB irradiation-induced pro-inflammatory cytokine production contribute to explain, at least in part, the reduction of skin inflammation (edema, mast cell counts, neutrophil recruitment, MMP-9 activity and collagen degradation).
RvD1 also reduced the production of IL-10 and TGF-β induced by UVB irradiation. IL-10 is an anti-inflammatory cytokine co-released with pro-inflammatory cytokines to achieve a fine tuning of inflammation (Verri et al., 2006). TGF-β as well, has an anti-inflammatory contribution and a tissue repair role since stimulates fibroblasts to produce collagen (Penn et al., 2012). These results also mean that UVB per se triggered the release of IL-10 and TGF-β as endogenous mechanisms to limit skin inflammation and orchestrate tissue repair. Thus, inhibiting IL-10 and TGF-β production may be a drawback effect of RvD1 and also an explanation for the partial effect of RvD1 on reestablishing collagen fiber density. However, there is also evidence that RvD1 delivery using neutrophil-derived nanoparticles accelerated keratinocyte healing and reduced inflammation, suggesting that selecting the adequate delivery system and even a mimetic delivery system may improve RvD1 therapeutic effects (Norling et al., 2011).
Cytokines such as TNF-α and IL-1β activate the phagocyte NADPH oxidase inducing the production of superoxide anion. This mechanism contributes to amplifying UVB tissue damage. However, it is also likely that the initial superoxide anion leaks from the mitochondria electron-transport chain Complex I and Complex III (Berneburg et al., 1999; Heck et al., 2003). Then, superoxide anion induces the production of cytokines including TNF-α and IL-1β triggering inflammation and amplifying tissue oxidative stress (Yamacita-Borin et al., 2015; Fattori et al., 2016). Cytokines chemoattract and activate phagocytes that will further produce superoxide anion (Garcia et al., 1999; Witko-Sarsat et al., 2000; Robinson et al., 2004) and RvD1 reduced gp91phox mRNA expression and superoxide anion production, indicating a reduction of phagocyte NADPH oxidase (NOX2) activity. Accumulated ROS act on the cell biological membrane and the availability of polyunsaturated fatty acids regulates the lipid peroxidation process. High peroxidation levels are associated with harmful effects on biological systems, such as loss of fluidity, inactivation of membrane enzymes and receptors, and ion permeability increase, leading to cell membrane rupture. In addition, peroxidation products can damage DNA. LOOH, which are the primary products of lipid oxidation (Linggnert et al., 1979), were used as markers of oxidative stress in the present study, and it was found that RvD1 treatment reduced production of LOOH induced by UVB irradiation exposure.
RvD1 not only reduced oxidative stress, but also improved the antioxidant capacity of the skin after exposure to UVB irradiation by maintaining GSH and catalase activities at basal levels. GSH plays an important role in protecting skin cells against oxidative damage through the direct elimination of ROS or acting as a coenzyme of glutathione peroxidase (Zaid et al., 2007; Halliwell, 2009). Catalase is an antioxidant enzyme that converts hydrogen peroxide in water and molecular oxygen (Schallreuter et al., 1999; D'Orazio et al., 2013). The loss of UVB-mediated cell viability is associated with a notable decrease in endogenous antioxidant defenses, therefore weakening the cellular antioxidant defense system (Zaid et al., 2007; Halliwell, 2009). GSH expression is controlled Nrf2 (Harvey et al., 2009). The data from several studies also showed that RvD1 reduced oxidative stress. RvD1 increased the levels of GSH and HO-1 mRNA expression in carbon tetrachloride (CCl4)-induced acute liver injury model (Chen et al., 2016). Also, RvD1 induced GSH release in human chondrocytes obtained from osteoarthritis patients. Our finding showed that RvD1 increases redox status as indicated by enhanced GSH levels. These data are consistent with those of the literature indicating that RvD1 inhibits GSH depletion (Cox et al., 2015). Lipid peroxidation is an endpoint oxidative stress reaction that generates aldehydes, such as 4-hydroxy-trans-2-nonenal (HNE). In turn, HNE reacts with glutathione generating GS-HNE conjugates that induce leukocyte recruitment, superoxide anion formation and production pro-inflammatory lipid mediators. RvD1 was shown to reduce GS-HNE-induced inflammation indicating RvD1 also targets the inflammatory response triggered by newly generated inducers of inflammation (Spite et al., 2009b).
Nrf2 modulates the expression of antioxidant and detoxifying enzymes, known as phase II enzymes. In this group of enzymes, HO-1, Nqo-1, and catalase (Kobayashi and Yamamoto, 2005; Choi et al., 2013) are also included. Exposure to UVB irradiation increased HO-1 mRNA expression and decrease Nrf2 and Nqo-1 mRNA expression. HO-1 is an antioxidant and anti-inflammatory enzyme that is responsive to different stress conditions, including the inflammation process; in this sense HO-1 is essential to main cellular resistance during oxidative stress conditions. Enhanced HO-1 mRNA expression was associated with the resolution of inflammation and natural killer (NK) cell-mediated cytotoxicity, which may explain why HO-1 production increases while other antioxidant enzymes are inhibited (Listopad et al., 2007; Martinez et al., 2016a,b). Corroborating with the current understanding of Nrf2, HO-1, and NQO-1 activities, treatment with RvD1 increased Nrf2 mRNA expression, which resulted in an enhancement of Ho-1 and Nqo-1mRNA expression, and maintenance the GSH levels and catalase activity. In other models, RvD1 contributed to the protection from the deterioration of tight junction proteins in a model of acute lung injury induced by lipopolysaccharide in mice by inducing HO-1 expression (Xie et al., 2013); increased GSH levels and HO-1 mRNA expression in carbon tetrachloride (CCl4)-induced acute liver injury model (Chen et al., 2016); and inhalation of CO accelerates inflammation resolution by inducing RvD1-dependent activation of HO-1 (Chiang et al., 2013). Therefore, RvD1 presents an active role in inducing Nrf2 that will orchestrate antioxidant responses.
In conclusion, the present study demonstrated for the first time, to our knowledge, that treatment with RvD1 inhibits oxidative and inflammatory damage induced by exposure to UVB irradiation in hairless mice. RvD1 inhibited the inflammatory cell counts in the skin, and since the migration and activation of inflammatory cells were damped by reducing cytokine production and inducing antioxidant and anti-inflammatory genes, as a result, RvD1 protected the skin from UVB irradiation-induced tissue alterations such as collagen degradation. Therefore, the present results suggest RvD1 as a potential therapy to control UVB-induced skin inflammation- and oxidative stress-related alterations.
Author contributions
PS, CM, RM, VF, TC, IP, and AB performed experiments. PS, RM, WV, and RC analyzed and interpreted data. WV and RC were responsible for conception and design of the study. PS organized the database. JV, SG, MB, WV, and RC provided research mentorship, supervision, received grants and provided essential reagentes. PS, RM, WV, and RC wrote the manuscript. All authors contributed to manuscript revision, read and approved the final version.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Universidade Estadual de Londrina, Pró-Reitoria de Pesquisa e Pós-Graduação (PROPPG), Escritório de Apoio ao Pesquisador; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil); Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil, agreement number 001); Financiadora de Estudos e Projetos and Secretaria de Estado da Ciência, Tecnologia e Ensino Superior do Paraná (FINEP/SETI-PR, Brazil); Programa de Apoio a Grupos de Excelência (PRONEX) grant supported by SETI/Fundação Araucária and MCTI/CNPq, and Governo do Estado do Paraná (agreement 014/2017, protocol 46.843). The authors also thank the support of CMLP-UEL and CEPPOS-UEL.
References
- Aebi H. (1984). Catalase in vitro. Methods Enzymol. 105, 121–126. 10.1016/S0076-6879(84)05016-3 [DOI] [PubMed] [Google Scholar]
- Afaq F., Adhami V. M., Mukhtar H. (2005). Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat. Res. 571, 153–173. 10.1016/j.mrfmmm.2004.07.019 [DOI] [PubMed] [Google Scholar]
- Anrather J., Racchumi G., Iadecola C. (2006). NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 281, 5657–5667. 10.1074/jbc.M506172200 [DOI] [PubMed] [Google Scholar]
- Balogh T. S., Pedriali C. A., Kaneko T. M. (2011). Proteção à radiação ultravioleta : recursos disponíveis na. Ann. Bras. Dermatol. 1, 732–742. 10.1590/S0365-05962011000400016 [DOI] [Google Scholar]
- Bayerl C., Taake S., Moll I., Jung E. G. (1995). Characterization of sunburn cells after exposure to ultraviolet light. Photodermatol. Photoimmunol. Photomed. 11, 149–154. 10.1111/j.1600-0781.1995.tb00157.x [DOI] [PubMed] [Google Scholar]
- Berneburg M., Grether-Beck S., Kürten V., Ruzicka T., Briviba K., Sies H., et al. (1999). Singlet oxygen mediates the UVA-induced generation of the photoaging- associated mitochondrial common deletion. J. Biol. Chem. 274, 15345–15349. 10.1074/jbc.274.22.15345 [DOI] [PubMed] [Google Scholar]
- Bowden G. T. (2004). Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat. Rev. Cancer 4, 23–35. 10.1038/nrc1253 [DOI] [PubMed] [Google Scholar]
- Brennan M., Bhatti H., Nerusu K. C., Bhagavathula N., Kang S., Fisher G. J., et al. (2003). Matrix metalloproteinase-1 is the major collagenolytic enzyme responsible for collagen damage in UV-irradiated human skin. Photochem. Photobiol. 78, 43–48. 10.1562/0031-8655 [DOI] [PubMed] [Google Scholar]
- Byrne S. N., Beaugie C., O'Sullivan C., Leighton S., Halliday G. M. (2011). The immune-modulating cytokine and endogenous alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am. J. Pathol. 179, 211–222. 10.1016/j.ajpath.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanini M. Z., Pinho-Ribeiro F. A., Ivan A. L. M., Ferreira V. S., Vilela F. M. P., Vicentini F. T. M. C., et al. (2013). Efficacy of topical formulations containing Pimenta pseudocaryophyllus extract against UVB-induced oxidative stress and inflammation in hairless mice. J. Photochem. Photobiol. B Biol. 127, 153–160. 10.1016/j.jphotobiol.2013.08.007 [DOI] [PubMed] [Google Scholar]
- Casagrande R., Georgetti S. R., Verri W. A., Dorta D. J., dos Santos A. C., Fonseca M. J. V. (2006). Protective effect of topical formulations containing quercetin against UVB-induced oxidative stress in hairless mice. J. Photochem. Photobiol. B Biol. 84, 21–27. 10.1016/j.jphotobiol.2006.01.006 [DOI] [PubMed] [Google Scholar]
- Chan M. M. Y., Moore A. R. (2010). Resolution of Inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 184, 6418–6426. 10.4049/jimmunol.0903816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Gong X., Jiang R., Wang B., Kuang G., Li K., et al. (2016). Resolvin D1 attenuates CCl4-induced acute liver injury involving up-regulation of HO-1 in mice. Immunopharmacol. Immunotoxicol. 38, 61–67. 10.3109/08923973.2015.1115517 [DOI] [PubMed] [Google Scholar]
- Chiang N., Arita M., Serhan C. N. (2005). Anti-inflammatory circuitry: Lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot. Essent. Fatty Acids 73, 163–77. 10.1016/j.plefa.2005.05.003 [DOI] [PubMed] [Google Scholar]
- Chiang N., Shinohara M., Dalli J., Mirakaj V., Kibi M., Choi A. M. K., et al. (2013). Inhaled carbon monoxide accelerates resolution of inflammation via unique pro-resolving mediator–heme oxygenase-1 circuits1. J. Immunol. 190, 6378–6388. 10.4049/jimmunol.1202969.Inhaled [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y., Choi D. I., Lee J. B., Yun S. J., Lee D. H., Eun J. B., et al. (2013). Ethanol extract of peanut sprout induces Nrf2 activation and expression of antioxidant and detoxifying enzymes in human dermal fibroblasts: implication for its protection against UVB-irradiated oxidative stress. Photochem. Photobiol. 89, 453–460. 10.1111/j.1751-1097.2012.01244.x [DOI] [PubMed] [Google Scholar]
- Cotran R., Collins T. (2009). Robbins Pathologic Basis of Disease. Philadelphia, PA: Saunders. [Google Scholar]
- Cox R., Phillips O., Fukumoto J., Fukumoto I., Parthasarathy P. T., Arias S., et al. (2015). Enhanced resolution of hyperoxic acute lung injury as a result of aspirin triggered resolvin D1 treatment. Am. J. Respir. Cell Mol. Biol. 53, 422–435. 10.1165/rcmb.2014-0339OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartt D. A., Hodges R. R., Li D., Shatos M. A., Lashkari K., Serhan C. N. (2011). Conjunctival goblet cell secretion stimulated by leukotrienes is reduced by resolvins D1 and E1 to promote resolution of inflammation. J. Immunol. 186, 4455–66. 10.4049/jimmunol.1000833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., Ediriwickrema A., Yang F., Lewis J., Girardi M., Saltzman W. M. (2015). A sunblock based on bioadhesive nanoparticles. Nat. Mater. 14, 1278–1285. 10.1038/nmat4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orazio J., Jarrett S., Amaro-Ortiz A., Scott T. (2013). UV radiation and the skin. Int. J. Mol. Sci. 14, 12222–12248. 10.3390/ijms140612222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusting G. J., Macdonald P. S. (1995). Endogenous nitric oxide in cardiovascular disease and transplantation. Ann. Med. 395–406. [DOI] [PubMed] [Google Scholar]
- Fattori V., Serafim K. G., Zarpelon A. C., Borghi S. M., Pinho-Ribeiro F. A., Alves-Filho J. C., et al. (2016). Differential regulation of oxidative stress and cytokine production by endothelin ETA and ETB receptors in superoxide anion-induced inflammation and pain in mice. J. Drug Target 25, 264–274. 10.1080/1061186X.2016.1245308 [DOI] [PubMed] [Google Scholar]
- Feldmeyer L., Keller M., Niklaus G., Hohl D., Werner S., Beer H. D. (2007). The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr. Biol. 17, 1140–1145. 10.1016/j.cub.2007.05.074 [DOI] [PubMed] [Google Scholar]
- Fernández-García E. (2014). Skin protection against UV light by dietary antioxidants. Food Funct. 5, 1994–2003. 10.1039/C4FO00280F [DOI] [PubMed] [Google Scholar]
- Figueiredo S. A., Vilela F. M. P., Da Silva C. A., Cunha T. M., Dos Santos M. H., Fonseca M. J. V. (2014). In vitro and in vivo photoprotective/photochemopreventive potential of Garcinia brasiliensis epicarp extract. J. Photochem. Photobiol. B Biol. 131, 65–73. 10.1016/j.jphotobiol.2014.01.004 [DOI] [PubMed] [Google Scholar]
- Fonseca M. J. V., Fonseca Y. M., Marquele-Oliveira F., Vicentini F. T. M. C., Furtado N. A. J. C., Sousa J. P. B., et al. (2011). Evaluation of the potential of Brazilian propolis against UV-induced oxidative stress. Evidence-based complement. Altern. Med. 2011:863917 10.1155/2011/863917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca Y. M., Catini C. D., Vicentini F. T. M. C., Nomizo A., Gerlach R. F., Fonseca M. J. V. (2010). Protective effect of Calendula officinalis extract against UVB-induced oxidative stress in skin: evaluation of reduced glutathione levels and matrix metalloproteinase secretion. J. Ethnopharmacol. 127, 596–601. 10.1016/j.jep.2009.12.019 [DOI] [PubMed] [Google Scholar]
- Garcia C., Pithon-Curi T. C., de Lourdes Firmano M., Pires de Melo M., Newsholme P., Curi R. (1999). Effects of adrenaline on glucose and glutamine metabolism and superoxide production by rat neutrophils. Clin. Sci. 96, 549–555. 10.1042/CS19980340 [DOI] [PubMed] [Google Scholar]
- Gonzalez Flecha B., Llesuy S., Boveris A. (1991). Hydroperoxide-initiated chemiluminescence: an assay for oxidative stress in biopsies of heart, liver, and muscle. Free Radic. Biol. Med. 10, 93–100. 10.1016/0891-5849(91)90002-K [DOI] [PubMed] [Google Scholar]
- Grabauskas G., Wu X., Owyang C. (2018). Resolvin D1 acting on G1 coupled Fpr2 receptor inhibits mast cell degranulation and prevents development of visceral hypersensitivity in Ibs. Gastroenterology 154, S−214. 10.1016/S0016-5085(18)31106-5 [DOI] [Google Scholar]
- Grady A. O., Dunne C., Kelly P. O., Murphy G. M., Leader M., Kay E. (2007). Differential expression of matrix metalloproteinase (MMP)−2, MMP-9 and tissue inhibitor of metalloproteinase (TIMP)−1 and TIMP-2 in non-melanoma skin cancer : implications for tumour progression. Histopathology 51, 793–804. 10.1111/j.1365-2559.2007.02885.x [DOI] [PubMed] [Google Scholar]
- Grimbaldeston M.a, Simpson A., Finlay-Jones J. J., Hart P. H. (2003). The effect of ultraviolet radiation exposure on the prevalence of mast cells in human skin. Br. J. Dermatol. 148, 300–306. 10.1046/j.1365-2133.2003.05113.x [DOI] [PubMed] [Google Scholar]
- Halliwell B. (2009). The wanderings of a free radical. Free Radic. Biol. Med. 46, 531–542. 10.1016/j.freeradbiomed.2008.11.008 [DOI] [PubMed] [Google Scholar]
- Hart P. H., Grimbaldeston M. A., Finlay-jones J. J. (2001). Brief review sunlight, immunosuppression and skin cancer: role of histamine and mast cells. Clin. Exp. Pharmacol. Physiol. 28, 1–8. 10.1046/j.1440-1681.2001.03392.x [DOI] [PubMed] [Google Scholar]
- Hart P. H., Grimbaldeston M. A., Swift G. J., Jaksic A., Noonan F. P., Finlay-Jones J. J. (1998). Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J. Exp. Med. 187, 2045–2053. 10.1084/jem.187.12.2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey C. J., Thimmulappa R. K., Singh A., Blake D. J., Ling G., Wakabayashi N., et al. (2009). Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 46, 443–453. 10.1016/j.freeradbiomed.2008.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvima I., Nilsson G. (2011). Mast cells as regulators of skin inflammation and immunity. Acta Derm. Venereol. 91, 644–650. 10.2340/00015555-1197 [DOI] [PubMed] [Google Scholar]
- Hasegawa T., Nakashima M., Suzuki Y. (2016). Nuclear DNA damage-triggered NLRP3 inflammasome activation promotes UVB-induced inflammatory responses in human keratinocytes. Biochem. Biophys. Res. Commun. 477, 329–335. 10.1016/j.bbrc.2016.06.106 [DOI] [PubMed] [Google Scholar]
- Haworth O., Levy B. D. (2007). Endogenous lipid mediators in the resolution of airway inflammation. Eur. Respir. J. 30, 980–992. 10.1183/09031936.00005807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck D. E., Vetrano A. M., Mariano T. M., Laskin J. D. (2003). UVB light stimulates production of reactive oxygen species: unexpected role for catalase. J. Biol. Chem. 278, 22432–22436. 10.1074/jbc.C300048200 [DOI] [PubMed] [Google Scholar]
- Hildesheim J., Awwad R. T., Fornace A. J. (2004). p38 mitogen-activated protein kinase inhibitor protects the epidermis against the acute damaging effects of ultraviolet irradiation by blocking apoptosis and inflammatory responses. J. Invest. Dermatol. 122, 497–502. 10.1111/j.1523-1747.2004.22229.x [DOI] [PubMed] [Google Scholar]
- Hsiao H. M., Sapinoro R. E., Thatcher T. H., Croasdell A., Levy E. P., Fulton R. A., et al. (2013). A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS ONE 8:e58258. 10.1371/journal.pone.0058258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S.-J., Tang S.-C., Liao P.-Y., Ge J.-S., Hsiao Y.-P., Yang J.-H. (2017). Photoprotective potential of glycolic acid by reducing NLRC4 and AIM2 inflammasome complex proteins in UVB radiation-induced normal human epidermal keratinocytes and mice. DNA Cell Biol. 36, 177–187. 10.1089/dna.2016.3471 [DOI] [PubMed] [Google Scholar]
- Hupel M., Poupart N., Ar Gall E. (2011). Development of a new in vitro method to evaluate the photoprotective sunscreen activity of plant extracts against high UV-B radiation. Talanta 86, 362–371. 10.1016/j.talanta.2011.09.029 [DOI] [PubMed] [Google Scholar]
- Ikai K., Danno K., Horio T., Narumiya S. (1985). Effect of ultraviolet irradiation on mast cell-deficient W/Wv mice. J. Invest. Dermatol. 85, 82–84. 10.1111/1523-1747.ep12275365 [DOI] [PubMed] [Google Scholar]
- Ivan A. L. M., Campanini M. Z., Martinez R. M., Ferreira V. S., Steffen V. S., Vicentini F. T. M. C., et al. (2014). Pyrrolidine dithiocarbamate inhibits UVB-induced skin inflammation and oxidative stress in hairless mice and exhibits antioxidant activity in vitro. J. Photochem. Photobiol. B Biol. 138, 124–133. 10.1016/j.jphotobiol.2014.05.010 [DOI] [PubMed] [Google Scholar]
- Jenkins G. (2002). Molecular mechanisms of skin ageing. Mech. Ageing Dev. 123, 801–810. 10.1016/S0047-6374(01)00425-0 [DOI] [PubMed] [Google Scholar]
- Joosten L. A. B., Netea M. G., Kim S.-H., Yoon D.-Y., Oppers-Walgreen B., Radstake T. R. D., et al. (2006). IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 103, 3298–3303. 10.1073/pnas.0511233103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Júnior L. R., Höehr N. F., Vellasco A. P., Kubota L. T. (2001). Sistema antioxidante envolvendo o ciclo metabólico da glutationa associado a métodos eletroanalíticos na avaliação do estresse oxidativo. Quim. Nova 24, 112–119. 10.1590/S0100-40422001000100019 [DOI] [Google Scholar]
- Katiyar S. K., Meeran S. M. (2007). Obesity increases the risk of UV radiation-induced oxidative stress and activation of MAPK and NF-κB signaling. Free Radic. Biol. Med. 42, 299–310. 10.1016/j.freeradbiomed.2006.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavkin J., Ellis D. A. F. (2011). Aging Skin: histology, physiology, and pathology. Facial Plast. Surg. Clin. North Am. 19, 229–234. 10.1016/j.fsc.2011.04.003 [DOI] [PubMed] [Google Scholar]
- Kim C. M., Kang S. M., Jeon H. J., Shin S. H. (2007). Production of Vibrio vulnificus metalloprotease VvpE begins during the early growth phase: usefulness of gelatin-zymography. J. Microbiol. Methods 70, 96–102. 10.1016/j.mimet.2007.03.016 [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Yamamoto M. (2005). Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 7, 385–394. 10.1089/ars.2005.7.385 [DOI] [PubMed] [Google Scholar]
- Kossodo S., Wong W.-R., Simon G., Kochevar I. E. (2004). Effects of UVR and UVR-induced cytokines on production of extracellular matrix proteins and proteases by dermal fibroblasts cultured in collagen gels%. Photochem. Photobiol. 79, 86–93. [DOI] [PubMed] [Google Scholar]
- Krystel-Whittemore M., Dileepan K. N., Wood J. G. (2016). Mast cell: a multi-functional master cell. Front. Immunol. 6:620. 10.3389/fimmu.2015.00620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Hodges R. R., Jiao J., Carozza R. B., Shatos M. A., Chiang N., et al. (2013). Resolvin D1 and aspirin-triggered resolvin D1 regulate histamine-stimulated conjunctival goblet cell secretion. Mucosal. Immunol. 6, 1119–1130. 10.1038/mi.2013.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linggnert H., Vallentin K., Eriksson C. E. (1979). Measurement of antioxidative effect in model system. J. Food Process. Preserv. 3, 87–103. 10.1111/j.1745-4549.1979.tb00574.x [DOI] [Google Scholar]
- Listopad J., Asadullah K., Sievers C., Ritter T., Meisel C., Sabat R., et al. (2007). Heme oxygenase-1 inhibits T cell-dependent skin inflammation and differentiation and function of antigen-presenting cells. Exp. Dermatol. 16, 661–670. 10.1111/j.1600-0625.2007.00581.x [DOI] [PubMed] [Google Scholar]
- Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J. (2016). Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 73, 3221–3247. 10.1007/s00018-016-2223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R. M., Fattori V., Saito P., Melo C. B. P., Borghi S. M., Pinto I. C., et al. (2018). Lipoxin A4 inhibits UV radiation-induced skin in fl ammation and oxidative stress in mice. J. Dermatol. Sci. 91, 164–174. 10.1016/j.jdermsci.2018.04.014 [DOI] [PubMed] [Google Scholar]
- Martinez R. M., Pinho-Ribeiro F. A., Steffen V. S., Caviglione C. V., Pala D., Baracat M. M., et al. (2016a). Topical formulation containing hesperidin methyl chalcone inhibits skin oxidative stress and inflammation induced by ultraviolet B irradiation. Photochem. Photobiol. Sci. 15, 554–563. 10.1039/C5PP00467E [DOI] [PubMed] [Google Scholar]
- Martinez R. M., Pinho-Ribeiro F. A., Steffen V. S., Caviglione C. V., Vignoli J. A., Baracat M. M., et al. (2015a). Hesperidin methyl chalcone inhibits oxidative stress and inflammation in a mouse model of ultraviolet B irradiation-induced skin damage. J. Photochem. Photobiol. B Biol. 148, 145–153. 10.1016/j.jphotobiol.2015.03.030 [DOI] [PubMed] [Google Scholar]
- Martinez R. M., Pinho-Ribeiro F. A., Steffen V. S., Caviglione C. V., Vignoli J. A., Barbosa D. S., et al. (2015b). Naringenin inhibits UVB irradiation-induced inflammation and oxidative stress in the skin of hairless mice. J. Nat. Prod. 78, 1647–1655. 10.1021/acs.jnatprod.5b00198 [DOI] [PubMed] [Google Scholar]
- Martinez R. M., Pinho-Ribeiro F. A., Steffen V. S., Silva T. C. C., Caviglione C. V., Bottura C., et al. (2016b). Topical formulation containing naringenin: efficacy against ultraviolet B irradiation-induced skin inflammation and oxidative stress in mice. PLoS ONE 11:e0146296. 10.1371/journal.pone.0146296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R. M., Pinho-Ribeiro F. A., Vale D. L., Steffen V. S., Vicentini F. T. M. C., Vignoli J. A., et al. (2017). Trans-chalcone added in topical formulation inhibits skin inflammation and oxidative stress in a model of ultraviolet B radiation skin damage in hairless mice. J. Photochem. Photobiol. B Biol. 171, 139–146. 10.1016/j.jphotobiol.2017.05.002 [DOI] [PubMed] [Google Scholar]
- Maverakis E., Miyamura Y., Bowen M. P., Correa G., Ono Y., Goodarzi H. (2010). Light, including ultraviolet. J. Autoimmun. 34, J247–J257. 10.1016/j.jaut.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Hoffert U., Wiedow O. (2011). Neutrophil serine proteases: Mediators of innate immune responses. Curr. Opin. Hematol. 18, 19–24. 10.1097/MOH.0b013e32834115d1 [DOI] [PubMed] [Google Scholar]
- Miyahara T., Runge S., Chatterjee A., Chen M., Mottola G., Fitzgerald J. M., et al. (2013). D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 27, 2220–2232. 10.1096/fj.12-225615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro K., Nagahashi M., Ramanathan R., Takabe K., Wakai T. (2016). Resolvins and omega three polyunsaturated fatty acids: clinical implications in inflammatory diseases and cancer. World J. Clin. Cases 4, 155–164. 10.12998/wjcc.v4.i7.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norling L. V., Spite M., Yang R., Roderick J., Perretti M., Serhan C. N. (2011). Cutting edge: humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J. Immunol. 186, 543–5547. 10.4049/jimmunol.1003865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros E., Maurette M., Orellana G., Cadet J., Douki T., Ravanat J., et al. (2009). Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation †. Photochem. Photobiol. Sci. 8, 903–911. 10.1039/b905343n [DOI] [PubMed] [Google Scholar]
- Onoue S., Kobayashi T., Takemoto Y., Sasaki I., Shinkai H. (2003). Induction of matrix metalloproteinase-9 secretion from human keratinocytes in culture by ultraviolet B irradiation. J. Dermatol. Sci. 33, 105–111. 10.1016/j.jdermsci.2003.08.002 [DOI] [PubMed] [Google Scholar]
- Penn J. W., Grobbelaar A. O., Rolfe K. J. (2012). The role of the TGF-β family in wound healing, burns and scarring: a review. Int. J. Burns Trauma 2, 18–28. [PMC free article] [PubMed] [Google Scholar]
- Recchiuti A., Serhan C. N. (2012). Pro-resolving lipid mediators (SPMs) and their actions in regulating miRNA in novel resolution circuits in inflammation. Front. Immunol. 3:298. 10.3389/fimmu.2012.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J. M., Ohira T., Badwey J. A. (2004). Regulation of the NADPH-oxidase complex of phagocytic leukocytes. Recent insights from structural biology, molecular genetics, and microscopy. Histochem. Cell Biol. 122, 293–304. 10.1007/s00418-004-0672-2 [DOI] [PubMed] [Google Scholar]
- Rogerio A. P., Haworth O., Croze R., Oh S. F., Uddin M., Carlo T., et al. (2012). Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J. Immunol. 189, 1983–1991. 10.4049/jimmunol.1101665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallreuter K. U., Moore J., Wood J. M., Beazley W. D., Gaze D. C., Tobin D. J., et al. (1999). In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a uvb-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 4, 91–96. 10.1038/sj.jidsp.5640189 [DOI] [PubMed] [Google Scholar]
- Schwarz A., Bhardwaj R., Aragane Y., Mahnke K., Riemann H., Metze D., et al. (1995). Ultraviolet-B-induced apoptosis of keratinocytes: evidence for partial involvement of tumor necrosis factor-α in the formation of sunburn cells. J. Invest. Dermatol. 104, 922–927. 10.1111/1523-1747.ep12606202 [DOI] [PubMed] [Google Scholar]
- Schwarz A., Mahnke K., Luger T. A., Schwarz T. (1997). Pentoxifylline reduces the formation of sunburn cells. Exp. Dermatol. 6, 1–5. 10.1111/j.1600-0625.1997.tb00138.x [DOI] [PubMed] [Google Scholar]
- Seki H., Sasaki T., Ueda T., Arita M. (2010). Resolvins as regulators of the immune system. Sci. World J. 10, 818–831. 10.1100/tsw.2010.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N., Chiang N., Van Dyke T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361. 10.1038/nri2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G., Strittmatter G. E., Grossi S., Garstkiewicz M., Auf Dem Keller U., French L. E., et al. (2015). Caspase-1 activity is required for UVB-induced apoptosis of human keratinocytes. J. Invest. Dermatol. 135, 1395–1404. 10.1038/jid.2014.551 [DOI] [PubMed] [Google Scholar]
- Song J. H., Piao M. J., Han X., Ah Kang K., Kang H. K., Yoon W. J., et al. (2016). t BA: a review of skin ageing and its medical therapy. Mol. Med. Rep. 14, 2937–2944. 10.3892/mmr.2016.5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M., Norling L. V., Summers L., Yang R., Cooper D., Petasis N. A., et al. (2009a). Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291. 10.1038/nature08541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M., Summers L., Porter T. F., Srivastava S., Bhatnagar A., Serhan C. N. (2009b). Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br. J. Pharmacol. 158, 1062–1073. 10.1111/j.1476-5381.2009.00234.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan P., Sabitha K. E., Shyamaladevi C. S. (2007). Attenuation of 4-Nitroquinoline 1-oxide induced in vitro lipid peroxidation by green tea polyphenols. Life Sci. 80, 1080–1086. 10.1016/j.lfs.2006.11.051 [DOI] [PubMed] [Google Scholar]
- Staurengo-Ferrari L., Trevelin S. C., Fattori V., Nascimento D. C., de Lima K. A., Pelayo J. S., et al. (2018). Interleukin-33 receptor (ST2) deficiency improves the outcome of staphylococcus aureus-induced septic arthritis. Front. Immunol. 9:962. 10.3389/fimmu.2018.00962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari A., Grage M. M. L., Harrison G. I., Sarkany R., Young A. R. (2013a). Human erythema and matrix metalloproteinase-1 mRNA induction, in vivo, share an action spectrum which suggests common chromophores. Photochem. Photobiol. Sci. 12, 95–103. 10.1002/jcp.21208 [DOI] [PubMed] [Google Scholar]
- Tewari A., Grage M. M. L., Harrison G. I., Sarkany R., Young A. R. (2013b). UVA1 is skin deep: molecular and clinical implications. Photochem. Photobiol. Sci. 12, 95–103. 10.1039/c2pp25323b [DOI] [PubMed] [Google Scholar]
- Verri W. A., Cunha T. M., Parada C. A., Poole S., Cunha F. Q., Ferreira S. H. (2006). Hypernociceptive role of cytokines and chemokines: targets for analgesic drug development? Pharmacol. Ther. 112, 116–138. 10.1016/j.pharmthera.2006.04.001 [DOI] [PubMed] [Google Scholar]
- Verri W. A., Guerrero A. T. G., Fukada S. Y., Valerio D. A., Cunha T. M., Xu D., et al. (2008). IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 2723–2728. 10.1073/pnas.0712116105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verri W. A., Souto F. O., Vieira S. M., Almeida S. C. L., Fukada S. Y., Xu D., et al. (2010). IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann. Rheum. Dis. 69, 1697–1703. 10.1136/ard.2009.122655 [DOI] [PubMed] [Google Scholar]
- Walsh L. J. (1995). Ultraviolet B irradiation of skin induces mast cell degranulation and release of tumour necrosis factor-α. Immunol. Cell Biol. 73, 226–233. 10.1111/j.1440-1711.1995.tb03862.x [DOI] [PubMed] [Google Scholar]
- Walz W., Cayabyab F. S. (2017). Neutrophil Infiltration and Matrix Metalloproteinase-9 in Lacunar Infarction. Neurochem. Res. 42, 2560–2565. 10.1007/s11064-017-2265-1 [DOI] [PubMed] [Google Scholar]
- Wang L., Yuan R., Yao C., Wu Q., Christelle M., Xie W., et al. (2014). Effects of resolvin D1 on inflammatory responses and oxidative stress of lipopolysaccharide-induced acute lung injury in mice. Chin. Med. J. 127, 803–809. 10.3760/cma.j.issn.0366-6999.20131044 [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V., Rieu P., Descamps-Latscha B., Lesavre P., Halbwachs-Mecarelli L. (2000). Neutrophils: molecules, functions and pathophysiological aspects. Lab. Investig. 80, 617–653. 10.1038/labinvest.3780067 [DOI] [PubMed] [Google Scholar]
- Xie W., Wang H., Wang L., Yao C., Yuan R., Wu Q. (2013). Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab. Investig. 93, 991–1000. 10.1038/labinvest.2013.80 [DOI] [PubMed] [Google Scholar]
- Yagi K. (1987). Lipid peroxides and human diseases. Chem. Phys. Lipids 45, 337–351. 10.1016/0009-3084(87)90071-5 [DOI] [PubMed] [Google Scholar]
- Yamacita-Borin F. Y., Zarpelon A. C., Pinho-Ribeiro F. A., Fattori V., Alves-Filho J. C., Cunha F. Q., et al. (2015). Superoxide anion-induced pain and inflammation depends on TNFα/TNFR1 signaling in mice. Neurosci. Lett. 605, 53–58. 10.1016/j.neulet.2015.08.015 [DOI] [PubMed] [Google Scholar]
- Zaid M. A., Afaq F., Syed D. N., Dreher M., Mukhtar H. (2007). Inhibition of UVB-mediated oxidative stress and markers of photoaging in immortalized HaCaT keratinocytes by pomegranate polyphenol extract POMx. Photochem. Photobiol. 83, 882–888. 10.1111/j.1751-1097.2007.00157.x [DOI] [PubMed] [Google Scholar]
- Zarpelon A. C., Cunha T. M., Alves-Filho J. C., Pinto L. G., Ferreira S. H., McInnes I. B., et al. (2013). IL-33/ST2 signalling contributes to carrageenin-induced innate inflammation and inflammatory pain: role of cytokines, endothelin-1 and prostaglandin E2. Br. J. Pharmacol. 169, 90–101. 10.1111/bph.12110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L., Wang B., Shinder G.a, Shivji G. M., Mak T. W., Sauder D. N. (1999). TNF receptor p55 plays a pivotal role in murine keratinocyte apoptosis induced by ultraviolet B irradiation. J. Immunol. 162, 1440–1447. [PubMed] [Google Scholar]