

Abstract
Breast carcinoma metastasis suppressor gene 1 (BRMS1) encodes an inhibitor of metastasis and is reported in many types of tumor metastasis. However, the mechanism of BRMS1-mediated inhibition of breast cancer metastasis at the transcriptional level remains elusive. Here, we identified using affinity purification and mass spectrometry (MS) that BRMS1 is an integral component of the LSD1/CoREST corepressor complex. Analysis of the BRMS1/LSD1 complex using high-throughput RNA deep sequencing (RNA-seq) identified a cohort of target genes such as VIM, INSIG2, KLK11, MRPL33, COL5A2, OLFML3 and SLC1A1, some of which are metastasis-related. Our results have showed that BRMS1 together with LSD1 are required for inhibition of breast cancer cell migration and invasion. Collectively, these findings demonstrate that BRMS1 executes transcriptional suppression of breast cancer metastasis by associating with the LSD1 and thus can be targeted for breast cancer therapy.
Keywords: BRMS1, LSD1, Vimentin, breast cancer, metastasis
Introduction
Breast cancer is currently the most frequently diagnosed cancer and a leading cause of death in women worldwide [1]. Metastasis is the most lethal attribute of malignant cancers, and invasion and migration are the markers of malignancy. The main causes of malignant tumors are genetic and epigenetic disorders [2]. To metastasize, a tumor cell requires the expression of particular genes and undergoes certain epigenetic changes that enable appropriate interactions with the changing microenvironment and promote continued survival and proliferation at secondary sites [3,4]. Metastasis suppressors inhibit a neoplastic cell’s metastasis while having a little effect on the primary tumor growth [5]. Metastasis suppressors affect multiple processes associated with metastasis, such as angiogenesis, extracellular matrix remodeling, and gene expression regulation [5]. Therefore, understanding the mechanism of tumor metastasis inhibition mediated by these suppressors at the molecular level is essential to develop new therapeutic avenues for preventing cancer metastasis.
BRMS1 is a potential metastasis suppressor gene that was reported to reduce the metastatic capacity of MDA-MB-435 human breast carcinoma cells by 70-90% without affecting tumorigenicity [6]. Subsequent studies indicated that BRMS1 suppresses metastasis not only in breast cancer, but also in melanoma, ovarian cancer, non-small cell lung cancer, and rectal cancer [7-10]. Aberrant BRMS1 promoter hypermethylation and gene silencing have been observed in advanced stages of cancer [11]. Further studies characterized that the BRMS1’s interaction with SIN3A/HDAC complexes and ARID4A resulting in transcriptional repression contributes to the metastasis inhibition [12-14]. A potential mechanism of transcriptional regulation by BRMS1 has been proposed through the inhibition of NFκB activity. BRMS1 interacts with RelA/p65 and recruits HDAC1 to NFκB binding regions [15]. BRMS1 suppresses tumor metastasis by regulating multiple transcriptional targets of NF-κB, such as OPN, uPA and Twist1 [15-19]. BRMS1 also alters specific cellular pathways associated with metastasis including the gap junctional intercellular communication [20,21], anoikis [15,22], and cell motility and invasion [7,8,23]. In addition, BRMS1 regulates the expression of many small non-coding RNAs that mediate the suppression of metastasis [24-26]. However, the molecular mechanism of BRMS1 mediated transcriptional regulation of metastasis has not been fully explored.
Lysine-specific demethylase 1 (LSD1) is the first histone demethylase that demethylates mono-methylated and di-methylated histone H3 lysine 4 (H3K4) [27]. LSD1 is found as a part of various transcriptional corepressor, such as CoREST, NuRD, CtBP, SIN3A, and a subset of HDAC complexes [28-31]. CoREST is a corepressor for the RE1 silencing transcription factor (REST), which represses neuronal genes in nonneuronal tissues [32]. CoREST enhances the binding of LSD1 to nucleosomal substrates and prevents LSD1 degradation [28,33]. The LSD1/CoREST complex play an important role in multiple tissue development and tumorigenesis [30,34,36]. It is reported that CtBP/LSD1/CoREST complex interacts with ZNF516 to inhibit the proliferation and invasive potential of breast cancer cells [36]. However, the molecular mechanism of LSD/CoREST complex in breast cancer cells metastasis is still largely unknown.
In this study, we investigated the role of BRMS1 in the epithelial-to-mesenchymal transition (EMT) and the inhibition of metastasis through epigenetic programming. We propose that BRMS1 is an integral component of LSD1/CoREST repressor complex, thus expanding the role of BRMS1 in epigenetic regulation.
Materials and methods
Cell culture and transfection
MCF-7, and MDA-MB-231 cell lines were purchased from the Chinese Academy of Medical Sciences, Shanghai, China. The cell bank routinely performed cell line authentication by short tandem repeat (STR) profiling and all of the cell lines were preserved in our laboratory for not more than 6 months after receipt. All the media were supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 mg/ml streptomycin (Gibco, BRL, Gaithersburg, MD, USA). Cells were cultured at 37°C in a humidified incubator equilibrated with 5% CO2. Transfections were performed using Lipofectamine 2000 or Lipofectamine® RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. All the experiments were performed in triplicate and repeated at least three times.
Antibodies and reagents
The antibodies and the sources from which these antibodies were procured were: anti-FLAG, anti-HDAC1, anti-HDAC2, anti-Fibronectin, and anti-Vimentin (Sigma-Aldrich); anti-SIN3A (Santa Cruz); anti-LSD1, anti-BRMS1, and anti-CoREST (Abcam); anti-α-Catenin, anti-γ-Catenin, anti-N-cadherin, and anti-E-cadherin (BD); anti-acetyl H3, anti-dimethyl H3-K4, and anti-monomethyl H3-K4 (Millipore). Dynabeads Protein G was purchased from Invitrogen (Thermo Fisher Scientific), and the protease inhibitor cocktail was obtained from Roche Applied Science. Glutathione SepharoseTM 4B beads were purchased from GE Healthcare.
Cloning
BRMS1 gene was PCR-amplified using MCF-7 cDNA as a template and was cloned in frame with the pCMV-Tag2B vector to construct pCMV-Tag2B-Flag-BRMS1. The pcDNA3.1-LSD1, pcDNA3.1-HDAC1, and pcDNA3.1-HDAC2 were constructed by standard recombinant DNA techniques. LSD1 and BRMS1 fragments were cloned following PCR in the pGEX4T3 plasmid (Clontech) for bacterial expression.
Immunopurification and mass spectrometry
Lysates from MCF-7 cells expressing FLAG-BRMS1 were added to an equilibrated anti-FLAG resin. The column was then washed and the bound proteins were eluted using FLAG peptide (Sigma-Aldrich). Fractions of the bed volume were collected, resolved on SDS-PAGE, and stained by silver staining. Gel bands were then subjected to LC-MS/MS analysis.
FPLC chromatography
MCF-7 nuclear extracts were prepared and dialyzed against buffer D (20 mM HEPES of pH 8.0, 10% glycerol, 0.1 mM EDTA, 300 mM NaCl) (Applygen Technologies). Approximately 6 mg of the nuclear protein extract was concentrated to 1 ml using a Millipore Ultrafree centrifugal filter apparatus (10 kDa nominal molecular mass limit), and applied to an 850 × 20 mm Superose 6 size exclusion column (Amersham Biosciences). The column had been pre-equilibrated with buffer D containing 1 mM dithiothreitol and calibrated with protein standards (blue dextran, 2000 kDa; thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 232 kDa; bovine serum albumin, 67 kDa; and RNase A, 13.7 kDa; all from Amersham Biosciences). The bound proteins were eluted from the column at a flow rate of 0.5 ml/min and the fractions were collected.
Immunoprecipitation
For immunoprecipitation assays, 500 mg of the cellular extracts were incubated either with appropriate primary antibodies or normal rabbit/mouse immunoglobin G control antibody (IgG) on a rotator at 4°C overnight, followed by the addition of Dynabeads Protein G for 2 h at 4°C. Beads were then washed four times with lysis buffer (50 mM Tris-Cl of pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate containing protease inhibitor cocktail). The immune complexes were subjected to SDS-PAGE and western blotting followed by immunoblotting with respective primary and secondary antibodies. Immunodetection was performed using enhanced chemiluminescence (ECL System, Amersham Biosciences) according to the manufacturer’s instructions.
Glutathione S-transferase pull-down
GST fusion constructs were expressed in BL21 Escherichia coli cells, and crude bacterial lysates were prepared by sonication in a buffer containing 50 mM Tris-HCl (pH 7.4), 1.5 mM EDTA, 1 mM dithiothreitol, 10% (v/v) glycerol, 0.4 M NaCl in the presence of the protease inhibitor cocktail. The in vitro transcription and translation experiments were performed with rabbit reticulocyte lysate (TNT Systems; Promega). In GST pull-down assays, approximately 10 µg of the appropriate GST fusion proteins were mixed with 5-8 µl of the in vitro transcribed/translated products and incubated in the binding buffer (75 mM NaCl, 50 mM HEPES, pH 7.9) containing protease inhibitor cocktail at room temperature for 30 min. The binding mix was then added to 30 µl of glutathione-Sepharose beads and incubated with rotation at 4°C for 2 h. The beads were washed three times with binding buffer, resuspended in 30 µl of 2 × SDS-PAGE loading buffer, and resolved on 10% gel. Protein bands were detected with specific antibodies using western blot analysis.
ChIP and Re-ChIP
ChIP and Re-ChIP assay was performed in MCF-7 cells as described previously [37,39]. The primer sequences are listed in Table S1.
Real-time quantitative RT-PCR
Total RNA was isolated from the samples with TRIzol reagent (Invitrogen). Any potential DNA contamination was removed by RNase-free DNase treatment (Promega). Relative quantitation was performed using the ABI PRISM 7500 sequence detection system (Applied Biosystems) that measures real-time SYBR green fluorescence and then calculated by the comparative Ct method (2-ΔΔCt) with the expression of GAPDH as an internal control. The primer sequences used are listed in Table S1.
Lentivirus production and infection
Recombinant lentiviruses expressing shSCR (control scrambled shRNA), shBRMS1, and shLSD1 were constructed according to the instructions by Shanghai GenePharma. The concentrated viruses were used to infect 5 × 105 cells in a 60 mm dish with 8 µg/ml polybrene. Infected cells were then subjected to the selection of target expression. The shRNA sequences are listed in Table S1.
In vitro wound-healing assay
MDA-MB-231 breast cancer cells in L-15 medium containing 10% FBS were seeded into wells of 24-well plates (Becton Dickinson). The cells were grown to confluence and wounds were made using sterile pipette tips. Cells were washed with PBS and incubated in the fresh medium without FBS. After 24 h of incubation at 37°C, the cells were images. Data shown are the mean ± SD for n = 6 wells per group.
Cell invasion assay
Transwell chamber filters (Becton Dickinson) were coated with Matrigel. MDA-MB-231 cells were suspended in serum-free L-15 media and seeded into the upper chamber at a density of 2 × 104 cells in a volume of 500 ml. The cells were then cultured in a well containing 500 ml of L-15 media with 10% fetal bovine serum at 37°C for 18 h. Cells on the upper side of the membrane were removed using cotton swabs and those on the other side were stained and counted. Four high-powered fields were counted for each membrane.
Sphere culture
MCF-7 cells were inoculated into a 1:1 mixture of DMEM/F12 medium (Hyclone) without serum containing B27 supplement (50 ×, Invitrogen), 0.4 % BSA, 5 mg/ml Insulin (Invitrogen), human recombinant epidermal growth factor (EGF; 10 ng/ml) and basic fibroblast growth factor (bFGF; 20 ng/ml) at a density of 10,000 cells/well in ultra-low attachment six-well plates (Corning, Inc., Corning, NY, USA). Fresh aliquots of stem cell medium were added every other day.
Statistical analysis
The results are reported as mean ± SD unless otherwise noted. Comparisons were performed using two-tailed paired t test based on a bi-directional hypothesis for continuous variables.
Results
BRMS1 is an integral component of the LSD1/CoREST complex
In an effort to better understand the mechanistic roles of BRMS1 in breast cancer, we employed affinity purification and mass spectrometry (MS) to identify the proteins that interact with BRMS1. In these experiments, FLAG-tagged BRMS1 (FLAG-BRMS1) was stably expressed in human breast carcinoma MCF-7 cells. Cellular extracts were prepared and subjected to affinity purification using an anti-FLAG affinity gel. Mass spectrometric analysis indicated that SIN3A, HDAC1, and HDAC2 co-purified with BRMS1 as reported previously [40]. Notably, we found that BRMS1 also co-purified LSD1, the first identified histone lysine demethylase, and the co-repressor CoREST (Figure 1A). Detailed results of the MS analysis are provided in Table S2. Previous studies have shown that LSD1 and CoREST are found in a complex [41,42]. The presence of BRMS1 in the LSD1/CoREST complex was further confirmed by detecting using their specific antibodies by western blot analysis (Figure 1B). This suggests that BRMS1 is associated with the LSD1/CoREST complex in vivo. To verify the presence of a BRMS1/LSD1/CoREST complex in vivo, protein fractionation experiments were performed with nuclear proteins by fast protein liquid chromatography (FPLC). Nuclear extracts were fractionated by superpose 6 gel filtration chromatography. Native BRMS1 was eluted with an apparent molecular mass much greater than that of the monomeric protein; western blotting revealed that a major peak at about 667-1000 kDa represented a complex of BRMS1, LSD1, HDAC1, HDAC2 and CoREST (Figure 1C). Significantly, the elution pattern of BRMS1 largely overlapped with that of the LSD1/CoREST complex proteins, further supporting the hypothesis that BRMS1 is associated with the LSD1/CoREST complex in vivo.
Figure 1.

BRMS1 interacts with the LSD1/CoREST complex. A. Immunoaffinity purification of BRMS1-containing protein complex. Cellular extracts from MCF-7 cells stably expressing FLAG-Vector or FLAG-BRMS1 were immunopurified with anti-FLAG affinity gel and eluted with the FLAG peptide. These eluates were resolved using SDS-PAGE and silver-stained. The protein bands were retrieved and subjected to MS. Peptide coverage of the indicated proteins is shown. B. Western blot analysis of the identified proteins in the purified fractions, using antibodies against the identified proteins. C. Co-fractionation of BRMS1 and the LSD1/CoREST complex by FPLC. Nuclear extracts of MCF-7 cells underwent fractionation on Superose 6 size exclusion columns. The fractions were subjected to western blot analysis. The elution profiles of the calibration proteins with known molecular masses (kDa) are indicated. An equal volume from each fraction was analyzed. D. Association of BRMS1 with the LSD1/CoREST complex in MCF-7 cells. Whole cell lysates were immunoprecipitated (IP) with antibodies against the indicated proteins. Immunocomplexes were then immunoblotted (IB) using antibodies against the indicated proteins. E. Reciprocal association of LSD1 with the SIN3A/HDAC complex in MDA-MB-231 cells.
To further explore the endogenous binding of BRMS1 and the LSD1/CoREST complex, immunoprecipitation (IP) was performed with antibodies against LSD1, CoREST, HDAC1, HDAC2, or SIN3A. The proteins were detected by immunoblotting (IB) with antibodies against BRMS1 in MCF-7 cells. This result demonstrates that BRMS1 co-immunoprecipitated with all of the LSD1/CoREST’s complex proteins. SIN3A was the positive control (Figure 1D, left panel). Reciprocally, IP with antibodies against BRMS1 and IB with antibodies against LSD1, CoREST, HDAC1, HDAC2, or SIN3A also revealed that the components of the LSD1/CoREST complex co-immunoprecipitated with BRMS1 (Figure 1D, right panel). In addition, the association between BRMS1 and the LSD1/CoREST complex was also detected in human breast carcinoma MDA-MB-231 cells (Figure 1E). Taken together, these data strongly suggest that BRMS1 interacts with the LSD1/CoREST complex in vivo and is an integral component of the LSD1/CoREST complex.
BRMS1 interacts directly with LSD1
In order to determine the molecular basis of the interaction of BRMS1 with the LSD1/CoREST complex, GST pull-down experiments were performed. Incubation of GST-fused BRMS1 with in vitro transcribed/translated individual components of the LSD1/CoREST complex revealed that BRMS1 interacts directly with LSD1, but not with the other components of the LSD1/CoREST complex (Figure 2A, Left panel). Reciprocal GST pull-down assay with GST-fused components of the LSD1/CoREST complex and in vitro transcribed/translated BRMS1 yielded similar results (Figure 2A, right panel). In order to map the interaction interface of LSD1 and BRMS1, GST pull-down assays were performed with the GST-fused LSD1 N-terminal fragment (1-166 aa), the SWIRM domain (167-260 aa), the AOD domain (280-852 aa), and the Tower domain (419-520 aa) using in vitro transcribed/translated BRMS1. Our results indicate that the SWIRM domain mediated the interaction of LSD1 with BRMS1 (Figure 2B). Analogously, BRMS1 has two coiled coil regions important for the protein-protein interactions. Mapping the interaction interface in BRMS1 using GST-fused BRMS1 domain constructs and in vitro transcribed/translated LSD1 revealed that the second coiled coil domain of the BRMS1 proteins is responsible for its interaction with LSD1 (Figure 2C). Taken together, these data not only provided further support of the specific interaction among BRMA1 and the LSD1/CoREST complex, but also delineated the molecular details involved in the formation of the BRMS1/LSD1/CoREST complex.
Figure 2.
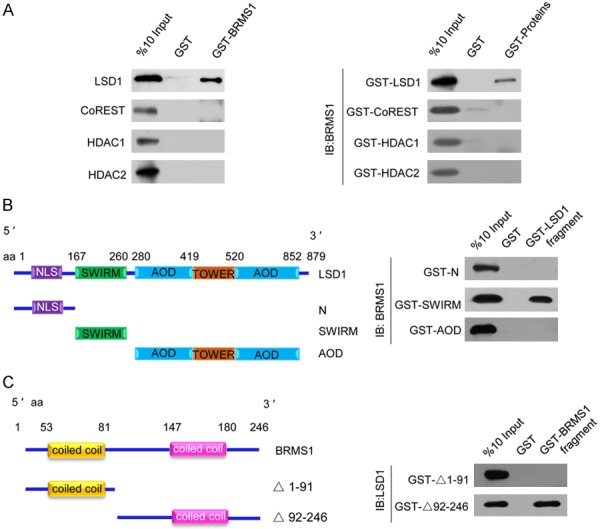
Molecular interaction between BRMS1 and the LSD1/CoREST complex. A. GST pull-down assays with GST-fused BRMS1 and in vitro transcribed/translated components of the LSD1/CoREST complex as indicated (left panel). GST pull-down assays with the indicated GST-fused proteins and in vitro transcribed/translated BRMS1 (right panel). B. GST pull-down assay with GST-fused LSD1 deletion constructs and in vitro transcribed/translated BRMS1. C. GST pull-down assay with the GST-fused BRMS1 deletion constructs and in vitro transcribed/translated LSD1.
Transcription target analysis for the BRMS1/LSD1 complex
To further investigate and understand the biological significance of BRMS1 and LSD1 interaction, we investigated the transcriptomes in BRMS1- or LSD1-deficient MCF-7 cells using high-throughput RNA deep sequencing approach (RNA-seq). Total RNA was extracted from MCF-7 cells transfected with control shRNA (shSCR) or shRNA-targeting BRMS1 or LSD1. RNA-seq analysis identified 967 and 2226 genes whose expressions were altered upon BRMS1 and LSD1 depletion, respectively. Cross-analysis of the transcriptomes from BRMS1- and LSD1-deficient cells identified 352 genes whose expressions were altered in both the BRMS1- and LSD1-depleted cells. These target genes were coregulated by BRMS1 and LSD1 (Figure 3A and 3B). To identify the putative functional processes associated with the targets that were coregulated by BRMS1 and LSD1, we performed Gene Ontology (GO) analysis. The top-ranked categories of the Biological Process (BP) analysis included the plasma membrane, cell cycle, chromosomal, biological adhesion, cell adhesion, etc. (Figure 3C), suggesting that the association of the BRMS1 and LSD1 may be related to the gene transcription regulation and epithelial-mesenchymal transition. Gene set enrichment analyses (GSEAs) showed an enrichment in the epithelial-mesenchymal transition pathways after BRMS1 knockdown (Figure 3D). To validate the RNA-seq analysis, we chose 7 representative genes implicated in the epithelial-mesenchymal transition regulation, VIM, INSIG2, KLK11, MRPL33, COL5A2, OLFML3 and SLC1A1, and validated their expression in MCF-7 cells using quantitative real-time PCR (qPCR). The results indicate that the mRNA levels of VIM, INSIG2, KLK11, MRPL33, COL5A2, OLFML3 and SLC1A1 increased upon the knockdown of either BRMS1 or LSD1 compared to shSCR or untreated MCF-7 cells (Figure 3E).
Figure 3.
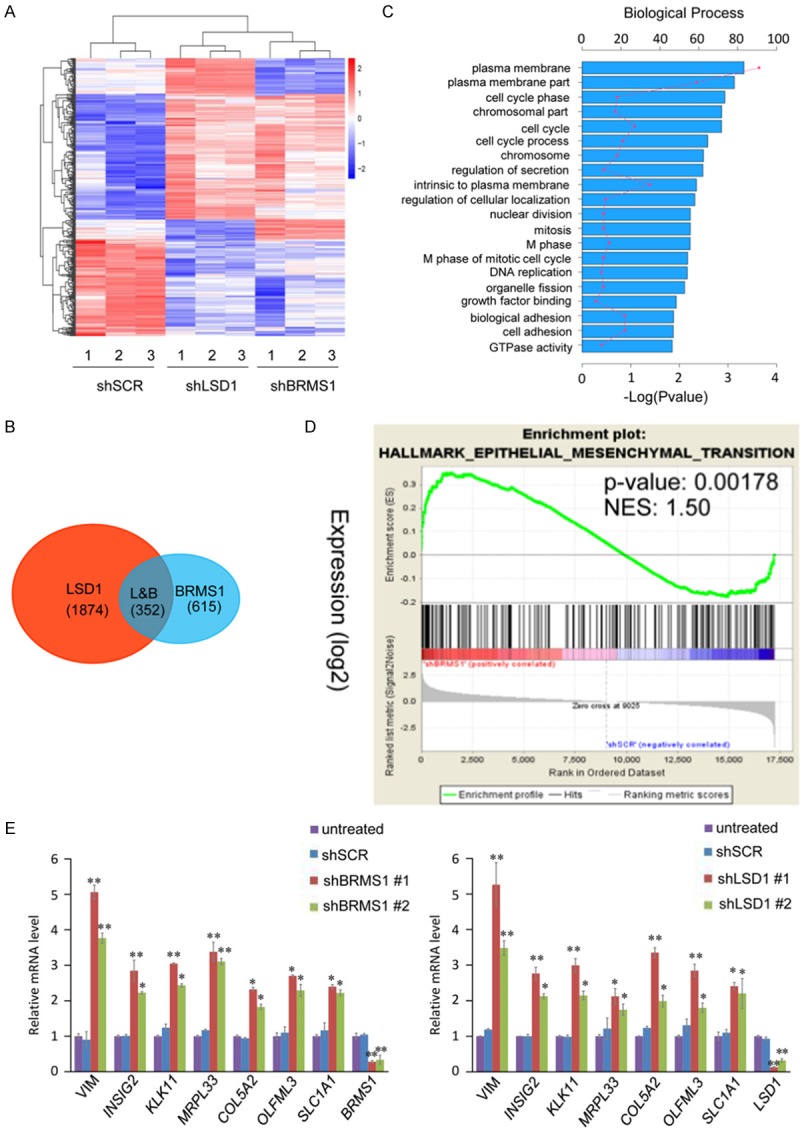
Genome-wide identification of transcriptional targets for the BRMS1/LSD1 complex. A. MCF-7 cells were transfected with shSCR, shBRMS1, and shLSD1 followed by RNA extraction and deep sequencing. Three independent samples were used in the RNA-seq analysis. mRNA expression data were clustered using Cluster 3.0 software. B. Intersection of BRMS1- and LSD1-regulated transcriptomes reveal 352 co-regulated genes with a fold change ≥ 2. C. GO analysis of the co-target genes of BRMS1 and LSD1 were performed to further explore the function of BRMS1/LSD1 complex. D. GSEA results showing enrichment gene signatures related to EMT in MCF-7 cells infected with shSCR or shBRMS1. E. MCF-7 cells were transfected with the indicated shRNAs, followed by RNA extraction and qRT-PCR analysis of the expression of indicated genes. The mRNA levels were normalized to those of GADPH. Since the shBRMS1 #1 and shLSD1 #1 are more effective, we chose them for the following experiments. *P < 0.05 and **P < 0.01 (two-tailed t-test).
Next, we wanted to study whether BRMS1 and LSD1 regulated the target gene expression by binding to their promoters. ChIP assays in MCF-7 cells with antibodies against BRMS1, LSD1, or control IgG revealed that BRMS1 and LSD1 co-occupied the promoters of VIM, INSIG2, KLK11, MRPL33, COL5A2, OLFML3 and SLC1A1. The ChIP results were quantitated using qPCR (Figure 4A) and visualized using conventional ChIP (Figure 4B). To further test our proposition that BRMS1 and LSD1 function in the same protein complex at target promoters, sequentially, we performed sequential ChIP/Re-ChIP with antibodies against BRMS1 and LSD1 for 7 target genes. The results show that the co-target gene promoters that were immunoprecipitated with antibodies against BRMS1 could be re-immunoprecipitated with the antibodies against LSD1 (Figure 4C, left panel). Similar results were obtained when the initial ChIP was performed with antibodies against LSD1 (Figure 4C, right panel). These results support the argument that BRMS1 and the LSD1 occupy the co-target gene promoters as one functionally collaborated protein complex. qChIP analyses showed that LSD1 knockdown led to a significant reduction in the binding of BRMS1 to the promoters of VIM, and vice-versa. Notably, the knockdown of either LSD1 or BRMS1 led to a severe increase in the H3K4me1/2 and H3 pan-acetylation (H3pan-ac) at the VIM promoter (Figure 4D). To determine the functional importance of BRMS1 association with LSD1/CoREST complex, we focused on the consequences of artificially recruiting LSD1 to the chromatin. MCF-7 cells containing the luciferase reporter gene under the control of the CMV promoter with five Gal4 DNA-binding sites (MCF-7-UAS-Luci) were transiently transfected with the plasmids expressing Gal4-DBD-LSD1 (Gal4-LSD1) or control Gal4-DBD vectors (Gal4). Gal4-LSD1 expression led to significantly decrease in the reporter gene expression as reported previously [43]. However, in BRMS1-depleted MCF-7-UAS-Luci cells, the transcriptional repression activity of LSD1 was partially impaired (Figure 4E). Similarly, the transcriptional repression activity of BRMS1 was also partially impaired in the LSD1-depleted MCF-7-UAS-Luci cells (Figure 4F). Thus, it appears that BRMS1 and LSD1 are both required for the optimal BRMS1/LSD1 complex transcriptional repression, suggesting that BRMS1 and LSD1 occupy the target promoters in a multiunit complex, and BRMS1 and LSD1’s functions are closely connected.
Figure 4.

BRMS1 and LSD1 coregulate the expression of VIM. A. qChIP analysis of the indicated genes in MCF-7 cells. Results are represented as fold change over the GAPDH control. Error bars represent mean ± SD for three independent experiments. *P < 0.05 and **P < 0.01 (two-tailed t-test). B. ChIP analysis of the indicated genes in MCF-7 cells by conventional DNA electrophoresis. IgG served as a negative control. C. Re-ChIP analysis of the seven selected co-targets in MCF-7 cells with antibodies against BRMS1 and LSD1 using conventional DNA electrophoresis. IgG served as a negative control. D. qChIP analysis of the recruitment of the indicated proteins on the VIM promoter in MCF-7 cells after infection with control lentivirus-mediated shRNA, or shRNAs-targeting BRMS1 or LSD1. Purified rabbit IgG was used as a negative control. Error bars represent mean ± SD of three independent experiments. *P < 0.05 and **P < 0.01 (two-tailed t-test). E and F. The control vector (containing Gal4-DBD only), Gal-4-LSD1, or Gal4-BRMS1 constructs was prepared and transfected alone or with the indicated specific lentivirus-mediated shRNAs into MCF-7 cells stably expressing Gal4-UAS reporter (MCF-7-Gal4-Luc cells). Gal4 luciferase reporter activity was measured. *P < 0.05 and **P < 0.01 (two-tailed t-test).
The BRMS1/LSD1 complex is essential for the breast cancer cell metastasis inhibition
We next wanted study the invasion-inhibitory effect of BRMS1/LSD1 complex. Of the common target genes identified earlier, VIM has been demonstrated to perform biological functions related to epithelial-mesenchymal transition. We therefore sought to investigate the possible role of the BRMS1/LSD1 complex in EMT and tumor metastasis. Western blots show that the expression of the mesenchymal markers (Vimentin and Fibronectin) increased significantly while that of the epithelial markers (E-cadherin and α-catenin) decreased remarkably upon LSD1/BRMS1 knockdown (Figure 5A, left bands). qPCR analysis yielded similar results (Figure 5A, right bands), indicating that the BRMS1 and LSD1 are required to maintain epithelial properties and dysregulation of BRMS1 and LSD1 may promote EMT.
Figure 5.

The BRMS1/LSD1 complex inhibits the invasion and migration potential of breast cancer cells. A. The expression of the indicated epithelial or mesenchymal markers was measured using western blot analysis (left) or real-time RT-PCR (right) in MCF-7 cells with BRMS1 or LSD1 depleted. *P < 0.05 and **P < 0.01 (two-tailed t-test). B. Wound-healing assay showed that the migration potential of BRMS1- or LSD1-depleted MDA-MB-231 cell was increased compared to the scrambled control shRNA infection group. *P < 0.05 (two-tailed t-test). C. BRMS1 and LSD1 repress the migration rate of breast cancer cells. MCF-7 cells with shSCR, shBRMS1, or shLSD1 were seeded on the Oris assay plates and allowed to adhere for 6 h. At this point, half of the stoppers were removed. Following a 48-h migration period, the remaining stoppers were also removed to provide pre-migration controls. Data shown are the mean per group (n = 6) ± SD. *P < 0.05 and **P < 0.01 (two-tailed t-test). D. Transwell invasion assays of MDA-MB-231 cells following transfection with the indicated specific shRNAs or siRNA. The invaded cells were stained and counted. The images represent one field under microscopy in each group. The efficiency of protein knockdown was verified by western blotting. *P < 0.05 and **P < 0.01 (two-tailed t-test). E. Immunoblot analysis of Cancer Stem Cell (CSC) markers of MCF-7 cells infected with shSCR, shBRMS1, or shLSD1 (left panel). MCF-7 cells were plated in the ultra-low attachment 24-well plate at a density of 10,000/well and cultured in tumor sphere medium containing shRNA against BRMS1 or LSD1. Tumor spheres were photographed (right panel). The data are presented as the mean ± SD with three independent experiments. *P < 0.05 and **P < 0.01 (two-tailed t-test).
Next, we investigated the role of BRMS1/LSD1 in tumor migration and invasion. BRMS1 or LSD1 was depleted in MDA-MB-231 cells via a lentivirus-mediated stable knockdown. The impact of loss-of-function of BRMS1 or LSD1 on migration potential was investigated using wound healing assay and Oris cell migration assay experiments (Platypus Technologies, Fitchburg, WI, USA). Compared to the control, BRMS1 or LSD1 depleted MDA-MB-231 cells had significantly increased migration, as determined by wound-healing assay (Figure 5B). In the Oris cell migration assay, the Harmony software was used to calculate the open areas of MDA-MB-231 cells pre- and post-migration and found that the BRMS1 or LSD1 knockdown in MDA-MB-231 cells was associated with an increased migration rate (Figure 5C). Furthermore, the impact of loss-of-function of BRMS1 or LSD1 on the invasive potential of these cells was assessed. In the transwell invasion assay, a significant increase in the invasiveness was observed upon BRMS1 or LSD1 knockdown, which could be partially restored by an additional Vimentin knockdown (Figure 5D). Collectively, these results indicate that the BRMS1 acts in conjunction with LSD1 and inhibits the migration and invasive potential of breast cancer cells, possibly through the transcriptional repression of VIM gene. Moreover, since the increase in CD44 and decrease in CD24 receptors mark the EMT-induced cancer stemness [44], we detected the CD44 and CD24 expression upon knockdown of BRMS1 or LSD1 in MCF-7 cells. Western blots show that while the CD24 was positively correlated with BRMS1 or LSD1’s expression levels, the CD44 was negatively correlated with BRMS1 or LSD1’s expression levels. Further, we evaluated the effect of BRMS1 and LSD1 on the sphere forming ability of MCF-7 cells. We found that the depletion of BRMS1 or LSD1 increased the number and size of spheres in MCF-7 cells compared to the control group (Figure 5E). Based on these results, it can be speculated that the low expression of BRMS1/LSD1 complex is required for the maintenance of breast cancer stem cell properties.
Through the analysis of data sourced from published clinical databases such as GSE20713, GSE48390 and GSE102484, the expression levels of BRMS1 and LSD1 were found to be negatively correlated with that of Vimentin, which supports our findings that Vimentin is transcriptionally repressed by BRMS1 and LSD1 (Figure 6A). Finally, in order to further extend our observations results to a clinicopathologically relevant context, we analyzed the expression of BRMS1 and its association with clinical behavior in breast cancer patients. Kaplan-Meier survival analysis (http://kmplot.com/analysis/) show that the overexpression of BRMS1/LSD1 was associated with improved overall survival in the breast cancer patients (Figure 6B).
Figure 6.

Clinicopathological significance of BRMS1/LSD1 in breast cancer. A. Analysis of the correlations between Vimentin and BRMS1/LSD1 in public datasets (GSE20713, GSE48390 and GSE102484). The relative level of Vimentin was plotted against that of BRMS1 or LSD1, respectively. B. Kaplan-Meier survival analysis of the relationship between survival time and BRMS1 or LSD1 signatures in breast cancer using the online tool (http://kmplot.com/analysis/). C. Graphic model as discussed in the text. DNA (black line); nucleosomes with single N terminus of H3 (blue ball).
Discussion
Breast cancer is a common cancer observed in women worldwide and significant amounts of research is being focused on understanding the pathogenesis of this disease. Here, we have demonstrated that BRMS1 recruits the LSD1/CoREST complex to inhibit the expression of a set of genes including VIM, INSIG2, KLK11, MRPL33, COL5A2, OLFML3 and SLC1A1, some of which are known to be critically involved in EMT, a hallmark of cancer metastasis [45,47]. Through a physical interaction of BRMS1 and LSD1, the transcriptional regulators BRMS1, LSD1, and CoREST cooperate to execute a transcriptional repression of the genes that induces breast cancer cell EMT and aggressiveness. As previously reported, BRMS1 can interact with the SIN3A complex and ARID4A, although the disruption of direct interaction between BRMS1 and ARID4A is not necessary for BRMS1-mediated metastasis suppression [12]. However, the transcriptional repression targets of BRMS1/SIN3A involved in the suppression of tumor metastasis are yet to be identified. Previous studies have shown that the mechanism of BRMS1 mediated transcriptional repression is mainly through the recruitment of HDAC1 to NFκB consensus binding regions resulting in the inhibition of NFκB-dependent transcriptional activation [18]. We propose that BRMS1 is an integral component of the LSD1/CoREST complex which mediates coordinated demethylation of H3K4me1/2 and histone acetylation on the target promoters for epigenetic repression of the VIM expression. We have shown that the abundance of the epigenetic modifications H3K4me1, H3K4me2, and H3 pan-ac on the promoters of the target genes is greatly elevated upon deletion of BRMS1 or LSD1. These findings support the hypothesis that BRMS1 and LSD1 act as an integrated complex. LSD1 has been reported to be an integral component of the NuRD complex, and that LSD1/NuRD (MTA3) can transcriptionally repress a series of EMT-promoting genes, such as TGFβ1, to inhibit breast cancer metastasis [29]. The current findings indicate that BRMS1 inhibits breast cancer metastasis, in part, through its interaction with the LSD1/CoREST complex and via the repression of VIM.
The LSD1/CoREST complex regulates many important biological functions through transcriptional regulation, including development and tumorigenesis [35,36,48]. Further, LSD1 has an essential role in development. For example, LSD1-containing co-repressor complex causes transcriptional regulation of the Gh gene during pituitary development [30]. Our findings suggest that the BRMS1/LSD1/COREST complex is involved in the EMT process and inhibits cancer metastasis. However, EMT also plays an important role in development, so future studies should clarify whether the BRMS1/LSD1/COREST complex can be involved in development.
EMT plays a crucial role in the developmental process adopted during tumorigenesis and promotes cancer cell metastatic capacity. During the EMT, epithelial cells lose their extensive adhesions to neighboring cells and apical-basal polarity, and differentiate into fibroblastic migratory cells with mesenchymal characteristics [49,50]. Importantly, Vimentin, the mesenchymal intermediate filament protein, and a hallmark of EMT, is overexpressed in malignant epithelial cancers, including breast cancer, and correlates with poor prognosis [51]. We detected the expression level of some of the invasion markers of EMT under the influence of loss-of-function of BRMS1 or LSD1. The expression of mesenchymal markers (Vimentin and Fibronectin) elevated significantly while the epithelial markers (E-cadherin and α-catenin) expression decreased remarkably. Our results indicate that the dysregulation of BRMS1/LSD1 complex may promote EMT process. Previous studies have found that EMT is a characteristic of cancer stem cells, in which EMTs generate stem cell-like cells and stem-like cells expressing markers associated with EMT [52]. We show that BRMS1 and LSD1 inhibited the ability of tumor sphere formation. In addition, BRMS1 and LSD1 are negatively correlated with the expression of CD44high/CD24low, which are markers of both human breast CSCs and normal mammary epithelial stem cells [53,54]. It is possible that the loss-of-function of BRMS1/LSD1 in epithelial breast cancer cells results in the acquisition of stem-cell characteristics following passage through an EMT.
In summary, our results demonstrate that the BRMS1/LSD1 complex inhibits breast cancer metastasis through transcriptional repression of VIM. Dysfunction of the BRMS1/LSD1 complex affects the fate of mammary epithelial cells and contributes to the EMT and metastasis of breast cancer (Figure 6C). Our data indicate that BRMS1 is a functional subunit of the LSD1/CoREST complex, thus expanding the role of BRMS1 in the epigenetic regulation and understanding the important requirements for the metastasis-suppressive function of BRMS1. Furthermore, our findings significantly add to the understanding of the complex hierarchical regulatory network of the EMT and support the pursuit of BRMS1 and LSD1 as potential prognostic indicators and/or targets for cancer therapy.
Acknowledgements
This work was supported by grants (81502446 to R. Q., 81402334 to Y. Y.) from the National Natural Science Foundation of China, the Major State Basic Research Development Program of China [grant number 2016YFA0102400 (to Y. Y. and R. Q.)], grants (2014M561192 and 2015T80224 to Y. Y.) from China Postdoctoral Science Foundation, grant (15JCQNJC11900 to Y. Y.) from the Tianjin Municipal Science and Technology Commission, and grant (20140105 to R. Q.) from the Science & Technology Development Fund of Tianjin Education Commission for Higher Education.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274:113–26. doi: 10.1111/joim.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Hurst DR, Welch DR. Metastasis suppressor genes at the interface between the environment and tumor cell growth. Int Rev Cell Mol Biol. 2011;286:107–80. doi: 10.1016/B978-0-12-385859-7.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langley RR, Fidler IJ. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- 5.Cook LM, Hurst DR, Welch DR. Metastasis suppressors and the tumor microenvironment. Semin Cancer Biol. 2011;21:113–22. doi: 10.1016/j.semcancer.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–9. [PubMed] [Google Scholar]
- 7.Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, Mauger DT, Welch DR. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–39. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006;16:522–31. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 9.Smith PW, Liu Y, Siefert SA, Moskaluk CA, Petroni GR, Jones DR. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Guan J, Sun Y, Chai J, Zou T, Gong W, Zhu Z, Liu X, Hou Q, Song X. Effect of BRMS1 on tumorigenicity and metastasis of human rectal cancer. Cell Biochem Biophys. 2014;70:505–9. doi: 10.1007/s12013-014-9948-x. [DOI] [PubMed] [Google Scholar]
- 11.Metge BJ, Frost AR, King JA, Dyess DL, Welch DR, Samant RS, Shevde LA. Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin Exp Metastasis. 2008;25:753–63. doi: 10.1007/s10585-008-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurst DR, Xie Y, Vaidya KS, Mehta A, Moore BP, Accavitti-Loper MA, Samant RS, Saxena R, Silveira AC, Welch DR. Alterations of BRMS1-ARID4A interaction modify gene expression but still suppress metastasis in human breast cancer cells. J Biol Chem. 2008;283:7438–44. doi: 10.1074/jbc.M709446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, Eckert KA, Verderame MF, Welch DR. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–9. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 14.Hurst DR. Metastasis suppression by BRMS1 associated with SIN3 chromatin remodeling complexes. Cancer Metastasis Rev. 2012;31:641–51. doi: 10.1007/s10555-012-9363-y. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–96. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 2005;65:3586–95. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 17.Cicek M, Fukuyama R, Cicek MS, Sizemore S, Welch DR, Sizemore N, Casey G. BRMS1 contributes to the negative regulation of uPA gene expression through recruitment of HDAC1 to the NF-kappaB binding site of the uPA promoter. Clin Exp Metastasis. 2009;26:229–37. doi: 10.1007/s10585-009-9235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Mayo MW, Xiao A, Hall EH, Amin EB, Kadota K, Adusumilli PS, Jones DR. Loss of BRMS1 promotes a mesenchymal phenotype through NF-kappaB-dependent regulation of Twist1. Mol Cell Biol. 2015;35:303–17. doi: 10.1128/MCB.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Jiang W, Wang Y, Wu J, Saiyin H, Qiao X, Mei X, Guo B, Fang X, Zhang L, Lou H, Wu C, Qiao S. Breast cancer metastasis suppressor 1 regulates hepatocellular carcinoma cell apoptosis via suppressing osteopontin expression. PLoS One. 2012;7:e42976. doi: 10.1371/journal.pone.0042976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapoor P, Saunders MM, Li Z, Zhou Z, Sheaffer N, Kunze EL, Samant RS, Welch DR, Donahue HJ. Breast cancer metastatic potential: correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. Int J Cancer. 2004;111:693–7. doi: 10.1002/ijc.20318. [DOI] [PubMed] [Google Scholar]
- 21.Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, Donahue HJ. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–7. [PubMed] [Google Scholar]
- 22.Phadke PA, Vaidya KS, Nash KT, Hurst DR, Welch DR. BRMS1 suppresses breast cancer experimental metastasis to multiple organs by inhibiting several steps of the metastatic process. Am J Pathol. 2008;172:809–17. doi: 10.2353/ajpath.2008.070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samant RS, Seraj MJ, Saunders MM, Sakamaki TS, Shevde LA, Harms JF, Leonard TO, Goldberg SF, Budgeon L, Meehan WJ, Winter CR, Christensen ND, Verderame MF, Donahue HJ, Welch DR. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metastasis. 2000;18:683–93. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 24.Edmonds MD, Hurst DR, Vaidya KS, Stafford LJ, Chen D, Welch DR. Breast cancer metastasis suppressor 1 coordinately regulates metastasis-associated microRNA expression. Clin Exp Metastasis. 2009;125:1778–85. doi: 10.1002/ijc.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–83. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 2009;69:7495–8. doi: 10.1158/0008-5472.CAN-09-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 28.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–5. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–72. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–7. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Huang W, Qiu R, Liu R, Zeng Y, Gao J, Zheng Y, Hou Y, Wang S, Yu W, Leng S, Feng D, Wang Y. LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J Mol Cell Biol. 2018;10:285–301. doi: 10.1093/jmcb/mjy021. [DOI] [PubMed] [Google Scholar]
- 32.Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–65. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- 33.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–64. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 34.Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27:562–72. doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 35.Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N, Bradley A, Cowley SM. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol. 2010;30:4851–63. doi: 10.1128/MCB.00521-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Liu X, He L, Yang J, Pei F, Li W, Liu S, Chen Z, Xie G, Xu B, Ting X, Zhang Z, Jin T, Liu X, Zhang W, Yuan S, Yang Z, Wu C, Zhang Y, Yang X, Yi X, Liang J, Shang Y, Sun L. ZNF516 suppresses EGFR by targeting the CtBP/LSD1/CoREST complex to chromatin. Nat Commun. 2017;8:691. doi: 10.1038/s41467-017-00702-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Sun L, Liang J, Yu W, Zhang Y, Wang Y, Chen Y, Li R, Sun X, Shang Y. The catalytic subunit of the proteasome is engaged in the entire process of estrogen receptor-regulated transcription. EMBO J. 2006;25:4223–33. doi: 10.1038/sj.emboj.7601306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Yi X, Sun X, Yin N, Shi B, Wu H, Wang D, Wu G, Shang Y. Differential gene regulation by the SRC family of coactivators. Genes Dev. 2004;18:1753–65. doi: 10.1101/gad.1194704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Zhang H, Liang J, Yu W, Shang Y. SIP, a novel ankyrin repeat containing protein, sequesters steroid receptor coactivators in the cytoplasm. EMBO J. 2007;26:2645–57. doi: 10.1038/sj.emboj.7601710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winter SF, Lukes L, Walker RC, Welch DR, Hunter KW. Allelic variation and differential expression of the mSIN3A histone deacetylase complex gene Arid4b promote mammary tumor growth and metastasis. PLoS Genet. 2012;8:e1002735. doi: 10.1371/journal.pgen.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–87. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc Natl Acad Sci U S A. 2006;103:13956–61. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai K, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. 2012;3:758. doi: 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 46.Sun S, Zhang G, Sun Q, Wu Z, Shi W, Yang B, Li Y. Insulin-induced gene 2 expression correlates with colorectal cancer metastasis and disease outcome. IUBMB Life. 2016;68:65–71. doi: 10.1002/iub.1461. [DOI] [PubMed] [Google Scholar]
- 47.Alexopoulou DK, Kontos CK, Christodoulou S, Papadopoulos IN, Scorilas A. KLK11 mRNA expression predicts poor disease-free and overall survival in colorectal adenocarcinoma patients. Biomark Med. 2014;8:671–85. doi: 10.2217/bmm.13.151. [DOI] [PubMed] [Google Scholar]
- 48.Lee MC, Spradling AC. The progenitor state is maintained by lysine-specific demethylase 1-mediated epigenetic plasticity during Drosophila follicle cell development. Genes Dev. 2014;28:2739–49. doi: 10.1101/gad.252692.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 50.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–46. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res. 2006;8:R7. doi: 10.1186/bcr1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.