

Abstract
Circadian rhythm disruption is intimately linked to atherosclerosis, and endothelial-to-mesenchymal transition (EndMT) is a major feature of atherosclerosis progression and unstable plaques. However, the mechanisms underlying the roles of Brain and Muscle ARNT-Like Protein-1 (BMAL1), an essential clock transcription activator, in EndMT and plaque instability have not been characterized. In the present study, we found a positive relationship among BMAL1 expression loss, EndMT, and plaque vulnerability in human carotid plaques. Furthermore, loss- and gain-of-function studies in human aortic endothelial cells (HAECs) revealed that BMAL1 inhibited oxidized low-density lipoprotein (oxLDL)-induced intracellular reactive oxygen species (ROS) accumulation and subsequent EndMT. Mechanistically, BMAL1 deficiency aggravated EndMT through BMP-mediated signaling. Collectively, our study demonstrates the underlying mechanism for the central role of BMAL1 loss in atherosclerosis progression and plaque stability transition promoted by oxidative stress, which can be targeted therapeutically to prevent the occurrence and progression of atherosclerosis.
Keywords: Atherosclerosis, endothelial-to-mesenchymal transition, BMAL1, plaque stability, BMP signaling
Introduction
Atherosclerosis is a progressive inflammatory disease characterized by leukocyte and lipid accumulation in lesions [1]. Although vessel wall inflammation, oxidative stress and endothelial dysfunction are believed to contribute to the progression of atherosclerotic plaques, the precise pathological mechanism remains unknown [2].
Circadian oscillation is a fundamental process that influences many physiological and biological functions [3]. Accumulating evidence suggests that circadian rhythm disruption is associated with atherosclerosis [4,5]; for example, exposure to shift work is a risk factor for atherosclerosis [6]. Furthermore, animal studies have shown that when aortae from Bmal1-/- or Per1/2-/- mice are transplanted into wild-type mice, significant atherosclerosis develops in the transplanted graft, indicating that cell-intrinsic molecular clocks function locally in the vessel wall independently of the central clock or systemic factor rhythms [7]. Additionally, Brain and Muscle ARNT-Like Protein-1 (BMAL1) loss in mouse endothelial cells increases chemokine expression, impairs endothelial integrity and barrier function, and subsequently increases leukocyte trafficking across the endothelial layer [8], demonstrating the protective roles of Bmal1 in the vascular endothelium [9].
Additional lines of evidence suggest a role for BMAL1 in suppressing reactive oxygen species (ROS) production. The endothelial cells of Bmal1-/- mice have significantly decreased endothelial nitric oxide synthase activation and, consequently, reduced nitric oxide production and increased superoxide levels [4,10]. Inflammatory signals and increased ROS production contribute to endothelial dysfunction [11,12] and facilitate endothelial-to-mesenchymal transition (EndMT), a major feature of atherosclerosis mediated by induction signals from TGF-β and BMP ligands [13] in atherosclerotic plaques [14,15]. However, the potential mechanism by which BMAL1 suppresses EndMT remains to be elucidated.
In the present study, we found a positive relationship among BMAL1 expression loss, EndMT, and plaque vulnerability in human carotid plaques. Furthermore, in vitro results revealed that BMAL1 inhibited oxidized low-density lipoprotein (oxLDL)-induced intracellular ROS accumulation and subsequent EndMT. Finally, we demonstrated that BMAL1 deficiency aggravated EndMT through BMP signaling. Therefore, our results suggested that oxLDL induces EndMT through BMAL1 downregulation, BMP signaling activation and subsequent ROS production, thus contributing to plaque vulnerability.
Materials and methods
Ethics statement
Carotid atherosclerotic plaque specimens were obtained from Zhongshan Hospital (Shanghai, China). Written informed consent was obtained from each patient, and the study was approved and supervised by the Ethics Committee of Zhongshan Hospital. All protocols were conducted in accordance with the ethical guidelines of the 1975 Declaration of Helsinki.
Cell culture and treatments
HAECs (ScienCell Research Laboratories, USA) were cultured using an Endothelial Cell Growth Medium (EGM)-2 BulletKit (Lonza Group AG, Switzerland) as detailed in our previous report [16]. Upon reaching 80% confluence, HAECs were serum-starved by incubation in EGM media containing 0.5% serum for 24 h. Then, the cells were treated with oxLDL (100 μg/mL) for 3 days in the presence or absence of a selective BMP type I receptor inhibitor, LDN-193189 (100 nM, Selleck Chemicals, USA), and an ROS scavenger, Tempol (100 μM, Sigma, USA).
Preparation of the retroviral vector
Human BMAL1 gain- and loss-of-function models were established as previously described [17]. To overexpress BMAL1 in HAECs, an expression construct was generated by subcloning PCR-amplified full-length human BMAL1 cDNA into an LvCP06 vector (Genechem, China). An empty vector was used as a negative control. To stably knock down BMAL1 in HAECs, a short hairpin RNA (shRNA) was designed and inserted into the pLKO.1 vector (shBMAL1) (Genechem, China), and scrambled shRNA was used as a negative control (shControl). All constructs used in this study were confirmed by DNA sequencing (Biosune Biotechnology, China). Viral particle production and cell transfection were carried out as previously described [18]. The final knockdown and overexpression efficiencies were determined with western blot analyses.
Western blot analysis
The relative protein expression levels of BMAL1, fibroblast-specific protein-1 (FSP-1), vimentin, phospho-Smad1/5 (p-SMAD1/5), and fibroblast activation protein alpha (FAP) were measured by western blotting using standard methods. The following antibodies were used: rabbit anti-BMAL1 (NB100-2288, Novus Biological, USA), rabbit anti-FSP-1 (ab27957, Abcam, USA), mouse anti-vimentin (ab8978, Abcam, USA), rabbit anti-Phospho-Smad1/5 (#9516, Cell Signaling Technology, USA), rabbit anti-VE-cadherin (#2500, Cell Signaling Technology, USA), rabbit anti-FAP (ab28244, Abcam, USA), mouse anti-GAPDH (G8795, Sigma, USA), horseradish peroxidase (HRP)-linked anti-mouse (#7076, Cell Signaling Technology, USA) and anti-rabbit (#7074, Cell Signaling Technology, USA) antibodies. The band intensities were quantified using ImageJ and normalized to GAPDH.
Histology staining
Human carotid artery plaque histological staining was performed using pre-existing tissue blocks derived from endarterectomy samples as previously reported [19]. In brief, the samples were formalin-fixed and paraffin-embedded. The blocks were sectioned at 5 μm intervals using a microtome. Then, the slides were deparaffinized in xylene and rehydrated with graded ethanol. Then, the sections were stained with hematoxylin and eosin (H&E) and Masson’s trichrome stain.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA from HAECs was harvested using TRIZOL reagent (Thermo Fisher Scientific, USA). First-strand complementary DNA was synthesized using an Omniscript RT kit (Qiagen). Quantitative RT-PCR using SYBR Green was used to quantify the mRNA changes. The expression levels were calculated using the 2-ΔΔCT method. All primers are listed in Table 1.
Table 1.
Primer sequences used for quantitative RT-PCR
| Gene | Primer sequence |
|---|---|
| GAPDH-F | 5’-ACGGATTTGGTCGTATTGGG-3’ |
| GAPDH-R | 5’-CGCTCCTGGAAGATGGTGAT-3’ |
| CDH5-F | 5’-AACCAGATGCACATTGATGAAGAG-3’ |
| CDH5-R | 5’-ATTCTTGCGACTCACGCTTGA-3’ |
| ACTA2-F | 5’-GGGTGATGGTGGGAATGG-3’ |
| ACTA2-R | 5’-GCAGGGTGGGATGCTCTT-3’ |
| OPN-F | 5’-CGAGGTGATAGTGTGGTTTATGG-3’ |
| OPN-R | 5’-GCACCATTCAACTCCTCGCTTTC-3’ |
| BMP2-F | 5’-TGCTAGTAACTTTTGGCCATGATG-3’ |
| BMP2-R | 5’-TTTGTGTTTGGCTTGACGTTTTT-3’ |
| BMP9-F | 5’-AGAACGTGAAGGTGGATTTCC-3’ |
| BMP9-R | 5’-CGCACAATGTTGGACGCTG-3’ |
| ID1-F | 5’-CTGCTCTACGACATGAACGG-3’ |
| ID1-R | 5’-GAAGGTCCCTGATGTAGTCGAT-3’ |
| FN1-F | 5’-CCCAGACTTATGGTGGCAATTC-3’ |
| FN1-R | 5’-AATTTCCGCCTCGAGTCTGA-3’ |
| BMP4-F | 5’-GCACTGGTCTTGAGTATCCT-3’ |
| BMP4-R | 5’-GAGGAAACGAAAAGCAGAGT-3’ |
| ALK2-F | 5’-ACGTGGAGTATGGCACTATC-3’ |
| ALK2-R | 5’-GATGTACACGAATGATCCAA-3’ |
| ALK3-F | 5’-ACATCTACAGCTTCGGCCTA-3’ |
| ALK3-R | 5’-GCATATCTTCGTATGACGGA-3’ |
Immunofluorescence (IF) and confocal microscopy
For immunofluorescence staining, sections were prepared according to standard protocols for H&E staining. Antigen retrieval was performed with citrate buffer (10 mM, pH 6.0) at 95°C. After cooling, the tissue slides were blocked with 5% bovine serum albumin (BSA) for 1 h and then incubated with primary antibodies overnight in a humidified chamber at 4°C. The primary antibodies used for IF were anti-BMAL1 (NB100-2288, Novus Biological, USA), anti-CD31 (ab28364, Abcam, USA), anti-vimentin (ab8978, Abcam, USA) and anti-FSP-1 (ab27957, Abcam, USA). The slides were then washed with Tris-buffered saline and incubated with Cy3- or FITC-conjugated secondary antibodies for 1 h at room temperature. All immunofluorescence micrographs were acquired using an Axiovert 200M microscopy system (Carl Zeiss, Germany). Image quantifications were performed using ImageJ software.
Measurement of ROS production
Intracellular ROS concentrations were measured using oxidant-sensing 2’, 7’-dichlorofluorescein diacetate (DCF-DA, 5 µM, Invitrogen, USA). The DCF fluorescence intensities were measured using a spectrophotometer (Leica, Germany) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was performed using SPSS software, version 19.0 (IBM SPSS, USA). The data are expressed as the mean ± standard deviation (SD). Comparisons were performed by one-way analysis of variance (ANOVA) among groups or by Student’s t test between two groups. P<0.05 was considered statistically significant.
Results
BMAL1 regulates EndMT in human carotid plaques
To determine whether BMAL1-induced EndMT occurs in human atherosclerotic plaques, we evaluated human carotid endarterectomy plaque samples. A neurologist and a pathologist assessed plaque stability based on the clinical manifestation and pathological results (Figure 1A and 1B) [20,21]. Disagreements between the two specialists were resolved by a third reviewer. Western blot analyses revealed that BMAL1 and VE-CADHERIN expression levels were significantly lower in vulnerable human carotid plaques than in stable human carotid plaques (Figure 1C). Mesenchymal markers (VIMENTIN, FSP-1 and FAP) and p-SMAD1/5 expression levels were significantly higher in vulnerable human carotid plaques and were negatively correlated with BMAL1 and VE-cad-herin (Figure 1C), and immunostaining showed similar results (Figure 1D-F). Furthermore, immunostaining results revealed that 19.7 ± 8.6% of the luminal endothelium of stable plaques coexpressed CD31 and VIMENTIN; this coexpression was significantly increased to 62.7 ± 11.6% in the luminal endothelium of vulnerable plaques (Figure 1D, P<0.05). In addition, compared to cells in stable plaques, more cells coexpressed CD31 and FSP-1 in the luminal endothelium of vulnerable plaques (15.0 ± 3.0% vs. 36.3 ± 16.1%, P<0.05, Figure 1E), while fewer cells coexpressed CD31 and BMAL1 in vulnerable plaques (51.7 ± 6.5% vs. 22.7 ± 6.8%, P<0.05, Figure 1F). These results from human samples reflected the participation of BMAL1-mediated EndMT in atherosclerotic progression.
Figure 1.
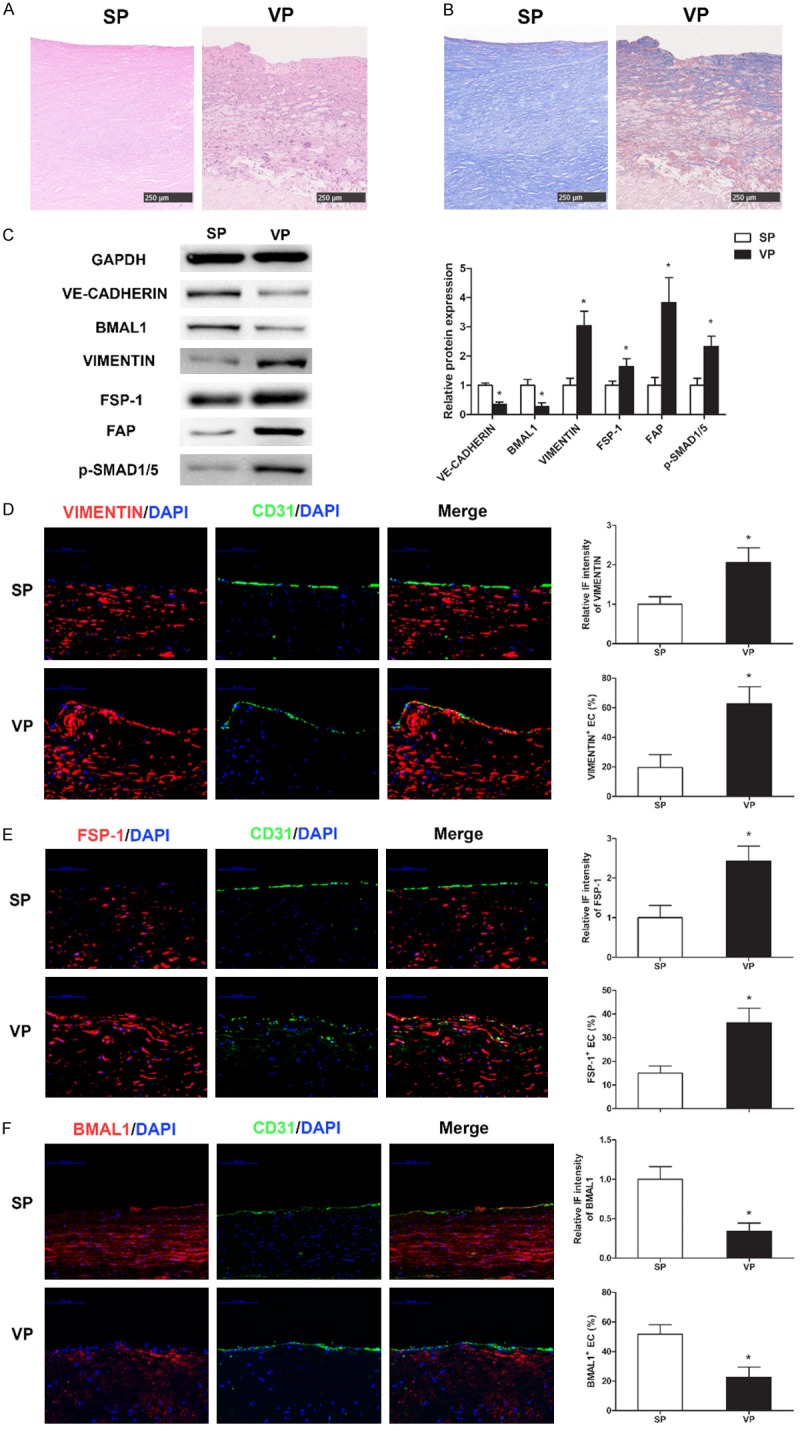
BMAL1 suppresses EndMT and restricts atherosclerotic plaque development. (A, B) Representative images of hematoxylin-eosin (A) and Masson (B) staining of stable plaques (SP) and vulnerable plaques (VP). Scale bar: 250 μm. (C) VE-CADHERIN, BMAL1, VIMENTIN, FSP-1, FAP and p-SMAD1/5 protein levels in SP and VP were assayed via western blotting. (D-F) Representative images of SP and VP after immunofluorescence staining for CD31 (green) and VIMENTIN (D) (red), FSP-1 (E) (red) and BMAL1 (F) (red). The nuclei were stained with 4’, 6-diamidino-2-phenylindole (DAPI) (blue). Scale bar: 100 μm. Immunofluorescence intensities of VIMENTIN, FSP-1 and BMAL1 in the plaque endothelium and the percentages of VIMENTIN+ ECs, FSP-1+ ECs and BMAL1+ ECs in the lumens carotid plaques were analyzed. The values represent the mean ± SD. *P<0.05 vs. stable plaques.
oxLDL induces EndMT and reduces BMAL1 expression in cultured HAECs
DCF analyses showed that the relative ROS levels were significantly higher in HAECs treated with 100 μg/mL oxLDL for 3 days than in normal control HAECs (Figure 2A). Western blot and qRT-PCR analyses demonstrated significantly decreased levels of BMAL1 and CDH5, an endothelial marker, and significantly increased levels of FSP-1, VIMENTIN, FAP, OPN and FN1 after 3 days of oxLDL treatment; however, the expression levels of ACTA2 were not significantly changed (Figure 2B and 2C). In addition, the expression levels of BMP2, BMP4, BMP9, ID1, ALK2, ALK3 and p-SMAD1/5 also increased significantly after 3 days of oxLDL treatment (Figure 2B and 2D).
Figure 2.
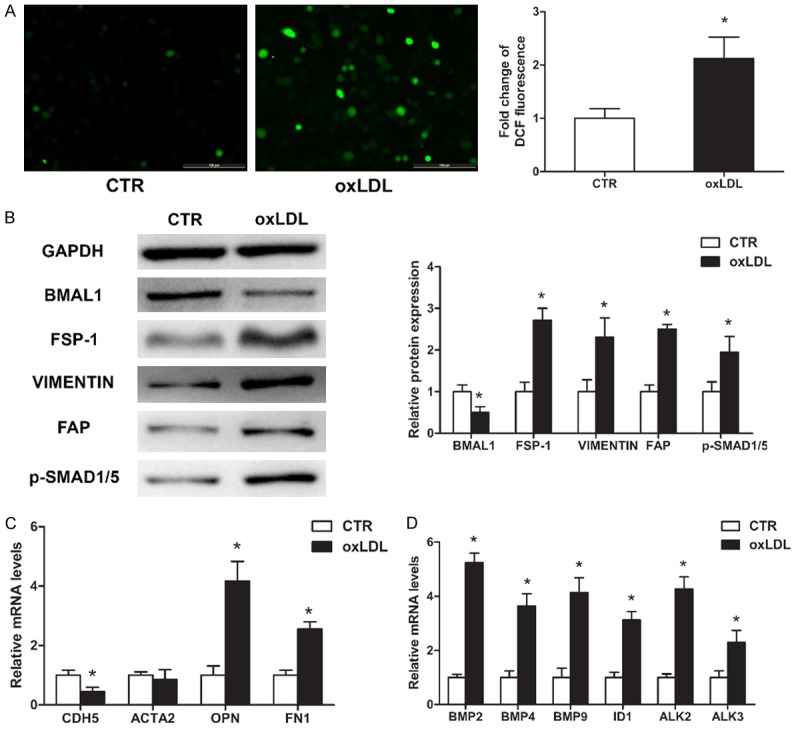
oxLDL induces EndMT and reduces BMAL1 expression in cultured HAECs. HAECs were cultured with or without 100 μg/mL oxLDL for 3 days. A. Intracellular ROS levels were measured by DCF fluorescence. Scale bar, 100 μm. B. BMAL1, FSP-1, VIMENTIN, FAP and p-SMAD1/5 protein levels were assayed by western blotting. C, D. CDH5, ACTA2, OPN, FN1, BMP2, BMP4, BMP9, ID1, ALK2 and ALK3 mRNA levels were assayed using qRT-PCR (n=3). The values represent the mean ± SD. *P<0.05 vs. the control group (CTR).
BMAL1 deficiency aggravates oxLDL-induced EndMT
To determine whether BMAL1 deficiency is closely related to EndMT progression, HAECs transduced with control or BMAL1-silencing vectors were treated with 100 µg/mL oxLDL for 3 days. BMAL1 deficiency significantly increased ROS accumulation and OPN, FN1, FSP-1, VIMENTIN and FAP expression but decreased CDH5 expression (Figure 3A-C). Interestingly, ACTA2, an EndMT marker, was downregulated slightly, although the difference was not significant (Figure 3C). In addition, BMAL1 deficiency significantly increased the expression levels of BMP2, BMP4, BMP9, ID1, ALK2, ALK3 and p-SMAD1/5 (Figure 3B and 3D).
Figure 3.
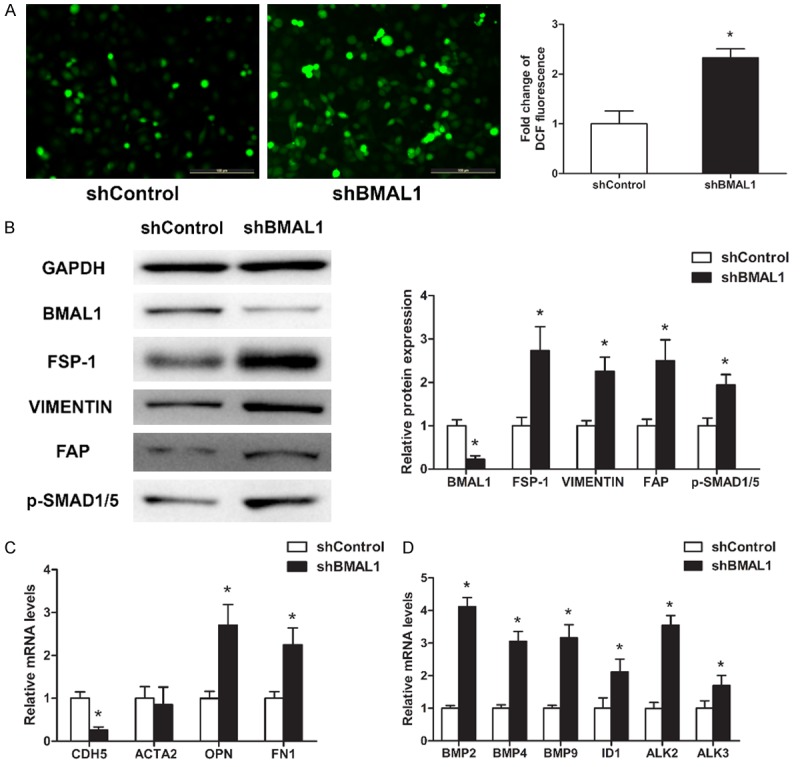
BMAL1 deficiency aggravates oxLDL-induced EndMT in HAECs. HAECs transduced with control (shControl) or BMAL1-silencing (shBMAL1) vectors were treated with 100 μg/mL oxLDL for 3 days. A. Intracellular ROS levels were measured by DCF fluorescence. Scale bar, 100 μm. B. BMAL1, FSP-1, VIMENTIN, FAP and p-SMAD1/5 protein levels were assayed by western blotting. C, D. CDH5, ACTA2, OPN, FN1, BMP2, BMP4, BMP9, ID1, ALK2 and ALK3 mRNA levels were assayed using qRT-PCR (n=3). The values represent the mean ± SD. *P<0.05 vs. scrambled control (shControl).
BMAL1 overexpression attenuates oxLDL-induced EndMT
To further confirm whether BMAL1 plays an important role in oxLDL-induced EndMT, HAECs transduced with control or BMAL1-overexpressing vectors were treated with 100 µg/mL oxLDL for 3 days. As expected, BMAL1 overexpression reduced ROS production (Figure 4A) and BMP2, BMP4, BMP9, ID1, ALK2, ALK3, p-SMAD1/5, OPN, FN1, FSP-1, VIMENTIN and FAP expression levels but significantly increased CDH5 expression (Figure 4B-D). ACTA2 was slightly upregulated, but the difference was not significant (Figure 4B). These results suggest that BMAL1 plays a protective role in oxLDL-induced EndMT.
Figure 4.
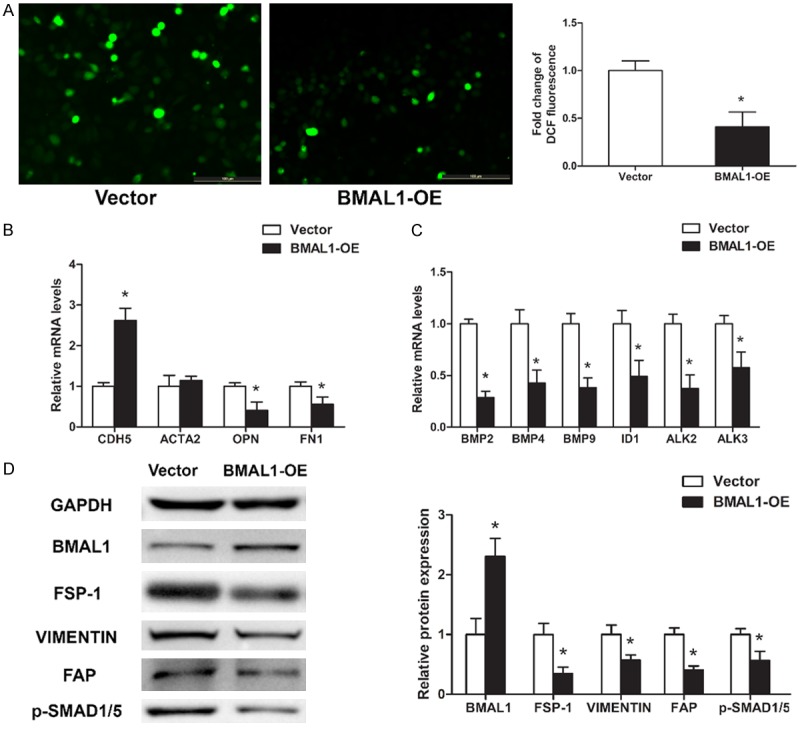
BMAL1 overexpression attenuates oxLDL-induced EndMT in HAECs. HAECs transduced with control (Vector) or BMAL1-overexpressing (BMAL1-OE) vectors were treated with 100 μg/mL oxLDL for 3 days. A. Intracellular ROS levels were measured by DCF fluorescence. Scale bar, 100 μm. B, C. CDH5, ACTA2, OPN, FN1, BMP2, BMP4, BMP9, ID1, ALK2 and ALK3 mRNA levels were assayed using qRT-PCR (n=3). D. BMAL1, FSP-1, VIMENTIN, FAP and p-SMAD1/5 protein levels were assayed by western blotting. The values represent the mean ± SD. *P<0.05 vs. Vector.
ROS scavengers inhibit BMP signaling-mediated EndMT
The interactions among ROS, BMAL1, BMP signaling and EndMT in HAECs were investigated by treating BMAL1-silenced HAECs cultured with 100 µg/mL oxLDL for 3 days with or without Tempol/LDN-193189.
When BMAL1-silenced HAECs were treated with Tempol or LDN-193189, FSP-1, VIMENTIN, FAP, OPN, FN1 expression levels and ROS production were decreased, while CDH5 expression levels were increased (Figure 5A-C). Notably, the expression level of CDH5 in cells treated with LDN-193189 was significantly higher than that in cells treated with Tempol (Figure 5C). In addition, BMAL1-silenced HAECs treated with LDN-193189 had significantly decreased p-SMAD1/5 and ID1 expression levels, while Tempol treatment had no significant effect on their expression levels (Figure 5A and 5C). These results indicated that BMAL1 deficiency leads to increased ROS production and subsequent EndMT through the BMP signaling pathway.
Figure 5.
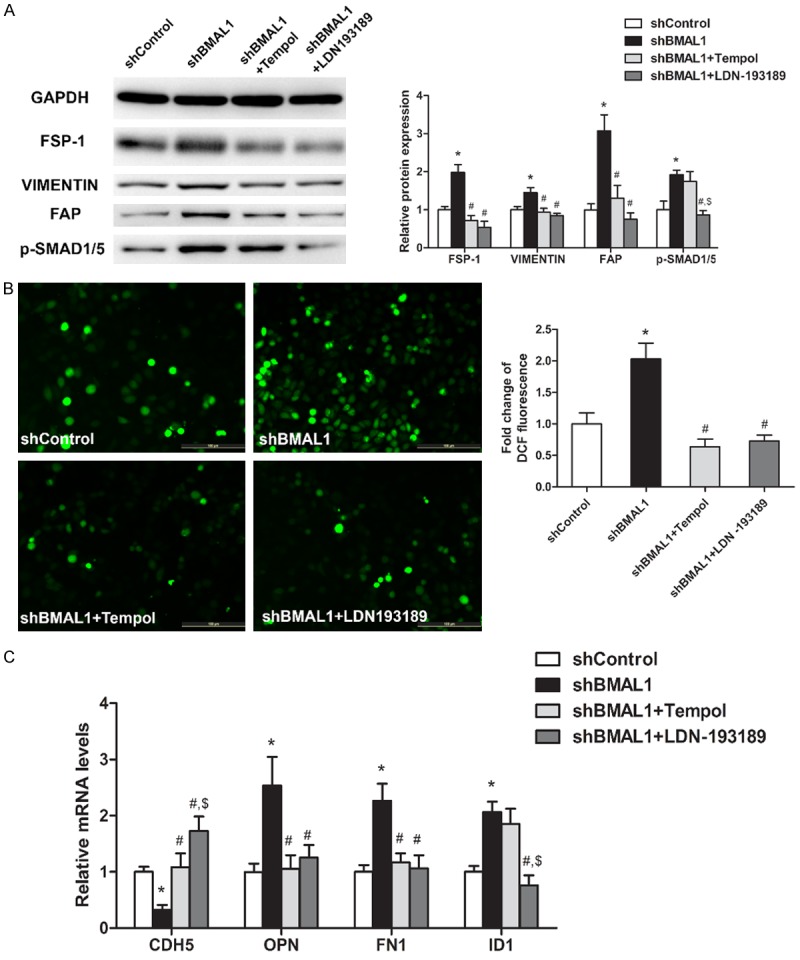
BMAL1 loss activates oxLDL-induced EndMT via BMP signaling in HAECs. HAECs transduced with control vectors (shControl) were treated with oxLDL (100 μg/mL), and HAECs transduced with BMAL1-silencing (shBMAL1) vectors were treated with oxLDL (100 μg/mL), oxLDL (100 μg/mL) + Tempol (100 μM) or oxLDL (100 μg/mL) + LDN-193189 (100 nM). A. FSP-1, VIMENTIN, FAP and p-SMAD1/5 protein levels were determined by western blotting. B. Intracellular ROS levels were measured by DCF fluorescence. C. CDH5, OPN, FN1 and ID1 mRNA levels were assayed using qRT-PCR (n=3). The values represent the mean ± SD. *P<0.05 vs. shControl HAECs treated with oxLDL. #P<0.05 vs. shBMAL1 HAECs treated with oxLDL. $P<0.05 vs. shBMAL1 HAECs treated with oxLDL + Tempol.
Discussion
Our results provide a novel mechanism by which BMAL1 suppresses EndMT and thus restricts atherosclerotic development. First, we showed that in HAECs, BMAL1 loss aggravated oxidative stress damage and subsequent EndMT, an important part of atherosclerotic plaque progression [14,22]. Consistent with the in vitro results, a positive relationship among BMAL1 expression loss, EndMT, and plaque vulnerability was also observed in carotid plaques of patients. Second, we demonstrated that BMAL1 deficiency aggravated intracellular ROS accumulation and EndMT through BMP-mediated signaling. Taken together, our results provide a mechanism to explain the central role of BMAL1 in atherosclerosis progression and the plaque phenotype switch.
EndMT plays a major role in various chronic fibrosis-type injuries and cardiovascular diseases [14], including pulmonary hypertension [23,24], vascular malformations [25], vascular calcification [26] and cardiac fibrosis [27]. In this study, EndMT was observed in human carotid plaques and was increased with plaque vulnerability. EndMT can be promoted by various stressors, including disturbed blood flow, hypoxia, hyperlipidemia and high glucose [13,28]. In our present study, oxLDL treatment caused HAECs to lose endothelial marker expression, including CDH5, but gain mesenchymal marker expression, including VIMENTIN, FSP-1, FAP, FN1 and OPN, verifying the induction of EndMT by oxLDL. Previous studies demonstrated that Bmal1 deficiency was closely related to hyperlipidemia and atherosclerosis [9]. We further demonstrated that BMAL1 loss was positively related to EndMT progression and plaque vulnerability in human atherosclerotic plaques. Furthermore, our in vitro studies confirmed the protective role of BMAL1 against EndMT induced by oxLDL in HAECs.
The endothelial cells of Bmal1-/- mice have been demonstrated to show significantly blunted endothelial nitric oxide synthase activation and, consequently, reduced nitric oxide production and increased superoxide levels [4,10]. Therefore, we further explored whether ROS was involved in BMAL1 deficiency-mediated EndMT. DCF fluorescence results demonstrated that silencing of BMAL1 expression increased ROS production in HAECs treated with oxLDL, while overexpression of BMAL1 alleviated ROS production, which was consistent with previous studies [29,30]. Moreover, we found that Tempol diminished ROS production and significantly inhibited EndMT, confirming the important role of ROS in BMAL1 deficiency-mediated EndMT. These findings suggested a vital role for BMAL1 loss-mediated ROS production in atherosclerosis.
EndMT is thought to be mediated by induction signals from TGF-β and BMP ligands [13]. Additionally, BMP signaling was found to be required for ROS induction by oxLDL in endothelial cells, a critical process in atherogenesis [31-33]. Therefore, we hypothesize that BMAL1 loss activates the BMP signaling pathway and leads to ROS accumulation, EndMT and subsequent plaque instability. In our present study, BMAL1 deficiency markedly increased BMP signaling pathway activity, intracellular ROS accumulation and EndMT compared to the control condition. In addition, LDN-193189, a selective BMP type I receptor inhibitor, could effectively rescue these changes, which supported our hypothesis. Notably, the expression level of CDH5 in cells treated with LDN-193189 was significantly higher than that in cells treated with Tempol, suggesting that BMP signaling pathway-mediated EndMT is partly dependent on ROS activation [34], which should be elucidated in a future study. Furthermore, BMAL1 loss and p-SMAD1/5 expression levels were positively related to the EndMT extent and plaque vulnerability in human carotid atherosclerotic plaque specimens, which added clinical significance to our study and provided a promising therapeutic target for atherosclerosis.
In conclusion, we elucidated that the loss of protective endothelial BMAL1 expression aggravates BMP-mediated intracellular ROS accumulation and EndMT progression and leads to subsequent human carotid plaque vulnerability (Figure 6). Controlling this process might represent feasible therapeutic avenues for atherosclerosis.
Figure 6.
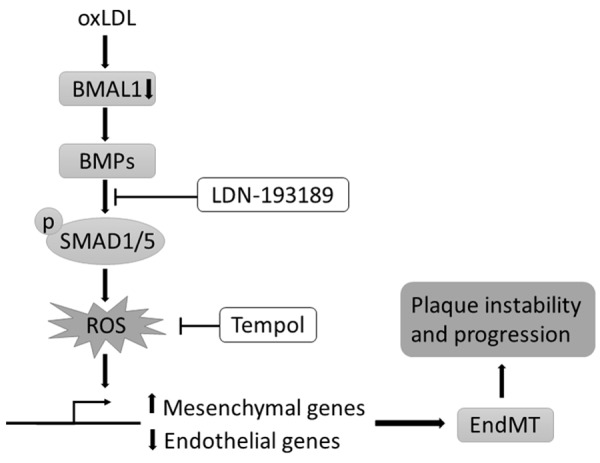
Diagram of the proposed mechanism for cellular contributions to atherosclerotic plaque progression. Loss of protective endothelial BMAL1 expression aggravates BMP-mediated intracellular ROS accumulation and EndMT progression and leads to subsequent plaque vulnerability.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grant nos. 81371129, 81670973, 81570433 and 81600371) and the Project of Shanghai Municipal Commission of Health and Family Planning (grant no. 20154Y0104).
Disclosure of conflict of interest
None.
References
- 1.Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. 2016;118:1808–1829. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Q, Qin L, Dai S, Zhang H, Pasula S, Zhou H, Chen H, Min W. AIP1 suppresses atherosclerosis by limiting hyperlipidemiainduced inflammation and vascular endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33:795–804. doi: 10.1161/ATVBAHA.113.301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC, Hsieh TH, Su FY, Lin LC, Abbondante S, Yang CY, Hsu HM, Yu JC, Chang KJ, Shew JY, Lee EY, Lee WH. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A. 2013;110:12331–12336. doi: 10.1073/pnas.1222684110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009;119:1510–1517. doi: 10.1161/CIRCULATIONAHA.108.827477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 6.Haupt CM, Alte D, Dörr M, Robinson DM, Felix SB, John U, Völzke H. The relation of exposure to shift work with atherosclerosis and myocardial infarction in a general population. Atherosclerosis. 2008;201:205–211. doi: 10.1016/j.atherosclerosis.2007.12.059. [DOI] [PubMed] [Google Scholar]
- 7.Casey BJ, Somerville LH, Gotlib IH, Ayduk O, Franklin NT, Askren MK, Jonides J, Berman MG, Wilson NL, Teslovich T, Glover G, Zayas V, Mischel W, Shoda Y. Behavioral and neural correlates of delay of gratification 40 years later. Proc Natl Acad Sci U S A. 2011;108:14998–15003. doi: 10.1073/pnas.1108561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, Saer B, Begley N, Poolman T, Pariollaud M, Farrow S, DeMayo F, Hussell T, Worthen GS, Ray D, Loudon A. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med. 2014;20:919–926. doi: 10.1038/nm.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAlpine CS, Swirski FK. Circadian influence on metabolism and inflammation in atherosclerosis. Circ Res. 2016;119:131–141. doi: 10.1161/CIRCRESAHA.116.308034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anea CB, Cheng B, Sharma S, Kumar S, Caldwell RW, Yao L, Ali MI, Merloiu AM, Stepp DW, Black SM, Fulton DJ, Rudic RD. Increased superoxide and endothelial NO synthase uncoupling in blood vessels of Bmal1-knockout mice. Circ Res. 2012;111:1157–1165. doi: 10.1161/CIRCRESAHA.111.261750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 12.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yung LM, Sánchez-Duffhues G, Ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. 2015;108:278–287. doi: 10.1093/cvr/cvv221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krenning G, Barauna VG, Krieger JE, Harmsen MC, Moonen JR. Endothelial plasticity: shifting phenotypes through force feedback. Stem Cells Int. 2016;2016:9762959. doi: 10.1155/2016/9762959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang X, Guo D, Lin C, Shi Z, Qian R, Fu W, Liu J, Li X, Fan L. hCLOCK causes Rho-kinasemediated endothelial dysfunction and NF-kappaB-mediated inflammatory responses. Oxid Med Cell Longev. 2015;2015:671839. doi: 10.1155/2015/671839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J, Zhou X, Tang Q, Yu R, Yu S, Long Y, Cao C, Han J, Shi A, Mao JJ, Chen X, Chen L. BMAL1 deficiency contributes to mandibular dysplasia by upregulating MMP3. Stem Cell Rep. 2018;10:180–195. doi: 10.1016/j.stemcr.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang X, Guo D, Lin C, Shi Z, Qian R, Fu W, Liu J, Li X, Fan L. hCLOCK induction by hypoxia promotes inflammatory responses by activating the NF-κB pathway. Mol Med Rep. 2017;15:1401–1406. doi: 10.3892/mmr.2017.6127. [DOI] [PubMed] [Google Scholar]
- 19.Tang H, Zhu M, Zhao G, Fu W, Shi Z, Ding Y, Tang X, Guo D. Loss of CLOCK under high glucose upregulates ROCK1-mediated endothelial to mesenchymal transition and aggravates plaque vulnerability. Atherosclerosis. 2018;275:58–67. doi: 10.1016/j.atherosclerosis.2018.05.046. [DOI] [PubMed] [Google Scholar]
- 20.Moreno PR. Vulnerable plaque: definition, diagnosis, and treatment. Cardiol Clin. 2010;28:1–30. doi: 10.1016/j.ccl.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Redgrave JN, Gallagher P, Lovett JK, Rothwell PM. Critical cap thickness and rupture in symptomatic carotid plaques: the oxford plaque study. Stroke. 2008;39:1722–1729. doi: 10.1161/STROKEAHA.107.507988. [DOI] [PubMed] [Google Scholar]
- 22.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–4528. doi: 10.1172/JCI82719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 24.Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129:692–703. doi: 10.1161/CIRCULATIONAHA.113.003734. [DOI] [PubMed] [Google Scholar]
- 25.Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492–496. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- 26.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Boström KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong D, Lee MA, Li Y, Yang DK, Kho C, Oh JG, Hong G, Lee A, Song MH, LaRocca TJ, Chen J, Liang L, Mitsuyama S, D’Escamard V, Kovacic C, Kwak TH, Hajjar RJ, Park WJ. Matricellular protein CCN5 reverses established cardiac fibrosis. J Am Coll Cardiol. 2016;67:1556–1568. doi: 10.1016/j.jacc.2016.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ, Krenning G, Harmsen MC. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res. 2015;108:377–386. doi: 10.1093/cvr/cvv175. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, Nelson DL, Ma K, Moore DD, Yechoor VK. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stressinduced beta-cell failure in mice. Mol Cell Biol. 2013;33:2327–2338. doi: 10.1128/MCB.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khapre RV, Kondratova AA, Susova O, Kondratov RV. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle. 2011;10:4162–4169. doi: 10.4161/cc.10.23.18381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14:70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Napoli C, Quehenberger O, De Nigris F, Abete P, Glass CK, Palinski W. Mildly oxidized low density lipoprotein activates multiple apoptotic signaling pathways in human coronary cells. FASEB J. 2000;14:1996–2007. doi: 10.1096/fj.99-0986com. [DOI] [PubMed] [Google Scholar]
- 33.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassègue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 34.Benn A, Bredow C, Casanova I, Vukičević S, Knaus P. VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J Cell Sci. 2016;129:206–218. doi: 10.1242/jcs.179960. [DOI] [PMC free article] [PubMed] [Google Scholar]