

Abstract
Heart failure caused by myocardial infarction is a common cardiovascular disease with high mortality rate. Myocardial mitophagy is involved in the process of occurrence and development of heart failure. In this study, we aimed to investigate the effects of Xin Fu Kang (XFK) oral liquid on myocardial mitophagy in a rat model of advanced heart failure. The rat model of advanced heart failure was established by ligating the left anterior descending (LAD) artery for eight weeks. Captopril and XFK were given by gavage separately. Cardiac function and myocardial mitochondrial ultrastructure were observed. Co-localization of mitophagy-related proteins was observed by fluorescence microscopy. Quantitative polymerase chain reaction (qPCR) and western blotting were performed for mRNA and protein level detection, respectively. Compared with the sham group, advanced heart failure group showed a significant reduction in cardiac function with destruction of myocardial mitochondrial structure. Co-localization between mitophagy-related proteins (parkin, p62, and LC3) and mitochondria increased significantly. The mRNA and protein levels of pink1, parkin, p62, and LC3 indicated that excessive mitophagy was observed in the rat model of advanced heart failure. XFK intervention could regulate pink/parkin pathway and inhibit excessive mitophagy.
Keywords: Mitophagy, pink1/parkin, myocardial infarction, heart failure, Xin Fu Kang oral liquid
Introduction
In recent years, the incidence of coronary heart disease has increased drastically. Myocardial infarction has become one of the major causes of heart failure. In the early stage of myocardial infarction, the body may regulate myocardial contractile function through neural-humoral regulation to maintain cardiac output and blood supplement. However, prolongation of the disease leads to gradual evolution into chronic heart failure, and the compensatory mechanism would evolve into decompensation, with decline in cardiac function and a series of ventricular remodeling phenomena [1], such as cardiomyocyte hypertrophy, apoptosis, myocardial fibrosis, and ventricular cavity expansion [2]. One important basis of ventricular remodeling is myocardial energy metabolism remodeling. It is especially important to ensure the quantity and quality of mitochondria, as they are the main sites for myocardial energy metabolism. Autophagy plays an important role in cell metabolism. Studies have shown that autophagy occurs when cells are under stress conditions, such as starvation, ischemia, hypoxia, oxidative stress, and digestion of useless or damaged organelles and macromolecules for cell survival [3,4]. Mitophagy is the process of selective removal of damaged mitochondria by maintaining the balance of mitochondrial mass and quantity [5]. It is closely related to mitochondrial fission. Damaged mitochondria produce more than 10 times reactive oxygen species (ROS) than healthy mitochondria, and large amounts of ROS can directly damage mitochondrial proteins and DNA and aggravate mitochondrial dysfunction. Mitochondrial fission allows the irreparable part of damaged mitochondrial proteins and DNA to be redistributed and eliminated by mitophagy so that the internal environment remains stable. Current research suggests that autophagy is a double-edged sword. A moderate stimulus within a certain period will enhance mitophagy, thereby recycling macromolecules, which subsequently ensures the fine quality of mitochondria and simultaneously reduces the level of oxidative stress. However, prolonged strong stimulus will inevitably induce excessive mitophagy leading to excessive consumption of mitochondria and macromolecules, thereby affecting energy metabolism and even inducing cell death (type II programmed death) [6,7]. Therefore, the regulation of mitochondria especially in mitophagy would be a new therapeutic approach for myocardial protection. Currently, some mitochondria-targeted drugs such as permeability transition pore inhibitors, nitric oxide analogs, antioxidants, potassium channel openers, and metabolic regulators have been studied in various clinical trials [8,9]. Nevertheless, many of them have been shown to fail, while other drugs are still under study [10,11].
Xin Fu Kang (XFK) is a traditional Chinese medicine that has advantages in the clinical treatment of heart failure. Our previous studies have confirmed that XFK could improve mitochondrial respiratory enzyme activity and myocardial energy metabolism in rat model of heart failure [12,13]. In addition, it plays important roles in mitochondrial integrity, mitochondrial respiratory function, mitochondrial proteome, and mitochondrial endometrial proteomics. Therefore, in this study, we selected XFK as the intervention drug to explore its effect on cardiac mitophagy in rats with heart failure after myocardial infarction. The findings of this study can provide evidence and new directions for mechanism research in advanced heart failure.
Materials and methods
XFK oral liquid analysis
XFK oral liquid was analyzed by using 1200 HPLC system (Agilent, California, US), consisting of a quaternary pump, a diode-array detector (DAD) detector, a thermostat column oven, and an Agilent SB C18 column. All the reference substances were purchased from the National Institutes for Food and Drug Control (Beijing, China) or Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). The purity of all references was more than 95%.
Animal experiments
One hundred and twenty male Sprague-Dawley (SD) rats (200-250 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The rats were housed in a temperature-controlled environment (22-24°C) with 12 hours light/dark cycles, relative humidity of 45-55%, and free access to rodent chow and purified water. All rats in this study received humane care according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and was supervised by the Ethics Committee of Guang’anmen Hospital of China Academy of Chinese Medical Sciences.
The rats were randomly divided into two experimental groups: sham and myocardial infarction (MI) operation groups. The rats in MI group were anesthetized with 1% pentobarbital sodium salt (3.5 mL/kg) by intraperitoneal injection. Tracheal intubation was performed to attach a breathing machine, and the respiration rate was maintained at 80-90/s. A horizontal incision was made between the third and fourth rib of the left chest to open the chest and expose the heart. The left anterior descending (LAD) artery was ligated with 6-0 absorbable suture at 2 mm below the left auricular appendage. Upon ligation, the anterior wall of the ventricle turned pale. In addition, the ECG lead II showed a significant uplift of ST segment. The operation of rats in sham group was performed in the same way, except that the suture was placed at the LAD without ligation. The above experimental procedure was performed based on a protocol described previously [14]. The rats in MI group were randomly divided into the following five groups: model; captopril; XFK-L; XFK-M; and XFK-S groups. The rats in captopril group were given captopril (100 mg/kg) by gavage, while those in XFK-L, XFK-M, and XFK-S groups were given XFK by gavage at doses of 10.8, 5.4, and 2.7 g/kg, respectively.
Echocardiography
Echocardiography was performed under anesthesia at 8 weeks after ligation according to a previous study [15]. The rats were placed in the proper posture after the thoracic walls were shaved clean. Two-dimensional and M-mode echocardiography was used to assess the morphology and function of the heart with an 8-MHz transducer connected to HP5500 color Doppler ultrasound imaging instrument (Agilent, California, US). The left ventricular end-diastolic diameter (LVIDd), left ventricular end-systolic diameter (LVIDs), fractional shortening (FS), and ejection fraction (EF) were recorded. All parameters were obtained from the mean values of three cardiac cycles.
Transmission electron microscopy
The left ventricular myocardium was cut into 1 mm3 cubes, treated with 2.5% glutaraldehyde for 24 h at 4°C, and immersed in 1% osmium tetroxide. Then, it was dehydrated in a graded ethanol series and then embedded. The specimens were cut into 60-nm ultrathin sections and double-stained with uranyl acetate and lead citrate. The microstructures of mitochondria and mitochondrial autophagosomes were photographed using H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
Immunofluorescence
The hearts were removed after thoracotomy and irrigated clean with cold saline, and then immediately made into frozen sections (4 mm). The frozen tissue section was fixed in 4% paraformaldehyde at 4°C for 15 min and then blocked with donkey serum for 1 h. Anti-COX IV (1:300; Abcam, Shanghai, China) was coupled with Anti-parkin (1:200; Abcam), Anti-p62 (1:200; Abcam), or Anti-LC3 (1:200; Abcam) primary antibody, respectively, and incubated overnight at 4°C. After washing with phosphate-buffered saline (PBS) solution, the sections were incubated with secondary antibodies (1:200; Life Technologies, California, US) for 1 h. Finally, the slides were mounted with mounting medium (containing DAPI) and visualized under epifluorescence microscope (Nikon, Tokyo, Japan). Three sections were randomly selected from three rats in each group for examination. Protein co-localization was analyzed by Image-Pro Plus Version 6.0 (IPP).
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA samples were extracted from heart tissues using Trizol Reagent (Invitrogen, California, US). Then, first-strand cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, California, US) according to the manufacturer’s protocols. The cDNA product was stored at -80°C before use. Quantitative RT-PCR was performed using the Step OneTM real-time PCR System (Applied Biosystems, California, US). For mRNA detection, Power SYBR® Green PCR Master Mix and Power SYBR® Green RT-PCR Reagents Kit (Life Technologies, California, US) were used according to the manufacturer’s instructions. All samples were run in triplicate, and the obtained values were represented by average. The relative expression levels of each gene were calculated using the 2-ΔΔCt method [16] relative to the reference expression of GAPDH. The primer sequences used for PCR are listed as follows: pink1 forward primer: 5’-CAAGCAAGTGTCTGACCCAC-3’; pink1 reverse primer: 5’-GCTTCATACACAGCGGCATT-3’; parkin forward primer: 5’-ACCCACCTA-CCACAGCTTTT-3’; parkin reverse primer: 5’-CAAGGTGAGGGTTGCTTGTC-3’; p62 forward primer: 5’-ATGGACATGGGGAGCTTCAA-3’; p62 reverse primer: 5’-GTGCTCTCTGTATGCTCCCT-3’; LC3 forward primer: 5’-AGAGCGATA-CAAGGGTGAGA-3’; LC3 reverse primer: 5’-CTTCAGAGATGGGTGTGGAC-3’; GAPDH forward primer: 5’-AAGGGCTCATGACCACAGTC-3’; GAPDH reverse primer: 5’-GGATGCAGGGATGATGTTCT-3’.
Western blotting
Total protein samples were extracted from heart tissues using RIPA lysis buffer (Solarbio, Beijing, China). Equal amounts of protein were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membrane was then blocked with 5% powdered milk at room temperature for 1 h, followed by incubation with primary antibodies (Abcam) at a ratio of 1:1000 (for pink1, parkin, p62, and GAPDH) and 1:500 (for LC3) overnight at 4°C. After washing and incubation with a goat-anti-rabbit secondary antibody conjugated to horseradish peroxidase (HRP) at a ratio of 1:10000, protein bands were detected by Bio-Rad Chemi-DocXRS (Bio-Rad Laboratories, California, US).
Statistical analysis
Data analysis was performed using GraphPad Prism 5.0 software. All data are expressed as mean ± standard error (SD). Statistical analysis was performed using Student’s t-test. A two tailed P<0.05 was considered significant.
Results
XFK analysis
The results are shown in Table 1. The contents of eight representative active reference substances, namely Astragaloside, Salvianic acid A sodium, Icariin, Ligustilide, Ligustrazine, Ginsenoside Rg1, Hesperidin, and Neohesperidin are listed.
Table 1.
The analyzed representative active reference substances contents for XFK oral liquid originated from each herbal medicine
| Latin name | Chinese name | Reference substance | Content (mg/10 ml) |
|---|---|---|---|
| Astragalusmembranaceus (Fisch.) Bge | Huang Qi | Astragaloside | 1.93 |
| Salvia miltiorrhiza Bge. | Dan Shen | Salvianic acid A sodium | 0.53 |
| Epimedium Linn. | Yin Yang Huo | Icariin | 14.35 |
| Angelica sinensis (Oliv.) Diels | Dang Gui | Ligustilide | 0.66 |
| Ligusticumchuanxiong Hort. | Chuan Xiong | Ligustrazine | 1.48 |
| Panax ginseng C. A. Mey. | Ren Shen | Ginsenoside Rg1 | 0.40 |
| Citrus sinensis (L.) Osbeck | Zhi Shi | Hesperidin | 16.11 |
| Neohesperidin | 1.00 |
Effects of XFK on cardiac function
Echocardiography was performed to evaluate cardiac function after MI or sham operation for 8 weeks. Figure 1A shows the results of LVIDd and LVIDs for all groups, and EF and FS are shown in Figure 1B. Compared with the sham group, the model group showed a significant decrease in EF and FS (P<0.01) and significant increase in LVIDd and LVIDs (P<0.01, P<0.05). Captopril and XFK reduced LVIDd (P>0.05) and LVIDs (P<0.05, P>0.05) and significantly increased EF (P<0.05, P<0.01) and FS (P<0.05, P>0.05). The above results show that XFK and captopril can effectively improve cardiac function and reduce left ventricular remodeling in rats with heart failure.
Figure 1.
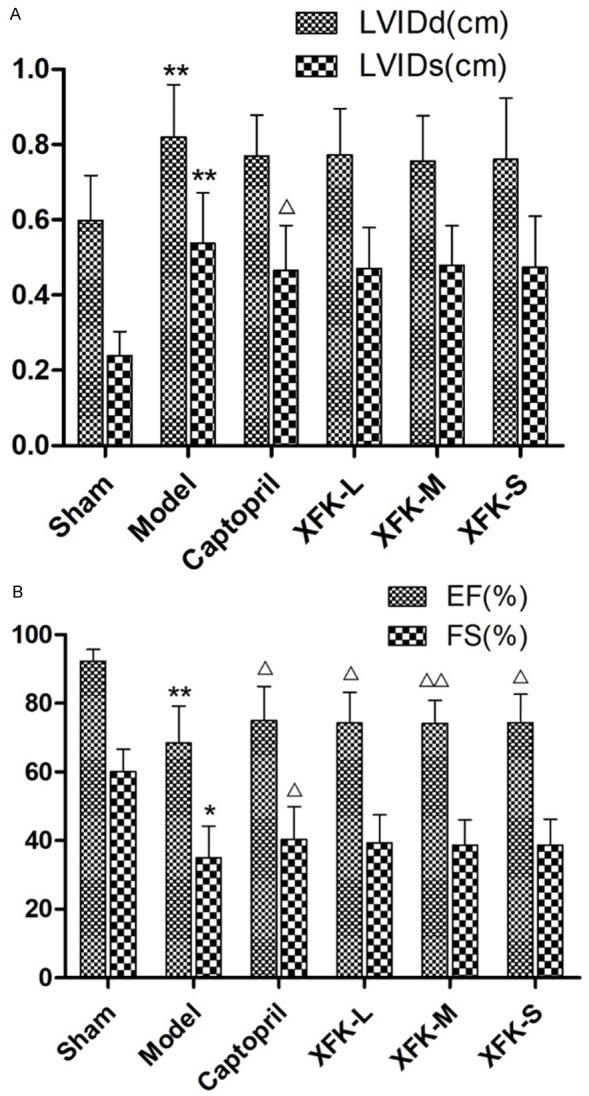
Echocardiography results after MI or sham operation for 8 weeks. A. LVIDd and LVIDs results for all groups. B. EF and FS measurements for all groups. Values are expressed as mean ± SD, n = 10. Versus Sham,**P<0.01 and versus Model, ΔP<0.05.
Effects of XFK on mitochondrial ultrastructure
Changes in mitochondrial ultrastructure are shown in Figure 2. Compared with the sham group, rats in the model group displayed myocardial myofibril disorder, mitochondrial swelling, cristae decrease or mitochondrial vacuolation, and more obvious mitochondrial autophagosomes. After drug intervention, these damages reduced to a certain degree. Mitochondrial damage was reduced and a relatively normal myocardial structure was observed (Figure 2). The results demonstrate the protective effects of captopril and XFK on myocardial ultrastructure.
Figure 2.

Mitochondrial ultrastructural changes in left ventricular tissue. Representative images (20000×) and (30000×) in each group are shown. Scale = 500 nm.
Effects of XFK on parkin, p62, and LC3 co-localization with mitochondria
Immunofluorescence microscopy was conducted to determine parkin, p62, and LC3 co-localization with mitochondria. The co-localization of parkin, p62, and LC3 with COX IV is shown in Figure 3. MI increased the co-localization of parkin, p62, and LC3 with COX IV significantly (P<0.05), while drug intervention reduced it to a certain degree (P<0.05, P<0.01, P>0.05). These results suggest that XFK and captopril can inhibit the formation of autophagosomes via pink1/parkin pathway.
Figure 3.

Detection of mitophagy-related protein co-localization with mitochondria in myocardium. Representative images (400×) are shown. Nuclei were stained with DAPI (blue). COX IV which is located in mitochondrial inner membrane was dyed in green while Parkin, p62 and LC3 were dyed in red. The co-located position turned yellow after being merged. (A) parkin co-located with COX IV, (B) p62 co-located with COX IV, (C) LC3 co-located with COX IV (D), quantitative analysis of the amount of co-localization presented by Pearson’s correlation. Values are expressed as mean ± SD, n = 9. Versus Sham, *P<0.05 and versus Model, ΔP<0.05, ΔΔP<0.01.
Effects of XFK on mitophagy-related gene expression
Based on the data in Figure 4, the mRNA level of pink1 reduced significantly (p<0.01) and the mRNA levels of parkin, p62, and LC3 increased significantly (p<0.01, p<0.05) in the model group compared with the sham group. Captopril and XFK can increase the mRNA level of pink1 (P<0.05, P>0.05) and decrease parkin, p62, and LC3 mRNA levels (P<0.01, P<0.05, P>0.05). Compared with the sham group, the model group showed a decrease in protein levels of pink1, parkin, and p62 (P<0.01) and a significant increase in the ratio of LC3II/LC3I (P<0.5). Captopril and XFK increased the protein levels of pink1, parkin, and p62 (P<0.01, P<0.05, P>0.05) and decreased LC3II/LC3I ratio (P<0.05, P>0.05). The above results confirm the results of immunofluorescence to a certain extent.
Figure 4.
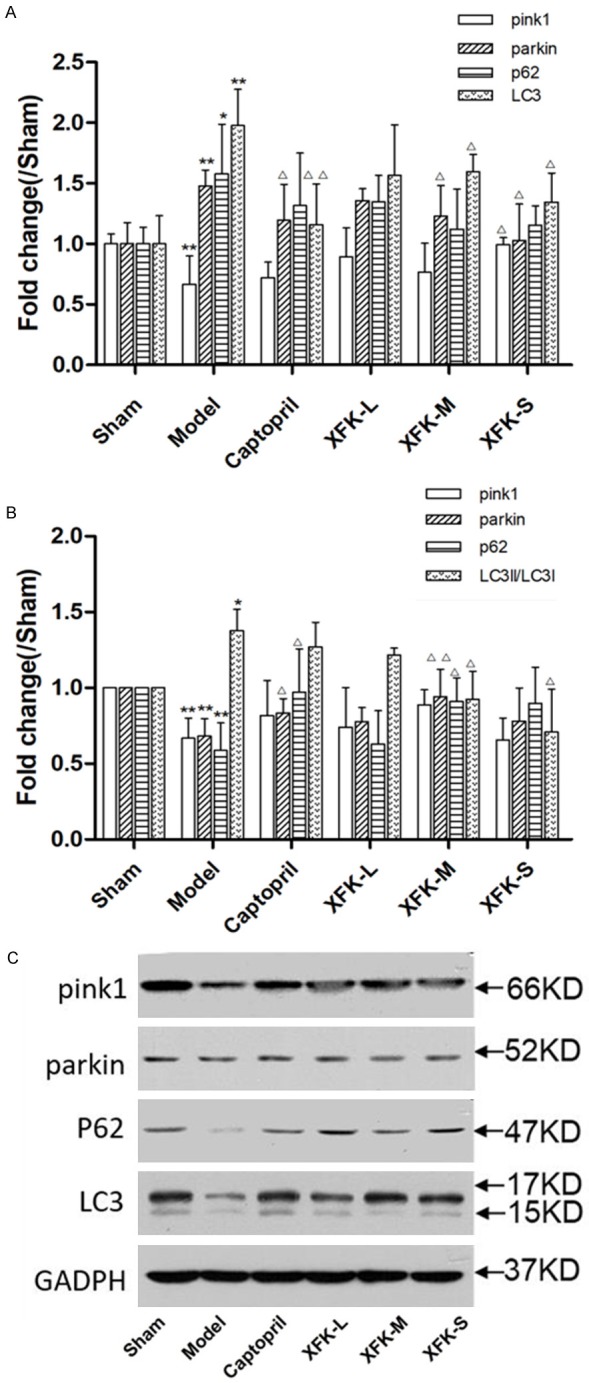
A. mRNA Levels of pink1, parkin, p62 and LC3 in each group; B and C. Expression of protein of pink1, parkin, p62 and LC3. Mean expression in each treated group is shown as increase/decrease compared with mean expression in Sham group which has been ascribed an arbitrary value of 1. The values are expressed as mean ± SD, n = 5. Versus Sham, **P<0.01, *P<0.05 and versus Model, ΔP<0.05, ΔΔP<0.01.
Discussion
In this study, we explored the effect of XFK on myocardial mitophagy in rats with heart failure after myocardial infarction. Following decompensation caused by myocardial infarction, ventricular remodeling increased around the infarct area and cardiac function reduced significantly. Left ventricular systolic function is a strong predictor of cardiovascular morbidity and mortality [17]. XFK and captopril show ameliorating effects on cardiac function in terms of EF%, FS%, and left ventricular cavity size (LVIDd, LVIDs).
Mitophagy is involved in various cardiovascular disease processes; however, the underlying mechanism is not yet clear. Some studies [18,19] have confirmed that mitophagy is relatively weakened and mitochondrial clearance is blocked in the early stages of heart failure (3-4 weeks after MI surgery), and these changes can be ameliorated by drug intervention. Nevertheless, there are only few studies investigating mitophagy in advanced heart failure (8 weeks after MI surgery). The findings of this study indicate excessive mitophagy in the myocardium of rats with advanced heart failure after myocardial infarction.
From the ultrastructure of myocardial tissue, it was observed that the number of mitochondria in the model group significantly reduced compared with that in the sham group, and there were more autophagosomes and monolayer membrane structures around the mitochondria. In each drug intervention group, various degrees of mitophagy occurred, but it was not as serious as the model group.
Since mitochondria are both a major source and major target of ROS [20,21], mitochondria are over-damaged by long-term excessive ischemia-hypoxia stimulation, thus triggering excessive mitophagy and causing an imbalance in the number of mitochondria. These findings are consistent with those of previous studies [7,22]. It has been shown that oxidative stress products, such as ROS can activate the pink1/parkin-dependent mitophagy pathway in degenerative and ischemic diseases [23,24]. Under healthy conditions, pink1 undergoes degradation after being transported from the mitochondrial outer membrane to the mitochondrial inner membrane, thus maintaining a low expression level. When mitochondria are damaged, the mitochondrial membrane potential changes, resulting in the blockade of pink1 transport. Excess pink1 accumulates on the mitochondrial outer membrane and recruits parkin from the cytosol to the mitochondrial outer membrane [25,26]. Parkin connects the receptor protein SQSTM1 (ubiquitin-binding adapter p62) to the mitochondria by ubiquitination of various substrates, and then binds to the phagocytic membrane surface protein LC3 to induce subsequent autophagosome formation [27,28]. In this study, immunofluorescence double labeling technique was used to observe the co-localization of parkin, p62, LC3, and mitochondrial inner membrane protein COX IV, respectively, to visually analyze the changes in pink1/parkin-dependent mitophagy pathway in the myocardium. From images and co-localization analysis, it was observed that co-localization of parkin, p62, and LC3 with mitochondria significantly increased in the model group. Drug intervention had an inhibitory effect on parkin, p62, and LC3 co-localization. Among them, the effect on LC3 was more significant, indicating that XFK significantly inhibits excessive myocardial mitophagy in rats with advanced heart failure through mechanisms other than pink1/parkin pathway.
The mRNA and protein levels of pink1, parkin, p62, and LC3 in the tissues were detected. The mRNA levels of pink1 significantly decreased, while the mRNA levels of parkin, p62, and LC3 significantly increased in model group compared with the sham group. Meanwhile, pink1, parkin, and p62 protein levels decreased and the ratio of LC3II/LC3I increased, indicating that pink1/parkin pathway plays an important role in excessive myocardial mitophagy and over-consumption of protein in rats with advanced heart failure. The transcription levels of parkin, p62, and LC3 genes increased significantly owing to the excessive demand of mitophagy. The decrease in transcription level of pink1 gene is linked to the negative feedback generated by overaccumulation of pink1 on the outer membrane of damaged mitochondria. In addition, some studies [29,30] showed that decrease in p62 expression and increase in LC3II/LC3I ratio, indicating unobstructed autophagy. However, some reports suggest that p62 protein expression is dependent on autophagic degradation, transcriptional upregulation, and availability of lysosomal-derived amino acids (synthetic materials), which are not always negatively correlated with autophagy [31].
In conclusion, the protein levels of pink1, parkin, p62, and LC3 decreased in the myocardium of rats with advanced heart failure, while the ratio of LC3II/LC3I increased. These findings are related to excessive autophagy and excessive protein degradation. Pharmacological interventions, especially XFK, could correct the transcriptional levels and protein expression levels of pink1, parkin, p62, and LC3 markedly, thereby playing a regulatory role through pink1/parkin pathway. However, further studies are warranted to elucidate the specific underlying mechanisms.
Acknowledgements
This research was supported by National Natural Science Foundation of China (81673971).
Disclosure of conflict of interest
None.
References
- 1.St John Sutton M, Linde C, Gold MR, Abraham WT, Ghio S, Cerkvenik J, Daubert JC REVERSE Study Group. Left ventricular architecture, long-term reverse remodeling, and clinical outcome in mild heart failure with cardiac resynchronization: results from the REVERSE trial. JACC Heart Fail. 2017;5:169–178. doi: 10.1016/j.jchf.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol. 2011;57:9–17. doi: 10.1016/j.jacc.2010.08.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho YY, Kim DJ, Lee HS, Jeong CH, Cho EJ, Kim MO, Byun S, Lee KY, Yao K, Carper A, Langfald A, Bode AM, Dong Z. Autophagy and cellular senescence mediated by Sox2 suppress malignancy of cancer cells. PLoS One. 2013;8:e57172. doi: 10.1371/journal.pone.0057172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 2017;284:183–195. doi: 10.1111/febs.13820. [DOI] [PubMed] [Google Scholar]
- 6.Zhang SW, Feng JN, Cao Y, Meng LP, Wang SL. Autophagy prevents autophagic cell death in Tetrahymena in response to oxidative stress. Dongwuxue Yanjiu. 2015;36:167–73. [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 8.Sharov VG, Todor AV, Imai M, Sabbah HN. Inhibition of mitochondrial permeability transition pores by cyclosporine a improves cytochrome C oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail Rev. 2005;10:305–310. doi: 10.1007/s10741-005-7545-1. [DOI] [PubMed] [Google Scholar]
- 9.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Walter L, Miyoshi H, Leverve X, Bernard P, Fontaine E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic Res. 2002;36:405–412. doi: 10.1080/10715760290021252. [DOI] [PubMed] [Google Scholar]
- 11.Novgorodov SA, Gudz TI, Obeid LM. Long-chain ceramide is a potent inhibitor of the mitochondrial permeability transition pore. J Biol Chem. 2008;283:24707–24717. doi: 10.1074/jbc.M801810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YF, Cao XB, Xu SL, Zhang G, He J, Hu Y, Cui Y, Yang M. Effects of Xinfukang oral liquid on myocardial energy metabolism in chronic pressure overload rats. Acta Academiae Medicinae Militaris Tertiae. 2009;31:1720–1723. [Google Scholar]
- 13.Wang W, Cao X, Xu S, He J, Hu Y. e0291 effects of Xinfukang oral liquid on the activities of respiratory enzyme in experimental congestive heart failure rats. Heart. 2010;96:A91. [Google Scholar]
- 14.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 15.Shang HH, Qian YQ, Bao JX, Zhou XD, Zhu YS. Detection of cardiac structure and function in rats by echocardiography. Chinese Heart Journal. 2000;12:87–89. [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Roberts AW, Clark AL, Witte KK. Review article: left ventricular dysfunction and heart failure in metabolic syndrome and diabetes without overt coronary artery disease--do we need to screen our patients? Diab Vasc Dis Res. 2009;6:153–163. doi: 10.1177/1479164109338774. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Xiemuziya M, Jiang Y, Liu L. Parkin regulates mitochondrial autophagy after myocardial infarction in rats. Med Sci Monit. 2016;22:1553–1559. doi: 10.12659/MSM.898722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aisa Z, Liao GC, Shen XL, Chen J, Li L, Jiang SB. Effect of autophagy on myocardial infarction and its mechanism. Eur Rev Med Pharmacol Sci. 2017;21:3705–3713. [PubMed] [Google Scholar]
- 20.Kim I, Rodriguezenriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherzshouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Qiao S, Xie H, Wang C, Wu X, Liu H, Liu C. Delayed anesthetic preconditioning protects against myocardial infarction via activation of nuclear factor-κB and upregulation of autophagy. J Anesth. 2013;27:251–260. doi: 10.1007/s00540-012-1494-3. [DOI] [PubMed] [Google Scholar]
- 23.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Autophagy. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32:431–436. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 30.Chen S, Le WD, Zhang XJ, Le WD. Why should autophagic flux be assessed? Acta Pharmacol Sin. 2013;34:595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]