

Abstract
Many cervical cancer (CC) patients suffer from cancer invasion and lymph node metastasis, resulting in poor therapeutic outcome. Evidence has indicated the involvement of misexpressed high-mobility group AT-hook 2 (HMGA2) in poor survival of cancer patients. This study hereby aims to investigate the role of HMGA2 in CC cell biological functions via the ATR/Chk1 signaling pathway. The cell line with the highest HMGA2 expression was selected to establish cell lines with wild-type and stable HMGA2 silencing. The underlying regulatory mechanisms of HMGA2 in CC cells were analyzed with the treatment of the ATR/Chk1 signaling pathway activator, inhibitor, shRNA against HMGA2 or pcDNA-HMGA2 plasmids, followed by quantification of expression levels of ATR, Chk1, Bcl-2, Bax, MMP-2, MMP-9, E-cadherin and N-cadherin. CC cell apoptosis, proliferation, migration, invasion and lymph node metastasis in nude mice were evaluated. The HeLa cell line with the highest HMGA2 expression was selected. HMGA2 inhibited the activation of the ATR/Chk1 signaling pathway. Notably, HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited epithelial mesenchymal transition (EMT), CC cell proliferation, invasion, migration, tumorigenicity and lymph node metastasis while promoting apoptosis, indicated by reduced expression of Bcl-2, MMP-2, MMP-9 and N-cadherin, with increased expression of E-cadherin and Bax. Collectively, our study provides evidence that HMGA2 gene silencing inhibits the activation of the ATR/Chk1 signaling pathway, whereby repressing EMT, proliferation, migration and invasion of CC cells and lymph node metastasis, and promoting CC cell apoptosis.
Keywords: HMGA2, ATR/Chk1 signaling pathway, cervical cancer, epithelial mesenchymal transition, lymph node metastasis
Introduction
Cervical cancer (CC) is the second most commonly occurring cancer among women across the world, with 500,000 new cases and 300,000 deaths reported in relation to CC worldwide on an annual basis [1]. CC usually arises in the uterine cervix transformation zone, where physiological metaplasia happens, furthermore, cervical intraepithelial neoplasia is caused commonly by human papillomavirus (HPV) infection in young female population after the set-about of sexual activity [2]. Radical hysterectomy, radiotherapy, or radiochemotherapy have been recommended as influential treatment choices to date for patients with CC [3]. Though patients with early-stage or locally advanced CC may be cured by means of radical surgery and/or chemoradiotherapy, those with metastatic cancers and those with persistent or recurrent CC after chemoradiotherapy have limited treatment options [4]. Invasion, metastasis and epithelial-mesenchymal transition (EMT) remain critical issues in CC treatment [5-7]. Recent studies have pointed out that molecular therapeutic targets offer potential in suppressing such biological activities of CC cells as invasion [8], migration [9] and lymph node metastasis [10].
High mobility group AT-hook 2 (HMGA2) is a chromatin remodeling factor that can bind to the region enriched with AT in DNA, playing a regulatory role in chromatin architecture which can either enhance or weaken the function of transcriptional enhancers [11]. HMGA2 has been found primarily expressed in mesenchyme prior to its differentiation, functions as a regulatory role to proliferation and differentiation of mesenchyme [12]. More advanced malignancy of breast cancer [13], decreased survival rates of patients with colorectal cancer [14], and poor prognosis in lung adenocarcinoma [15] have been demonstrated in relation to mis-expression of HMGA2. Ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) are crucial members of the phosphatidylinositol 3-kinase-related kinase family of serine/threonine protein kinases, compassing mammalian target of rapamycin, DNA-dependent protein kinase catalytic subunit, and suppressor of morphogenesis in genitalia [16]. These protein kinases have been revealed to regulate the DNA damage response to cell survival, differentiation, metabolism, proliferation, motility as well as nonsense-mediated mRNA decay [17]. The ATR and Chk1 inhibitors have been proved by Manic et al. to kill tumor cells [18]. Correspondingly, a study has reported the use of inhibitors to ATR or Chk1 and HMGA2 in the treatment of HMGA2-positive human CC cells [19]. Thus, in this present study, we determined to investigate the role of HMGA2 in CC cell biological functions through regulating the ATR/Chk1 signaling pathway.
Methods and materials
Cell line selection
Human CC cell lines Hela, SiHa, C33a and Caski, and normal cervical cell line, Ect1/E6E7, (Medical and Science Research Center of Guangxi Medical University, Nanning, Guangxi, China) were cultured in a RPMI 1640 medium (Gibco, Glen Waverly, Victoria, Australia) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 mg/mL streptomycin in an incubator (Invitrogen Inc., Carlsbad, CA, USA) under a humidified atmosphere of 5% CO2 at 37°C. When cell confluence had reached 90%, cell passage began. The culture fluid was removed, and the specimen was washed by phosphate buffer saline (PBS) for 2 times and detached by 0.25% trypsin until the cells were in round shape and space appeared among cells. Then certain amount of FBS was added to terminate the detachment. A single cell suspension was prepared using a pipette gently. The expression of HMGA2 in CC cell lines was detected by reverse transcription quantitative polymerase chain reaction (RT-qPCR) and the cell line with the highest expression was selected for further research.
RT-qPCR
The total RNA was extracted by Trizol (15596026, Invitrogen, Carlsbad, CA, USA), and the integrity of RNA was verified by 1% agarose gel electrophoresis, while the concentration and purity of RNA were measured by Nano-Drop ND-1000 Spectrophotometer. The PrimeScript RT reagent Kit (RR047A, Takara, Tokyo, Japan) was adopted to reversely transcribe RNA into cDNA with reference to the instructions. The reaction condition was as follows: at 37°C for 15 min and at 85°C for 5 s; the system was set as 20 μL. The primers of HMGA2, ATR, Chk1, B-cell lymphoma-2 (Bcl-2), Bcl-2 associated protein X (Bax), matrix metalloproteinases-2 (MMP-2), MMP-9 and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were designed and synthesized by Shanghai Sangon Biotech Co., Ltd., (Shanghai, China) (Table 1). The reverse transcription system was 20 μL and conducted in accordance with the instructions of EasyScript First-Strand cDNA Synthesis SuperMix (AE301-02, Beijing TransGen Biotech Co., Ltd., Beijing, China). Reaction liquid was collected to perform RT-PCR in accordance with the instruction of SYBR®Premix Ex TaqTM II kit (TaKaRa, Dalian, Liaoning, China), and the reaction system (20 μL) consisted of 10 μL SYBR Premix, 2 μL cDNA, 0.6 μL forward primers, 0.6 μL reverse primer, and 6.8 μL Diethyl phosphorocyanidate (DEPC) water. The 7500 fluorescent quantitative PCR (ABI, Oyster Bay, NY, USA) was used for real-time quantitative RT-PCR and the reaction conditions were as follows: predenaturation at 95°C for 30 s, 40 cycles of denaturation at 95°C for 30 s, annealing for 20 s, and extension at 72°C for 30 s. The ratio of the expression of the target gene in the experimental group to the control group was expressed as 2-ΔΔCt, and the formula was as follows: ΔCt = CT (target gene) - CT (internal reference); ΔΔCT = ΔCtexperiment group - ΔCtcontrol group. Ct represented the number of amplification cycles when the real time fluorescence intensity of the reaction reached the preset threshold and when the amplification was in logarithmic growth. The expression of HMGA2, ATR, Chk1, Bcl-2, Bax, MMP-2, MMP-9 and GAPDH were determined and the experiment was repeated 3 times independently.
Table 1.
Primer sequence for RT-qPCR
| Target gene | Forward primer (5’-3’) | Reverse primer (5’-3’) |
|---|---|---|
| HMGA2 | ACCCAGGGGAAGACCCAAA | CCTCTTGGCCGTTTTTCTCCA |
| ATR | GGCCAAAGGCAGTTGTATTGA | GTGAGTACCCCAAAAATAGCAGG |
| Chk1 | ATATGAAGCGTGCCGTAGACT | TGCCTATGTCTGGCTCTATTCTG |
| Bcl-2 | GGTGGGGTCATGTGTGTGG | CGGTTCAGGTACTCAGTCATCC |
| Bax | CCCGAGAGGTCTTTTTCCGAG | CCAGCCCATGATGGTTCTGAT |
| MMP-2 | TACAGGATCATTGGCTACACACC | GGTCACATCGCTCCAGACT |
| MMP-9 | TGTACCGCTATGGTTACACTCG | GGCAGGGACAGTTGCTTCT |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
Notes: HMGA2, high-mobility group AT-hook 2; ATR, ATM- and Rad3-related; Chk1, checkpoint kinase 1; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2 associated protein X; MMP-2, matrix metalloproteinases-2; MMP-9, matrix metalloproteinases-9; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase.
Western blot analysis
Cells of each group were collected and added with RIPA lysate (P0013B, Shanghai Beyotime Biotechnology Co., Ltd., Shanghai, China), phenylmethanesulfonyl fluoride (PMSF) and phosphatase inhibitors. Following resuspension over gentle shaking, the cells were incubated on ice for 30 min and centrifuged for 10 min at 4°C (12000 r/min), and the supernatant was taken as the total protein of tissue or cell. The bicinchoninic acid (BCA) kit (Nanjing Beyotime Biotech Co., Ltd., Nanjing, Jiangsu, China) was employed to determine the protein concentration, and the concentration was adjusted into 4 µg/µL using PBS. Then, 30 µg cell total protein was taken to undergo sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to the nitrocellulose membrane using wet transfer method. The membrane was sealed in 5% skim milk powder (formulated with Tris-buffered Saline with Tween 20 (TBST)) for 1.5 h. In accordance with the instruction, corresponding antibodies were diluted. The primary rabbit anti-human antibodies were HMGA2 (ab97276, 1:500), ATR (ab2905, 1:1000), p-ATR (ab227851, 1:3000), Chk1 (ab40866, 1:10000), p-Chk1 (ab58576, 1:1000), Bcl-2 (ab32124, 1:1000), Bax (ab32503, 1:1000), MMP-2 (ab92536, 1:1000), MMP-9 (ab73734, 1:1000), and GAPDH (ab9485, 1:2500). All above antibodies were purchased from Abcam (Cambridge, MA, USA). The sealed nitrocellulose membrane was put into a plastic dish containing the antibodies above, and incubated overnight at 4°C over shaking. On the next day, the membrane was washed by TBST for 15 min × 3 times, and then incubated with diluted horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG (ab205718, 1:2000~1:50000) as the secondary antibody at room temperature for 2 h. The membrane was washed by TBST for 15 min × 3 times. Electrochemiluminescence (ECL) was added to develop color, and the sample was later photographed by SmartView Pro 2000 (UVCI-2100, Major Science, Saratoga, CA, USA). Quantity One Software was adopted to analyze the gray level of protein band [20].
Establishment of cell line with stable silencing of HMGA2
PLKO.1-purowas used as the lentiviral vector, and the synthesis of siRNA-HMGA2 was guided by the U6 promoter, and siRNA-HMGA2 had a puromycin resistance marker. The sense strand and anti-sense strands were annealed into double-strand, which was then connected with the lentivirus vector using T4 ligase. Next, 4.5 μL product was transformed into DH5α competent cell (TIANGEN Biotechnology, Co., Ltd., Beijing, China), which then underwent resistance screening on a Luria-Bertani (LB) plate containing purinomicin in an incubator at 37°C. The plasmid was extracted to be sequenced by Beijing Genomics Institute (Beijing, China) to verify whether the inserted sequence was right or not. Lentivirus was packed and produced at the virus titre of 106 TU (Sigma, St. Louis, MO, USA). One day before transfection, 2 × 105 HeLa cells were paved on a six-well plate to 60%~70%. Transfection was performed with 8 μL/mL coagel (Sigma, St. Louis, MO, USA) and 50 μL lentivirus added in each well. After 48 h of transfection, the plate was replaced by a RPMI 1640 medium containing 1 μg/mL puromycin. Every 2~3 days, the cells were observed and the medium was replaced. The process was terminated until the un-transfected cells were killed completely. When clone formed, the cells were amplified and cultured. The clone was isolated by limiting dilution method, and thus to acquire stable cell line. The cell line with stable silencing of HMGA2 was named as HMGA2-KD-HeLa.
Cell grouping and transfection
The wildtype HeLa cell line was classified into 5 groups: blank (cells pretreated with PBS but without any transfection), negative control (NC) (cells transfected with NC plasmid), 1,3-bis (2-chloroethyl)-1-nitrosourea (BCNU) (cells transfected with BCNU, the activator of ATR/Chk1 signaling pathway, which was dissolved in dimethyl sulfoxide to the concentration of 20 μM/L) (Sigma, St. Louis, MO, USA), caffeine (cells transfected with caffeine the inhibitor of ATR/Chk1 signaling pathway, which was dissolved in phosphate buffer to the concentration of 2 mM/L) (Sigma, St. Louis, MO, USA), and the sh-HMGA2 + BCNU (cells transfected with sh-HMGA2 plasmid + BCNU, the activator of ATR/Chk1 signaling pathway). The cell line with stable silencing of HMGA2 was classified into 4 groups: blank (cells pretreated with PBS but without any transfection), NC (cells transfected with NC plasmid), pcDNA-HMGA2 (cells transfected with pcDNA-HMGA2 plasmid) (Shanghai Hanbio Biotechnology Co., Ltd., Shanghai, China) and pcDNA-HMGA2 + caffeine (cells transfected with pcDNA-HMGA2 plasmid + caffeine 2 mM/L). All above plasmids were purchased from Dharmacon Co., Ltd. (Lafayette, CO, USA). The HeLa cells were inoculated in a six-well plate at the density of 3 × 105 cells/well and cultured in a new complete medium. When the cell confluence reached 50%~80%, the cells were transfected using lipofectamin 2000 (Invitrogen, Carlsbad, CA, USA) kit. Then, 250 μL serum-free Opti-MEM (Gibco, NY, USA) was used to dilute 4 μg target plasmid and 10 μL Lipofectamine 2000 for 5 min at the room temperature, respectively. The above two were mixed and incubated at room temperature for 20 min. The mixture was then added into the wells. Following incubation at 37°C and 5% CO2 for 6 h, the cells were further cultured in complete medium for another 48 h. Afterwards, the cells were collected.
Immunofluorescence
Trypsin was used to detach the tissue cells in logarithmic growth phase, followed by centrifugation. The sterilized cover slip (24 mm × 24 mm) was put in the medium for the inoculation of cell lines in each group. After 24 h, when the cell confluence reached 70%, the cells were allowed to crawl on the slip. The cells were washed by pre-cooled PBS and fixed with 4% formaldehyde for 20 min at room temperature. Following PBS washing for 5 min × 3 times, cells were sealed in 10% goat serum for 15 min, and added with rabbit anti-human E-cad-herin antibody (ab40772, 1:500) and N-cad-herin antibody (ab18203, 1:5000), both of which were purchased from Abcam, Cambri-dge, MA, USA. After incubation overnight in a 4°C refrigerator, the samples were put into a incubator at 37°C to rewarm for 45 min, and washed by PBS for 5 min × 3 times. Fluorescence labeled goat anti-rabbit IgG (ab205718, 1:2000~1:50000) was added for incubation as the secondary antibody avoiding the exposure to light at room temperature for 2 h. The cells were washed with PBS for 5 min × 3 times, and then stained with 4,6-diamidino-2-phenylindole (DAPI, 1:10, Vector Laboratories, Burlingam, CA) away from light for 5 min, followed by 2 times of PBS washing, 2 min per time. And then the cells were dehydrated with gradient ethanol, cleared with xylene, sealed with neutral gum and then detected under the microscope.
Uranyl acetate-lead citrate staining observed under transmission electron microscope (TEM)
Cells in each group were inoculated in a 24-well plate, and cultured for 72 h in medium containing 4 μM DDP, 8 μM UA and 4 μM DDP + 8 μM UA at 37°C with 5% CO2. The cells were centrifuged for 10 min at 1000 r/min, collected, immediately fixed with 2.5% glutaraldehyde for 1 h, and dissolved in 1% osmium oxide PBS at 4°C for 1 h. Later, the deposit of cells were embedded in water containing 2% agar and dehydrated with gradient ethanol. Afterwards, the cells were embedded in LR White gum and polymerized for 24 h at 60°C. The ultrathin sections were double stained with uranyl acetate and lead citrate. When dried out, the slices were observed under a TEM (Leica Microsystems, Wetzlar, Germany) and pictured [21].
5-ethynyl-2’-deoxyuridine (EdU) assay
Cells were inoculated in a 96-well plate for 48 h at the density of 1.6 × 105 cells/well, afterwards each well was added with 50 mM EdU to culture the cells for 4 h at 37°C. Cells were fixed with 4% formaldehyde for 15 min at room temperature, and cleared with 0.5% Triton X-100 at room temperature for 20 min. Following 3 times of PBS washing, each well was added with 100 mL Apollo® mixture, and the cells were further incubated for another 30 min at room temperature. Another 100 mL Hoechst33342 dye was added to stain the cells for 30 min, which was later observed and pictured under a fluorescence microscope (Olympus, Tokyo, Japan). The number of EdU positive cells (red blood cells) was measured by Image-Pro Plus (IPP) 6.0 software (Media Cybernetics, Bethesda, MD, USA). The incorporation rate of EdU represents the proportion of EdU positive cells in blue stained HeLa positive cells. The experiment was repeated 3 times to calculate the average value [22].
Scratch test
The back of the 6-well plate was marked with lines using a mark pen. Cells from each group were detached by 0.25% trypsin and the cell suspension was prepared. Then the number of cells was counted and the cells were inoculated into the 6-well plate at the density of 1 × 106 cells/well over gentle shaking. After inoculation of 24 h in a complete medium, the cells were further inoculated in a RPM1640 medium containing 10% FBS. A scratch was created using a 200 µL sterilized micropipette perpendicular to the mark at the back of 6-well plate. And then the cells were washed for 3 times and those damaged by the liquid transfer gun were removed. The rest of cells were moved into a serum-free medium and incubated in a 5% CO2 incubator at 37°C. And then the cells were observed at the time point of 0 h and 24 h under the microscope [20].
Transwell assay
Matrigel (Sigma, St. Louis, MO, USA) was allowed to stay overnight at 4°C to melt and diluted with precooled 4°C serum-free RPMI-1640 medium to the final concentration of 1 mg/mL on ice. And then diluted Matrigel was added into the bottom of upper chamber of Transwell (diameter: 8 µm) (80 μL/well) perpendicular to the center to make the matrigel distribute evenly. Following incubation for 4 h at 37°C, the cells were detached and washed with PBS and the serum-free RPMI-1640 medium, respectively. The serum-free RPMI-1640 medium was used to culture the suspended cells, the number of which was counted later and the concentration of which was adapted to 1 × 106 cells/mL. The lower chamber, i.e. the bottom of the 24-well plate, was added with 700 µL RPMI-1640 medium containing 10% FBS, while the upper chamber was added with cell suspension, followed by incubation for 24 h in an incubator. The chamber was taken out using a tweezers with care and liquid in the upper chamber was absorbed. The cells were fixed with 4% polyoxymethylene for 30 min at room temperature, and stained with 0.05% crystal violet for 30 min. The upper chamber was rinsed and immersed in water for several times and the cells in the upper chamber were removed with a wet cotton swab. After dried out, 10 fields from the upper chamber were randomly selected, of which the number of cells was counted under a microscope (× 200) (XSP-8CA, Shanghai Optical Instrument Factory, Shanghai, China), and the result was analyzed. The experiment was repeated 3 times.
Xenograft tumor in nude mice
A total of 30 male BALB/c nude mice (age: 4~6 weeks, weight: 18~20 g, Shanghai Laboratory Animal Center of Chinese Academy of Sciences, Shanghai, China) were randomly classified into 5 groups, 6 mice each group: blank, NC, BCNU, caffeine and sh-HMGA2 + BCNU. After transfection, the HeLa cells in logarithmic growth phase were detached by trypsin and washed by PBS for 2 times while centrifugation. And then cell suspension was prepared with cool PBS and the cell concentration was adjusted to 1 × 107 cells/mL. Then the cells were stained with 0.4% trypan blue. The number of living cells was set >98%. Then, 0.1 mL cell suspension liquid was injected into the subcutaneous part of the right axillary artery of the nude mice in each group. The animals were fed in specific pathogen free (SPF) experiment room at 25°C with the humidity between 45% and 50%. Mice were exposed to light and darkness 12 h respectively per day with access to autoclaved standard feed and aseptic water. After successful inoculation, observation of the tumor formation in nude mice was employed on the 7th, 14th, 21st, 28th and 35th day. The maximum diameter (a) and the minimum diameter (b) of the tumor that appeared in mice were measured by a vernier caliper, and the approximate volume of tumor was calculated according to the formula, V (mm3) = (a × b2)/2, which was later used to drawa growth curve of the tumor. On the 35th day, the mice were killed using cervical dislocation method and then the skin was cut open, the intact tumor was resected and weighed using an electronic scale. All animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MA, USA).
Hematoxylin-eosin (HE) staining
The tissues were collected within 30 min after the tumor was resected. The intact transplantable tumor tissue was taken to measure the volume and weight of the tumor. The main locations of lymph nodes in body, including neck, axilla, groin, iliac vessels and visceral organs, were used to observe lymph node metastasis in transplantable tumor. All of these tissues were fixed with 10% formaldehyde and embedded in paraffin, then sliced and stained with hematoxylin-eosin. Then the changes of the morphology and number of tumor cells in the transplantable tumor and lymph node were observed. The metastasis number of positive cells in the lymph node was recorded.
The tumor tissue was collected. After fixed, the tissue was embedded in paraffin, sliced into slices with the thickness of 4 µm, dewaxed with xylene, then dehydrated with gradient ethanol (xylene (I) for 5 min, toluene (II) for 5 min, 100% ethanol for 2 min, 95% ethanol for 1 min, 80% ethanol for 1 min, 75% ethanol for 1 min), and finally washed with distilled water for 2 min. Hematoxylin was used to stain slices for 5 min, followed by a rinse with running water. Next, hydrochloric acid and ethanol was used for differentiation for 30 s. The slices were then immersed in water for 15 min or in warm water at approximately 50°C for 5 min. Eosin was then applied to the slices for 2 min. Then the slices were dehydrated, cleared: 95% ethanol (I) for 1 min, 95% ethanol (II) for 1 min, 100% ethanol (I) for 1 min, 100% ethanol (II) for 1 min, toluene and carbolic acid (3:1) for 1 min, toluene (I) for 1 min, xylene (II) for 1 min, and sealed in neutral gum. The specimen was put under the inverted microscope (XSP-8CA, Shanghai Optical Instrument Factory, Shanghai, China) for observation and photography.
Statistical analysis
The data were processed by SPSS 22.0 statistical software (IBM Corp. Armonk, NY, USA). The measurement data were expressed by mean ± standard deviation. All data were tested for normality with Shapiro-Wilk’s test. One-way analysis of variance (ANOVA) was used for variance analysis and significance test among multiple groups, and least-significant difference (LSD) test was used for pairwise comparison of mean values among multiple groups. Data with skewed distributions were evaluated with Kruskal-Wallis H non-parametric test. Repeated-measures analysis of variance was applied to comparisons of values at different time points. P<0.05 is of significant statistical difference.
Results
HeLa cell line has the highest expression of HMGA2
The selection process of the qualified cell line was performed based on the expression levels of HMGA2 in each group using RT-qPCR. As the result (Figure 1) demonstrated, in comparison to the normal cervical cells, Ect1/E6E7, HMGA2 was highly expressed in the CC cell lines: HeLa, SiHa, C33a and Caski, among which the expression of HMGA2 in SiHa, C33a and Caski cell lines was lower than that in HeLa cell line (all P<0.05). Therefore, the HeLa cell of CC was selected as the study subject for further study.
Figure 1.
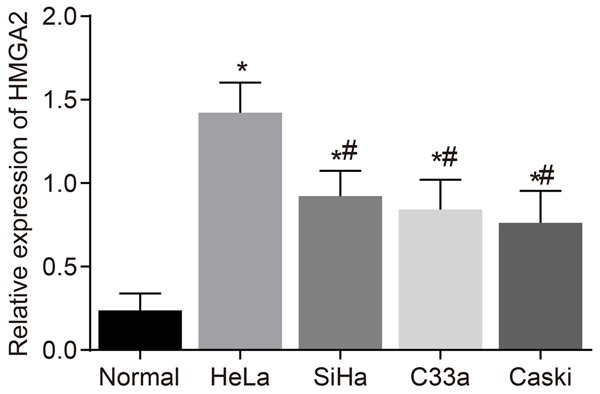
HeLa cell line was identified with the highest HMGA2 expression after RT-qPCR detection. Notes: *, P<0.05 vs. the control group; #, P<0.05 vs. the HeLa cell line; the data of RT-qPCR were measurement data, expressed by mean ± standard deviation. The comparison of data among multiple groups were analyzed by one-way ANOVA; the experiment was repeated 3 times; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
HMGA2 silencing suppresses the activation of ATR/Chk1 signaling pathway
To assess the effect of HMGA2 on the activation of the ATR/Chk1 signaling pathway-related genes, ATR (p-ATR) and Chk1 (p-Chk1), RT-qPCR and western blot analysis were employed. The expression of ATR/Chk1 signaling pathway-related genes in the blank and NC groups of both the HeLa and HMGA2-KD-HeLacell lines showed no significant difference (P>0.05). In the HeLa cell line (Figure 2A-C), the expression of ATR (p-ATR) and Chk1 (p-Chk1) was lower in the caffeine group than that in the blank and NC groups, while the expression of HMGA2 showed no obvious difference (P>0.05). In the BCNU group, the expression of ATR (p-ATR) and Chk1 (p-Chk1) was upregulated while the expression of HMGA2 remained unchanged at large (P>0.05). The expression of HMGA2 in the sh-HMGA2 + BCNU group was down regulated while the expression of ATR (p-ATR) and Chk1 (p-Chk1) remained unchanged. In the HMGA2-KD-HeLa cell line (Figure 2D-F), the expression of HMGA2, ATR (p-ATR) and Chk1 (p-Chk1) in the pcDNA-HMGA2 group was up-regulated in comparison to the blank and NC groups (P<0.05). In the pcDNA-HMGA2 + caffeine group, the expression of HMGA2 increased (P<0.05), while the expression of ATR (p-ATR) and Chk1 (p-Chk1) showed little difference (P>0.05). The results above suggest that HMGA2 silencing or inhibitor of the ATR/Chk1 signaling pathway suppresses the activation of the ATR/Chk1 signaling pathway-related genes, ATR (p-ATR) and Chk (p-Chk1).
Figure 2.
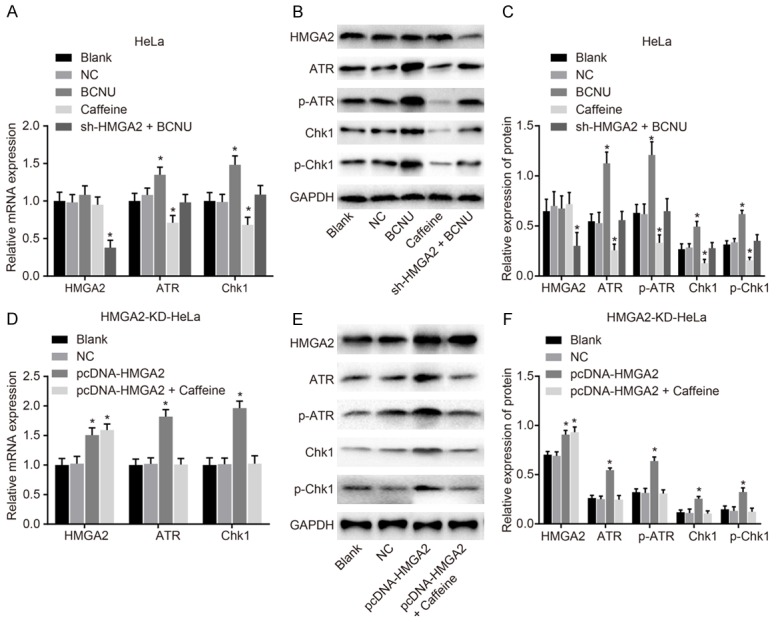
HMGA2 silencing or inhibitor of the ATR/Chk1 signaling pathway suppresses the activation of ATR (p-ATR) and Chk1 (p-Chk1). Notes: Based on the analyses of RT-qPCR (A) and western blot analysis (B, C) in the HeLa cell line, the expression of ATR (p-ATR) and Chk1 (p-Chk1) was decreased/increased in response to caffeine/BCNU transfection; Furthermore, in the HMGA2-KD-HeLa cell line, the detection of RT-qPCR (D) and western blot analysis (E, F) the expression of ATR (p-ATR) and Chk1 (p-Chk1) was increased after transfection of pcDNA-HMGA2; *, P<0.05 vs. the blank group; the data of RT-qPCR and western blotting analysis were measurement data, expressed by mean ± standard deviation. The comparison of data among multiple groups was analyzed by one-way ANOVA; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibits EMT in CC cells
For investigation on the function of HMGA2 and the ATR/Chk1 signaling pathway on EMT in CC cells, immunofluorescence staining was adopted. There was no significant difference regarding the positive expression rate of EMT-related protein (N-cadherin and E-cadherin), between the blank and NC groups in the HeLa and HM-GA2-KD-HeLa cell lines (P>0.05). In the HeLa cell line (Figure 3A, 3C), the positive expression rate of E-cadherin elevated and that of N-cadherin was reduced in the caffeine group in comparison to the blank and NC groups (P<0.05). On the contrary, the positive expression rate of E-cadherin in the BCNU group went down and that of N-cadherin went up (P<0.05). In the sh-HMGA2 + BCNU group, the positive expression rates of E-cadherin and N-cadherin exhibited no remarkable difference (P>0.05). In the HMGA2-KD-HeLa cell line (Figure 3B, 3D), the positive expression rate of E-cadherin decreased and that of N-cadherin increased in the pcDNA-HMGA2 group in comparison to the blank group and NC group (P<0.05). The protein expression of E-cadherin and N-cadherin in the pcDNA-HMGA2 + caffeine group was not obviously different (P>0.05). The results above indicate that HMGA2 gene silencing or inhibition of the ATR/Chk1 signaling pathway inhibited EMT in CC cells.
Figure 3.

HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway repressed epithelial-to-mesenchymal transition in cervical cancer cells, according to the findings of immunofluorescence staining. Notes: Based on the immunofluorescence staining (A, 400 ×, scale bar: 25 μm) and corresponding quantitative analysis (C) in the HeLa cell line, N-cadherin positive expression was decreased/increased, and E-cadherin positive expression was increased/decreased in response to caffeine/BCNU transfection; In the HMGA2-KD-HeLa cell line, immunofluorescence staining (B, 400 ×, scale bar: 25 μm) and corresponding quantitative analysis (D) indicated that N-cadherin positive expression was increased, and E-cadherin positive expression was decreased in response to pcDNA-HMGA2; *, P<0.05 vs. the blank group; red-stained cells are positive cells, and the data were measurement data, expressed by mean ± standard deviation and analyzed by student t test; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway enhances apoptosis of CC cells
Furthermore, the influence of HMGA2 and the ATR/Chk1 signaling pathway on apoptosis of CC cells was analyzed by means of TEM observation following uranyl acetate-lead citrate staining (Figure 4A-D) and the detection of RT-qPCR and western blot analysis (Figure 4E-J) for apoptosis-related genes. HeLa cells in the blank group manifested slight apoptosis characteristics, such as cell membrane contraction. There was no significant difference between the NC and blank groups (P>0.05). When the ATR/Chk1 signaling pathway was suppressed in the caffeine group, obvious apoptotic characteristics appeared, such as nuclear contraction, cell nuclear membrane contraction, the aggregation of chromosomes beneath the cell membrane, nuclear cleavage, vesicular or dental follicle-shaped protuberance on the surface of cells and individual apoptotic body; the expression of anti-apoptotic gene Bcl-2 reduced, and the expression of pro-apoptotic gene Bax elevated. When the ATR/Chk1 signaling pathway was activated in the BCNU group, no early apoptotic cells were detected, and the cell membrane remained intact; the expression of anti-apoptotic gene Bcl-2 increased, and the expression of pro-apoptotic gene Bax decreased (P<0.05). In comparison to the blank group, the change of the cells in the sh-HMGA2 + BCNU group was not obvious (P>0.05). In the HMGA2-KD-HeLacell line, the apoptosis condition was not notably different between the blank and NC groups (P>0.05). In comparison to the blank and NC groups, the early apoptosis of cells in the pcDNA-HMGA2 group was suppressed, indicated by higher expression of anti-apoptotic gene Bcl-2 and lower expression of pro-apoptotic gene Bax (P<0.05). The cell apoptosis in the pcDNA-HMGA2 + caffeine group was not noticeably different (P>0.05). According to the aforementioned results, HMGA2 gene silencing or inhibition of the ATR/Chk1 signaling pathway facilitated apoptosis of CC cells.
Figure 4.

HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway accelerated apoptosis of cervical cancer cells, according to the findings of TEM examination, RT-qPCR and western blot analysis. Notes: (A) The TEM observation (A, B, 10,000 ×) and quantitative analysis for apoptosis rate (C, D) in the HeLa cell line and the HMGA2-KD-HeLa cell line suggested that the inhibition of the ATR/Chk1 signaling pathway led to obvious apoptotic characteristics and promoted apoptosis rate, such as nuclear contraction, cell nuclear membrane contraction, the aggregation of chromosomes beneath the cell membrane, nuclear cleavage, vesicular or dental follicle-shaped protuberance on the surface of cells and individual apoptotic body; Following the RT-qPCR (E, H) and western blot analysis (F, G, I, J), caffeine reduced Bcl-2 expression and elevated Bax expression, while BCNU reversed the tendency in the HeLa cell line (E-G); whereas, the pcDNA-HMGA2 increased Bcl-2 expression and decreased Bax expression in the HMGA2-KD-HeLa cell line (H-J); *, P<0.05 vs. the blank group; TEM, transmission electron microscope; RT-qPCR, reverse transcription quantitative polymerase chain reaction; the data of apoptotic cells after transfection were measurement data, expressed by mean ± standard deviation; the data of different groups were analyzed by one-way ANOVA; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway suppresses proliferation of CC cells
Next, EdU staining (Figure 5) was utilized to detect CC cell proliferation affected by HMGA2 and the ATR/Chk1 signaling pathway. The cell proliferation between the blank and NC groups of the HeLa cell line and the HMGA2-KD-HeLa cell line was of no significant difference (P>0.05). In the HeLa cell line, compared with the blank group, the ratio of EdU positive cells was lower in the caffeine group, suggesting that the cell proliferation was suppressed, while the ratio of EdU positive cells in the BCNU group was higher, indicating the enhanced cell proliferation (P<0.05). The proliferation rate of cells in the sh-HMGA2 + BCNU group did not change significantly (P>0.05). In comparison to the blank and NC groups, cell proliferation was promoted in the pcDNA-HMGA2 group, indicated by higher ratio of EdU positive cells (P<0.05), but the cell proliferation in the pcDNA-HMGA2 + caffeine group displayed no significant difference (P>0.05). The above data suggest that HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited proliferation of CC cells.
Figure 5.
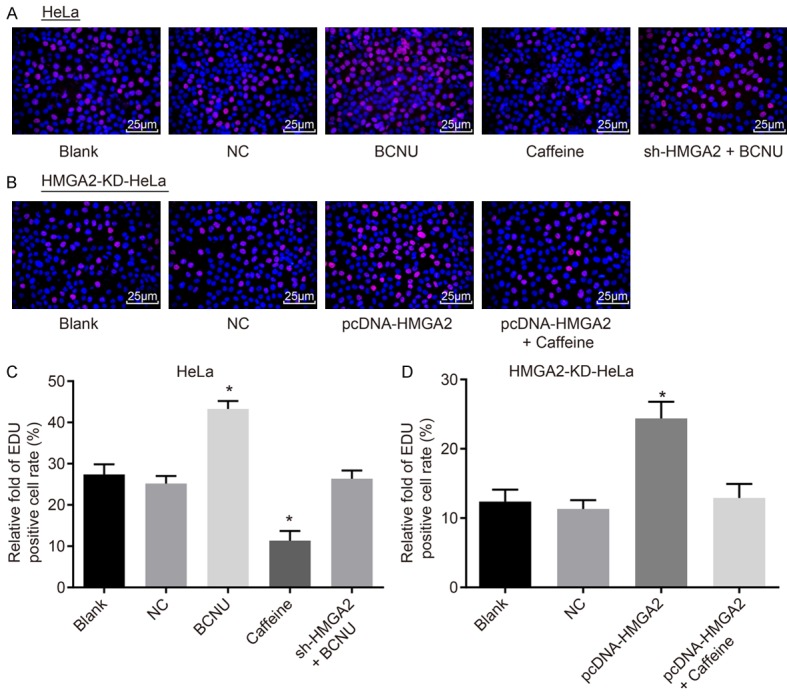
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited proliferation of cervical cancer cells, according to the findings of EdU staining. Notes: Based on EdU staining (A, 400 ×) and relative EdU-positive cell rate (C) in the HeLa cell line, transfection of caffeine repressed cervical cancer cell proliferation and EdU-positive cell rate, while transfection of BCNU reversed the tendency; In the HMGA2-KD-HeLa cell line, EdU staining (B, 400 ×) and relative EdU-positive cell rate (D) indicated that transfection of pcDNA-HMGA2 promoted cervical cancer cell proliferation and EdU-positive cell rate; *, P<0.05 vs. the blank group; the red-stained cells are EdU-positive cells, the data of which were measurement data, expressed by mean ± standard deviation; the data of different groups were analyzed by one-way ANOVA; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway suppresses migration and invasion of CC cells
Scratch test and Transwell assay were carried out to evaluate the effects of HMGA2 and the ATR/Chk1 signaling pathway on migration and invasion of CC cells. As the result shown (Figure 6), the migration and invasion ability of cells in the HeLa cell line and HMGA2-KD-HeLa cell line was of no obvious difference between the blank and NC groups (P>0.05). In the HeLa cell line, the cell migration and invasion rates as well as the expression of migration-related genes (MMP-2 and MMP-9) decreased obviously in the caffeine group, while opposite changing tendency was observed in the BCNU group (P<0.05), and the cell migration and invasion rates in the sh-HMGA2 + BCNU group showed no significant difference (P>0.05). In the HMGA2-KD-HeLa cell line, the cell migration and invasion rates in the pcDNA-HMGA2 group were elevated in comparison to the blank group and NC group, and the expression of MMP-2 and MMP-9 was upregulated (P<0.05). There was no evident significance regarding the cell migration and invasion in the pcDNA-HMGA2 + Caffeine group (P>0.05). These findings elucidate that HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited migration and invasion of CC cells.
Figure 6.
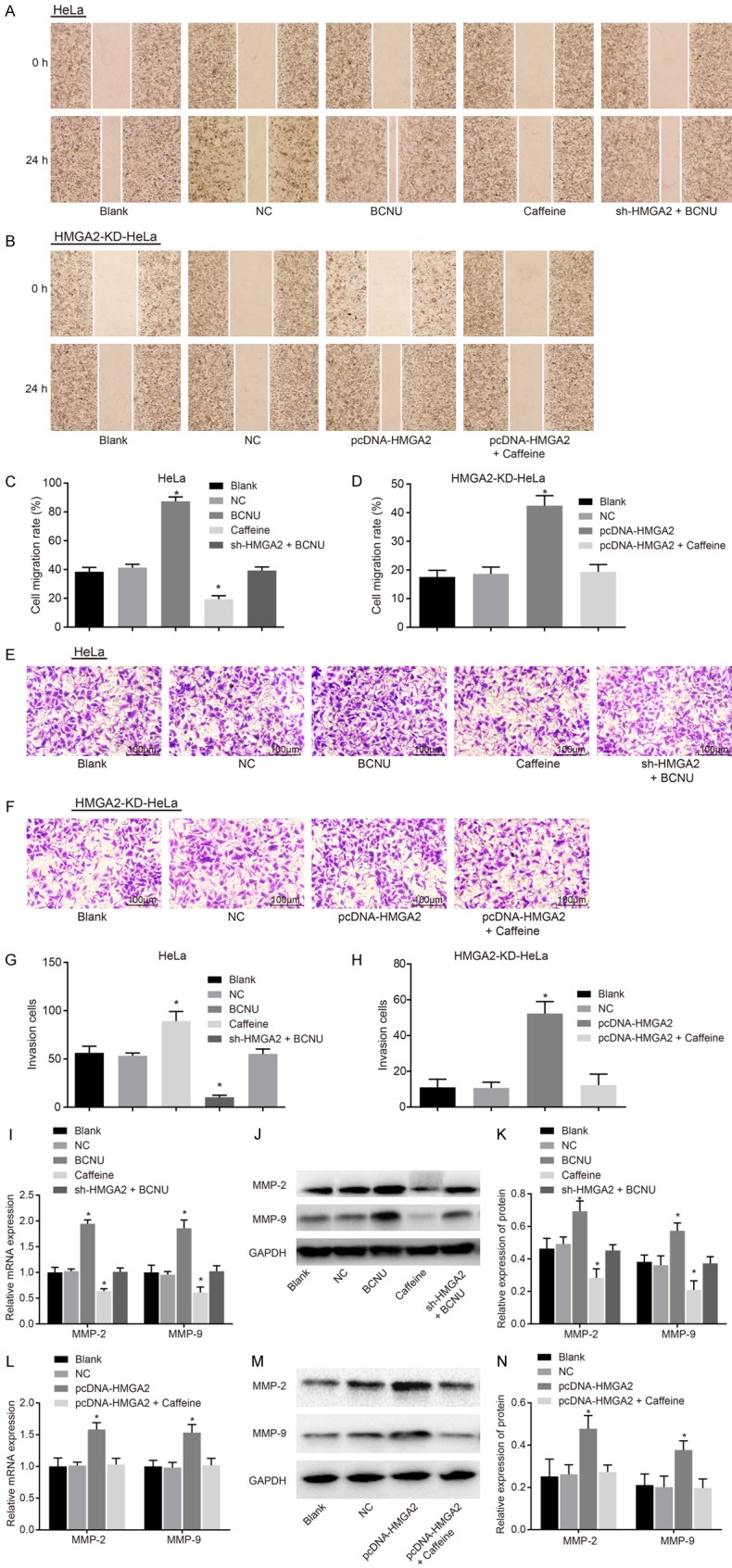
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited migration and invasion of cervical cancer cells. Notes: In the HeLa cell line, the scratch test (A) and migration rate (C) indicated that caffeine reduced cervical cancer cell migration ability; moreover, Transwell assay (E) and number of invasion cells (G) demonstrated reduced cervical cancer cell invasion in response to caffeine; RT-qPCR (I) and western blot analysis (J, K) suggested that expression of metastasis-related genes (MMP-2, MMP-9) was decreased; In the HMGA2-KD-HeLacell line, the scratch test (B) and migration rate (D) indicated that pcDNA-HMGA2 promoted cervical cancer cell migration ability; moreover, Transwell assay (F) and number of invasion cells (H) demonstrated increased cervical cancer cell invasion; RT-qPCR (L) and western blot analysis (M, N) suggested that expression of metastasis-related genes (MMP-2, MMP-9) was increased; *, P<0.05 vs. the blank group; the migration distance is measurement data, expressed by mean ± standard deviation; the number of cell invasion is enumeration data; comparison in the migration distance and invasion ability was performed by one-way ANOVA; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway suppresses tumorigenicity of CC in nude mice
The tumor xenograft in nude mice was conducted in order to measure CC cell tumorigenicity. As is shown by the results (Figure 7), the volume and weight of tumor in the blank and NC groups showed no significant difference (P>0.05). In comparison to the blank group, the volume of tumor in the caffeine group generally reduced from 7 d, 14 d, 21 d, 28 d and 35 dafter inoculation, so did the tumor weight after 35 d (P<0.05), and the tumor volume and weight in the BCNU group increased notably (P<0.05), but did not differ significantly in the sh-HMGA2 + BCNU group (P>0.05). The results above demonstrate that HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited tumorigenicity of nude mice with CC.
Figure 7.

HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited tumorigenicity of cervical cancer in nude mice. Notes: (A) Pictures of resected tumors from nude mice of different groups; the tumor volume (B) and weight (C) of nude mice was decreased in response to caffeine transfection and increased in response to BCNU; *, P<0.05 vs. the blank group; the data were expressed as mean ± standard deviation; the data among multiple groups were compared using one-way ANOVA; values obtained at different time points were compared using repeated-measures analysis of variance; the experiment was repeated 3 times.
HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway suppresses lymph node metastasis in xenografts of nude mice
In order to identify whether HMGA2 and the ATR/Chk1 signaling pathway could influence lymph node metastasis in xenografts of nude mice, HE staining method was performed. The tumor cells in the lymph nodes arranged in disorder, with no exact morphological structure, in round shape; the nuclei were large and in abnormal shape; the chromatin were in deep color, and the heterogeneity was obvious. It was confirmed that the tumor cells of the lymph nodes were derived from the transplanted HeLa cells, and cells of such type were regarded as positive metastasis of lymph node and the number of positive lymph node was recorded (Figure 8): there was no significant difference regarding the number of lymph node metastasis between the blank and NC groups (P>0.05); in comparison to the blank group, the metastasis number of lymph node was reduced in the caffeine group (P<0.05), but elevated in the BCNU group (P<0.05), and there was no remarkable difference in terms of the number of lymph node metastasis in the sh-HMGA2 + BCNU group (P>0.05). According to the above findings, HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited lymph node metastasis of CC cells.
Figure 8.

HMGA2 silencing or inhibition of the ATR/Chk1 signaling pathway inhibited lymph node metastasis in xenografts of nude mice. Notes: (A) Based on HE staining (A, 400 ×) and the number of positive lymph nodes (B) indicated that lymph node metastasis in xenografts of nude mice was reduced in response to caffeine transfection and increased in response to BCNU; *, P<0.05 vs. the blank group; the data of positive lymph nodes were were measurement data, expressed by mean ± standard deviation; the data of different groups were analyzed by one-way ANOVA; the experiment was repeated 3 times.
Discussion
CC has been considered as one of the major causes of cancer-related death in female across the world [23], and many CC patients suffer from early cancer metastasis, leading to poor treatment outcomes after resection of primary cancer [24]. Evidence has been provided that HMGB1 is regulatory for the viability and proliferation of CC cell lines, and HMGB1 silencing reduces the clonogenic capacity of HPV-positive CC cell lines [23]. Thus, we took the aim at the role of HMGA2 in CC cell biological functions through regulating the ATR/Chk1 signaling pathway, and proposed that repression of HMGA2 is conducive to inhibiting the progression of CC through the inhibition of the ATR/Chk1 signaling pathway.
In this study, we found that HMGA2 was in high expression in CC. HMGA2, forming a part in the high mobility group (HMG) protein family, is a non-histone chromosomal protein [25,26]. HMGA regulates gene transcription via interaction with different transcription factors and the chromatin structure [27], and is a well-known regulator of cell differentiation, apoptosis, growth, and DNA repair [28,29]. Abnormal expression of HMGA2 enhances cancer invasion, metastasis, and EMT by triggering the transforming growth factor beta (TGFβ) and Wnt/β-catenin signaling pathways [30]. HMGA2 has been found not only upregulated in both the early and late stages of high-grade serous ovarian carcinoma, but also overexpressed in ovarian cancer, and its overexpression is interrelated with advanced stage tumors, distant metastasis and undesirable results in various carcinomas [29]. A study reported that the repression of HMGA2 expression inhibits cell proliferation, and results in apoptosis in thoroughly-differentiated liposarcoma cells; additionally, HMGA2 mRNA expression from primary tumors shows an upward change in primary lung cancers, indicating that HMGA2 can serve as an influential therapeutic target in the lung and other cancers featured with upregulated HMGA2 expression [31].
Another significant finding of the current study is that suppression of ATR/Chk1 signaling pathway repressed cell apoptosis, invasion and migration. ATR shares similarity in sequence to lipid kinases of thePI3K family, and phosphorylates protein substrates [32]. ATR functions as a key role in the DNA damage response pathways [19]. ATR and its correspondent downstream targets are activated in response to UV and agents that suppress DNA replication forks [33,34]. Likewise, Chk2 is regarded as a tumor suppressor in relation to apoptosis regulation with or without the involvement of ATM in that when phosphorylated, activated Chk2 phosphorylates the tumor suppressor gene p53 and multiple Cdc25 molecules that can negatively regulate the activation of kinases depending on cyclin, which may help to explain the potential mechanism [35]. The ATR/Chk1 signaling pathway is commonly upregulated in cancers and promotes tumor growth; furthermore, ATR and CHK1 inhibitors are able to kill tumor cells [18]. Through inhibiting Chk1 by multiple genotoxic agents, DNA damage and replication checkpoint responses are repressed, which leads to enhanced tumor cell killing [36].
In addition, available evidence was presented in our study, suggesting that HMGA2 activated the ATR/Chk1 signaling pathway, by which mechanism CC cell proliferation, migration, invasion, EMT and lymph node metastasis were promoted. HMGA2 overexpression has been recognized to be closely associated with various malignant tumors [26] and tightly linked to poor metastasis and prognosis [14,37]. Nevertheless, HMGA2 is not indispensable for ATR-Chk1 interaction in human cancer cells, increased and sustained ATR and Chk1 phosphorylation depends on HMGA2, indicating a crucial role for HMGA2 in modulating the activity of the ATR-Chk1 signaling complex [19]. Chk1 is a potent target for anticancer therapy and many Chk1-selective inhibitors are involved in various stages of clinical treatments [38,39].
Collectively, in the present study, we indicated that silencing of HMGA2 gene inhibited the ATR/Chk1 signaling pathway, so as to suppress EMT, proliferation, migration and invasion of CC cells as well as the lymph node metastasis. Based on the in-vitro results demonstrated in this study, further studies are needed to investigate the expression profile of HMGA2 in patient samples, in order to find the correlation of HMGA24 with tumorigenesis, metastasis and prognosis and reveal the potential therapeutic target of HMGA2.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81100412), the 59th General Program of China Postdoctoral Science Foundation (No. 2016M592038) and Key Projects of Outstanding Young Talents Program in 2016 (No. gxyqZD2016059). The authors want to show their appreciation to the reviewers for their helpful comments.
Disclosure of conflict of interest
None.
References
- 1.Iwata T, Miyauchi A, Suga Y, Nishio H, Nakamura M, Ohno A, Hirao N, Morisada T, Tanaka K, Ueyama H, Watari H, Aoki D. Neoadjuvant chemotherapy for locally advanced cervical cancer. Chin J Cancer Res. 2016;28:235–240. doi: 10.21147/j.issn.1000-9604.2016.02.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuzick J, Arbyn M, Sankaranarayanan R, Tsu V, Ronco G, Mayrand MH, Dillner J, Meijer CJ. Overview of human papillomavirus-based and other novel options for cervical cancer screening in developed and developing countries. Vaccine. 2008;26(Suppl 10):K29–41. doi: 10.1016/j.vaccine.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 3.Greimel ER, Winter R, Kapp KS, Haas J. Quality of life and sexual functioning after cervical cancer treatment: a long-term follow-up study. Psychooncology. 2009;18:476–482. doi: 10.1002/pon.1426. [DOI] [PubMed] [Google Scholar]
- 4.Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao MM, Michael HE, Monk BJ. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–743. doi: 10.1056/NEJMoa1309748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Ou J, Guo Y, Dai T, Li X, Liu J, Xia M, Liu L, He M. TBLR1 is a novel prognostic marker and promotes epithelial-mesenchymal transition in cervical cancer. Br J Cancer. 2014;111:112–124. doi: 10.1038/bjc.2014.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu X, Xi X, Yan Q, Zhang Z, Cai B, Lu W, Wan X. MicroRNA-361-5p facilitates cervical cancer progression through mediation of epithelial-tomesenchymal transition. Med Oncol. 2013;30:751. doi: 10.1007/s12032-013-0751-0. [DOI] [PubMed] [Google Scholar]
- 7.Yan S, Wang Y, Yang Q, Li X, Kong X, Zhang N, Yuan C, Yang N, Kong B. Low-dose radiationinduced epithelial-mesenchymal transition through NF-kappaB in cervical cancer cells. Int J Oncol. 2013;42:1801–1806. doi: 10.3892/ijo.2013.1852. [DOI] [PubMed] [Google Scholar]
- 8.Ramachandran I, Thavathiru E, Ramalingam S, Natarajan G, Mills WK, Benbrook DM, Zuna R, Lightfoot S, Reis A, Anant S, Queimado L. Wnt inhibitory factor 1 induces apoptosis and inhibits cervical cancer growth, invasion and angiogenesis in vivo. Oncogene. 2012;31:2725–2737. doi: 10.1038/onc.2011.455. [DOI] [PubMed] [Google Scholar]
- 9.Wang F, Li Y, Zhou J, Xu J, Peng C, Ye F, Shen Y, Lu W, Wan X, Xie X. miR-375 is down-regulated in squamous cervical cancer and inhibits cell migration and invasion via targeting transcription factor SP1. Am J Pathol. 2011;179:2580–2588. doi: 10.1016/j.ajpath.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kodama J, Hasengaowa , Kusumoto T, Seki N, Matsuo T, Ojima Y, Nakamura K, Hongo A Hasengaowa. Association of CXCR4 and CCR7 chemokine receptor expression and lymph node metastasis in human cervical cancer. Ann Oncol. 2007;18:70–76. doi: 10.1093/annonc/mdl342. [DOI] [PubMed] [Google Scholar]
- 11.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, Chen P, Chen J, Chada KK, He C, Rosner MR. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013;110:9920–9925. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, D’Armiento J, Chada K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013;73:4289–4299. doi: 10.1158/0008-5472.CAN-12-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Zhang S, Shan J, Hu Z, Liu X, Chen L, Ren X, Yao L, Sheng H, Li L, Ann D, Yen Y, Wang J, Wang X. Elevated HMGA2 expression is associated with cancer aggressiveness and predicts poor outcome in breast cancer. Cancer Lett. 2016;376:284–292. doi: 10.1016/j.canlet.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Liu X, Li AY, Chen L, Lai L, Lin HH, Hu S, Yao L, Peng J, Loera S, Xue L, Zhou B, Zhou L, Zheng S, Chu P, Zhang S, Ann DK, Yen Y. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin Cancer Res. 2011;17:2570–2580. doi: 10.1158/1078-0432.CCR-10-2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao X, Dai M, Li Q, Wang Z, Lu Y, Song Z. HMGA2 regulates lung cancer proliferation and metastasis. Thorac Cancer. 2017;8:501–510. doi: 10.1111/1759-7714.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mordes DA, Cortez D. Activation of ATR and related PIKKs. Cell Cycle. 2008;7:2809–2812. doi: 10.4161/cc.7.18.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Manic G, Obrist F, Sistigu A, Vitale I. Trial watch: targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2:e1012976. doi: 10.1080/23723556.2015.1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natarajan S, Hombach-Klonisch S, Dröge P, Klonisch T. HMGA2 inhibits apoptosis through interaction with ATR-CHK1 signaling complex in human cancer cells. Neoplasia. 2013;15:263–280. doi: 10.1593/neo.121988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexopoulou AN, Leao M, Caballero OL, Da Silva L, Reid L, Lakhani SR, Simpson AJ, Marshall JF, Neville AM, Jat PS. Dissecting the transcriptional networks underlying breast cancer: NR4A1 reduces the migration of normal and breast cancer cell lines. Breast Cancer Res. 2010;12:R51. doi: 10.1186/bcr2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Hou Y, Yu J, Lu Y, Chang L, Jiang M, Wu X. Synergism of ursolic acid and cisplatin promotes apoptosis and enhances growth inhibition of cervical cancer cells via suppressing NF-kappaB p65. Oncotarget. 2017;8:97416–97427. doi: 10.18632/oncotarget.22133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo T, Wang W, Zhang H, Liu Y, Chen P, Ma K, Zhou C. ISL1 promotes pancreatic islet cell proliferation. PLoS One. 2011;6:e22387. doi: 10.1371/journal.pone.0022387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morale MG, da Silva Abjaude W, Silva AM, Villa LL, Boccardo E. HPV-transformed cells exhibit altered HMGB1-TLR4/MyD88-SARM1 signaling axis. Sci Rep. 2018;8:3476. doi: 10.1038/s41598-018-21416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen B, Hou Z, Li C, Tong Y. MiRNA-494 inhibits metastasis of cervical cancer through Pttg1. Tumour Biol. 2015;36:7143–7149. doi: 10.1007/s13277-015-3440-0. [DOI] [PubMed] [Google Scholar]
- 25.Young AR, Narita M. Oncogenic HMGA2: short or small? Genes Dev. 2007;21:1005–1009. doi: 10.1101/gad.1554707. [DOI] [PubMed] [Google Scholar]
- 26.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 27.Cleynen I, Van de Ven WJ. The HMGA proteins: a myriad of functions (review) Int J Oncol. 2008;32:289–305. [PubMed] [Google Scholar]
- 28.Fedele M, Fusco A. HMGA and cancer. Biochim Biophys Acta. 2010;1799:48–54. doi: 10.1016/j.bbagrm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Wei JJ. HMGA2 and high-grade serous ovarian carcinoma. J Mol Med (Berl) 2013;91:1155–1165. doi: 10.1007/s00109-013-1055-8. [DOI] [PubMed] [Google Scholar]
- 30.Nie D, Zhang L, Guo Q, Mao X. High mobility group protein A2 overexpression indicates poor prognosis for cancer patients: a metaanalysis. Oncotarget. 2018;9:1237–1247. doi: 10.18632/oncotarget.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, Schuldenfrei A, Kowalski J, Bhattacharya R, Ashfaq R, Resar LM. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res. 2008;6:743–750. doi: 10.1158/1541-7786.MCR-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA, Mills GB. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003;278:40057–40066. doi: 10.1074/jbc.M303621200. [DOI] [PubMed] [Google Scholar]
- 33.Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- 34.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Cimprich. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, De Stanchina E, Bristow RG, Suda T, Lowe SW, Jeggo PA, Elledge SJ, Mak TW. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002;22:6521–6532. doi: 10.1128/MCB.22.18.6521-6532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith J, Tho LM, Xu N, Gillespie DA. The ATMChk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 37.Langelotz C, Schmid P, Jakob C, Heider U, Wernecke KD, Possinger K, Sezer O. Expression of high-mobility-group-protein HMGI-C mRNA in the peripheral blood is an independent poor prognostic indicator for survival in metastatic breast cancer. Br J Cancer. 2003;88:1406–1410. doi: 10.1038/sj.bjc.6600935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16:376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–316. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]