

Abstract
Numerous studies have demonstrated that short-term air pollution exposure causes cardiac autonomic imbalance as measured by heart rate variability (HRV). We previously showed that a single exposure to acrolein, a ubiquitous gaseous component of air pollution, not only causes autonomic imbalance, but also increases arrhythmia through transient receptor potential A1 (TRPA1) cation channels. Thus, the goal of this study was to characterize acrolein-induced autonomic changes in both normal and TRPA1-knockout mice (KO). Conscious, unrestrained C57BL/6 (WT) and KO mice were exposed to 3 ppm acrolein for 3 hours. Separate groups were treated with either atenolol (sympathetic blocker), atropine (parasympathetic blocker) or hexamethonium (autonomic neurotransmission blocker), immediately before exposure. Electrocardiogram (ECG) and heart rate (HR) were recorded continuously before, during and after exposure. Exposure to acrolein produced significant increases in standard deviation of normal-to-normal R-R intervals (SDNN), Root Mean Square of the Successive Differences (RMSSD) and Low-Frequency (LF), as well as an increase in arrhythmia in WT mice. Treatment with atenolol reduced this response while atropine enhanced it, and both drugs blocked the acrolein-induced increase in arrhythmia; hexamethonium had no effect. On the other hand, neither acrolein nor any drug had an effect in the KO mice. Thus, acrolein-induced HRV responses appear to be mediated by a combined parasympathetic and sympathetic modulation. KO mice did not demonstrate any increases in HRV with exposure to acrolein. These data demonstrate that the cardiac effects of irritant air pollutants likely involve disruption of homeostatic balance and altered regulation even in healthy animals.
Keywords: Acrolein, cardiac, autonomic nervous system, heart rate variability, arrhythmia, whole body plethysmography
INTRODUCTION
Numerous biological pathways link air pollution with risk for cardiovascular disease and adverse clinical outcomes, including systemic inflammation and oxidative stress, vasoconstriction, enhanced coagulation/thrombosis, autonomic effects due to sensory receptor activation, and the direct effects of translocated particles on the myocardium (Lee and Widdicombe 2001; Simkhovich et al. 2008). Of these, airway sensory activation resulting in changes in autonomic nervous system function may be the most relevant when considering the short-term, reversible, and often latent effects of an acute exposure. In fact, clinical panel studies (He et al. 2011) and epidemiological studies have repeatedly shown that exposure to air pollution causes changes in heart rate variability (HRV) (Gold et al. 2000; Pope et al. 1999), which is an indicator of autonomic influence on the heart. Yet, experimental data characterizing HRV changes during exposure are limited, particularly with regard to focused examinations of the relative influence of the parasympathetic and sympathetic branches of the autonomic nervous system, as both branches dynamically control cardiac function on a beat-by-beat basis. Results from this study fill data gaps linking specific changes in cardiac autonomic modulation with HRV responses during air pollution exposure; particularly as they relate to TRPA1 and irritant-induced activation in the airways.
Autonomic reflex arcs activated by sensory receptors in the airways are believed to mediate some of the first cardiovascular effects measured after exposure to air pollution. In relation to this, there is evidence to suggest that (1) nociceptive nerve fibers in the airways are activated by irritant gaseous components of ambient air pollution (Bessac and Jordt 2010), (2) activation of these fibers produces afferent signaling to the brainstem and activates specific areas of the brain that play a role in the regulation of cardiovascular and ventilatory function (Bonham et al. 2006; Mutoh et al. 2000), and (3) modulation of efferent autonomic activity in the brainstem occurs as a result of neuropeptide release and can persist for some time after the initial stimulation (i.e. plasticity) (Bonham et al. 2001; Carr and Undem 2003). Consistent with such experimental results, our group previously showed that the airway irritant receptor TRPA1 cation channel mediates cardiac arrhythmia through autonomic modulation 24 hours after diesel exhaust exposure (Hazari et al. 2012). The link between sensory activation and altered autonomic modulation was further confirmed by the abrogation of altered HRV and cardiac function during acrolein exposure in TRPA1 knockout mice (Kurhanewicz et al. 2016).
HRV is often used to understand the influence of the autonomic nervous system on the heart. It is an indirect measure, representing input from homeostatic control mechanisms that regulate, among other things, cardiovascular and respiratory function (Task force of European Society of Cardiology and the North American Society of Pacing Electrophysiology 1996). As such, the cardiac cycle and R-R interval variability are continuously adjusted by multiple mechanisms including the modulation of autonomic outflow at the sinoatrial (SA) node, the dynamic regulation of the vasculature, as well as endocrine/paracrine, endothelial and mechanical factors. Additionally, dynamic mechanisms like baroreflex, which monitors and maintains blood pressure, chemoreflex, which responds to changes in oxygen and carbon dioxide in the blood, and respiratory sinus arrhythmia, which originates from the cyclical changes in thoracic pressure during breathing, can also drive changes in HRV. Despite this complexity, several measures of HRV have been shown to correlate with modulation of one or both arms of the autonomic nervous system and thus indicate changes in the neural inputs to the heart. In this sense, the prognostic value of HRV is evident for a number of diseases, exposures and conditions. However, it is premature to assume that changes in HRV in rodents, up or down, can be regarded as a negative prognostic indicator or an endpoint of toxicity following exposure to air pollutants without associated dysfunction (e.g. arrhythmia).
Thus, the current study focuses on the role of the parasympathetic and sympathetic branches of the autonomic nervous system in HRV, which has not been completely characterized in mice. In particular, we address whether a given response is related to the normal dynamic tendency of HRV or if it is an exposure-induced effect. In addition, the fluctuation of autonomic influences on the heart during an air pollution exposure have not been delineated. This is particularly important given certain changes in HRV have been strongly associated with increased cardiovascular risk. Whether this relates to an increased level of sympathetic influence due to the environmental stressor or parasympathetic (i.e. vagal) dominance because of irritant activation in the airways remains to be determined. Hence, in this study, pharmacological blockade was used to determine the relative contributions of the parasympathetic and sympathetic branches to HRV changes induced by acrolein exposure. Ventilatory function was also measured during exposure to better understand acrolein effects on breathing patterns.
MATERIALS AND METHODS
Animals –
Female C57BL/6 (21 ± 1.1 g) and TRPA1 −/− mice (27 ± 3.8 g) between 15 and 30 weeks old were used in this study (Jackson Laboratory - Bar Harbor, ME). Mice were initially housed five per cage and maintained on a 12-hr light/dark cycle at approximately 22°C and 50% relative humidity in an AAALAC–approved facility. Food (Prolab RMH 3000; PMI Nutrition International, St. Louis, MO) and water were provided ad libitum. All protocols were approved by the Institutional Animal Care and Use Committee of the U.S. Environmental Protection Agency and are in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The animals were treated humanely and with regard for alleviation of suffering. Background controls were used as appropriate.
Experimental Groups and Pharmacological Agents –
Six to eight wild-type (WT) or TRPA1 knockout (KO) mice were randomly assigned to one of four pharmacological treatment groups: (1) Vehicle (0.9% sodium chloride), (2) Atropine (2 mg/kg), (3) Atenolol (2 mg/kg), or (3) Hexamethonium chloride (80 mg/kg). Atenolol is a β−1 adrenergic receptor antagonist and blocks sympathetic neurotransmission in the heart. Atropine is a muscarinic receptor antagonist and blocks parasympathetic neurotransmission in the heart. Hexamethonium blocks both sympathetic and parasympathetic peripheral neurotransmissions. All drugs were purchased from Sigma-Aldrich (St. Louis MO). Animals were pre-treated (intraperitoneal, i.p.) with the pharmacological agents three times prior to exposure (days −14, −10, −7). On day 0 animals were treated immediately prior to filtered air (FA) exposure, and on day 1 animals were treated immediately prior to acrolein exposure.
Surgical implantation of radiotelemeters –
Animals were weighed and then anesthetized using inhaled isoflurane (Isothesia, Butler Animal Health Supply, Dublin OH). Anesthesia was induced by spontaneous breathing of 2.5% isoflurane in pure oxygen at a flow rate of 1 L/min and then maintained by 1.5% isoflurane in pure oxygen at a flow rate of 0.5 L/min; all animals received the analgesic Buprenorphrine hydrochloride (0.03 mg/kg, i.p. Reckitt Benckiser Ltd, Hull, England) immediately prior to the procedure. Using aseptic techniques, each animal was implanted subcutaneously, to the right of the midline on the dorsal side, with a radiotelemeter (ETA-F10, Data Sciences International, St Paul, MN). The two electrode leads were then tunneled subcutaneously across the lateral dorsal sides and the distal portions were fixed in positions that approximated those of the lead II of a standard ECG. Body heat was maintained both during and immediately after the surgery. Animals were given food and water post-surgery and were housed individually. Prior to exposure animals were allowed two weeks to recover from the surgery and reestablish circadian rhythms; this was verified by a complete recovery of normal body temperature, heart rate and normal sleep cycles.
Radiotelemetry data acquisition –
Radiotelemetry methodology (Data Sciences International, Inc., St. Paul, MN) was used to track changes in cardiovascular function by monitoring heart rate (HR), and ECG waveforms immediately following telemeter implantation, during exposure, and up to 24 hours post-exposure. This methodology provided continuous monitoring and collection of physiologic data from individual mice to a remote receiver. Sixty-second ECG segments were recorded every 5 minutes during the pre- and post-exposure periods. ECG was recorded continuously during exposure (baseline and hours 1–4); HR was automatically obtained from the waveforms (Dataquest ART Software, version 3.01, Data Sciences International, St. Paul, MN, USA). Non-conducted p-wave arrhythmias, which indicate an temporary atrioventricular block, were counted as previously described (Kurhanewicz et al. 2014); the mice in our studies do not experience a significant number of atrial or ventricular premature beats or other types of arrhythmias.
HRV Analysis –
Heart rate variability (HRV) was measured and calculated as previously described (Kurhanewicz et al. 2014). Briefly, the mean of the differences between sequential RRs for the complete set of ECG waveforms were calculated using ECGAuto (v 2.8.1.26, EMKA technologies S.A.S, Paris, France). The time-domain measures included standard deviation of the time between normal-to-normal beats (SDNN), and root mean squared of successive differences (RMSSD). Analysis of HRV was also conducted in the frequency domain using a Fast-Fourier transform; the spectrum was divided into low-frequency (LF, 0.15–1.5 Hz) and high-frequency (HF, 1.5–5 Hz) regions. All ECG streams with less than 1 minute of identifiable RR intervals were excluded from HRV analysis. Thorough visual inspection was conducted to identify and exclude arrhythmias and artifacts.
Whole-Body Plethysmography –
Ventilatory function (e.g. enhanced pause, tidal volume and minute ventilation) was assessed in awake, unrestrained mice using a whole-body plethysmograph (Buxco, Wilmington, NC). Measurements were made at baseline during a sham air exposure as well as during acrolein. The plethysmograph pressure was monitored using Biosystems XA software (Buxco Electronics Inc., Wilmington, NC). Using respiratory-induced fluctuations in ambient pressure, ventilatory parameters including tidal volume (Vt), breathing frequency (f), inspiratory time (Ti), expiratory time (Te), minute volume (MV), peak inspiratory flow (PIF), peak expiratory flow (PEF) and enhanced pause (Penh), which is a measure of ventilatory timing and can indicate airway irritation, were calculated and recorded on a breath-by-breath basis.
Exposure –
Exposure to acrolein was performed as previously described (Kurhanewicz et al. 2017). Briefly, the study protocol included two days of animal-to-chamber acclimatization prior to exposure, and 30 mins of additional chamber acclimatization immediately before the 3-hour exposures to FA or acrolein. Each mouse was first exposed to FA and then acrolein 24 hours later; hence, each mouse served as its own control. Acrolein exposures took place in whole-body plethysmography chambers (Model PLY3213, Buxco Electronics, Inc., Wilmington, NC, USA) from which ventilatory parameters were measured as described above. Acrolein gas was metered from a 1,000 ppm cylinder into a glass mixing chamber where the gas was mixed and diluted with dry filtered air to achieve a final concentration of 3 ppm of acrolein with a total flow of 6 L/min. The actual chamber concentration was measured once per hour using an HP5890 gas chromatograph (GMI Inc., Ramsey, MN, USA) equipped with manual injection, a flame ionization detector and a DB-VRX capillary column.
Statistics –
All data are expressed as means ± SEM. Statistical analyses of the data were performed with GraphPad Prism 6 (GraphPad software, San Diego CA). The sample size of 6 provided sufficient power (minimum power of 0.8) in the statistical analysis of various biological/molecular (lung lavage indicators of inflammation and toxicity, serum and plasma indicators of coagulation, inflammation, and toxicity, heart and lung tissue indicators of inflammation and toxicity, and heart and lung pathology) and physiological (electrocardiogram, blood pressure, heart rate, ventilation) endpoints. For HR, ECG intervals and HRV, two-way analysis of variance (ANOVA) for repeated-measures and Bonferroni post hoc tests were used to determine statistical differences. A one-way ANOVA was used to analyze arrhythmia counts. Comparisons were made across all groups taking into account the multiple endpoints, exposure groups and time points as well as any interactions.
RESULTS
Characterization of heart rate and heart rate variability in WT and TRPA1 KO mice during air exposure.
HR and HRV were characterized in WT and TRPA1 KO mice injected with vehicle and exposed to FA during pre-exposure (−24hr), exposure (1–3hr), and post-exposure (6hr and 24hr) time periods (Supplemental Figure 1). HR initially increased in both WT and KO mice during exposure to FA in the chamber. HR decreased steadily in WT mice before returning to baseline levels around mid-exposure, whereas HR returned to baseline more rapidly during the first hour of exposure in KO mice. Concurrently, KO mice had a significant increase in SDNN and RMSSD when compared to WT, and showed a higher trend in both parameters for the next 24hrs. Other measures of HRV and arrhythmia counts were similar between the strains.
Effect of pharmacological blockade of autonomic function on HR and HRV in WT and TRPA1 KO mice.
Supplemental Figure 2 shows HR and HRV in WT and TRPA1 KO mice injected with either atenolol, atropine, hexamethonium or vehicle and exposed to FA during pre-exposure, exposure, and post-exposure. Treatment with atenolol decreased HR in both strains but had no effect on any measure of HRV. In contrast, treatment with atropine significantly increased HR in KO mice and caused SDNN to decrease in both strains. Treatment of WT mice with hexamethonium had no effect on HR and caused a decreasing trend in SDNN; however, it caused HR and SDNN to decrease significantly in KO mice. There were no other pharmacological effects on any measures of HR or HRV in either strain (Supplemental Figure 2).
Effect of acrolein exposure on HR, HRV and arrhythmia in WT and TRPA1 KO mice.
Figure 1 shows HR and HRV in WT and TRPA1 KO mice injected with vehicle and exposed to either FA or acrolein, during pre-exposure, exposure, and post-exposure time periods. Acrolein did not affect HR in WT mice; however, it caused significant increases in SDNN, RMSSD, and LF (Figure 1A). Furthermore, arrhythmia counts were higher in WT mice during acrolein exposure (6.3 ± 2.6) when compared to FA (1.3 ± 1.0) (Figure 1B). On the other hand, there were no changes in HR or HRV in KO mice during acrolein; yet, there was a small but significant increase in arrhythmia (3.7 ± 3.5) in these mice when compared to pre-exposure.
Figure 1.
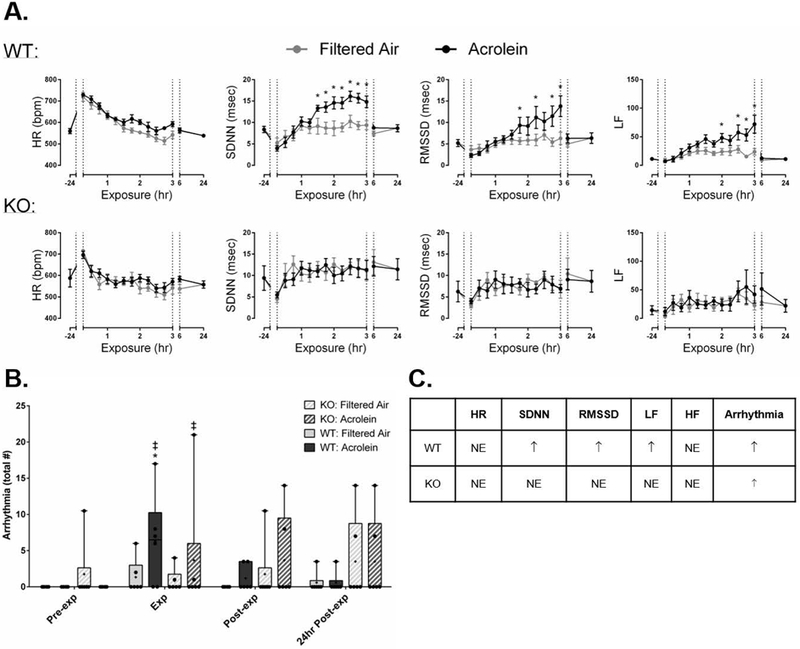
TRPA1 mediates acrolein-induced increases in HRV and arrhythmia. A. Acrolein did not affect HR in WT mice; however, it caused significant increases in SDNN, RMSSD, and LF. B. Arrhythmia counts were higher in WT mice during acrolein when compared to FA, and KO mice had a significant increase when compared to pre-exposure. C. Summary of acrolein exposure effects in WT and KO mice compared with controls. Data points are means ± SEM. p ≤ 0.05; * significantly different from FA; ‡ significantly different from pre-exposure (n=7–8).
Effect of pharmacological blockade of autonomic function on acrolein-induced changes in HR, HRV and arrhythmia in WT and TRPA1 KO mice.
Atenolol blocked acrolein-induced increases in SDNN and RMSSD in WT mice but did not affect the increase in LF (Figure 2A and B). Conversely, atropine enhanced increases in HR, SDNN, RMSSD and LF, and increased HF (Figure 2A and C). Hexamethonium potentiated the immediate decrease in HR due to acrolein and partially blunted the increases in SDNN and RMSSD. Hexamethonium treatment also increased HF during the final half hour of exposure (Figure 2A and D). There was no effect of any drug during acrolein exposure in KO mice (Figure 3). Percent change during acrolein exposure relative to the control group for both WT and KO mice is shown in Supplemental Figure 3). Treatment with all three pharmacological agents, atropine particularly, blocked acrolein-induced increase in arrhythmia in WT and KO mice (Supplemental Figure 4).
Figure 2.
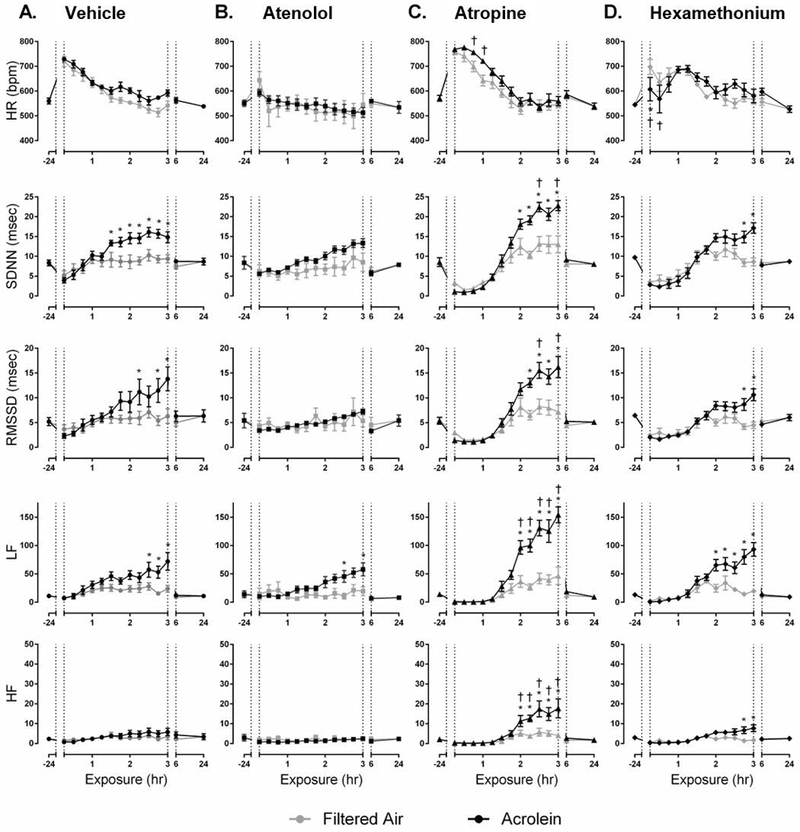
Pharmacological blockade of cardiac autonomic function in WT mice alters HR and HRV response to acrolein exposure. Acrolein-induced increases in SDNN and RMSSD, but not LF, were blocked by atenolol (A. and B.). In contrast, atropine potentiated the acrolein effect and increased HF (C.), whereas hexamethonium only increased HF (D.). Exposure periods are defined as pre-exposure period (−24hr), exposure (1–3hr), immediately post-exposure (6hr), and one-day post-exposure (24hr). Data points are means ± SEM. p ≤ 0.05; * significantly different from FA; † significantly different from vehicle-treated controls (n=7–8).
Figure 3.
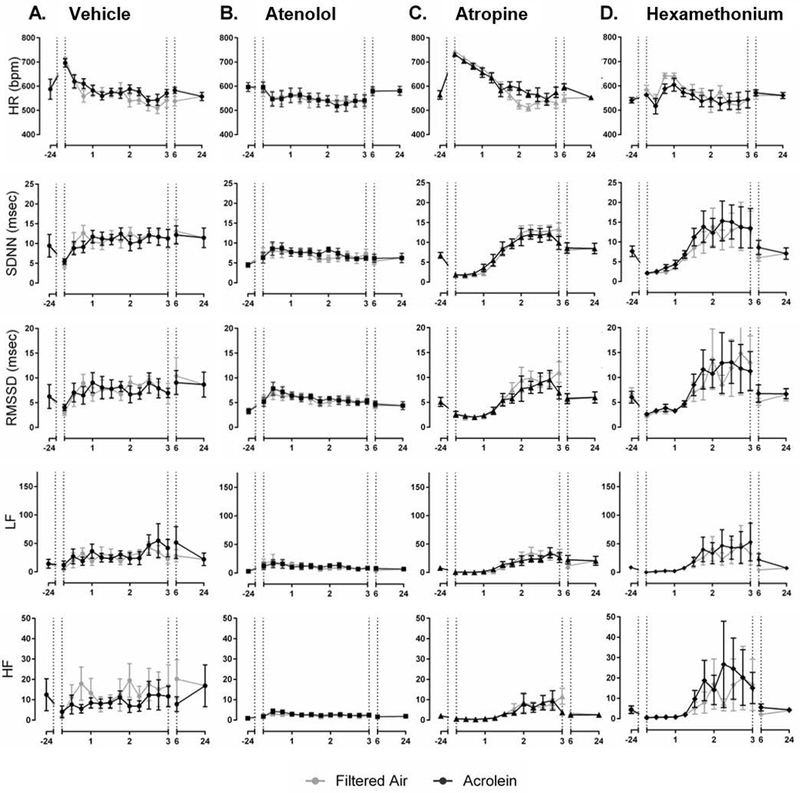
Pharmacological blockade of cardiac autonomic function in TRPA1 KO mice does not change the HR and HRV response to acrolein exposure. There was no effect of atenolol (B.), atropine (C.) or hexamethonium (D.) on any of the parameters during acrolein exposure in KO mice. Exposure periods are defined as pre-exposure period (−24hr), exposure (1–3hr), immediately post-exposure (6hr), and one-day post-exposure (24hr). Data points are means ± SEM (n=7–8).
Ventilatory parameters in WT and TRPA1 KO mice during air (baseline) exposure and after pharmacological blockade of autonomic function.
Compared with WT mice, TRPA1 KO mice had increased Ti and Te, and decreased f during a sham exposure to FA (Supplemental Figure 5). Enhanced pause, which is a measure of ventilatory timing was also increased in KO mice. WT and KO mice did not have significant ventilatory responses to either atenolol or atropine. However, hexamethonium caused a decrease in Ti, f, Vt, MV and penh in KO mice (Supplemental Figure 6).
Effect of acrolein exposure on ventilatory function in WT and TRPA1 KO mice.
Ventilatory parameters were measured in WT and TRPA1 KO mice injected with vehicle and exposed to FA or acrolein (Figure 4). Exposure to acrolein caused increased Te, PEF, Vt, penh and decreased f in WT mice (Figure 4A) when compared to FA, whereas KO mice did not experience any changes in ventilation during acrolein exposure (Figure 4A). Ventilatory parameters were measured on a breath-by-breath basis and analyzed accordingly (Figure 4B).
Figure 4.
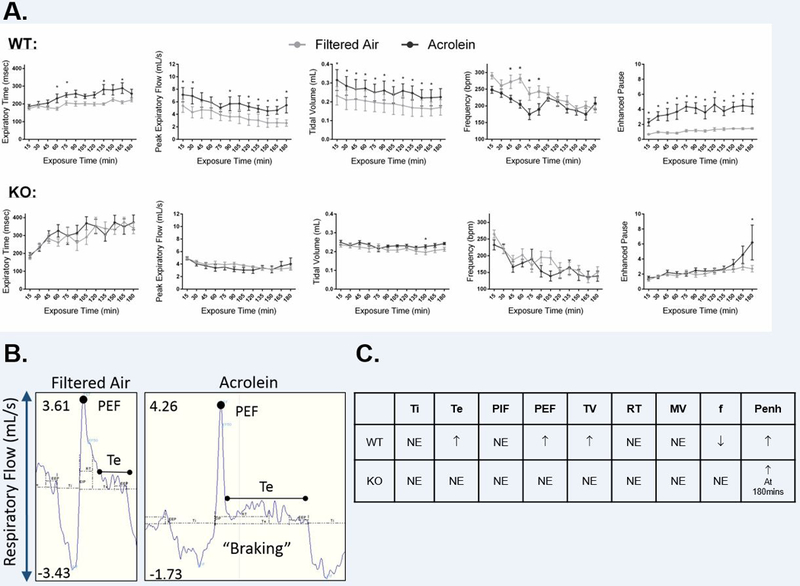
TRPA1 mediates acrolein-induced changes in ventilation. A. Exposure to acrolein caused a significant increase in Te, PEF, Vt, penh and a decrease in f in vehicle-injected WT mice. There were no ventilatory effects in KO mice during acrolein. B. Representative traces from WT mice exposed to either FA or acrolein showing ventilatory “braking” in response to acrolein exposure. C. Summary of ventilatory response to acrolein in WT and KO mice compared with FA. Data points are means ± SEM. p ≤ 0.05; * significantly different from FA (n=7–8).
Effect of pharmacological treatment on acrolein-induced ventilatory changes in WT and TRPA1 KO mice.
Ventilatory parameters were measured in WT and TRPA1 KO mice treated with one of three pharmacological agents or vehicle and exposed to acrolein. Atenolol blocked the acrolein-induced increase in Te and decrease in f in WT mice (Figure 5A). Treatment with atropine blunted the acrolein-induced decrease in f as well as increases in Te and Vt (Figure 5C). Hexamethonium produced a biphasic response during which Ti and Te were only increased during early exposure to acrolein, while peak expiratory flow, and Vt were further elevated. Hexamethonium also reduced f early during exposure and elevated it later (Figure 6D). TRPA1 KO animals exposed to acrolein had an increase in penh only during the final fifteen minutes of exposure; this was blocked by treatment with atropine only (Figure 6C). Percent change during acrolein exposure relative to the control group for both WT and KO mice is shown in Supplemental Figure 7. Table 2 provides a summary of all the ventilatory responses for both WT and KO mice.
Figure 5.
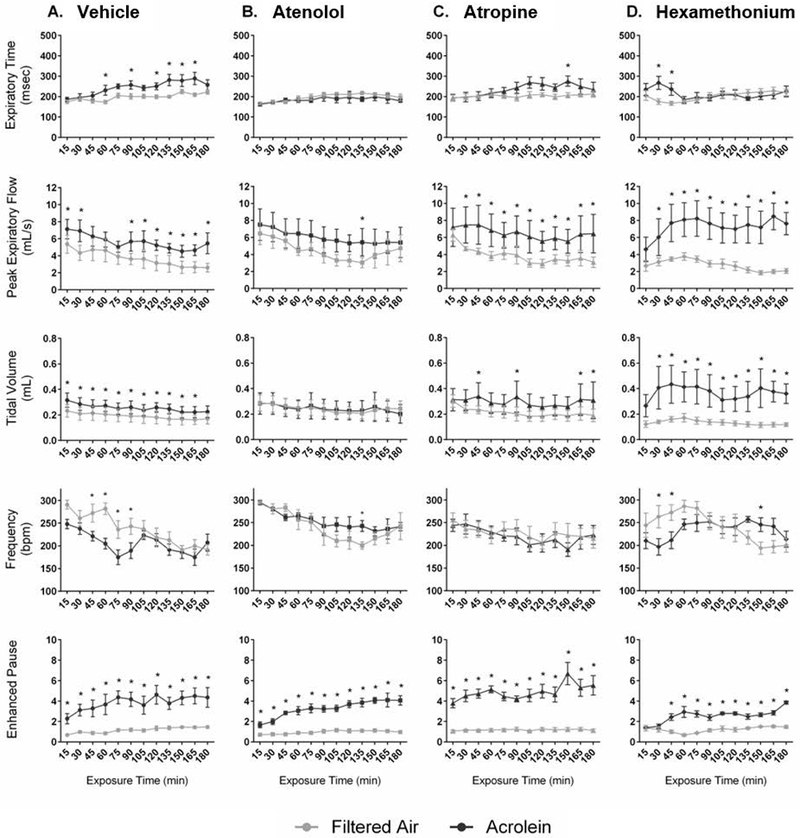
Blockade of cardiac autonomic function alters the ventilatory response to acrolein exposure in WT mice. Treatment of WT mice with atenolol blocked the acrolein-induced increase in Te and decrease in f, and reduced PEF (B.). Similar effects were observed with atropine except for the PEF (C.). In contrast, hexamethonium blocked increases in Te during the 2nd and 3rd hours of exposure, but caused Vt and PEF to increase (D.). Data points are means ± SEM. p ≤ 0.05; * significantly different from FA; † significantly different from vehicle-treated controls (n=7–8).
Figure 6.
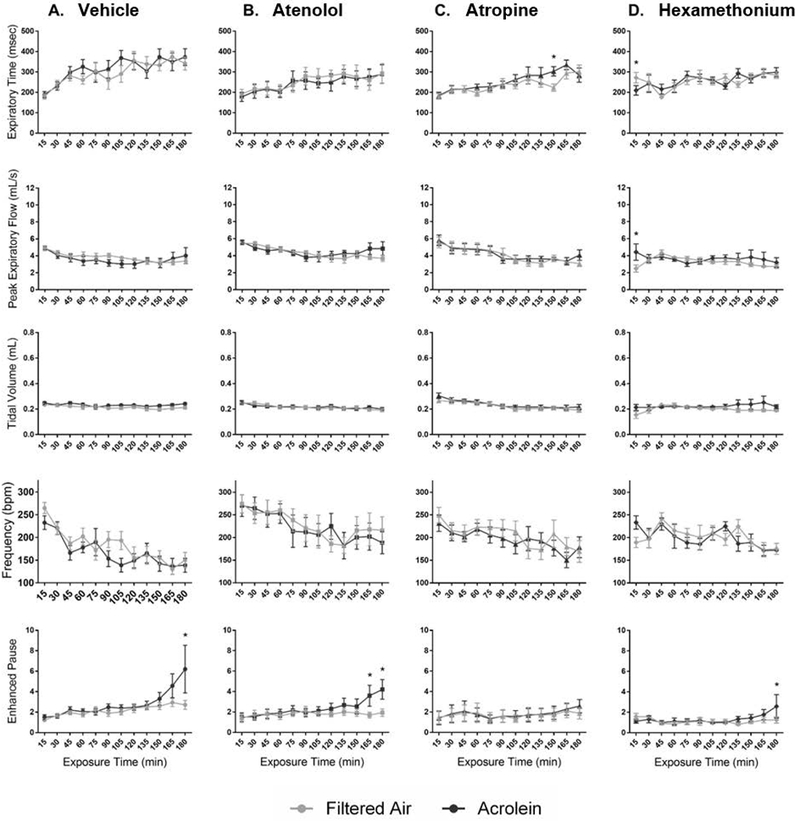
Blockade of cardiac autonomic function does not alter the ventilatory response of KO mice to acrolein. Acrolein caused an increase in penh in the last hour of exposure in KO mice; this was blocked by atropine only (C.). Data points are means ± SEM. p ≤ 0.05; * significantly different from FA; † significantly different from vehicle-treated controls (n=7–8).
Table 2.
Summary of acrolein exposure and pharmacological blockade of autonomic function on ventilatory parameters in WT and TRPA1 KO mice; compared with FA-exposed controls
| WT: | Treatment | Ti | Te | PIF | PEF | TV | RT | MV | Freq | PenH |
|---|---|---|---|---|---|---|---|---|---|---|
| Saline + Acrolein | NE | ↑ | NE | ↑ | ↑ | NE | NE | ↓ | ↑ | |
| Atenolol + Acrolein | NE | NE | NE | ↑ | NE | ↓ | NE | NE | ↑ | |
| Atropine + Acrolein | NE | ↑ | NE | ↑ | ↑ | ↓ | NE | NE | ↑ | |
| Hexametdonium + Acrolein | ↑ early |
↑ early |
↑ | ↑ | ↑↑ | ↑ early |
↑ | ↓↑ | ↑ | |
| KO: | Treatment | Ti | Te | PIF | PEF | TV | RT | MV | Freq | PenH |
|---|---|---|---|---|---|---|---|---|---|---|
| Saline + Acrolein | NE | NE | NE | NE | NE | NE | NE | NE | ↑at 180min |
|
| Atenolol + Acrolein | NE | NE | NE | NE | NE | NE | NE | NE | ↑at 180min |
|
| Atropine + Acrolein | NE | NE | NE | NE | NE | NE | NE | NE | NE | |
| Hexametdonium + Acrolein | NE | NE | NE | NE | NE | NE | NE | NE | ↑at 180min |
|
Ti = inspiratory time; Te = expiratory time; PIF = peak inspiratory time; PEF = peak expiratory time; TV = tidal volume; RT = relaxation time; MV = minute ventilation; Freq. = breatding frequency; PenH = enhanced pause.
DISCUSSION
Building on our previous findings, this study demonstrates that TRPA1 mediates acrolein-induced effects through mechanisms involving both the parasympathetic and sympathetic branches of the autonomic nervous system. In addition, TRPA1 also plays a role in the immediate breathing changes which indicate airway irritation due to acrolein. Although, one of the proposed pathways for the cardiovascular effects of air pollution includes the activation of irritant reflex arcs and modulation of autonomic function, data characterizing this phenomenon are scarce. The cardiovascular system is regulated by an internal sensory apparatus (e.g. baro- and chemoreceptors) that continuously monitors the blood and vasculature, and sends information to the parasympathetic and sympathetic branches, which in turn alter the electrical and mechanical function of the heart and vasculature. It is also modulated by external “sensors” which respond to environmental stimuli by activating cardiopulmonary reflex arcs and resetting autonomic modulation of the heart (Perez et al., 2015).
We previously showed that TRPA1 mediates acrolein-induced increased HRV (i.e. increased SDNN, RMSSD and LF) and incidence of brady-arrhythmia, which together suggest parasympathetic modulation (Kurhanewicz et. al 2017); those results were confirmed in this study. However, here we further show that sympathetic blockade prior to exposure inhibits the acrolein HRV response. Atenolol, which blocks β1-adrenergic receptors and sympathetic neurotransmission to the heart, significantly reduced both the increased SDNN and RMSSD due to acrolein. Conversely, atropine, which prevents parasympathetic neurotransmission by blocking muscarinic receptors on the heart, potentiated all of the acrolein-induced HRV responses and in particular HF, which was not affected by acrolein in untreated mice. Thus, even though an increase in HRV, specifically RMSSD and HF, generally reflects parasympathetic modulation, sympathetic influence also appears to play a role.
As such, the response of air-exposed mice to the drugs provided some insight into the fluctuation of autonomic cardiac influence during acrolein exposure. Blockade of parasympathetic, sympathetic or both branches confirmed two things in general: (1) baseline HR in mice is greatly influenced by the sympathetic branch, hence the significant reduction in HR with atenolol and similar trend with hexamethonium, and minimal effect with atropine, and (2) although short-term HRV is under the control of both branches it is predominantly influenced by parasympathetic modulation (Gehrmann et al., 2000; Pham et al., 2009). This was seen by the atropine-induced decrease in SDNN and a similar trend in RMSSD and LF, and a lack of effect of atenolol on any HRV parameter. These characteristics need to be carefully considered when assessing the HRV effects of acrolein exposure as described herein, particularly when combined with pharmacological blockade of the autonomic branches.
In this study, exposure to acrolein produced a bi-phasic response characterized by an immediate and transient sympathetic shift followed by enhanced parasympathetic modulation. That we observed an increase in HR and a shift towards greater sympathetic dominance early during exposure in both air- and acrolein-exposed mice suggests a generalized stress response, most likely caused by handling, injection and placement in the exposure chamber. This response appears to be irresolvable, even with acclimatization (Andreev-Andrievskiy et al., 2014). However, midway through the exposure acrolein prevents the recovery of the initial increase in HR, an effect that coincided with a significant increase in SDNN, RMSSD and LF. Furthermore, it is apparent from our pharmacological pretreatments that most of the physiological changes due to the drugs occurred within the first half of the exposure period. Therefore, the effects observed in the second half of the exposure likely reflected a compensatory response to the dissipation of the drug. This physiological compensation when the drug washes out has been reported by others (Farah et al., 2006; Fuder and Muscholl, 1995) and in our case can most readily be seen in the opposite shift in SDNN, RMSSD and LF in controls (i.e. during the second half of exposure), and which likely contributes to blocking the HRV effects of acrolein.
It is notable that during the first half of the exposure, atenolol blocked the generalized stress-induced increase in HR and likely prevented any blood pressure increases as well, although the latter was not measured directly. On the other hand, atenolol does not block the stress-induced increased sympathetic nerve activity given it blocks neurotransmission at the level of the heart. Thus increased sympathetic nerve activity would continue to inhibit acetylcholine release from parasympathetic nerves keeping SDNN and RMSSD low. This has been observed with other models of stress in the same strain of mice (Andreev-Andrievskiy et al., 2014; Farah et al., 2006; Fuder and Muscholl, 1995). Considering the second half of exposure, atenolol’s ability to block the acrolein-induced increase in SDNN and RMSSD might be due to the recovery of sympathetic influence on the heart as the drug washes out. This may have balanced the increased parasympathetic shift which occurred due to irritant inhalation (Carll et al., 2015; Farraj et al., 2015; Hazari et al., 2014). To some degree this is likely related to baroreflex influence on autonomic balance, since the atenolol-induced effects (i.e. decreased HR and BP) in the first half of exposure likely result in reflex sympathetic activation later (Just et al., 2000).
The aforementioned conclusion is also supported by the fact that cholinergic blockade resulted in a further increase in HR and minimal-to-no effect on SDNN and RMSSD in the first half of acrolein exposure. These results are in accordance with the concept that the cardiac stress response in mice is due to activation of sympathetic influence and withdrawal of parasympathetic activity. This also reiterates the point that HRV in mice is dominated by parasympathetic tone especially when considered with atenolol’s lack of SDNN and RMSSD effects. Subsequently, as atropine washed out in the second half of the exposure, recovery of parasympathetic influence further increased SDNN, RMSSD and even the frequency domain measures. In fact, the increase in LF during the second half of exposure, which was not blocked by any treatment, may in part indicate the respective recovery of sympathetic and parasympathetic influence following atenolol and atropine treatments. This is a reasonable assertion given LF indicates not only sympathetic and parasympathetic modulation but baroreflex input as well (Reyes del Paso et al., 2013).
Hexamethonium blocked both braches of the autonomic nervous system at the ganglion and this appeared to reduce the acrolein-induced HRV response overall. However, a small exposure effect remained, confirming that parasympathetic activity dominates mouse HRV and indicating homeostatic control mechanisms which function outside of direct autonomic modulation of heart rate still contribute to HRV measures (Parati et al., 1995; Sayers, 1973). These include baroreflex and respiratory sinus arrhythmia, which can also drive changes in HRV (Reyes del Paso et al., 2013; Sayers, 1973). In fact, early work on upper airway irritants suggested a biphasic response to agents, such as acrolein, whereby there is early sympathetic modulation followed by a shift to parasympathetic dominance (Alarie, 1973). It is this later phase of parasympathetic dominance that was suppressed by atenolol and enhanced by atropine, possibly not by the direct action of the drugs, but rather due to physiological compensation by the cardiovascular system.
When compared to WT, pharmacological blockade produced different results in naïve TRPA1 KO mice indicating that basal autonomic function, particularly parasympathetic modulation, is sensitive to the activity of TRPA1. Unlike WT, HR and HRV returned to baseline more quickly in TRPA1 KO mice, irrespective of exposure, indicating enhanced cardiac parasympathetic modulation (Stein et al., 1994). Furthermore, TRPA1 KO mice had a greater increase in HR and decrease in SDNN due to atropine than WT mice; not to mention hexamethonium only decreased SDNN in the KO’s. Once again, this suggests that parasympathetic influence might play a more dominant role in TRPA1 KO mice and points to the fact that the mechanism of acrolein effects are mediated through TRPA1 and autonomic modulation involving both the parasympathetic and sympathetic branches. It is worthwhile to note that vehicle-treated TRPA1 KO mice had a trend towards higher SDNN after the sham exposure and if they were treated with atropine the increase in HR became comparable to WT.
Considering the baseline differences in HRV between the WT and TRPA1 KO mice, drawing conclusions from strain comparison is not straightforward. In addition, there are a number of other elements in this study which contribute to the difficulty/complexity in analyzing the results, chief of which is the temporal natures of the stress response, the exposure response and the pharmacodynamics of the agents used to block cardiac autonomic neurotransmission. These were not fully appreciated until after the completion of this study. In retrospect, it may have been more suitable to allow the animals to acclimate in the chamber, begin exposure and then infuse the pharmacological agents rather than rely on a single injection immediately prior to exposure. In this way the stress response, exposure response and pharmacological interventions would have been parsed into distinct stages rather than overlaid on each other.
Wildtype and TRPA1 KO mice had differences at baseline in not only cardiac function, but ventilatory function as well. TRPA1 KO mice generally had lower f and increased Ti and Te (i.e. slower breathing) than WT. This further supports the conclusion that the presence of TRPA1 has a direct impact on intrinsic homeostatic mechanisms, even beyond the cardiovascular and respiratory systems (Holzer 2011). On the other hand, aside from a general decrease in breathing rate (e.g. Vt, MV) due to hexamethonium, neither strain exhibited significant changes due to atenolol or atropine. These data reflect the fact that hexamethonium’s mode of action is different from atenolol and atropine in that the former impacts all autonomic nerves at the level of the ganglion whereas the latter affect the target tissue.
Exposure to acrolein produced striking changes in the quality and rhythm of ventilation in WT mice, but not in TRPA1 mice. The changes are all hallmarks of an upper airway irritant response which is mediated by stimulation of the trigeminal nerve (Alarie, 1973; Morris et al., 2003; Vijayaraghavan et al., 1993). The decrease in respiratory rate and elongated expiratory phase specifically are linked with the phenomena of ventilatory “braking” whereby reflexive glottal closure and inhibition of the phrenic nerve impair expiratory flow at the onset of expiration (Alarie, 1973; Vijayaraghavan et al., 1993). These results corroborate findings by others (Bessac and Jordt, 2008) that TRPA1 mediates these responses. Interestingly, blockade with atenolol only inhibited the frequency and tidal volume changes due to acrolein; these effects were likely due to bradycardia and hypotension. On the other hand, hexamethonium worsened the acrolein-induced changes in frequency and tidal volume. Although the role of the autonomic branches here is unclear, it appears the activity of one or the other is necessary to maintain normal breathing. Yet, any effects observed due to the drugs may also be secondary to the cardiovascular changes. Further work connecting irritant ventilatory responses to cardiovascular changes is needed.
CONCLUSIONS
These data demonstrate that exposure to acrolein, a gaseous air pollutant and an important constituent of combustion can disrupt normal autonomic balance in healthy young mice, producing marked changes in both cardiac and ventilatory function. Both the sympathetic and parasympathetic branches of the autonomic nervous system appear to play a role in the physiological responses observed, likely in a biphasic manner characterized by early sympathetic activation followed by a prolonged period of vagal dominance. This study highlights the inextricable link between the cardiac and pulmonary systems and begins to describe how a complex interplay between sympathetic and parasympathetic arms of the autonomic nervous system may modulate their function during exposure to air pollution. The increase in risk inherent to modulation of autonomic balance is not obvious. Rather these changes may represent a latent homeostatic shift in which autonomic imbalance may predispose to adverse cardiovascular events if a subsequent stressor is encountered.
Supplementary Material
Table 1.
Summary of acrolein exposure and pharmacological treatment effects (compared with FA-exposed controls) on measures of HRV and occurrence of arrhythmia in WT and TRPA1 KO mice.
| WT: | Treatment | Heart Rate | SDNN | RMSSD | LF | HF | Arrhytdmia |
|---|---|---|---|---|---|---|---|
| Saline + Acrolein | NE | ↑ | ↑ | ↑ | NE | ↑ | |
| Atenolol + Acrolein | NE | NE | NE | ↑ | NE | NE | |
| Atropine + Acrolein | NE | ↑↑ | ↑↑ | ↑↑ | ↑ | NE | |
| Hexametdonium + Acrolein | ↓↑ | ↑ | ↑ | ↑ | ↑ | NE | |
| KO: | Treatment | Heart Rate | SDNN | RMSSD | LF | HF | Arrhytdmia |
|---|---|---|---|---|---|---|---|
| Saline + Acrolein | NE | NE | NE | NE | NE | ↑ | |
| Atenolol + Acrolein | NE | NE | NE | NE | NE | NE | |
| Atropine + Acrolein | NE | NE | NE | NE | NE | NE | |
| Hexametdonium + Acrolein | NE | NE | NE | NE | NE | NE | |
SDNN = standard deviation of normal-to-normal R-R intervals
RMSSD = Root Mean Square of tde Successive Differences
LF = Low-Frequency
HF = High-Frequency
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Najwa Haykal Coates and Kimberly Stratford for their technical assistance in carrying out these experiments. We thank Drs. Colette Miller, Elizabeth Chan, Ian Gilmour, Wayne Cascio and Daniel Costa for their careful review of this manuscript. This work was funded by the U.S. Environmental Protection Agency. NAK was funded by the joint UNC Curriculum in Toxicology-EPA Training Agreement, CR83515201 and EPA Cooperative Agreement, CR83578501.
ABBREVIATIONS
- WT
Wildtype
- KO
Knockout
- HR
Heart Rate
- HRV
Heart Rate Variability
- ECG
Electrocardiogram
- SDNN
Standard deviation of successive N-N intervals
- RMSSD
Root mean square differences of successive N-N intervals
- LF
Low frequency
- HF
High frequency
- Ti
inspiratory time
- Te
expiratory time
- TV
tidal volume
- F
frequency
- PEF
peak expiratory flow
- PIF
peak inspiratory flow
- TRPA1
Transient receptor potential cation channel A1
- SA
Sinoatrial
- ANOVA
Analysis of variance
- BAL
Bronchoalveolar lavage
Footnotes
Disclaimer: This paper has been reviewed and approved for release by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency. Approval does not signify that the contents necessarily reflect the views and policies of the U.S. EPA, nor does mention of trade names.
REFERENCES
- Alarie Y, 1973. Sensory irritation by airborne chemicals. CRC Crit. Rev. Toxicol. 2, 299–363. doi: 10.3109/10408447309082020 [DOI] [PubMed] [Google Scholar]
- Andreev-Andrievskiy AA, Popova AS, Borovik AS, Dolgov ON, Tsvirkun DV, Custaud M, Vinogradova OL, 2014. Stress-associated cardiovascular reaction masks heart rate dependence on physical load in mice. Physiol. Behav. 132, 1–9. doi: 10.1016/j.physbeh.2014.03.033 [DOI] [PubMed] [Google Scholar]
- Bessac BF, Jordt S-E, 2010. Sensory detection and responses to toxic gases: mechanisms, health effects, and countermeasures. Proc. Am. Thorac. Soc. 7, 269–277. doi: 10.1513/pats.201001-004SM [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessac BF, Jordt S-E, 2008. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda). 23, 360–370. doi: 10.1152/physiol.00026.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonham AC, Chen CY, Mutoh T, Joad JP, 2001. Lung C-fiber CNS reflex: role in the respiratory consequences of extended environmental tobacco smoke exposure in young guinea pigs. Environ. Health Perspect. 109 Suppl , 573–578. doi:sc271_5_1835 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonham AC, Chen C-Y, Sekizawa S-I, Joad JP, 2006. Plasticity in the nucleus tractus solitarius and its influence on lung and airway reflexes. J. Appl. Physiol. 101, 322–7. doi: 10.1152/japplphysiol.00143.2006 [DOI] [PubMed] [Google Scholar]
- Carll AP, Haykal-Coates N, Winsett DW, Hazari MS, Ledbetter AD, Richards JH, Cascio WE, Costa DL, Farraj AK, 2015. Cardiomyopathy confers susceptibility to particulate matter-induced oxidative stress, vagal dominance, arrhythmia and pulmonary inflammation in heart failure-prone rats. Inhal. Toxicol. 27, 100–12. doi: 10.3109/08958378.2014.995387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MJ, Undem BJ, 2003. Bronchopulmonary afferent nerves. Respirology 8, 291–301. [DOI] [PubMed] [Google Scholar]
- Farah VMA, Joaquim LF, Morris M, 2006. Stress cardiovascular/autonomic interactions in mice. Physiol. Behav. 89, 569–75. doi: 10.1016/j.physbeh.2006.07.015 [DOI] [PubMed] [Google Scholar]
- Farraj AK, Walsh L, Haykal-Coates N, Malik F, McGee J, Winsett D, Duvall R, Kovalcik K, Cascio WE, Higuchi M, Hazari MS, 2015. Cardiac effects of seasonal ambient particulate matter and ozone co-exposure in rats. Part. Fibre Toxicol. 12, 12. doi: 10.1186/s12989-015-0087-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuder H, Muscholl E, 1995. Heteroreceptor-mediated modulation of noradrenaline and acetylcholine release from peripheral nerves. Rev. Physiol. Biochem. Pharmacol. 126, 265–412. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI, 2000. Phenotypic screening for heart rate variability in the mouse. Am. J. Physiol. Heart Circ. Physiol. 279, H733–40. [DOI] [PubMed] [Google Scholar]
- Gold DR, Litonjua A, Schwartz J, Lovett E, Larson A, Nearing B, Allen G, Verrier M, Cherry R, Verrier R, 2000. Ambient pollution and heart rate variability. Circulation 101, 1267–73. [DOI] [PubMed] [Google Scholar]
- Hazari MS, Callaway J, Winsett DW, Lamb C, Haykal-coates N, Krantz QT, King C, Costa DL, Farraj AK, 2012. Environmental health perspectives. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazari MS, Griggs J, Winsett DW, Haykal-Coates N, Ledbetter A, Costa DL, Farraj AK, 2014. A single exposure to acrolein desensitizes baroreflex responsiveness and increases cardiac arrhythmias in normotensive and hypertensive rats. Cardiovasc. Toxicol. 14, 52–63. doi: 10.1007/s12012-013-9228-9 [DOI] [PubMed] [Google Scholar]
- He F, Shaffer ML, Li X, Rodriguez-Colon S, Wolbrette DL, Williams R, Cascio WE, Liao D, 2011. Individual-level PM₂.₅ exposure and the time course of impaired heart rate variability: the APACR Study. J Expo Sci Environ Epidemiol. 21(1):65–73. doi: 10.1038/jes.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P 2011. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol. Ther. 131(1):142–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just A, Faulhaber J, Ehmke H, 2000. Autonomic cardiovascular control in conscious mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R2214–21. [DOI] [PubMed] [Google Scholar]
- Kurhanewicz N, McIntosh-Kastrinsky R, Tong H, Walsh L, Farraj AK, Hazari MS, 2014. Ozone co-exposure modifies cardiac responses to fine and ultrafine ambient particulate matter in mice: concordance of electrocardiogram and mechanical responses. Part Fibre Toxicol. 11:54–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurhanewicz N, McIntosh-Kastrinsky R, Tong H, Ledbetter A, Walsh L, Farraj A, Hazari MS, 2017. TRPA1 mediates changes in heart rate variability and cardiac mechanical function in mice exposed to acrolein. Toxicol Appl Pharmacol. 324:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, Widdicombe JG, 2001. Modulation of airway sensitivity to inhaled irritants: role of inflammatory mediators. Environ. Health Perspect. 109 Suppl , 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JB, Symanowicz PT, Olsen JE, Thrall RS, Cloutier MM, Hubbard AK, 2003. Immediate sensory nerve-mediated respiratory responses to irritants in healthy and allergic airway-diseased mice. J. Appl. Physiol. 94, 1563–71. doi: 10.1152/japplphysiol.00572.2002 [DOI] [PubMed] [Google Scholar]
- Mutoh T, Bonham AC, Joad JP, 2000. Substance P in the nucleus of the solitary tract augments bronchopulmonary C fiber reflex output. Am. J. Physiol. Regul. Integr. Comp. Physiol 279, R1215–23. [DOI] [PubMed] [Google Scholar]
- Parati G, Saul JP, Di Rienzo M, Mancia G, 1995. Spectral analysis of blood pressure and heart rate variability in evaluating cardiovascular regulation. A critical appraisal. Hypertension 25, 1276–86. [DOI] [PubMed] [Google Scholar]
- Perez CM, Hazari MS, Farraj AK, 2015. Role of autonomic reflex arcs in cardiovascular responses to air pollution exposure. Cardiovasc. Toxicol. 15, 69–78. doi: 10.1007/s12012-014-9272-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham H, Bonham AC, Pinkerton KE, Chen C-Y, 2009. Central neuroplasticity and decreased heart rate variability after particulate matter exposure in mice. Environ. Health Perspect. 117, 1448–53. doi: 10.1289/ehp.0900674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA, Verrier RL, Lovett EG, Larson AC, Raizenne ME, Kanner RE, Schwartz J, Villegas GM, Gold DR, Dockery DW, 1999. Heart rate variability associated with particulate air pollution. Am. Heart J. 138, 890–9. [DOI] [PubMed] [Google Scholar]
- Reyes del Paso GA, Langewitz W, Mulder LJM, van Roon A, Duschek S, 2013. The utility of low frequency heart rate variability as an index of sympathetic cardiac tone: a review with emphasis on a reanalysis of previous studies. Psychophysiology 50, 477–87. doi: 10.1111/psyp.12027 [DOI] [PubMed] [Google Scholar]
- Sayers BM, 1973. Analysis of heart rate variability. Ergonomics 16, 17–32. doi: 10.1080/00140137308924479 [DOI] [PubMed] [Google Scholar]
- Simkhovich BZ, Kleinman MT, Kloner R a, 2008. Air pollution and cardiovascular injury epidemiology, toxicology, and mechanisms. J. Am. Coll. Cardiol. 52, 719–726. doi: 10.1016/j.jacc.2008.05.029 [DOI] [PubMed] [Google Scholar]
- Stein PK, Bosner MS, Kleiger RE, Conger BM, 1994. Heart rate variability: a measure of cardiac autonomic tone. Am. Heart J. 127, 1376–81. [DOI] [PubMed] [Google Scholar]
- Task force of European Society of Cardiology and the North American Society of Pacing Electrophysiology, 1996. Heart rate variability. Standards of measurement, physiological interpretation, and clinical use. Eur. Heart J. 17, 354–81. [PubMed] [Google Scholar]
- Vijayaraghavan R, Schaper M, Thompson R, Stock MF, Alarie Y, 1993. Characteristic modifications of the breathing pattern of mice to evaluate the effects of airborne chemicals on the respiratory tract. Arch. Toxicol. 67, 478–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.