

Abstract

A series of polymer–drug conjugates based on 2-methacryloyloxyethyl phosphorylcholine (MPC) was prepared with the glioblastoma drug temozolomide (TMZ) as pendent groups. Random and block copolymers were synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization using a TMZ-containing methacrylate monomer. The solution properties of the polyMPC–TMZ copolymers were investigated by dynamic light scattering and transmission electron microscopy, revealing well-defined nanostructures from the block copolymers. Conjugation of TMZ to polyMPC enhanced drug stability, with decomposition half-life values ranging from 2- to 19-times longer than that of free TMZ. The cytotoxicity of polyMPC–TMZ was evaluated in both chemosensitive (U87MG) and chemoresistant (T98G) glioblastoma cell lines. Furthermore, the polyMPC–TMZ platform was expanded considerably by the preparation of redox-sensitive polyMPC–TMZ copolymers utilizing disulfides as the polymer-to-drug linker.
Keywords: glioblastoma, temozolomide, drug delivery, polymer-drug conjugate, poly(methacryloyloxyethyl phosphorylcholine)
Introduction
Glioblastoma is a solid neoplasm that originates from non-neuronal glial cells of the brain and represents the most commonly diagnosed central nervous system tumor in the United States.1 Due to its highly aggressive and lethal nature, glioblastoma is treated clinically using a regimen of surgery, radiation, and chemotherapy.2,3 Despite the demonstrated benefits of this combination therapy, recurrence is overwhelmingly inevitable even in patients who respond positively to initial treatment.2 For complete eradication of infiltrative cancer cells in surrounding healthy tissue, chemotherapy is necessary. However, selection of appropriate drugs (typically alkylating agents such as temozolomide, carmustine, lomustine, and procarbazine) is challenging, as the blood–brain barrier (BBB) generally only allows passage of lipophilic small molecules and essential nutrients from the bloodstream to the brain.4,5 This, combined with issues typical of most small molecule chemotherapeutics (i.e., off-target toxicity, rapid clearance from the bloodstream, and poor tumor selectivity), drives the development of new strategies for efficacious treatment of glioblastoma.
Local treatment immediately following surgical resection represents a potential alternative to systemic tumor treatment as a means of bypassing the BBB and delivering chemotherapeutics directly to invasive cells in a sustained manner. Gliadel, a biodegradable carmustine-loaded polymer wafer, remains the only therapeutic approved for local glioblastoma treatment to date. However, the clinical use of Gliadel is limited by side effects, including seizures, cerebral edemas, and intracranial bleeding as well as incomplete wafer degradation and wafer migration.6 However, while “softer” matrices, such as drug-loaded hydrogels,7 may prove to be safe alternatives, noninvasive systemic treatments that can cross the BBB hold potential for improving glioblastoma chemotherapy without the complications inherent to surgical implants.
Temozolomide (TMZ), a bis(imidazotetrazine) heterocycle, is the first-line drug used for treating glioblastoma, and as such, research is devoted to enhancing its delivery and efficacy. Administered orally, TMZ is a prodrug that releases a DNA alkylating methyldiazonium cation upon decomposition at physiological pH, as shown in Figure 1.8,9 Alkylation occurs primarily at O6 positions of guanine residues, generating DNA mismatch errors, which lead to apoptosis.9 While TMZ crosses the BBB, its efficacy is impeded by enzyme-mediated chemoresistance and hydrolytic instability: TMZ rapidly degrades in vivo and exhibits a plasma half-life of <2 h.10 As such, frequent dosing is required to maintain suitable antitumor activity.8,9,11 Furthermore, its hydrolytic instability complicates dosing, as TMZ degrades over time ex vivo when stored improperly.12−14 As such, recent advances have focused on enhancing the stability of TMZ in solution and improving its overall efficacy in glioblastoma tumors.
Figure 1.

Decomposition of TMZ to the cytotoxic methyldiazonium cation.
Reported strategies to stabilize TMZ include its cocrystallization with organic acids,14 encapsulation in injectable nanomaterials,15,16 and conjugation to biocompatible polymer backbones.17,18 Covalent conjugation to polymers offers significant advantages, including extending TMZ lifetime in vivo, improving drug pharamacokinetics, allowing for high drug loading while retaining aqueous solubility, and masking drug-related toxicity.19−21 Moreover, unlike conventional polymer prodrugs, TMZ may retain therapeutic activity when attached to polymers, irrespective of the conjugation method. We previously described the preparation of polymer–TMZ conjugates using a novel TMZ–methacrylate monomer that directly incorporates TMZ into a biocompatible polymer by controlled free radical polymerization.22 Our initial investigation focused on polymer–drug conjugates using zwitterionic poly(2-methacryloyloxyethyl phosphorylcholine) (polyMPC), a polymer that has been investigated as a biomimetic scaffold for chemotherapeutic conjugation and delivery due to its excellent biocompatibility and water solubility.23−27 We demonstrated that polymerizations yielded well-defined random copolymers containing pendent TMZ moieties at tunable and high drug loadings (>50 mol percent) while preserving the fidelity of the TMZ structure. Furthermore, these copolymers enhanced TMZ solution stability, indicating the potential advantages of this simple and reproducible polymerization strategy.
Here, we describe the synthesis and cell culture evaluation of polyMPC–TMZ conjugates. Random and block copolymers bearing pendent TMZ moieties were prepared using a TMZ–methacrylate monomer, and the effect of polymer architecture on TMZ stability was evaluated. The antitumor activity of the copolymers was tested in chemosenstive and chemoresistant glioblastoma cell lines, and the mechanism and extent of cellular uptake was assessed using confocal microscopy and flow cytometry, respectively. Additionally, this polymer–drug platform was enhanced by incorporating redox-sensitive linkers between the polymer and drug.
Materials and Methods
Materials
2-Methacryloyloxyethyl phosphorylcholine (MPC), 2-hydroxyethyl methacrylate (HEMA), 4-(dimethylamino)pyridine (DMAP), sodium nitrite, dimethyl sulfoxide (DMSO), sodium trifluoroacetate, 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid, 4,4′-azobis(4-cyanovaleric acid) (ACVA), cystamine dihydrochloride, cysteamine, trityl chloride, methacryloyl chloride, reduced glutathione (GSH), triisopropylsilane (TIPS), and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich. Anhydrous diethyl ether, dichloromethane (DCM), ethyl acetate, hexanes, methanol (MeOH), acetonitrile, concentrated sulfuric acid, tetrahydrofuran (THF), hydrochloric acid (HCl), monobasic sodium phosphate (NaH2PO4), and sodium sulfate anhydrous (Na2SO4) were purchased from Fisher Chemical. 2,2,2-Trifluoroethanol (TFE) was purchased from Oakwood Products, Inc. Temozolomide (TMZ) and 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDC) were purchased from TCI America. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc. Amicon Ultra-15 regenerated cellulose centrifugal filters with a nominal molecular weight cutoff of 10 kDa were purchased from Merck Millipore Ltd. Unless otherwise noted, all chemicals were used as received without further purification. Inhibitor was removed from MPC prior to polymerizations following a previously described procedure.23 HEMA was purified by short-path vacuum distillation using a Kugelrohr apparatus. Human glioblastoma (U87MG and T98G) cells were purchased from American Type Culture Collection (ATCC). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals. Gibco antibiotic–antimycotic (100X) was purchased from Fisher Scientific. Cell viability was measured using CellTiter-Glo luminescent cell viability assays (Promega). LysoTracker Red was purchased from Life Technologies. Fluorescent 4′,6-diamidino-2-phenylindole (DAPI) stain was purchased from Molecular Probes. TMZ–methacrylate 1 and random copolymers R1–R4 were synthesized as described previously.22 TMZ–carboxylic acid was prepared following a literature procedure.28
Instrumentation
1H NMR (500 MHz), 13C NMR (125 MHz), and 31P NMR (202 MHz) spectra were collected using a Bruker Ascend 500 spectrometer equipped with a Prodigy cryoprobe. High-resolution mass spectroscopy (HRMS) data were obtained using a JEOL-700 MStation spectrometer equipped with electron impact (EI) and fast atom bombardment (FAB) sources. UV–vis absorption measurements were made on a Shimadzu UV-2600 spectrophotometer. Variable temperature UV–vis spectroscopy was performed using a Quantum Northwest dual temperature-controlled cuvette holder and a TC 1 temperature controller. Molecular weight and dispersity (Đ) values of all polymers were estimated by gel permeation chromatography (GPC) using TFE with 0.02 M sodium trifluoroacetate as eluent. Narrow dispersity poly(methyl methacrylate) (PMMA) samples were employed to construct a calibration curve. GPC was operated at 1.0 mL/min and 40 °C with an Agilent 1200 system equipped with an isocratic pump, a degasser, an autosampler, one 50 × 8 mm2 Polymer Standards Service (PSS) PFG guard column, three 300 × 7.5 mm2 PSS PFG analytical linear M columns with 7 μm particle size, and Agilent 1200 refractive index and UV detectors. DLS was performed using a Malvern Zetasizer Nano ZS. Cryo-TEM was performed on an FEI TecnaiT12 electron microscope using samples prepared on 400 square mesh carbon-coated copper grids (Electron Microscopy Sciences). Confocal microscopy was performed using a Nikon A1 resonant scanning confocal microscope with structured illumination super resolution (A1R-SIMe).
Synthesis of PolyMPC Macro-CTA (2)
In a 20 mL vial charged with a stir bar, MPC (3.90 g, 13.2 mmol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (74.3 mg, 0.270 mmol), and ACVA (15.6 mg, 0.0560 mmol) were dissolved in MeOH (13.2 mL). The solution was degassed with bubbling nitrogen at 0 °C for approximately 20 min and then was heated at 70 °C. Upon achieving monomer conversion of ∼80%, as estimated by 1H NMR spectroscopy, the polymerization was terminated by exposure to air, and the solution was precipitated in THF. The crude polymer was isolated by centrifugation (2000 rpm) and was dialyzed against water. Lyophilization afforded 2 as a pink solid in yields >80%. Mn of the polyMPC macro-CTA was estimated using 1H NMR spectroscopy by comparing signal intensities at 3.66–3.82 ppm (CH2—N) (PC methylene protons) to that of the terminal phenyl protons (7.40–7.96 ppm). 1H NMR (500 MHz, MeOD-d4, δ, ppm): 0.73–1.22 (br, 3H), 1.68–2.17 (br, 2H), 3.23–3.38 (s, 9H), 3.66–3.82 (br, 2H), 4.00–4.15 (br, 2H), 4.15–4.29 (br, 2H), 4.29–4.41 (br, 2H), 7.40–7.96. 31P NMR (202 MHz, MeOD-d4, δ, ppm): −0.45. Mn, NMR = 15 100–19 800 g/mol, Mn, TFE GPC = 20 900–21 900 g/mol, Đ = 1.05–1.13.
General Procedure for the Synthesis of PolyMPC–TMZ Block Copolymers (B1–B3)
In a 7 mL vial charged with a stir bar, polyMPC macro-CTA 2, TMZ–methacrylate 1, and ACVA were dissolved in TFE ([1]0 = 0.25 M), targeting [ACVA]0:[2]0 = 0.2:1 and [1]0:[2]0 ranging from 17 to 37. The mixture was purged with nitrogen gas at 0 °C for 15 min and then stirred at 70 °C to initiate polymerization, with monomer conversion judged by 1H NMR spectroscopy. At monomer conversion of >90%, the mixture was quenched in liquid nitrogen and exposed to air. The crude reaction mixture was precipitated three times from TFE into THF, and the polymer was isolated by centrifugation. The isolated polymer was dissolved in aqueous 0.1 M HCl (10 mL), added to a centrifugal dialysis filter with a nominal molecular weight cutoff of 10 kDa, and centrifuged (4000g, 30 min, room temperature). The filtrate was discarded, and centrifugal dialysis was repeated twice. The concentrated polymer was dissolved in aqueous 0.1 M HCl, and lyophilization afforded polymers B1–B3 as pink solids in yields >50%. Incorporation of monomer 1 into the copolymer structure was estimated by 1H NMR spectroscopy by comparing relative signal intensities at 8.72 ppm (C—H in TMZ) and 2.78–3.22 ppm (N—(CH3)3 in MPC). 1H NMR (500 MHz, TFE-d3, δ, ppm): 0.00–1.07 (br, 6H), 1.25–2.13 (br, 4H), 2.78–3.22 (s, 9H), 3.53 (br, 2H), 3.88 (s, 3H), 3.93–4.26 (br, 4H), 4.34 (br, 2H), 4.43–4.75 (br, 4H), 8.72 (s, 1H). 13C NMR (125 MHz, TFE-d3, δ, ppm): 18.47, 20.23, 38.51, 46.48, 46.95, 55.89, 62.81, 64.91, 65.94, 67.36, 67.88, 126.34, 131.34, 138.27, 140.01, 161.35, 179.79, 180.62. 31P NMR (202 MHz, TFE-d3, δ, ppm): −2.79.
Synthesis of TMZ–Methacrylamide (6)
Compound 3 was synthesized following a modified literature procedure.29 In a 250 mL round-bottom flask charged with a stir bar, cystamine hydrochloride (5.10 g, 22.6 mmol) and triethylamine (9.5 mL, 68.2 mmol) were combined in methanol (70 mL). To this mixture was slowly added a solution of di-tert-butyl dicarbonate (2.44 g, 11.2 mmol) in MeOH (45 mL) via addition funnel at 0 °C. The reaction mixture was stirred at room temperature for 16 h, after which the solvent was removed under reduced pressure. The white residue was resuspended in 40 mL of aqueous 1 M NaH2PO4, and the solution was washed with diethyl ether (3 × 40 mL). The pH of the aqueous phase was adjusted to approximately pH 9 using aqueous 1 M NaOH, and the product was extracted into ethyl acetate (3 × 40 mL). The organic fractions were combined, dried with anhydrous Na2SO4, and concentrated under reduced pressure to give 3 as a viscous yellow oil (1.97 g, 55% yield). 1H NMR (500 MHz, CDCl3, δ, ppm): 1.34 (br, 2H), 1.43 (s, 9H), 2.76 (m, 4H), 3.00 (t, 2H), 3.44 (q, 2H), 4.96 (br, 1H). 13C NMR (125 MHz, CDCl3, δ, ppm): 28.40, 38.39, 40.61, 45.52, 79.52, 155.75. Compound 4 was synthesized following a modified literature procedure.29 To a dry 500 mL round-bottom flask charged with a stir bar and flushed with nitrogen were added 3 (1.97 g, 7.79 mmol) and anhydrous DCM (100 mL). The mixture was cooled to 0 °C, and triethylamine (2.2 mL, 15.8 mmol) was added. To this was slowly added a solution of methacryloyl chloride (1 mL, 10.1 mmol) in anhydrous DCM (30 mL) via addition funnel, and the reaction was warmed to room temperature. After 16 h, the reaction mixture was washed with saturated brine (3 × 45 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to give an off-white solid. The crude product was purified by column chromatography on silica gel, eluting with ethyl acetate:hexanes (1:1), to give 4 as a white solid (1.34 g, 54% yield). 1H NMR (500 MHz, CDCl3, δ, ppm): 1.44 (s, 9H), 1.98 (s, 3H), 2.80 (t, 2H), 2.88 (t, 2H), 3.45 (q, 2H), 3.64 (q, 2H), 4.96 (br, 1H), 5.35 (s, 1H), 6.45 (br, 1H). 13C NMR (125 MHz, CDCl3, δ, ppm): 18.66, 28.39, 38.01, 38.23, 38.52, 39.39, 119.89, 139.81, 155.86, 168.58. In a 20 mL vial charged with a stir bar, 4 (1.34 g, 4.18 mmol) was dissolved in DCM (6 mL). To this solution was added trifluoroacetic acid (5.65 mL), and the reaction mixture was stirred at room temperature. After 16 h, the solvent was removed under reduced pressure to give the crude product as a viscous oil, which was then washed with diethyl ether, yielding 5 as a white solid (1.0588 g, 76% yield). 1H NMR (500 MHz, CDCl3, δ, ppm): 1.85 (s, 3H), 2.84 (t, 2H), 2.92 (t, 2H), 3.10 (t, 2H), 3.41 (q, 2H), 5.35 (s, 1H), 5.66 (s, 1H), 7.88 (s, 3H), 8.13 (t, 1H). 13C NMR (125 MHz, CDCl3, δ, ppm): 19.05, 34.65, 37.18, 38.29, 38.85, 119.79, 140.21, 168.04. In a 20 mL scintillation vial charged with a stir bar, TMZ–carboxylic acid (352 mg, 1.81 mmol) was suspended in DCM (4 mL). To this suspension was added 5 (449 mg, 1.34 mmol), catalytic DMAP (16.6 mg, 0.134 mmol), and EDC (393 mg, 1.5 eq., 2.05 mmol), giving a red homogeneous reaction mixture. This solution was stirred at room temperature for 16 h, after which the reaction had turned opaque and orange. Sufficient DCM was added to the suspension to completely dissolve the precipitate, and the organic phase was washed with aqueous 0.1 M HCl (3 × 40 mL). The organic layer was then dried over Na2SO4 and concentrated under reduced pressure, giving TMZ–methacrylamide 6 as a white solid (454 mg, 85% yield). 1H NMR (500 MHz, DMSO, δ, ppm): 1.84 (s, 3H), 2.85 (t, 2H), 2.94 (t, 2H), 3.41 (q, 2H), 3.61 (q, 2H), 3.86 (s, 3H), 5.33 (s, 1H), 5.66 (s, 1H), 8.09 (t, 1H), 8.65 (t, 1H), 8.85 (s, 1H). 13C NMR (125 MHz, DMSO, δ, ppm): 19.05, 36.63, 37.49, 38.49, 38.93, 119.72. 128.96, 130.58, 135.05, 139.67, 140.23, 160.22, 167.96. HRMS-FAB (m/z): [M + H]+ calculated for C14H22N7O3S2: 398.4899; found: 398.1069.
Synthesis of TMZ–Thiol (7)
In a 50 mL round-bottom flask charged with a stir bar, cysteamine (3.34 g, 43.3 mmol) was dissolved in TFA (30 mL). To this was added trityl chloride (12.1 g, 43.3 mmol), and the reaction mixture was stirred overnight at room temperature. After 16 h, the solution was concentrated by rotary evaporation. The crude product was suspended in ethyl acetate (50 mL) and washed with aqueous 3 M NaOH (3 × 15 mL), water (15 mL), saturated NaHCO3 (2 × 15 mL), and brine (3 × 15 mL). The organic layer was dried over Na2SO4, and a precipitate was observed. To the suspension was added chloroform, and the suspension was heated to dissolve the precipitant. The solution was concentrated by rotary evaporation, and the crude product was recrystallized from hexanes:DCM (1:1) to give trityl-protected cysteamine as a white solid (2.64, 14% yield). Then, in a 20 mL vial charged with a stir bar, TMZ–carboxylic acid (893 mg, 4.66 mmol), trityl-protected cysteamine (1.66 g, 3.80 mmol), and DMAP (49.4 mg, 0.400 mmol) were suspended in 10 mL of DCM. To this suspension was added TEA (585 μL, 4.20 mmol), and the vial was cooled to 0 °C. EDC (1.16 g, 6.10 mmol) was added to the suspension; the mixture became homogeneous and red. After the solution was stirred at room temperature for 20 h, it was diluted with DCM (20 mL) and washed with aqueous 0.1 M HCl (3 × 50 mL). The organic layer was dried over Na2SO4 and concentrated by rotary evaporation. To a 20 mL vial charged with a stir bar, the crude product was combined with TFA (2 mL) and dissolved in DCM (2 mL). To this was added triisopropylsilane (1 mL), and the mixture was allowed to stir at room temperature. After 1 h, the solution was concentrated by rotary evaporation and purified by column chromatography on silica gel, eluting with ethyl acetate, to give TMZ–thiol 7 as a white solid (487.5 mg, 42% yield). 1H NMR (400 MHz, DMSO, δ, ppm): 2.43 (t, 1H), 2.67 (q, 2H), 3.46 (q, 2H), 3.87 (s, 3H), 8.64 (t, 1H), 8.85 (s, 1H). 13C NMR (100 MHz, DMSO, δ, ppm): 23.33, 36.15, 41.94, 128.47, 130.18, 134.51, 139.19, 159.65.
General Procedure for the Synthesis of Disulfide-Containing PolyMPC–TMZ Copolymers (D1–D2)
In a 7 mL vial charged with a stir bar, MPC and TMZ–methacrylamide 6 were dissolved in TFE at a total monomer concentration of 0.75 M. ACVA and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid were added as the radical initiator (I) and chain-transfer agent (CTA), respectively, targeting [CTA]0:[I]0 of 5:1 and [MPC]0:[6]0 ranging from 85:15 to 55:55. The solution was degassed with bubbling nitrogen at room temperature for approximately 15 min and then was heated to 70 °C to initiate polymerization. Upon achieving monomer conversion >80%, as estimated by 1H NMR spectroscopy, the polymerization was quenched into a liquid nitrogen bath and exposed to air. The mixture was precipitated three times from TFE into THF, and the polymer was isolated by centrifugation. The isolated polymer was dissolved in aqueous 0.1 M HCl (10 mL), added to a centrifugal dialysis filter with a nominal molecular weight cutoff of 10 kDa, and centrifuged (4000g, 45 min, room temperature). The filtrate was discarded, and centrifugal dialysis was repeated twice. The concentrated polymer was dissolved in aqueous 0.1 M HCl, and lyophilization afforded polymers D1–D2 as pink solids in yields >60%. Incorporation of monomer 6 into the copolymer structure was estimated by 1H NMR spectroscopy by comparing the relative signal intensities at 8.50 ppm (C—H in TMZ) and 3.16–3.49 ppm (N—(CH3)3 in MPC). 1H NMR (500 MHz, TFE-d3, δ, ppm): 0.05–1.28 (br, 6H), 1.29–2.33 (br, 4H), 2.53–2.93 (br, 4H), 3.08 (s, 9H), 3.25–3.48 (br, 2H), 3.48–3.65 (br, 3H), 3.77–3.95 (br, 4H), 3.96–4.30 (br, 4H), 4.33–4.50 (br, 2H), 8.42 (s, 1H). 13C NMR (125 MHz, TFE-d3, δ, ppm): 17.18, 18.53, 35.99, 38.22, 39.28, 45.25, 53.92, 54.75, 63.83, 65.50, 65.92, 134.30, 138.40, 160.38, 177.86, 178.83. 31P NMR (202 MHz, TFE-d3, δ, ppm): −2.68.
General Procedure for UV–vis Degradation Studies
The degradation of TMZ and polyMPC–TMZ conjugates under physiological conditions was assessed using UV–vis spectroscopy. Pure TMZ and polyMPC–TMZ copolymers were prepared as 1 mg/mL solutions in phosphate-buffered saline (PBS) at pH 7.4. The solutions were diluted to an approximate TMZ-equivalent concentration of 0.013 mg/mL and then transferred to quartz cuvettes containing a magnetic stir bar. Solutions were incubated at 37 °C while stirring at 500 rpm in the UV–vis spectrometer, and absorbance spectra (λ = 200–450 nm) were measured at predetermined intervals over a total of 6, 23, 23, and 33 h for TMZ, R1–R4, D1–D2, and B1–B3 respectively. The decrease in absorbance intensity at λ = 328–330 nm, corresponding to the urea of intact TMZ, was measured, and exponential decay curves were generated from plotting the normalized absorbance (A/A0) as a function of time. Fitting these curves gave the decay constant, which was used to determine the t1/2 of free TMZ and polyMPC–TMZ copolymers.
DLS of PolyMPC–TMZ Copolymers R1–R4 and B1–B3
Suspensions of polyMPC–TMZ copolymers R1–R4 and B1–B3 (1 mg/mL) were prepared in pH 7.4 PBS. The suspensions were passed through 0.45 μm cellulose acetate filters into disposable cuvettes and analyzed by DLS at 37 °C. Three measurements were made for each suspension, with measurements consisting of 10 runs each.
Cell Culture
U87MG and T98G glioblastoma cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and antibiotic–antimycotic solution (1×). All cells were grown in 5% CO2 incubators at 37 °C. For all cytotoxicity assays, cell viability was measured post-treatment using CellTiter-Glo luminescent viability assays following manufacturer instructions (Promega) on a FLUOstar OPTIMA plate reader (BMG LABTECH). The percentage of TMZ-mediated toxicity was calculated relative to untreated cells and plotted to give dose–response curves. IC50 values for each treatment were calculated using GraphPad Prism 7 software. In vitro cytotoxicity assays were performed by seeding U87MG and T98G cells in 96-well plates (∼1–2 × 103 cells per plate). The cells were then incubated for 6 days with a range of TMZ-equivalent concentrations (1–20 000 μM) of free TMZ, polyMPC–TMZ conjugates R1–R4 and B1–B3, and polyMPC as a control (20 000 μM). To evaluate cytotoxicity of disulfide-containing conjugates, U87MG cells were seeded in 96-well plates (∼2 × 103 cells per plate). The cells were then incubated for 6 days with a range of TMZ-equivalent concentrations (1–10 000 μM) of free TMZ, TMZ–thiol 7, disulfide-containing polyMPC–TMZ copolymers D1–D2, and polyMPC as a control (20 000 μM) in the presence of 0 or 1 mM GSH.
Evaluation of Cellular Uptake
U87MG cells were placed on 35 mm dishes with 10 mM glass microwells (Mattek) overnight. The following day, cells were treated with fluorescein-labeled polyMPC and polyMPC-TMZ conjugates RF1, RF2, BF1, and BF2 at polymer concentrations of 200 μM for 2 h. Following incubation, cells were washed three times with PBS and incubated with a 50 mM solution of LysoTracker Red for 10 min. Cells were then washed three times with PBS and fixed with 4% paraformaldehyde in PBS for 5 min. Cells were then washed three times with PBS, incubated with a DAPI solution, and examined by confocal microscopy. In addition to confocal microscopy, flow cytometry was performed to quantify cellular uptake of fluorescently labeled polyMPC and polyMPC–TMZ conjugates. U87MG cells were grown in T-25 tissue culture flasks and treated with fluorescein-labeled polyMPC and polyMPC–TMZ conjugates RF1, RF2, BF1, and BF2 at polymer concentrations of 200 μM. After 2 h, the cells were trypsinized and washed with PBS. Fluorescence was detected using a BD DUAL LSRFortessa flow cytometer, and the data were analyzed using FlowJo software. The mean fluorescence intensity was calculated using the relative incorporation of fluorophore in each conjugate, as estimated by a fluorescein calibration curve.
Results and Discussion
Synthesis of PolyMPC–TMZ Copolymers
PolyMPC–TMZ copolymers with random and block copolymer architectures were prepared by RAFT polymerization. As shown in Figure 2, random copolymers R1–R4 were prepared using a previously reported procedure by copolymerizing MPC with TMZ–methacrylate 1, utilizing 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid and 4,4′-azobis(4-cyanovaleric acid) as chain-transfer agent (CTA) and radical initiator species, respectively.22 Polymerizations were performed at 70 °C in 2,2,2-trifluoroethanol (TFE), which was found to effectively solubilize both monomers and preserve the TMZ chemical structure under the polymerization conditions.22 PolyMPC–TMZ random copolymers R1–R4 were prepared, targeting TMZ incorporations of 20, 25, 35, and 50 mol percent, respectively, and number-average molecular weight (Mn) values of approximately 30 kDa. Copolymers R1–R4 were purified by repeated precipitation from TFE into tetrahydrofuran (THF), followed by centrifugal dialysis against aqueous 0.1 M HCl, a dialysis solution selected to prevent TMZ degradation. Lyophilization of the product gave random copolymers R1–R4 as pink solids (the color attributed to the dithioester chain-end) in 70–80% yields, after accounting for monomer conversion (85–90%).
Figure 2.

Synthesis of polyMPC–TMZ copolymers with random (a) and block (b) copolymer architectures by RAFT polymerization of TMZ methacrylate 1.
PolyMPC–TMZ conjugates were synthesized as diblock copolymers starting from a polyMPC macro-CTA bearing a dithioester chain-end suitable for RAFT chain extension with TMZ–methacrylate 1 (Figure 2). PolyMPC macro-CTA 2 was prepared by the homopolymerization of MPC in TFE, targeting an Mn of 15 kDa and quenching polymerization at <80% monomer conversion, as estimated by 1H NMR spectroscopy. The polymer product was precipitated into THF, dialyzed in water, and isolated by lyophilization in 80% yield, after accounting for monomer conversion. 1H NMR spectroscopy of polymer 2 showed resonances corresponding to chain-end phenyl protons at 7.40–7.96 ppm (Figure S1), confirming retention of the dithioester chain-ends. Chain-end analysis, integrating the aromatic protons of the phenyl end groups vs. the PC methylene (δ = 3.66–3.82 ppm) resonances, gave an estimated Mn of polymer 2 ranging from 15.1 to 19.8 kDa with impressively low dispersity (Đ) values of 1.05–1.13 (as estimated using GPC eluting with TFE relative to poly(methyl methacrylate) (PMMA) standards). TMZ-containing block copolymers B1–B3 targeting drug loadings of 20, 26, and 36 mol percent, respectively, were prepared by polymerizing 1 in TFE at 70 °C using macro-CTA 2 and ACVA as the radical initiator. Monomer conversions of >90% were achieved in approximately 4–8 h, and the block copolymers were purified in similar fashion to that of the random copolymers and isolated as pink solids in >50% yield.
The structural integrity of the pendent TMZ moieties, a crucial feature for preserving drug efficacy, was confirmed by NMR spectroscopy of copolymers R1–R4 and B1–B3. In the 1H NMR spectra of polyMPC–TMZ random and block copolymers, resonances corresponding to TMZ imidazole protons were observed at 8.53 and 8.72 ppm, respectively (Figures S2 and S3). Additional resonances at 3.92 (random) and 3.88 (block) ppm are attributed to the TMZ methyltriazene protons. Importantly, these signals for the polymer-bound TMZ are single, clean resonances for both copolymer architectures, with no sign of degradation to the 5-aminoimidazole-4-carboxamide (AIC) byproduct. 13C NMR spectroscopy further confirmed retention of the TMZ structure, with characteristic resonances of the methyltriazene moiety appearing at 38.2 and 38.5 ppm for the random (Figure S4) and block (Figure S5) architectures, respectively. Moreover, UV–vis spectra of the copolymers in TFE, shown in Figure 3, exhibited an absorbance maximum at λ = 323 nm, characteristic of the urea group of intact TMZ.
Figure 3.
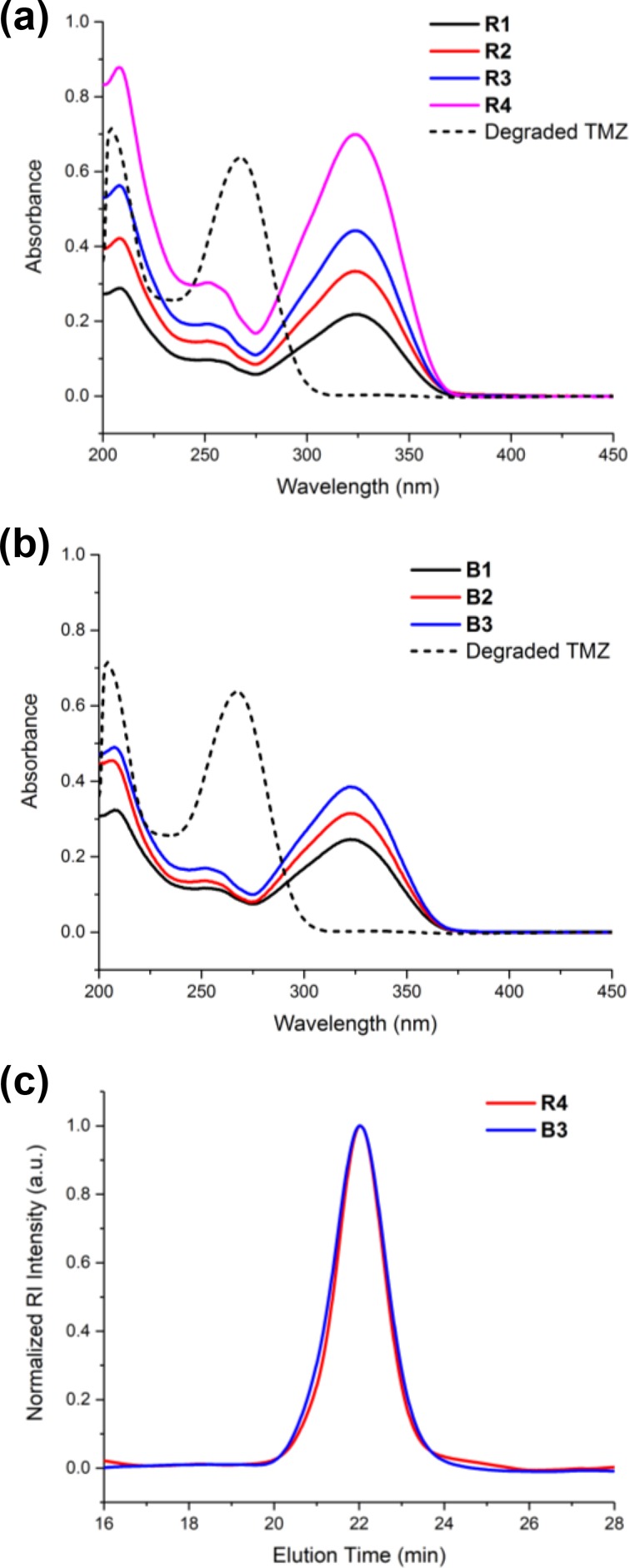
UV–vis spectra of polyMPC–TMZ random (R) (a) and block (B) (b) copolymers in TFE at a concentration of 0.05 mg/mL. A representative spectrum of degraded TMZ is shown in each compilation. Representative GPC chromatograms of polyMPC–TMZ copolymers R4 and B3 eluting in TFE with RI detection (c).
TMZ incorporations into the polyMPC–TMZ copolymers R1–R4 and B1–B3 were estimated using 1H NMR spectroscopy, integrating signals from the TMZ imidazole and PC trimethylammonium (δrandom = 2.86–3.29 ppm, δblock = 2.78–3.22 ppm) protons. For each copolymer, numerous samples were prepared for in vitro evaluation in glioblastoma cell lines. Characterized drug loading in these samples, summarized in Table 1, was in excellent agreement with targeted values and exhibited minimal compositional variation. Additional syntheses targeted TMZ loadings of 65 and 50 mol percent for copolymers with random and block architectures, respectively. While copolymers with such high drug loadings were synthetically accessible, their low water solubility made them unsuitable for further evaluation.
Table 1. TMZ Drug Loading and Molecular Weight Characterization of PolyMPC–TMZ Copolymers R1–R4 and B1–B3.
| polymer | targeted TMZ incorporation (mol %) | measured TMZ incorporationa (mol %) | theoretical Mnb (kDa) | Mnc (kDa) | Đc |
|---|---|---|---|---|---|
| R1 | 20 | 15–17 | 25.5–27.3 | 34.3–42.8 | 1.14–1.22 |
| R2 | 25 | 23–26 | 26.8–29.7 | 39.4–42.1 | 1.21 |
| R3 | 35 | 32–33 | 27.6–27.8 | 36.2–40.5 | 1.14–1.20 |
| R4 | 50 | 47–50 | 28.3–29.2 | 40.6–46.4 | 1.15–1.28 |
| B1 | 20 | 14–16 | 18.1–25.0 | 23.6–24.8 | 1.14–1.15 |
| B2 | 26 | 24–25 | 19.4–26.9 | 22.6–29.6 | 1.09–1.19 |
| B3 | 36 | 31–35 | 22.5–30.4 | 24.9–31.5 | 1.09–1.21 |
Estimated by 1H NMR.
Determined from percent monomer conversion, which was estimated by 1H NMR spectroscopy.
Estimated by GPC eluting with TFE, calibrated against PMMA standards.
The molecular weights of polyMPC–TMZ copolymers were estimated by GPC, eluting in TFE and calibrated against PMMA standards. Representative chromatograms for random and block copolymers R4 and B3, shown in Figure 3, were relatively narrow and distinctly monomodal. As shown in Table 1, polyMPC–TMZ random and block copolymers were isolated with Mn values of 36.2–46.4 and 22.6–31.5 kDa, respectively, and Đ values of 1.09–1.28. Copolymers of the same architecture were found to possess similar estimated Mn values across multiple samples, demonstrating the excellent reproducibility of this synthetic approach. Importantly, GPC chromatograms obtained with UV detection at λ = 323 nm showed no evidence of residual TMZ methacrylate 1 in any of the copolymers (Figure S6).
Aqueous Assembly of PolyMPC–TMZ Copolymers
Possessing both hydrophlic PC zwitterions and hydrophobic TMZ moieties, polyMPC–TMZ copolymers were anticipated to form aqueous assemblies of much larger hydrodynamic sizes than TMZ itself. Critical aggregation concentration (CAC) values of polyMPC–TMZ copolymers were determined in pH 7.4 phosphate-buffered saline (PBS) at 37 °C using dynamic light scattering (DLS);30 the absorption characteristics of TMZ preclude using fluorescent (e.g., pyrene)31 or absorption (e.g., 1,6-diphenyl-1,3,5-hexatriene)32 encapsulation assays to obtain CAC values. Representative plots of DLS scattering intensity for solutions of B3 and R4 in PBS at polymer concentrations ranging from 0.005 to 5 mg/mL are shown in Figure 4. Notably, block copolymer B3 exhibited a sharp increase in scattering intensity at a critical concentration of approximately 0.48 mg/mL, indicating the onset of copolymer aggregation. Similar behavior was observed for polymers B1 and B2 at concentrations of 0.70 and 0.45 mg/mL, respectively (Figure S7). CAC values for B1–B3, summarized in Figure 4c, were estimated as the onset of increasing scattering intensity. In contrast, scattering intensity values for solutions of R4 remained relatively constant at each polymer concentration, pointing to an indiscernible CAC for polyMPC–TMZ random copolymers.
Figure 4.

Aqueous assembly of polyMPC–TMZ copolymers: (a) representative scattering intensities for suspensions of polymers R4 and B3 with varying polymer concentrations in pH 7.4 PBS at 37 °C measured by DLS. For block copolymers B1–B3, the CAC was estimated as the onset of increasing scattering intensity; (b) DLS plots of polymers B1–B3 and R4 in pH 7.4 PBS (37 °C) at a concentration of 1 mg/mL; (c) CAC values and hydrodynamic diameters for B1–B3 and R4 measured using DLS at 37 °C in pH 7.4 PBS; (d) representative cryo-TEM image of nanoparticles formed from polymer B3 in water at a concentration of 1 mg/mL.
The hydrodynamic diameters of structures formed from B1–B3 in PBS at 37 °C were estimated using DLS at a concentration of 1 mg/mL, above the CAC of each polymer. The DLS plots in Figure 4b show that polyMPC–TMZ block copolymers form monomodal nanoparticles (dh = 24–38 nm) with no evidence of particle aggregation. Remarkably, these block copolymers readily assembled in water into well-defined and narrowly dispersed aggregates without the need for intricate solution-assembly procedures (e.g., solvent-directed assembly or thin film hydration).33−35 In contrast, a trimodal size distribution was observed for random copolymer R4, which was dominated by the smaller structures (dh ≈ 7 nm). As expected, the hydrodynamic diameters of R1–R3 were similarily small (Figure S8). Suspensions of block copolymer B3 in pure water were cast on carbon-coated copper grids, vitrified in liquid ethane, and visualized by cryogenic transmission electron microscopy (cryo-TEM). As shown in Figure 4d, B3 nanoparticles were observed as spherical aggregates with discrete coronae composed of polyMPC. These nanoparticles exhibited a narrow size distribution, with a mean diameter of 12.6 ± 2.8 nm. The larger diameter obtained by DLS relative to cryo-TEM is attributed to the hydration layer of the hydrophilic nanoparticle corona; similar overestimation by DLS relative to EM observations has been reported for other polymer nanoparticles.36
Hydrolytic Stability of TZ in polyMPC Conjugates
The hydrolytic instability of TMZ leads to rapid decomposition under physiological conditions, with reported half-lives in pH 7 PBS and blood plasma of only 1 and 1.8 h, respectively.10 This premature TMZ degradation leads to off-target hematoxicity and reduces the concentration of TMZ available to cancer cells. Developing TMZ formulations that stabilize the drug and prolong its half-life in solution is critical to augmenting efficacy. The decomposition of TMZ was easily monitored by UV–vis spectroscopy: as TMZ degraded, the absorption corresponding to the urea moiety of pristine TMZ (λ = 328–330 nm) decreased in intensity, as shown in Figure 5a, and a new absorption attributed to the amide group of the AIC byproduct was observed at λ = 265–267 nm.14,37
Figure 5.

(a) Representative UV–vis cascade curves showing evidence of TMZ degradation by a decrease in peak intensity at λ = 328–330 nm, corresponding to the TMZ urea, to the AIC byproduct, seen as an increase in peak intensity at λ = 265–267 nm; (b) degradation profiles for TMZ, R1–R4, and B1–B3 incubated in pH 7.4 PBS at 37 °C; (c) values of t1/2 estimated for TMZ, R1–R4, and B1–B3 from exponential fitting of decay curves as well as corresponding experimental timeframes.
We previously showed that conjugation of TMZ to the polyMPC backbone increased the drug half-life (t1/2) in aqueous solution more than 2-fold for polyMPC–TMZ random copolymers containing approximately 50 mol percent TMZ.22 To further investigate the impact of polymer conjugation, drug loading, and architecture on TMZ stability, polyMPC–TMZ copolymers R1–R4 and B1–B3 were incubated at 37 °C in pH 7.4 PBS and compared to TMZ itself. UV–vis absorption spectra were recorded over time, and TMZ degradation profiles were constructed from the absorption decrease at λ = 328–330 nm (Figure 5b). Half-life values for TMZ and the polyMPC–TMZ copolymers, summarized in Figure 5c, were obtained from exponential decay curves generated by plotting the normalized absorbance (A/A0) as a function of incubation time. As shown in Figure 5, TMZ degraded quickly in PBS, exhibiting a t1/2 of only 1.0 h. The polyMPC–TMZ random copolymers lengthened TMZ half-life 2- to 3-fold, a finding that proved independent of the mole percent TMZ incorporation for the random copolymers. Polymer architecture had a marked effect on TMZ solution stability, with block copolymers B1, B2, and B3 raising t1/2 values to 12.8, 18.9, and 19.1 h, respectively. In contrast to the random copolymers, increasing TMZ incorporation from 16−17 to 23–26 mol percent led to a dramatic increase of t1/2 values; however, additional TMZ incorporation beyond 26 mol percent did not further improve solution stability. While TMZ in the R1–R4 polymers degraded almost entirely within 24 h, significant amounts of TMZ (18–37%) incorporated into the block copolymers remained intact, even after incubation at pH 7.4 and 37 °C for 33 h. These results highlight the role of polymer architecture in enhancing TMZ stability in an aqueous environment. Such improvements in TMZ stability enabled by the polyMPC–TMZ platform are comparable, if not superior, to other delivery systems,17,18 while remaining synthetically accessible.
In addition to stabilizing TMZ by polymer conjugation, we demonstrated TMZ uptake into block copolymers. Aqueous suspensions of block copolymer amphiphiles, polyMPC–TMZ block copolymer B2 and a poly(MPC-b-butyl methacrylate) (polyMPC–BMA) copolymer with 28 mol percent BMA, were used to solubilize free TMZ in pH 7.4 PBS at polymer concentrations of 5 mg/mL. By eye, TMZ-contaning polymer B2 dissolved TMZ more rapidly than the polyMPC–BMA surfactant. Furthermore, the polyMPC–TMZ block copolymer suspended free TMZ completely at a drug concentration of 4 mg/mL, while polyMPC–BMA, under the same conditions, left residual (undissolved) TMZ. These TMZ–block copolymer suspensions were diluted approximately 400 times and incubated at 37 °C, with TMZ degradation monitored by UV–vis spectroscopy. Under these conditions, the solution stability was not significantly extended in comparison to free TMZ—half-lives of 1.3 and 1.4 h were measured for TMZ suspensions prepared using B2 and polyMPC–BMA, respectively. TMZ stability was also examined at a polyMPC–BMA concentration well-above the CAC (1 mg/mL), as estimated using a pyrene fluoresence assay.31 Despite solubilization and encapsulation of TMZ into the hydrophobic core of the block copolymer aggregates, the TMZ t1/2 was extended minimally to 1.8–2.7 h. A comparable experiment was not feasible for the polyMPC–TMZ conjugates, as TMZ groups pendent to the polymer backbone saturated the UV–vis detector at this concentration (i.e., 1 mg/mL) and prevented absorbance measurements of encapsulated TMZ. These results suggest that polymer–TMZ conjugation is superior to encapsulation for significantly enhancing TMZ solution stability.
Cellular Uptake of PolyMPC–TMZ Copolymers
The polyMPC–TMZ conjugate platform, unlike those of conventional prodrugs, does not require triggered release of covalently conjugated TMZ moieties for antitumor activity. As such, copolymer internalization is likely necessary for efficacious treatment, by enabling release of methyldiazonium cations near cellular DNA. To investigate cellular uptake and intracellular accumulation, fluorescently labeled random copolymers with 16 and 51 mol percent TMZ (RF1 and RF2, respectively) and block copolymers with 11 and 33 mol percent TMZ (BF1 and BF2, respectively), were synthesized, incorporating ∼1 mol percent of fluorescein methacrylate to allow microscopic visualization. U87MG cells were incubated for 2 h with fluorescein-labeled polyMPC or polyMPC–TMZ copolymers, then visualized by confocal microscopy at a set camera exposure time. polyMPC–TMZ copolymers exhibited increased intracellular accumulation compared to polyMPC alone, suggesting that the pendent hydrophobic TMZ moieties promoted cellular uptake into the cytoplasm (Figure S9). Similar behavior has been reported previously: Goda et al. demonstrated that while polyMPC has low cellular permeability on its own, the incorporation of hydrophobic units (e.g., butyl methacrylate) to form amphiphilic MPC-based copolymers increased cell penetration.38 While polyMPC and polyMPC–TMZ conjugates did not localize in the nucleus, as evidenced by the lack of overlay with DAPI-stained regions, the polymer–drug conjugates did exhibit perinuclear localization. Furthermore, both the fluorescently labeled polyMPC copolymer and polyMPC–TMZ conjugates colocalized in lysosomes, marked by significant overlay with red-stained regions, as shown in Figure 6. We hypothesize that polymer–drug conjugates, following endosome-mediated uptake into the cell, localize in the lysosomes and efflux into the cytosol, allowing for methyldiazonium cation release near the nucleus for DNA methylation.
Figure 6.
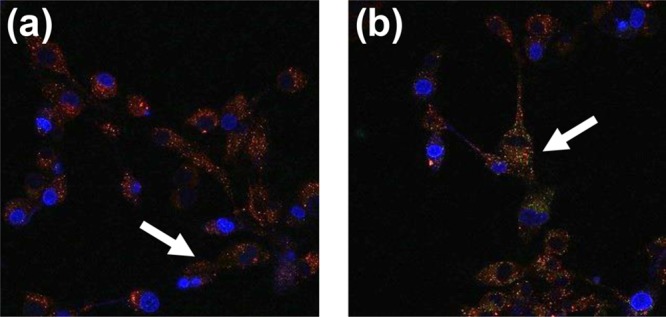
Fluorescence micrographs of U87MG cells incubated for 2 h with (a) RF1 and (b) BF1. Cell nuclei were stained with DAPI (blue), lysosomes were stained with LysoTracker Red (red), and the fluorescein-labeled copolymer appear as green. White arrows indicate overlay of red and green fluorescence, suggesting copolymer localization in lysosomes. Micrographs for all tested copolymers, compared to controls, are shown in the SI (Figure S9).
Cellular uptake of polymer–drug conjugates was quantified by flow cytometry: fluorescently labeled polyMPC, RF1, RF2, BF1, and BF2 were incubated at fluorescein-equivalent concentrations in U87MG cells for 2 h, after which the relative intracellular fluorescence intensities were determined on a fluorescence plate reader, as compiled in Figure 7. In agreement with qualitative fluorescence microscopy observations, polyMPC–TMZ intracellular accumulation was far greater than that of polyMPC itself, indicating that pendent TMZ moieties enhanced polymer uptake into glioblastoma cells. Interestingly, block copolymers BF1 and BF2 achieved markedly higher uptake than the random copolymer analogues, despite the segregation of TMZ into core domains that have minimal interaction with cell membranes. While this result was unexpected, previous reports on the effect of monomer distribution on cellular internalization have shown that uptake is largely influenced by polymer chemistry and not copolymer architecture itself.39−41
Figure 7.
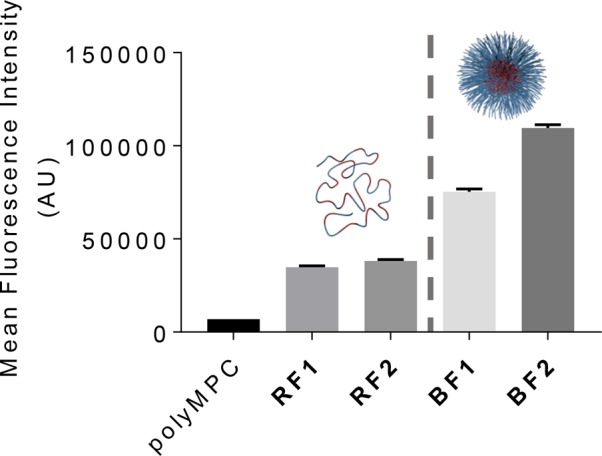
Intracellular abundance of polyMPC and polyMPC–TMZ conjugates (random copolymers RF1 and RF2; block copolymers BF1 and BF2) after 2 h incubation at fluorescein-equivalent concentrations in U87MG cells.
In Vitro Evaluation of PolyMPC–TMZ Copolymers in Glioblastoma Cell Lines
The antitumor activity of polyMPC–TMZ copolymers was investigated in TMZ-sensitive (i.e., U87MG) and TMZ-resistant (i.e., T98G) glioblastoma cells. These cell lines have been utilized extensively for testing TMZ cytotoxicity42−46 as well as TMZ-containing delivery systems.18,47,48 In dose–response assays in the literature, an unusually broad range of IC50 values have been reported for TMZ, spanning from 10 to 500 μM for U87MG cells and 250–1600 μM for T98G cells.46 Other small molecule chemotherapeutics (e.g., doxorubicin)25,49−52 have comparatively narrow ranges of reported IC50 values; the wide variation seen with TMZ likely arises from differences in experimental protocols. Moreover, the poor aqueous solubility and hydrolytic instability of TMZ add complexity and likely increase variability in cell culture experiments, as aqueous TMZ solutions must be added to cells immediately after preparation. Recognizing the importance of handling TMZ in such a way as to accurately measure its cytotoxicity, aqueous exposure was kept to a minimum prior to cell culture experiments.
To evaluate cytotoxicity in glioblastoma cells, free TMZ and polyMPC–TMZ copolymers were incubated at TMZ-equivalent concentrations with U87MG or T98G cells for 6 days, with polyMPC serving as a negative control. Cell viability was determined using a CellTiter-Glo assay to generate dose–response curves (Figure 8). Free TMZ exhibited IC50 values of 192 ± 72 and 418 ± 116 μM in U87MG and T98G cells, respectively. The IC50 values for the polyMPC–TMZ copolymers, summarized in Figure 8, were 7- to 10-fold higher than the free TMZ values in both cell lines. While polyMPC–TMZ lacks a responsive polymer-to-drug linker and, as such, is not a typical polymer prodrug, reduced cytotoxicity of polymer-bound drugs vs. free drugs has been noted in many examples of polymer–drug conjugates and is advantageous for achieving higher maximum tolerated doses in vivo.25−27,53,54 Thus, as expected, higher polyMPC–TMZ copolymer concentrations were necessary to induce cytotoxicity in both cell lines. Moreover, we noted a plateau in IC50 values once a critical TMZ incorporation was achieved (>20 mol percent): R1 and B1 had significantly higher IC50 values, as compared to R2–R4 and B2–B3, respectively. Polymer architecture appeared to have no effect on the cytotoxicity of polyMPC–TMZ copolymers in chemosensitive U87MG cells. However, block copolymers with sufficiently high TMZ loading (>20 mol percent) exhibited significant (i.e., 54–82%) reduction in IC50 values in chemoresistant T98G cells, relative to the random copolymer conjugates with comparable drug loadings. This improvement in antiglioblastoma activity is likely due to the enhanced TMZ solution stability and the increased cellular uptake afforded by the block copolymer architecture relative to the random structures. While these copolymers showed higher IC50 values than free TMZ, they remained efficacious in both chemosensitive and chemoresistant cell lines. This, coupled with their favorable solution stability properties, makes polyMPC–TMZ conjugates promising for allowing higher dosing and increased antitumor efficiency in future in vivo work.
Figure 8.

Cell viability of polyMPC–TMZ random (a) and block (b) copolymers in U87MG (top) and T98G (bottom) glioblastoma cells; (c) IC50 values for free TMZ and polyMPC–TMZ copolymers in both cell lines (± indicates standard deviation).
Redox-Responsive PolyMPC–TMZ Conjugates
While the polyMPC–TMZ copolymers demonstrated enhanced TMZ solution stability and efficacious in vitro antitumor activity, we sought to further expand this delivery platform by preparing responsive polymer–drug conjugates containing redox-sensitive disulfides as the polymer-to-TMZ linkages. Polymer prodrugs utilizing disulfide linkers have been examined for chemotherapeutics,27,55,56 taking advantage of the high reducing potential of intracellular environments, relative to extracellular space,57 to promote triggered and localized drug release. Such degradable linkers will provide a mechanism for releasing intact TMZ from the polymer backbone in environments with high concentrations of reducing agents.
Seeking to produce releasable TMZ prodrugs, disulfide-containing TMZ monomer 6 was synthesized using the strategy shown in Figure 9. Methacrylamide precursor 5 was prepared following a modified literature procedure29 and then coupled to TMZ–carboxylic acid using carbodiimide-mediated conditions to give TMZ–methacrylamide 6 as a white powder in 80–85% yield. We note that attempts to synthesize a comparable disulfide-containing TMZ–methacrylate monomer were unsuccessful due to low coupling efficiency between the carboxylic acid and the corresponding disulfide-containing hydroxyethyl methacrylate. Redox-responsive polyMPC–TMZ random copolymers D1 and D2 were synthesized by RAFT polymerization, using conditions similar to those described for polymers R1–R4, targeting 15 and 50 mol percent TMZ incorporation, respectively. Notably, the lower solubility of 6 in TFE required more dilute polymerization conditions (0.75 M) relative to comparable polymers without disulfide linkers (1.0 M). Redox-responsive copolymers D1 and D2 were isolated in 73 and 60% yields, respectively. TMZ incorporations were characterized by 1H NMR spectroscopy, and copolymer molecular weights were estimated using GPC eluting in TFE, calibrated against PMMA standards (Table 2). The observation of an absorption maxima at λ = 326 nm in the UV–vis spectra of polymers D1 and D2 confirmed that pendent TMZ moieties did not degrade during polymerization (Figure S10). While chain extension from macro-CTA 2 was possible, the poor solubility of disulfide-containing TMZ–methacrylamide 6 in aqueous and organic solvents precluded purification and isolation of the resulting block copolymers in appreciable yields. As such, our investigation of redox-sensitive polyMPC–TMZ prodrugs focused on random copolymers D1 and D2.
Figure 9.

(a) Synthesis of TMZ–methacrylamide 6: (i) di-tert-butyl dicarbonate, triethylamine, MeOH; (ii) methacryloyl chloride, triethylamine, dichloromethane; (iii) trifluoroacetic acid, dichloromethane; (iv) TMZ–carboxylic acid 4, N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride, 4-(dimethylamino)pyridine, triethylamine, dichloromethane; see Materials and Methods section for detailed procedures. (b) Chemical structures of redox-sensitive polyMPC–TMZ random copolymers D1 and D2, and a proposed mechanism for triggered release of TMZ in the presence of in vivo reducing agents, such as glutathione, giving TMZ–thiol (7).
Table 2. TMZ Drug Loading and Molecular Weight Characterization of Redox-Responsive PolyMPC–TMZ Copolymers D1–D2.
| polymer | target TMZ (mol %) | measured TMZa (mol %) | theoretical Mnb (kDa) | Mnc (kDa) | Đc |
|---|---|---|---|---|---|
| D1 | 15 | 12 | 27.6 | 36.8 | 1.17 |
| D2 | 50 | 43 | 31.2 | 39.3 | 1.14 |
Estimated by 1H NMR spectroscopy.
Theoretical Mn determined from percent monomer conversion, as estimated by 1H NMR spectroscopy.
Estimated by GPC eluting with TFE, calibrated against PMMA standards.
The hydrolytic stability of the disulfide-containing polyMPC–TMZ random copolymer D1 was investigated by UV–vis spectroscopy at varying concentrations of glutathione (GSH) in pH 7.4 PBS. We specifically selected 2 μM GSH and 1 mM GSH to reflect the redox environments of human blood plasma and glioblastoma tumor tissue, respectively.58−60Copolymer D1 was incubated either in pure PBS buffer or GSH-containing PBS buffer at 37 °C, and the UV–vis absorption spectra were collected over 24 h. As previously described, degradation profiles were generated from the absorption decrease at λ = 328–330 nm, corresponding to the TMZ urea, and exponential decay curves were constructed by plotting the normalized absorbance (A/Ao) against incubation time. As with R1 and R4 (the random copolymers without disulfide linkers), polymer conjugation extended the TMZ solution stability at physiological pH and temperature: the t1/2 for D1 was 3.7 h in pure PBS buffer, compared to 3.1 h for R1. The TMZ solution stability did not change significantly at 2 μM GSH, with D1 exhibiting a t1/2 of 3.6 h, suggesting that the disulfide-containing copolymers will retain slow TMZ decomposition in circulation relative to TMZ itself. Notably, increasing the GSH concentration to 1 mM halved the t1/2 to 1.7 h, which is attributed to the glutathione-mediated reduction of disulfide polymer-to-drug linkers and subsequent release of small molecule TMZ from the polymer backbone.
The cytotoxicity of disulfide-containing polyMPC–TMZ copolymers D1 and D2, at TMZ-equivalent concentrations, was evaluated in U87MG cells under GSH-free (0 mM GSH) and GSH-rich conditions (1 mM GSH), to mimic glioblastoma tumor tissue environments. As controls, cells were incubated for 6 days with free TMZ and TMZ–thiol 7, one of the possible byproducts of glutathione-mediated disulfide exchange. As shown in Figure 10, dose–response curves were generated for each glutathione concentration, giving IC50 values for free TMZ, TMZ–thiol, and disulfide-containing polyMPC–TMZ conjugates D1 and D2. The cytotoxicity of copolymers D1 and D2 was dependent on the amount of GSH in the media: the IC50 values decreased, respectively, from 1999 and 2592 μM in nonglutathione-enriched media to 1308 and 756 μM in 1 mM GSH media, indicating that TMZ release from the polymer backbone led to a potentiation of cytotoxicity. These results suggest the potential benefit of disulfide-containing polyMPC–TMZ copolymers with built-in polymer-to-drug linkers for redox-responsive release to treat glioblastoma tumor cells.
Figure 10.

Dose–response curves for U87MG cells treated with TMZ, TMZ–thiol, and polyMPC–TMZ conjugates (D1 and D2) in unmodified (a) and GSH-enriched (b) media; (c) IC50 values for free TMZ, TMZ–thiol, and polyMPC–TMZ copolymers at different glutathione concentrations (± indicates standard deviation).
Conclusions
In summary, we have demonstrated the preparation of well-defined and versatile polyMPC–TMZ random and block copolymers using controlled free radical polymerization. The direct conjugation of TMZ to the polymer backbone, through a TMZ-containing methacrylate, allowed for the preparation of polyMPC–TMZ conjugates having a wide range of drug loadings, which exhibited enhanced hydrolytic stability compared to free TMZ. Copolymer architecture played a significant role in drug efficacy, with the block copolymers showing the formation of well-defined nanostructures, higher intracellular abundance, and lower IC50 values in glioblastoma cells, compared to the polyMPC–TMZ random copolymers. Degradable polyMPC–TMZ copolymers, prepared with a disulfide polymer-to-drug linker for redox-triggered TMZ release, likewise demonstrated favorable aqueous TMZ stability and cytotoxicity against U87MG cells. The enhanced hydrodynamic sizes and solution stability, as well as the demonstrated antiglioblastoma activity, of these polyMPC–TMZ conjugates suggest their suitability for future in vivo experiments.
Acknowledgments
The authors thank Ryan Selhorst and Alex Ribbe for their assistance in electron microscopy measurements. This work was supported by the National Institutes of Health under Award Number R21 CA167674 and the National Science Foundation Graduate Research Fellowship under Grant Number 1451512 (S.M.W.). Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Center. Electron microscopy visualization was performed at the University of Massachusetts Electron Microscopy Center. Confocal microscopy visualization was performed at the University of Massachusetts Light Microscopy Core facility.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00766.
NMR, GPC, DLS, UV–vis, and fluorescence micrographs (PDF)
Author Contributions
† S.M.W. and M.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Ostrom Q. T.; Gittleman H.; Fulop J.; Liu M.; Blanda R.; Kromer C.; Wolinsky Y.; Kruchko C.; Barnholtz-Sloan J. S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro. Oncol. 2015, 17, iv1–iv62. 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omuro A.; Deangelis L. M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842–1850. 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- Stupp R.; Mason W. P.; van den Bent M. J.; Weller M.; Fisher B.; Taphoorn M. J. B.; Belanger K.; Brandes A. A.; Marosi C.; Bogdahn U.; et al Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Karim R.; Palazzo C.; Evrard B.; Piel G. Nanocarriers for the Treatment of Glioblastoma Multiforme: Current State-of-the-Art. J. Controlled Release 2016, 227, 23–37. 10.1016/j.jconrel.2016.02.026. [DOI] [PubMed] [Google Scholar]
- Ballabh P.; Braun A.; Nedergaard M. The Blood-Brain Barrier: An Overview: Structure, Regulation, and Clinical Implications. Neurobiol. Dis. 2004, 16, 1–13. 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- De Bonis P.; Anile C.; Pompucci A.; Fiorentino A.; Balducci M.; Chiesa S.; Maira G.; Mangiola A. Safety and Efficacy of Gliadel Wafers for Newly Diagnosed and Recurrent Glioblastoma. Acta Neurochir. 2012, 154, 1371–1378. 10.1007/s00701-012-1413-2. [DOI] [PubMed] [Google Scholar]
- Fourniols T.; Randolph L. D.; Staub A.; Vanvarenberg K.; Leprince J. G.; Préat V.; Des Rieux A.; Danhier F. Temozolomide-Loaded Photopolymerizable PEG-DMA-Based Hydrogel for the Treatment of Glioblastoma. J. Controlled Release 2015, 210, 95–104. 10.1016/j.jconrel.2015.05.272. [DOI] [PubMed] [Google Scholar]
- Newlands E. S.; Stevens M. F. G.; Wedge S. R.; Wheelhouse R. T.; Brock C. Temozolomide: A Review of Its Discovery, Chemical Properties, Pre-Clinical Development, and Clinical Trials. Cancer Treat. Rev. 1997, 23, 35–61. 10.1016/S0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Stevens M. F. G.; Bradshaw T. D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. 10.2174/1874-470211205010102. [DOI] [PubMed] [Google Scholar]
- Newlands E. S.; Blackledge G. R.; Slack J. A.; Rustin G. J.; Smith D. B.; Stuart N. S.; Quarterman C. P.; Hoffman R.; Stevens M. F.; Brampton M. H. Phase I Trial of Temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. 10.1038/bjc.1992.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M. F. G.; Hickman J. A.; Langdon S. P.; Chubb D.; Vickers L.; Stone R.; Baig G.; Goddard C.; Gibson N. W.; Slack J. A.; et al Antitumor Activity and Pharmacokinetics in Mice of 8-Carbamoyl-3-Methyl-Imidazo[5,1,d]-1,2,3,5-Tetrazin-4(3H)-One (CCRG 81045; M & B 39831), a Novel Drug with Potential as an Alternative to Dacarbazine. Cancer Res. 1987, 47, 5846–5852. [PubMed] [Google Scholar]
- Braverman O.; Feinshtein R.; Weisman A.; Kaspi J.. Temozolomide Storage System. US20060222792A1, 2006.
- Mirzaei S.; Khalilian M. H.; Taherpour A. A. Mechanistic Study of the Hydrolytic Degradation and Protonation of Temozolomide. RSC Adv. 2015, 5, 41112–41119. 10.1039/C5RA04680G. [DOI] [Google Scholar]
- Babu N. J.; Sanphui P.; Nangia A. Crystal Engineering of Stable Temozolomide Cocrystals. Chem. - Asian J. 2012, 7, 2274–2285. 10.1002/asia.201200205. [DOI] [PubMed] [Google Scholar]
- Appel E. A.; Rowland M. J.; Loh X. J.; Heywood R. M.; Watts C.; Scherman O. A. Enhanced Stability and Activity of Temozolomide in Primary Glioblastoma Multiforme Cells with Cucurbit[n]uril. Chem. Commun. 2012, 48, 9843–9845. 10.1039/c2cc35131e. [DOI] [PubMed] [Google Scholar]
- Di Martino A.; Kucharczyk P.; Capakova Z.; Humpolicek P.; Sedlarik V. Enhancement of Temozolomide Stability by Loading in Chitosan-Carboxylated Polylactide-Based Nanoparticles. J. Nanopart. Res. 2017, 19, 71. 10.1007/s11051-017-3756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil R.; Portilla-Arias J.; Ding H.; Inoue S.; Konda B.; Hu J.; Wawrowsky K. A.; Shin P. K.; Black K. L.; Holler E.; et al Temozolomide Delivery to Tumor Cells by a Multifunctional Nano Vehicle Based on Poly(β-L-Malic Acid). Pharm. Res. 2010, 27, 2317–2329. 10.1007/s11095-010-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.; Wang K.; Stephen Z. R.; Mu Q.; Kievit F. M.; Chiu D. T.; Press O. W.; Zhang M. Temozolomide Nanoparticles for Targeted Glioblastoma Therapy. ACS Appl. Mater. Interfaces 2015, 7, 6674–6682. 10.1021/am5092165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson N.; Ghandehari H. Polymeric Conjugates for Drug Delivery. Chem. Mater. 2012, 24, 840–853. 10.1021/cm2031569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M. E.; Szoka F. C.; Fréchet J. M. J. Soluble Polymer Carriers for the Treatment of Cancer: The Importance of Molecular Architecture. Acc. Chem. Res. 2009, 42, 1141–1151. 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in Vivo. Adv. Drug Delivery Rev. 2013, 65, 71–79. 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Skinner M.; Ward S. M.; Emrick T. Versatile Synthesis of Polymer-Temozolomide Conjugates. ACS Macro Lett. 2017, 6, 215–218. 10.1021/acsmacrolett.7b00007. [DOI] [PubMed] [Google Scholar]
- Wong K. E.; Mora M. C.; Skinner M.; McRae Page S.; Crisi G. M.; Arenas R. B.; Schneider S. S.; Emrick T. Evaluation of PolyMPC-Dox Prodrugs in a Human Ovarian Tumor Model. Mol. Pharmaceutics 2016, 13, 1679–1687. 10.1021/acs.molpharmaceut.6b00092. [DOI] [PubMed] [Google Scholar]
- McRae Page S.; Henchey E.; Chen X.; Schneider S.; Emrick T. Efficacy of PolyMPC-DOX Prodrugs in 4T1 Tumor-Bearing Mice. Mol. Pharmaceutics 2014, 11, 1715–1720. 10.1021/mp500009r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Parelkar S. S.; Henchey E.; Schneider S.; Emrick T. PolyMPC–Doxorubicin Prodrugs. Bioconjugate Chem. 2012, 23, 1753–1763. 10.1021/bc200667s. [DOI] [PubMed] [Google Scholar]
- Chen X.; McRae S.; Parelkar S. S.; Emrick T. Polymeric Phosphorylcholine-Camptothecin Conjugates Prepared by Controlled Free Radical Polymerization and Click Chemistry. Bioconjugate Chem. 2009, 20, 2331–2341. 10.1021/bc900339x. [DOI] [PubMed] [Google Scholar]
- McRae Page S.; Martorella M.; Parelkar S. S.; Kosif I.; Emrick T. Disulfide Cross-Linked Phosphorylcholine Micelles for Triggered Release of Camptothecin. Mol. Pharmaceutics 2013, 10, 2684–2692. 10.1021/mp400114n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith J.; Jennings S. A.; Clark A. S.; Stevens M. F. G. Antitumor Imidazotetrazines. 41.1 Conjugation of the Antitumor Agents Mitozolomide and Temozolomide to Peptides and Lexitropsins Bearing DNA Major and Minor Groove-Binding Structural Motifs. J. Med. Chem. 2002, 45, 5458–5470. 10.1021/jm020936d. [DOI] [PubMed] [Google Scholar]
- Suga Y.; Sunayama H.; Ooya T.; Takeuchi T. Molecularly Imprinted Polymers Prepared Using Protein-Conjugated Cleavable Monomers Followed by Site-Specific Post-Imprinting Introduction of Fluorescent Reporter Molecules. Chem. Commun. (Cambridge, U. K.) 2013, 49, 8450–8452. 10.1039/c3cc40484f. [DOI] [PubMed] [Google Scholar]
- Topel Ö.; Çakir A.; Budama L.; Hoda N. Determination of Critical Micelle Concentration of Polybutadiene-Block-Poly(ethyleneoxide) Diblock Copolymer by Fluorescence Spectroscopy and Dynamic Light Scattering. J. Mol. Liq. 2013, 177, 40–43. 10.1016/j.molliq.2012.10.013. [DOI] [Google Scholar]
- Kalyanasundaram K.; Thomas J. K. Environmental Effects on Vibronic Band Intensities in Pyrene Monomer Fluorescence and Their Application in Studies of Micellar Systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. 10.1021/ja00449a004. [DOI] [Google Scholar]
- Ko D. Y.; Patel M.; Jung B. K.; Park J. H.; Jeong B. Phosphorylcholine-Based Zwitterionic Biocompatible Thermogel. Biomacromolecules 2015, 16, 3853–3862. 10.1021/acs.biomac.5b01169. [DOI] [PubMed] [Google Scholar]
- Kita-Tokarczyk K.; Grumelard J.; Haefele T.; Meier W. Block Copolymer Vesicles—using Concepts from Polymer Chemistry to Mimic Biomembranes. Polymer 2005, 46, 3540–3563. 10.1016/j.polymer.2005.02.083. [DOI] [Google Scholar]
- Du J.; O’Reilly R. K. Advances and Challenges in Smart and Functional Polymer Vesicles. Soft Matter 2009, 5, 3544–3561. 10.1039/b905635a. [DOI] [Google Scholar]
- Zhang L.; Eisenberg A. Multiple Morphologies and Characteristics of “Crew-Cut” Micelle-like Aggregates of Polystyrene-B-Poly(acrylic Acid) Diblock Copolymers in Aqueous Solutions. J. Am. Chem. Soc. 1996, 118, 3168–3181. 10.1021/ja953709s. [DOI] [Google Scholar]
- Patil N.; Falentin-Daudré C.; Jérôme C.; Detrembleur C. Mussel-Inspired Protein-Repelling Ambivalent Block Copolymers: Controlled Synthesis and Characterization. Polym. Chem. 2015, 6, 2919–2933. 10.1039/C5PY00127G. [DOI] [Google Scholar]
- Arrowsmith J.; Jennings S. A.; Langnel D. A. F.; Wheelhouse R. T.; Stevens M. F. G. Antitumour Imidazotetrazines. Part 39. Synthesis of Bis(imidazotetrazine)s with Saturated Spacer Groups. J. Chem. Soc., Perkin Trans. 2000, 1, 4432–4438. 10.1039/b005652i. [DOI] [Google Scholar]
- Goda T.; Goto Y.; Ishihara K. Cell-Penetrating Macromolecules : Direct Penetration of Amphipathic Phospholipid Polymers across Plasma Membrane of Living Cells. Biomaterials 2010, 31, 2380–2387. 10.1016/j.biomaterials.2009.11.095. [DOI] [PubMed] [Google Scholar]
- Barz M.; Luxenhofer R.; Zentel R.; Kabanov A. The Uptake of N-(2-Hydroxypropyl)-Methacrylamide Based Homo, Random, and Block Copolymers by Human Multi-Drug Resistant Breat Adenocarcinoma Cells. Biomaterials 2009, 30, 5682–5690. 10.1016/j.biomaterials.2009.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes J.; Peltier R.; Gody G.; Blum M.; Recalcati S.; Klok H.-A.; Perrier S. Influence of Block versus Random Monomer Distribution on the Cellular Uptake of Hydrophilic Copolymers. ACS Macro Lett. 2016, 5, 1416–1420. 10.1021/acsmacrolett.6b00652. [DOI] [PubMed] [Google Scholar]
- Tong J.; Luxenhofer R.; Yi X.; Jordan R.; Kabanov A. Protein Modification with Amphiphilic Block Copoly(2-Oxazoline)s as a New Platform for Enhanced Cellular Delivery. Mol. Pharmaceutics 2010, 7, 984–992. 10.1021/mp100102p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M. M.; Gomez-Manzano C.; Bekele B. N.; Yung W. K.; Fueyo J. Adenovirus-Based Strategies Overcome Temozolomide Resistance by Silencing the O6-Methylguanine-DNA Methyltransferase Promoter. Cancer Res. 2007, 67, 11499–11504. 10.1158/0008-5472.CAN-07-5312. [DOI] [PubMed] [Google Scholar]
- Baer J. C.; Freeman A. A.; Newlands E. S.; Watson A. J.; Rafferty J. A.; Margison G. P. Depletion of O6-Alkylguanine-DNA Alkyltransferase Correlates with Potentiation of Temozolomide and CCNU Toxicity in Human Tumour Cells. Br. J. Cancer 1993, 67, 1299–1302. 10.1038/bjc.1993.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T.; Bedwell J.; Kondo Y.; Kondo S.; Germano I. M. Inhibition of DNA Repair for Sensitizing Resistant Glioma Cells to Temozolomide. J. Neurosurg. 2003, 99, 1047–1052. 10.3171/jns.2003.99.6.1047. [DOI] [PubMed] [Google Scholar]
- Kanzawa T.; Germano I. M.; Kondo Y.; Ito H.; Kyo S.; Kondo S. Inhibition of Telomerase Activity in Malignant Glioma Cells Correlates with Their Sensitivity to Temozolomide. Br. J. Cancer 2003, 89, 922–929. 10.1038/sj.bjc.6601193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210. 10.1016/j.gendis.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-S.; Rait A.; Kim E.; DeMarco J.; Pirollo K. F.; Chang E. H. Encapsulation of Temozolomide in a Tumor-Targeting Nanocomplex Enhances Anti-Cancer Efficacy and Reduces Toxicity in a Mouse Model of Glioblastoma. Cancer Lett. 2015, 369, 250–258. 10.1016/j.canlet.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci A.; Prasetyanto E. A.; Septiadi D.; Manicardi A.; Brognara E.; Gambari R.; Corradini R.; De Cola L. Combined Delivery of Temozolomide and Anti-miR221 PNA Using Mesoporous Silica Nanoparticles Induces Apoptosis in Resistant Glioma Cells. Small 2015, 11, 5687–5695. 10.1002/smll.201500540. [DOI] [PubMed] [Google Scholar]
- Wu A.; Loutherback K.; Lambert G.; Estévez-Salmerón L.; Tlsty T. D.; Austin R. H.; Sturm J. C. Cell Motility and Drug Gradients in the Emergence of Resistance to Chemotherapy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16103–16108. 10.1073/pnas.1314385110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.; Watson M. B.; O’Kane S. L.; Drew P. J.; Lind M. J.; Cawkwell L. The Analysis of Doxorubicin Resistance in Human Breast Cancer Cells Using Antibody Microarrays. Mol. Cancer Ther. 2006, 5, 2115–2120. 10.1158/1535-7163.MCT-06-0190. [DOI] [PubMed] [Google Scholar]
- Gouazé-Andersson V.; Yu J. Y.; Kreitenberg A. J.; Bielawska A.; Giuliano A. E.; Cabot M. C. Ceramide and Glucosylceramide Upregular Expression of the Multidrug Resistance Gene MDR1 in Cancer Cells. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2007, 1771, 1407–1417. 10.1016/j.bbalip.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroui S.; Ram N.; Appaix F.; Ronjat M.; Kenani A.; Pirollet F.; De Waard M. Maurocalcin as a Non Toxic Drug Carrier Overcomes Doxorubicin Resistance in the Cancer Cell Line MDA-MB 231. Pharm. Res. 2009, 26, 836–845. 10.1007/s11095-008-9782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Kopečková P.; Minko T.; Nanayakkara V.; Kopeček J. Synthesis of Starlike N-(2-Hydroxypropyl)methacrylamide Copolymers: Potential Drug Carriers. Biomacromolecules 2000, 1, 313–319. 10.1021/bm0000236. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Schiavon O.; Pasut G.; Mendichi R.; Andersson L.; Tsirk A.; Ford J.; Wu G.; Kneller S.; Davies J.; et al PEG-Doxorubicin Conjugates: Influence of Polymer Structure on Drug Release, In Vitro Cytotoxicity, Biodistribution, and Antitumor Activity. Bioconjugate Chem. 2005, 16, 775–784. 10.1021/bc040241m. [DOI] [PubMed] [Google Scholar]
- Hu X.; Hu J.; Tian J.; Ge Z.; Zhang G.; Luo K.; Liu S. Polyprodrug Amphiphiles: Hierarchical Assemblies for Shape-Regulated Cellular Internalization, Trafficking, and Drug Delivery. J. Am. Chem. Soc. 2013, 135, 17617–17629. 10.1021/ja409686x. [DOI] [PubMed] [Google Scholar]
- Liu P.; Shi B.; Yue C.; Gao G.; Li P.; Yi H.; Li M.; Wang B.; Ma Y.; Cai L. Dextran-Based Redox-Responsive Doxorubicin Prodrug Micelles for Overcoming Multidrug Resistance. Polym. Chem. 2013, 4, 5793–5799. 10.1039/c3py00830d. [DOI] [Google Scholar]
- Schafer F. Q.; Buettner G. R. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radical Biol. Med. 2001, 30, 1191–1212. 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Jones D. P.; Carlson J. L.; Samiec P. S.; Sternberg P.; Mody V. C.; Reed R. L.; Brown L. A. S. Glutathione Measurement in Human Plasma Evaluation of Sample Collection, Storage and Devatization Conditions for Analysis of Densyl Derivatives by HPLC. Clin. Chim. Acta 1998, 275, 175–184. 10.1016/S0009-8981(98)00089-8. [DOI] [PubMed] [Google Scholar]
- Wright A. J.; Fellows G. A.; Griffiths J. R.; Wilson M.; Bell B. A.; Howe F. A. Ex-Vivo HRMAS of Adult Brain Tumors: Metabolite Quantification and Assignment of Tumor Biomarkers. Mol. Cancer 2010, 9, 66. 10.1186/1476-4598-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo H.; Mio T.; Kokunai T.; Tamaki N.; Sumino K.; Matsumoto S. Quantitative Analysis of Glutathione in Human Brain Tumors. J. Neurosurg. 1990, 72, 610–615. 10.3171/jns.1990.72.4.0610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.