

Abstract
Background
The insulin/insulin‐like growth factor/relaxin family represents a group of structurally related but functionally diverse proteins. The family member relaxin‐2 has been evaluated in clinical trials for its efficacy in the treatment of acute heart failure. In this study, we assessed the role of insulin‐like peptide 6 (INSL6), another member of this protein family, in murine heart failure models using genetic loss‐of‐function and protein delivery methods.
Methods and Results
Insl6‐deficient and wild‐type (C57BL/6N) mice were administered angiotensin II or isoproterenol via continuous infusion with an osmotic pump or via intraperitoneal injection once a day, respectively, for 2 weeks. In both models, Insl6‐knockout mice exhibited greater cardiac systolic dysfunction and left ventricular dilatation. Cardiac dysfunction in the Insl6‐knockout mice was associated with more extensive cardiac fibrosis and greater expression of fibrosis‐associated genes. The continuous infusion of chemically synthesized INSL6 significantly attenuated left ventricular systolic dysfunction and cardiac fibrosis induced by isoproterenol infusion. Gene expression profiling suggests liver X receptor/retinoid X receptor signaling is activated in the isoproterenol‐challenged hearts treated with INSL6 protein.
Conclusions
Endogenous Insl6 protein inhibits cardiac systolic dysfunction and cardiac fibrosis in angiotensin II– and isoproterenol‐induced cardiac stress models. The administration of recombinant INSL6 protein could have utility for the treatment of heart failure and cardiac fibrosis.
Keywords: anti–cardiac remodeling, anti‐fibrosis, heart failure, relaxin family protein
Subject Categories: Heart Failure
Clinical Perspective
What Is New?
Mice deficient in insulin‐like peptide 6 (INSL6), a member of the relaxin protein family, exhibit greater cardiac pathological features after the continuous infusion of angiotensin II or isoproterenol.
Administration of a chemically synthesized INSL6 peptide protects murine heart from pathological cardiac remodeling induced by isoproterenol.
Insl6/INSL6 functions in part through the modulation of cardiac fibrosis.
What Are the Clinical Implications?
INSL6, as well as other members of the relaxin family of proteins, may be useful for the treatment of heart failure.
Introduction
Insulin‐like peptide 6 (INSL6) is a hormone that belongs to the insulin/insulin‐like growth factor/relaxin peptide superfamily. This family is characterized by a common 3‐domain structure, and it plays crucial roles for metabolic regulation, growth, and reproduction.1 Proteins in this family are composed of a variable‐length C‐peptide that separates A‐ and B‐chain peptides and a signal peptide that is linked to a B‐chain peptide. The A‐ and B‐chain peptides are relatively invariant within the family, and they contain highly conserved cysteine motifs. For most family members, mature hormones are processed by proteolytic cleavage of C‐peptide and signal peptide, and the A‐ and B‐chain peptides are connected by 2 interchain and 1 intrachain disulfide bonds.2, 3
The relaxin/insulin‐like peptide subfamily can be distinguished from the insulin/insulin‐like growth factor peptides by differences in their signaling mechanisms. Although insulin and insulin‐like growth factors use tyrosine kinase receptors, most members of the relaxin/INSL subfamily are believed to act on G‐protein coupled receptors. Humans encode 7 relaxin/INSL proteins.2, 4 They include ovarian relaxin, also referred to as human relaxin‐2, human relaxin‐1, human relaxin‐3, and INSL3‐6. The common ancestral gene appears to be INSL3, producing other subfamily members through evolutionary gene duplication mechanisms.2 The relaxin/INSL family proteins tend to be expressed in reproductive tissues,4 although relaxin‐3 and INSL5 are highly expressed in nongonadal tissues, including the brain and gut, respectively.
Among the relaxin/INSL subfamily of proteins, relaxin‐2 has been investigated to the greatest extent. Relaxin‐2 was originally identified as a peptide hormone that is elevated in the ovary, placenta, and blood of pregnant females.5 Relaxin‐2 is also expressed by the brain, kidney, and heart at lower levels,6 where it is believed to have a fundamental role in conditioning the reproductive organs and, perhaps, the adaptation of the cardiovascular system to pregnancy.7, 8, 9, 10 RLN2 gene expression is detected in human heart and is downregulated in chronic heart failure.11 The relaxin family peptide receptor 1, the receptor of human relaxin‐2, is also expressed in the reproductive organs as well as the heart, arteries, kidney, lung, liver, and brain.6 On the basis of these findings, the cardiovascular functions of relaxin‐2 have been explored in various models involving both female and male animals. Several cardioprotective roles have been assigned to relaxin‐2, including vasodilatation, inhibition of fibrosis, inhibition of apoptosis, promotion of angiogenesis, and anti‐inflammation actions.12 Among these, the antifibrotic actions of relaxin‐2 are commonly reported. For example, aging relaxin‐2–deficient male mice display increased cardiac collagen deposition and diastolic dysfunction.13 Relaxin‐2 overexpression by systemic adenovirus delivery attenuates cardiac collagen deposition in mice that overexpress the β2‐adrenergic receptor.14 Moreover, the antifibrotic actions of recombinant human relaxin‐2 have been demonstrated in various cardiac disease models, including myocardial infarction,15 diabetic cardiomyopathy,16 and isoproterenol‐induced cardiac injury,17 and in the spontaneously hypertensive rat.18
In contrast to relaxin‐2, characterization of other members of the relaxin/INSL subfamily is relatively limited. INSL6 is predominantly expressed in testis, and homozygous INSL6‐deficient male mice are infertile.19 Previously, we documented the upregulation of INSL6 in the growing skeletal muscle of mice that were genetically modified to express constitutively active protein kinase B in this tissue.20, 21 INSL6 expression is also upregulated in mouse skeletal muscle after cardiotoxin‐induced skeletal muscle injury, and adenovirus‐mediated overexpression of INSL6 facilitates muscle regeneration in this model.20 In a model of experimental autoimmune myositis, INSL6‐deficient mice display greater motor function impairment, whereas INSL6 overexpression protects muscle from dysfunction.22
Although similar to relaxin‐2 in overall structure, INSL6 shares only 43.1% homology to relaxin‐2 at the amino acid level. Despite this low level of homology, clinical and experimental investigations into the cardioprotective roles of relaxin‐2/serelaxin led us to assess the role of INSL6 in murine cardiac injury models. Using a genetic loss‐of‐function model and the delivery of recombinant Insl6 protein, we find that Insl6 protects against pathological cardiac remodeling and fibrosis. These data suggest that INSL6 and possibly other members of the relaxin/Insl family can have utility in the treatment of heart failure.
Methods
The data, analytical methods, and study materials will be/have been made available to other researchers for the purposes of reproducing the results or replicating the procedures. The authors declare that all supporting data are available within the article and its online supplementary files.
Experimental Animals
Insl6‐knockout mice were generated as previously described.19 Briefly, exon 1 of the Insl6 gene is replaced by the PGK‐neomycin cassette. Insl6 knockout mice were backcrossed with C57BL/6N >6 times in the animal facility at Boston University (Boston, MA), and a nearly homogeneous strain background (99.5% for C57BL/6) was confirmed by Jackson Laboratory's Genome Scanning Service. Because male infertility has been reported,19 Insl6+/− heterozygous male and Insl6−/− female mice were used for breeding. Insl6+/+ male and Insl6+/+ female mice of the same background were bred as control groups. C57BL/6N mice were purchased from Charles River Laboratories. All animal studies were performed with 8‐ to 12‐week‐old male mice. Study protocols were approved by the Institutional Animal Care and Use Committee at Boston University.
Cardiac Injury Models
The isoproterenol‐induced cardiac injury model was used, as previously reported.23, 24, 25 Briefly, isoprenaline hydrochloride (I5627; Sigma‐Aldrich) dissolved in sterile saline at 50 mg/mL was filtered via a 20‐μm filter. Aliquots of stock solution were stored in the dark in a −20°C freezer. The working solution was freshly prepared with diluting stock solution containing sterile saline. Isoproterenol (50 mg/kg per day; total, 100 μL) was injected IP once a day for 14 days. The control group received same amount of sterile saline. The angiotensin II (AngII)–induced cardiac injury model was performed, as previously reported.26, 27, 28, 29, 30, 31 Human AngII protein (A9525; Sigma‐Aldrich) was dissolved with sterile saline. The AngII solution (2 mg/kg per day) was administrated to mice subcutaneously for 14 days via an ALZET osmotic pump (model 1002; Durect). A pump with sterile saline was implanted in the control group. All mice were euthanized at 14 days. Recombinant human INSL6 protein was chemically produced using fluorenylmethyloxycarbonyl‐based solid‐phase peptide chemistry and a regioselective disulfide bond construction protocol, as previously described.32 INSL6 protein (50–70 nmol/kg per day) was infused into mice SC using an ALZET osmotic pump (model 2002; Durect). The osmotic pump with vehicle (pH 8.5, ammonium bicarbonate) was implanted in the control group. All isoproterenol model studies in Insl6‐knockout and INSL6 protein were performed in a blinded manner by ≥2 experienced researchers. Specifically, the assignment of mice to experimental groups and pump preparation was performed by one unblinded researcher, and a different blinded researcher obtained data from each experimental group. Group identities were revealed after all analyses were completed.
Echocardiography and Blood Pressure Measurements
Cardiac function of mice was assessed at day 13 after isoproterenol or AngII administration by echocardiography using a Vevo2100 machine with a 550 probe and a Vevo770 machine using a 707B probe (VisualSonics). Mice were lightly anesthetized with isoflurane. An examination was performed with heart rate at 450 to 550 beats per minute, and we excluded mice that were significantly affected by anesthesia. Left ventricular (LV) internal diastolic dimension and LV internal systolic dimension were measured from M‐mode images obtained by LV short‐axis view. LV systolic function was assessed by fractional shortening (FS) and ejection fraction (EF). FS was obtained from M‐mode by short‐axis view, and EF was obtained from B‐mode by tracing of LV long‐axis view. Except for the EF measurements, 3 measured values per mouse were used for statistical analysis. Mouse blood pressure was measured at 12 days after isoproterenol or AngII administration by tail cuff method using BP‐2000 Blood Pressure Analysis System (Visitech Systems, Inc). The median systolic blood pressure was calculated from 15 to 20 measured values for each mouse.
Histological Features
Heart samples were obtained at 14 days after isoproterenol or AngII administration or 11 days after coadministration of recombinant INSL6 protein and isoproterenol. Cardiac fibrosis was assessed by Picrosirius Red staining, as previously reported.33 Briefly, paraffin‐embedded hearts were sectioned by microtome for a 6‐μm thickness. The sections were deparaffinized, rehydrated, and incubated with freshly prepared Picrosirius Red staining buffer (1.2%/w picric acid in water, 0.1%/w Fast Green FCF, and 0.1%/w Direct Red 80 solved in PBS) for 1 hour at room temperature. Then, sections were washed with distilled water and dehydrated. The sections were mounted by coverslip using Permount mounting medium (Fisher Scientific). The images were produced by a light microscope (BZ‐9000; Keyence). Cardiac fibrosis was assessed from the 3 most severe images per mouse in the isoproterenol model. Fibrosis in the AngII model was assessed with whole heart sections to measure perivascular fibrosis and interstitial fibrosis patterns. Fibrosis was quantified using Photoshop software.
RNA Extraction and Analysis
Heart LV samples harvested at 14 days from the different experimental groups were snap frozen. An RNeasy Lipid Tissue Mini kit (Qiagen) was used to extract mRNA with a Qiacube machine, according to the manufacturer's protocol. Alternatively, samples were homogenized in 1 mL of TRIzol reagent with stainless steel beads using TissueLyser II (all from Qiagen) and extracted, as described previously.34 The concentration of RNA was measured using a NanoDrop 1000 (Thermo Scientific). Extracted RNA (1 μg) was reverse transcribed to cDNA by QuantiTect Reverse Transcription kit (Qiagen), according to the manufacturer's protocol. Real‐time quantitative reverse transcription–polymerase chain reaction was performed by ViiA TM7 Real Time PCR system (Applied Biosystems) with Power SYBR Green PCR Master Mix (Applied Biosystems). The relative levels of transcript were determined by using Δ‐Δ Cycle threshold methods. Each target gene level was normalized by 18S and 36B4. Primer sequences are shown in Table 1. For gene expression profiling, 3 pooled RNA samples for sham, isoproterenol with vehicle, and isoproterenol with INSL6 protein were sent to the Boston University core facility for processing. Briefly, RNA samples were amplified, labelled, and hybridized on Affymetrix mouse Gene Expression Array 2.0, per the manufacturer's instructions. Microarray data processing for gene‐level expression values was performed, as previously described,35 and deposited into the Minimum information about a microarray experiment (MIAME)‐compliant National Center for Biotechnology Information Gene Expression Omnibus with accession number GSE102612 (Reviewer Token: ezahmywmlvwprsr). Genes with a fold change >1.5 or <−1.5 by isoproterenol (sham versus isoproterenol with vehicle) or INSL6 protein treatment (isoproterenol with vehicle versus isoproterenol with INSL6) were uploaded to Ingenuity Pathway Analysis (Qiagen) for canonical pathway analysis and comparison analysis. Pathways with –log(Benjamini‐Hochberg multiple testing correction P value) ≥10 and absolute Z score ≥1 were considered significantly enriched pathways.
Table 1.
Primer Sequences
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| nppa | 5′‐AAG AAC CTG CTA GAC CAC CTG‐3′ | 5′‐TGC TTC CTC AGT CTG CTC AC‐3′ |
| nppb | 5′‐CAA GGC CTC ACA AAA GAA CA‐3′ | 5′‐ATC CGA TCC GGT CTA TCT TG‐3′ |
| Tgfb1 | 5′‐GTG CGG CAG CTG TAC ATT GAC TTT‐3′ | 5′‐TGT ACT GTG TGT CCA GGC TCC AAA‐3′ |
| Col1a1 | 5′‐GGG TCT AGA CAT GTT CAG CTT TGT G‐3′ | 5′‐ACC CTT AGG CCA TTG TGT ATG C‐3′ |
| Col1a3 | 5′‐AGG CTG AAG GAA ACA GCA AA‐3′ | 5′‐TAG TCT CAT TGC CTT GCG TG‐3′ |
| 36b4 | 5′‐GCT CCA AGC AGA TGC AGC A‐3′ | 5′‐CCG GAT GTG AGG CAG CAG‐3′ |
| 18S | 5′‐CTT AGA GGG ACA AGT GGC G‐3′ | 5′‐GGA CAT CTA AGG GCA TCA CA‐3′ |
Statistical Analysis
All results are presented as mean±SD using GraphPad Prism 5 and 6 (GraphPad Software). Statistical analyses were performed using SPSS Statistics 20 (IBM) and GraphPad prisms. Distribution of data (data of normality) was analyzed using a Shapiro‐Wilk test. Then, data with a normal distribution were analyzed using parametric analysis. For treatment and genotype effect comparison, 2‐way ANOVA was performed, and post hoc multiple comparison was performed by Sidak's test or Tukey's test (Figures 1C through 1E, 2, 3B, 3C, 4C through 4E, 5B, and 5C, Tables 2 and 3. For 2‐group comparison, the unpaired t tests with Welch's correction were performed (Figures 6C through 6E, 7B, and 7C, Table 4). P<0.05 was considered statistically significant.
Figure 1.
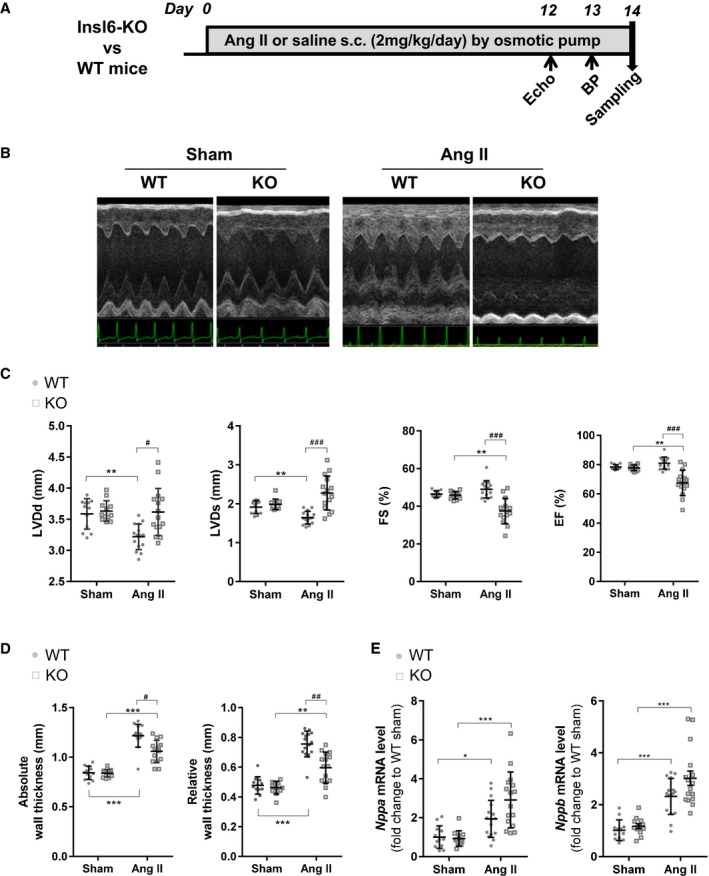
Exacerbated cardiac systolic dysfunction in insulin‐like peptide 6–knockout (Insl6‐KO) mice in the angiotensin II (AngII) infusion model compared with wild type (WT). A, Timeline for AngII infusion model for Insl6‐KO (KO) mice and littermate control WT mice. B, Representative M‐mode images of left ventricle by echocardiography at 13 days of saline (Sham) or AngII infusion. C, Cardiac function and morphological parameters by echocardiography measurements; left ventricular diastolic dimension (LVDd), left ventricular systolic dimension (LVDs), fractional shortening (FS), and ejection fraction (EF) are shown. D, Absolute wall thickness and relative wall thickness are calculated from echocardiogram measurements. E, Relative transcript levels of Nppa and Nppb in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=12 for each mouse genotype in sham group, n=16 for each mouse genotype in AngII group). BP indicates blood pressure. *P<0.05, **P<0.005, ***P<0.001 indicate significant differences between sham and AngII group in the same mouse strain. # P<0.05, ## P<0.005, ### P<0.001 indicate significant difference between WT and KO.
Figure 2.
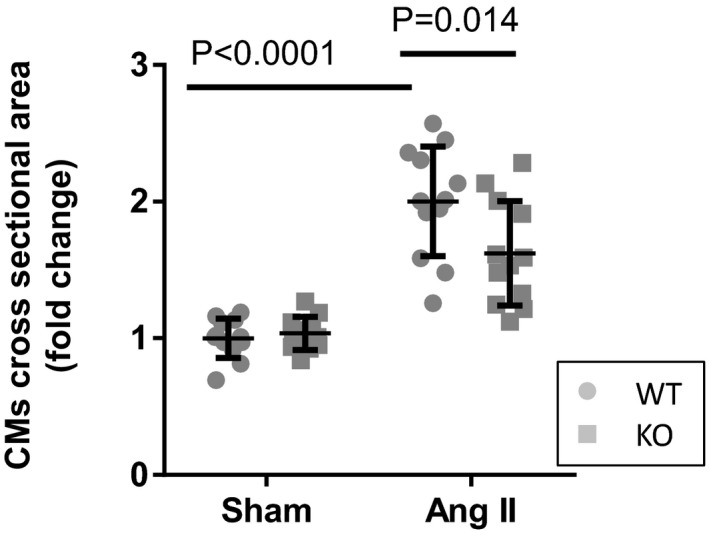
Decreased angiotensin II (AngII)–induced cardiac myocyte (CM) hypertrophy in insulin‐like peptide 6–knockout (KO) mice. Relative increase (fold change) of cross‐sectional area of CMs in left ventricle. The hearts were harvested at day 14. Error bars represent mean±SD (n=12 per group). WT indicates wild type.
Figure 3.
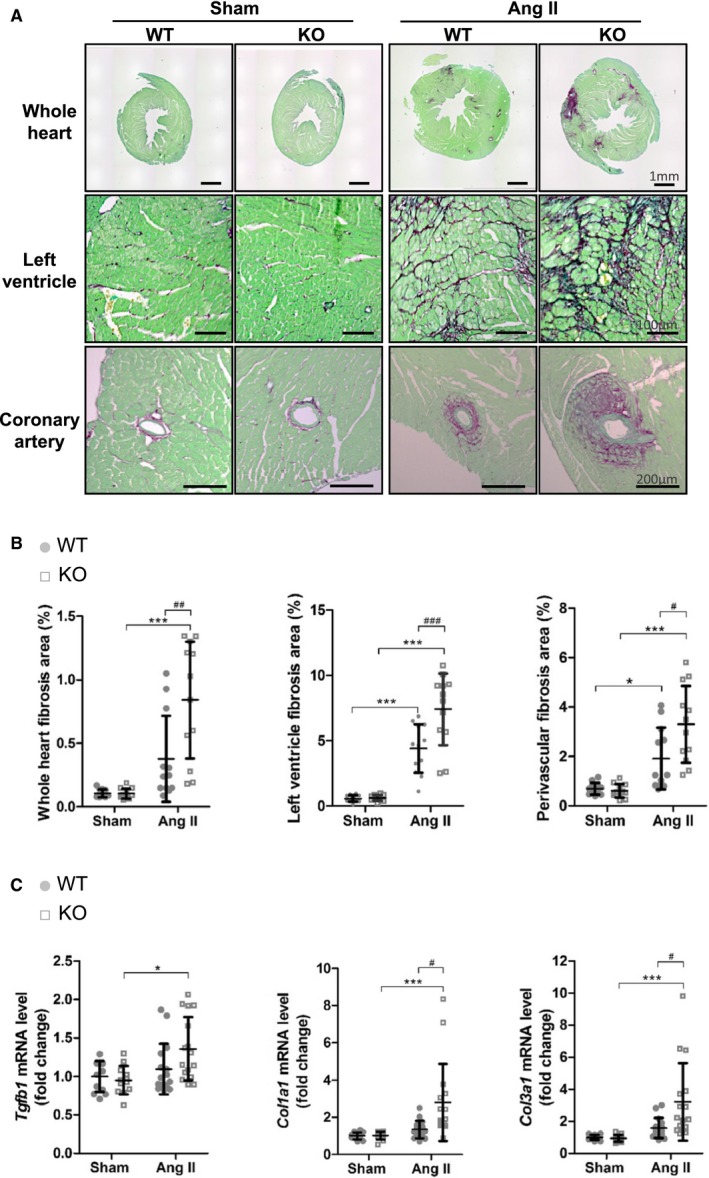
Increased cardiac fibrosis in insulin‐like peptide 6–knockout (KO) mice in the angiotensin II (AngII) infusion model compared with wild‐type (WT) mice. A, Fibrosis of the heart in the AngII model is shown by Picrosirius Red staining. Representative images of fibrosis in the entire heart section (top), left ventricle (middle), and perivascular area (bottom). The scale bar indicates 1 mm (top), 100 μm (middle), and 200 μm (bottom). B, Fibrosis quantification for each categorized field in heart histological features. C, Relative transcript level of Tgfb1, Col1a1, and Col3a1 in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=12 for each WT and KO experimental group in sham conditions, and n=16 for each experimental AngII condition). *P<0.05, ***P<0.001 indicate significant differences between sham and AngII group in the same mouse strain. # P<0.05, ## P<0.005, ### P<0.001 indicate significant difference between WT and KO.
Figure 4.
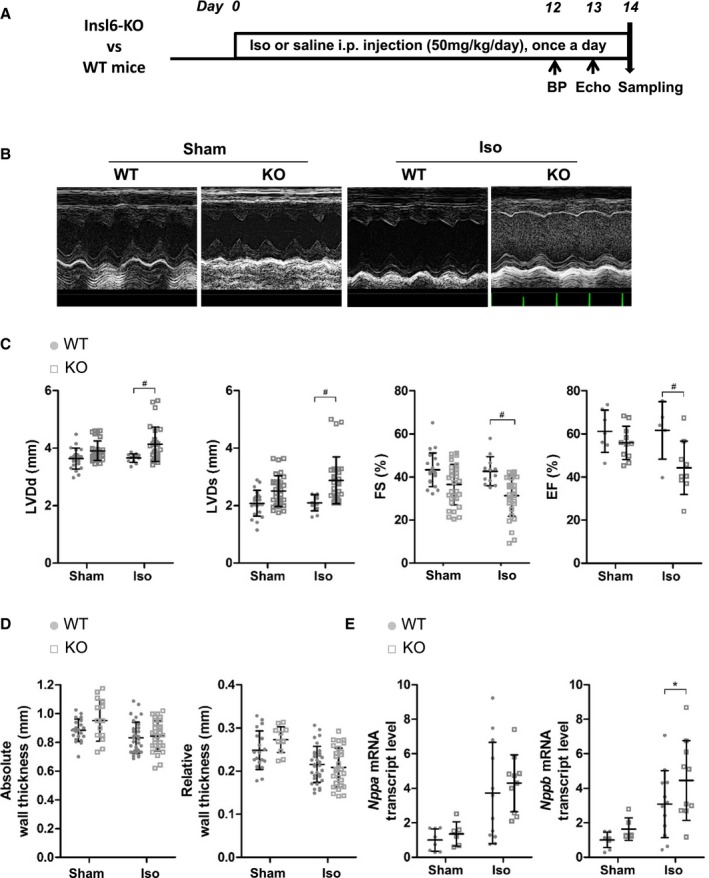
Exacerbated left ventricular systolic dysfunction in insulin‐like peptide 6–knockout (Insl6‐KO) mice in the isoproterenol (Iso) injection model compared with wild‐type (WT) mice. A, Timeline for Iso‐induced cardiac remodeling in Insl6‐KO (KO) mice and littermate control WT mice. B, Representative M‐mode images of left ventricle (LV) by echocardiography at 13 days of Iso injection. C, Cardiac function and morphological parameters from echocardiography measurements; LV diastolic dimension (LVDd), LV systolic dimension (LVDs), fractional shortening (FS), and ejection fraction (EF) are shown. D, Absolute wall thickness and relative wall thickness are calculated from echocardiogram measurements. Echocardiographic parameters were performed in triplicate for each mouse, except for the determination of EF. E, Relative transcript levels of Nppa and Nppb in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=8, 6, 12, and 10 for WT‐sham, KO‐sham, WT‐Iso, and KO‐Iso, respectively). BP indicates blood pressure. *P<0.05 indicates significant differences between sham and isoproterenol group in the same mouse strain. # P<0.05 indicates significant difference between WT and KO.
Figure 5.

Advanced cardiac fibrosis in insulin‐like peptide 6–knockout (KO) mice in the isoproterenol (Iso) model compared with wild‐type (WT) mice. A, Heart sections stained by Picrosirius Red staining. Representative images of interstitial left ventricular fibrosis of endocardium are shown. B, Cardiac fibrosis area was quantified as pixel count. C, Relative transcript level of Tgfb1, Col1a1, and Col3a1 in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=8, 6, 12, and 10 for WT‐sham, KO‐sham, WT‐Iso, and KO‐Iso, respectively). *P<0.05, **P<0.005 indicate significant differences between sham and isoproterenol group in the same mouse strain. # P<0.05, ## P<0.005 indicate significant differences between WT and KO.
Table 2.
Physiological Findings of WT and Insl6‐Knockout Mice Induced With or Without AngII
| Variable | Sham | AngII | ||||
|---|---|---|---|---|---|---|
| WT (n=12) | Knockout (n=12) | P Value Sham Groupa | WT (n=16) | Knockout (n=16) | P Value AngII Groupa | |
| BW, g | 27.44±2.09 | 27.65±2.40 | 0.9642 | 24.48±2.01 | 24.43±1.97 | 0.9976 |
| HW/TL, mg/mmb | 5.87±0.59 | 5.69±0.55 | 0.8972 | 7.50±1.32 | 8.42±1.40 | 0.0430 |
| HW/BW, mg/gb | 4.91±0.51 | 4.71±0.41 | 0.8040 | 6.87±0.93 | 7.903±1.10 | 0.0019 |
| LW/TL, mg/mmc | 7.14±1.42 | 6.81±1.45 | 0.8773 | 6.89±1.73 | 8.40±2.30 | 0.0430 |
| LW/BW, mg/gc | 5.94±1.00 | 5.61±0.99 | 0.8253 | 6.40±1.38 | 7.80±2.06 | 0.0190 |
| Systolic BP, mm Hg | 106.4±5.21 | 109.3±8.72 | 0.9746 | 156.3±10.41 | 156.2±11.64 | >0.999 |
Data are shown as mean±SD. AngII indicates angiotensin II; BP, blood pressure; BW, body weight; HW, heart weight; Insl6, insulin‐like peptide 6; LW, lung weight; TL, tibia length; WT, wild type.
Ordinary 2‐way ANOVA, Sidak's multiple comparison test between WT and knockout in the same experimental group.
HW is normalized by TL (HW/TL) or BW (HW/BW).
LW is normalized by TL (LW/TL) or BW (LW/BW).
Table 3.
Organ Weight and BP at End Points: WT and Insl6‐Knockout Mice Induced With or Without Isoproterenol Challenge
| Variable | Sham | Isoproterenol | ||||
|---|---|---|---|---|---|---|
| WT (n=10) | Knockout (n=6) | P Value Sham Groupa | WT (n=14) | Knockout (n=11) | P Value Isoproterenol Groupa | |
| BW, g | 27.30±0.870 | 27.33±0.954 | 0.9996 | 27.43±0.635 | 28.64±0.472 | 0.3554 |
| HW/TL, mg/mmb | 5.378±0.164 | 5.419±0.181 | 0.9925 | 5.862±0.150 | 6.399±0.332 | 0.1444 |
| HW/BW, mg/gb | 4.483±0.146 | 4.515±0.087 | 0.9930 | 4.901±0.107 | 5.070±0.279 | 0.7555 |
| LW/TL, mg/mmc | 6.026±0.140 | 6.491±0.191 | 0.8317 | 6.855±0.125 | 8.123±0.933 | 0.1260 |
| LW/BW, mg/gc | 5.016±0.088 | 5.435±0.257 | 0.8016 | 5.740±0.065 | 6.444±0.769 | 0.3713 |
| Systolic BP, mm Hg | 125.7±1.452 | 120.0±3.100 | 0.8499 | 117.4±7.115 | 114.4±8.28 | 0.9609 |
BP indicates blood pressure; BW, body weight; HW, heart weight; Insl6, insulin‐like peptide 6; LW, lung weight; TL, tibia length; WT, wild type.
Ordinary 2‐way ANOVA, Sidak's multiple comparison test between WT and knockout in the same experimental group.
HW is normalized by TL (HW/TL) or BW (HW/BW).
LW is normalized by TL (LW/TL) or BW (LW/BW).
Figure 6.

Recombinant insulin‐like peptide 6 (INSL6) protein attenuated left ventricular (LV) systolic dysfunction caused by isoproterenol (Iso) in wild‐type (WT) mice. A, Timeline for administration of human recombinant INSL6 protein (INSL6, 70 nmol/kg per day) or vehicle to Iso‐injected C57BL/6N WT mice. B, Representative M‐mode images of LV by echocardiography at 10 days of Iso injection. C, Echocardiography‐measured LV diastolic dimension (LVDd), LV systolic dimension (LVDs), fractional shortening (FS), and ejection fraction (EF) were shown. D, Absolute wall thickness and relative wall thickness are calculated from echocardiogram measurements. Echocardiographic parameters were measured in triplicate for each mouse, except for the determination of EF. E, Relative transcript levels of Nppa and Nppb in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=9 for Iso+vehicle, n=8 for Iso+INSL6). BP indicates blood pressure; and KO, knockout. *P<0.05 indicates significant differences found between 2 groups.
Figure 7.

Recombinant insulin‐like peptide 6 (INSL6) protein attenuated isoproterenol (Iso)–induced cardiac fibrosis in wild‐type (WT) mice. A, Representative Picrosirius Red staining images for hearts from C57BL6/N mice injected with 10 days of Iso with continuous vehicle or INSL6 protein administration. Whole heart (top) and zoomed‐in left ventricle (LV; bottom) images were shown. B, Cardiac fibrosis area was quantified by pixel count from zoomed‐in LV images. C, Relative transcript level of Tgfb1, Col1a1, and Col3a1 in hearts assessed by real‐time quantitative polymerase chain reaction. Error bars represent mean±SD (n=9 for Iso+vehicle, n=8 for Iso+INSL6). KO indicates knockout. *P<0.05 indicates significant differences found between 2 groups.
Table 4.
Organ Weight and BP at the Terminal End Point: C57BL/6 WT Mice Treated With Isoproterenol and Recombinant INSL6 Protein or Vehicle
| Variable | Isoproterenol | ||
|---|---|---|---|
| Vehicle (n=8) | INSL6 (n=9) | P Valuea | |
| BW, g | 29.00±0.5774 | 29.38±0.6529 | 0.6725 |
| HW/TL, mg/mmb | 6.281±0.1688 | 6.104±0.1297 | 0.4130 |
| HW/BW, mg/gb | 4.899±0.1015 | 4.679±0.0729 | 0.0936 |
| LW/TL, mg/mmc | 7.024±0.1883 | 6.319±0.157 | 0.0110 |
| LW/BW, mg/gc | 5.49±0.1737 | 4.842±0.0857 | 0.0034 |
| Systolic BP, mm Hg | 111.6±2.511 | 112.8±4.221 | 0.8163 |
BP indicates blood pressure; BW, body weight; HW, heart weight; INSL6, insulin‐like peptide 6; LW, lung weight; TL, tibia length; WT, wild type.
Unpaired t test, 2 tailed.
HW is normalized by TL (HW/TL) or BW (HW/BW).
LW is normalized by TL (LW/TL) or BW (LW/BW).
Results
Impaired Cardiac Systolic Function in Insl6‐Knockout Mice in the AngII Model
To investigate the role of endogenous INSL6 in a murine model of heart failure, wild‐type (WT) and Insl6‐knockout mice were infused with AngII or vehicle for 2 weeks (Figure 1A). In the sham state, there were no significant differences with respect to body weight (BW), systolic blood pressure, or cardiac function between WT and knockout mice (Table 2). With AngII infusion, heart weight (HW) normalized to tibia length (TL) or BW was significantly increased in Insl6‐knockout mice compared with WT mice group (HW/TL, P=0.043; and HW/BW, P=0.0019; Table 2). Similarly, the lung weight (LW) of AngII‐infused Insl6‐knockout mice was significantly greater than that of AngII‐infused WT mice (LW/TL, P=0.043; and LW/BW, P=0.019; Table 2), indicative of lung congestion.
Echocardiographic analysis showed that AngII infusion led to significant concentric hypertrophy in both strains (Figure 1B). Under these conditions, WT mice treated with AngII maintained cardiac systolic function, as represented by lack of changes in FS and EF that was accompanied by increased wall thickness and reduced LV chamber size (Figure 1C and 1D). In contrast, Insl6‐knockout mice infused with this dose of AngII displayed exacerbated cardiac dysfunction compared with WT mice treated with AngII (22.6% [P=0.001] and 16.5% [P=0.001] reductions in FS and EF, respectively) (Figure 1C). This was accompanied by greater LV chamber dilatation and reductions in wall thickness (Figure 1C and 1D). In histological analyses, cardiomyocyte cross‐sectional area was significantly increased in AngII group compared with sham group in WT mice, and this increase of cardiomyocyte size was significantly attenuated in Insl6‐knockout mice compared with WT mice (P=0.014), suggesting chamber dilatation under these conditions (Figure 2). We also investigated the expression of cardiac natriuretic peptides that serve as biomarkers for predicting the onset of heart failure.36 Although AngII infusion upregulated Nppa and Nppb mRNA expression in hearts from both strains of mice, Insl6‐knockout mice treated with AngII showed increases in Nppa and Nppb mRNA expression compared with WT mice (P=0.0314 and 0.0408, respectively, when performing unpaired t tests on the 2 AngII‐infused groups) (Figure 1E). Collectively, these data suggest that Insl6 deficiency exacerbates pathological cardiac remodeling in response to continuous AngII stimulation.
Exacerbated Cardiac Fibrosis in AngII‐Infused Insl6‐Knockout Mice
To investigate the role of INSL6 on fibrosis associated with cardiac dysfunction, sections of hearts from WT and Insl6‐knockout mice, treated with AngII or vehicle infusion for 2 weeks, were stained with Picrosirius Red and quantified. AngII infusion induced significant cardiac fibrosis in WT mice in the interstitium and perivascular area of the LV (P<0.0001 and P=0.0255, respectively; Figure 3A and 3B). Greater AngII‐induced fibrosis was observed in the hearts of Insl6‐knockout mice compared with WT mice (P=0.0015, P=0.0004, and P=0.0089, respectively; Figure 3A and 3B). The expression of Tgfb1, a key mediator of fibrosis, was also significantly increased in Insl6‐knockout compared with WT mice (P=0.0058; Figure 3C). Correspondingly, there was significant upregulation in Col1a1 and Col3a1 mRNA expression in the Insl6‐knockout hearts treated with AngII compared with WT (P=0.042 and P=0.0066, respectively; Figure 3C). Cytokine expression was analyzed because it is widely recognized that inflammation triggers tissue fibrosis. There was a significant increase in IL6 mRNA expression level and a trend toward increase in Tnfα in Insl6‐knockout mice compared with WT mice (P<0.05 and P=0.149, respectively) (data not shown). However, an increase in the infiltration of inflammatory cells (lymphocytes and macrophage) in Insl6‐knockout mice hearts was not detected by real‐time quantitative polymerase chain reaction and histological analysis (data not shown). Collectively, these results indicate that endogenous INSL6 plays a central role in limiting cardiac fibrosis in the model of AngII‐induced cardiac dysfunction.
Isoproterenol‐Induced LV Systolic Dysfunction Is Exacerbated in Insl6‐Knockout Mice
A moderate‐dose isoproterenol administration to WT and Insl6‐knockout mice was used to investigate cardiac dysfunction and fibrosis in a model that displays more uniform cardiac fibrosis than the AngII infusion model (Figure 4A). In WT mice, this dose of isoproterenol (50 mg/kg per day IP) led to an increase in HW (HW/TL and HW/BW; unpaired t test P=0.0413 and P=0.0300, respectively; Table 3) but had little or no effect on cardiac function (Figure 4B and 4C). In contrast, Insl6‐knockout mice displayed significantly worsened cardiac systolic function after treatment with isoproterenol. Specifically, Insl6‐knockout hearts displayed a more dilated LV systolic dimension (P=0.0306; Figure 4C), and parameters for cardiac systolic function, FS and EF, were also significantly reduced in Insl6‐knockout mice compared with WT mice (P=0.0493 and P=0.0405, respectively; Figure 4C). In isoproterenol‐treated mice, a significant upregulation in Nppb mRNA level was observed in the Insl6‐knockout strain (P=0.0152) despite the lack of changes in wall thickness or Nppa expression (Figure 4D and 4E).
Isoproterenol‐Induced LV Interstitial Fibrosis Is Exacerbated in Insl6‐Knockout Mice
Compared with AngII, isoproterenol administration induced a diffuse and more uniform pattern of interstitial LV fibrosis primarily in the endocardium of both WT and Insl6‐knockout mice (Figure 5A). Quantification by Picrosirius Red staining of sections of the LV revealed significantly increased isoproterenol‐induced cardiac fibrosis in Insl6‐knockout mice compared with WT mice (P=0.0073; Figure 5B). Similarly, isoproterenol treatment led to significant increases in Tgfb1 and Col3a1 mRNA expression in Insl6‐knockout mice (P=0.0167 and P=0.0483, respectively; Figure 5C), whereas a trend of increased Col1a1 mRNA was observed in Insl6‐knockout mice (unpaired t test P=0.1148; Figure 5C).
Recombinant Human INSL6 Protein Protects the Heart From Isoproterenol‐Induced Cardiac Systolic Dysfunction and Fibrosis
To investigate whether INSL6 protein protects against cardiac remodeling, a recombinant human INSL6 protein formulation (70 nmol/kg per day) was administered continuously via SC osmotic pump in WT C57BL/6N male mice 2 days before the initiation of isoproterenol treatment (Figure 6A). INSL6 protein treatment significantly reduced LW (LW/TL and LW/BW, P=0.011 and P=0.0034, respectively; Table 4). Isoproterenol‐induced changes in LV systolic and diastolic dimensions were significantly attenuated by INSL6 protein treatment compared with vehicle control group (P=0.0236 and P=0.0068, respectively; Figure 6B and 6C, left panel). Isoproterenol‐induced LV systolic dysfunction was also attenuated by INSL6 protein treatment (P=0.0395 for FS and P=0.0391 for EF; Figure 6C, right panel). Although there was no detectable effect on LV wall thickness (Figure 6D), INSL6 protein treatment significantly attenuated isoproterenol‐induced Nppb mRNA expression compared with vehicle control (P=0.0104; Figure 6E).
The INSL6‐treated group displayed significantly attenuated interstitial fibrosis compared with the vehicle control group (P=0.00473; Figure 7A and 7B). Correspondingly, INSL6 protein treatment resulted in a reduction of Tgfb1 mRNA expression (P=0.0411) and trends toward reduction in Col1a1 and Col3a1 mRNA expression compared with the vehicle control condition (Figure 7C). Moreover, INSL6 protein administration led to the upregulated expression of matrix metalloproteinase 2 mRNA compared with vehicle control (P=0.0003) (data not shown), suggesting that INSL6 protein administration can also suppress cardiac fibrosis by promoting its degradation.
Transcriptome Signature of INSL6 Treatment in Response to Isoproterenol‐Induced Heart Failure
To develop a nonbiased assessment of gene regulatory alterations associated with INSL6‐mediated cardioprotection, Affymetrix microarray transcriptome profiling was performed on sham hearts, isoproterenol‐challenged hearts treated with vehicle, and isoproterenol‐challenged hearts treated with INSL6 protein. Principle component analysis showed that both isoproterenol and INSL6 infusion administration had significant impacts in gene expression profiling in hearts from C56BL/6N male mice. Isoproterenol administration significantly altered the gene expression profile along the Principle component 1 axis (ie, x axis on Figure 8A), which contributes to 51% of the total gene expression variance. INSL6 protein also significantly alters the gene expression along PC2 axis (ie, y axis on Figure 8A), which contributes to 49% of the total variance. Using a 1.5‐fold expression change as a cutoff, 455 genes were identified whose expression was altered by isoproterenol (sham versus isoproterenol) and 529 genes were identified whose expression was altered by INSL6 protein (isoproterenol versus isoproterenol with INSL6 protein) (Figure 8B and 8C). Among these, expression of 160 genes was altered by both isoproterenol and INSL6 protein treatment, as shown by the Venn diagram in Figure 8B. Of these 160 genes, a large majority (ie, 147 genes) were regulated in an opposing manner; 74 genes were upregulated by isoproterenol challenge and downregulated by INSL6 protein treatment, and 73 genes were downregulated by isoproterenol challenge and upregulated by INSL6 protein treatment. A list of these genes can be found in Table S1.
Figure 8.
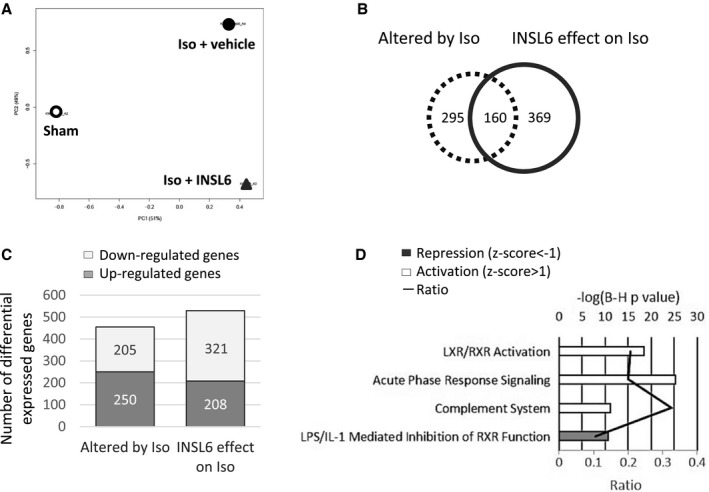
Global gene expression analysis for hearts from C57BL6/N mice treated with or without recombinant insulin‐like peptide 6 (INSL6) protein in isoproterenol (Iso) model. Three heart samples were pooled and profiled by Affymetrix Mouse Gene 2.0ST array. A, Principle component analysis of global gene expression in hearts from saline‐injected mice (Sham, open circle), Iso‐injected and vehicle‐administrated mice (Iso+vehicle, filled circle), and Iso‐injected and recombinant INSL6 protein–administrated mice (Iso+INSL6, triangle). B, Venn diagram depicts the number of differentially expressed genes that were altered by Iso injection (dotted circle, comparison between Sham and Iso+vehicle group) alone, by recombinant INSL6 protein administration (solid line circle, comparison between Iso+vehicle and Iso+INSL6 group) alone, or by both (overlapped area between dotted and solid line circle). Differentially expressed genes were defined as a fold change >1.5‐fold between 2 groups. C, Histogram depicts the number of upregulated and downregulated genes with fold change >1.5‐fold. D, Top 4 enriched canonical signaling pathways in genes differentially expressed by INSL6 protein on Iso‐treated heart. A multiple‐testing corrected P value was calculated using the Benjamini‐Hochberg method to control the rate of false discoveries. The ratio value represents the number of molecules that belong to the Ingenuity pathway analysis database‐defined pathway. Z score predicts activation or repression of pathway. IL‐1 indicates interleukin‐1; LPS, lipopolysaccharide; LXR, liver X receptor; and RXR, retinoid X receptor.
To identify the pathways altered by INSL6 protein treatment, canonical pathways represented by the differentially expressed genes were identified using the literature‐based Ingenuity Pathway Analysis using a Z‐score criterion ≥1.0 (Figure 8D). INSL6 protein treatment induced the activation of genes associated with “liver X receptor (Lxr)/retinoid X receptor (Rxr) signaling” (P<0.001, Z score=4.8) and downregulated genes classified as “lipopolysaccharide–interleukin‐1 mediated inhibition of RXR function” (P<0.001, Z score=−3.3). INSL6 treatment also modulated genes associated with “acute‐phase response signaling” (33 upregulated and 1 downregulated; P<0.001, Z score=1.8) and “complement system” (11 upregulated and 1 downregulated; P<0.001, Z score=1.1).
Discussion
Heart failure is a progressive syndrome in which structural and functional abnormalities of the heart lead to the systemic failure to sufficiently deliver oxygen and nutrients to metabolically active tissues.37 The treatment of heart failure is challenging, involving prolonged in‐hospital recovery times and frequent readmission. It is predicted that the prevalence of heart failure will increase 46% between 2012 and 2030.38 Thus, the development of novel treatments for heart failure is warranted. Recently, a recombinant human relaxin‐2 formulation, referred to as serelaxin, has been assessed as a potential therapeutic agent for acute heart failure, with mixed success.39, 40 In the phase 3 RELAX‐AHF (Serelaxin, Recombinant Human Relaxin‐2, for Treatment of Acute Heart Failure) trial of patients with acute heart failure, those who received a 16‐hour infusion of serelaxin showed improvements in biomarker expression patterns reflecting cardiac, renal, and hepatic damage, and these improvements correlated with reductions in all‐cause mortality by day 180.39, 41 In animal models, serelaxin and other formulations of recombinant relaxin‐2 have been shown to be protective in experimental models of cardiac injury.15, 16, 17, 18, 42, 43
Although the relaxin/INSL family displays limited sequence homology between members, we examined whether INSL6 can affect cardiac remodeling in 2 pharmacological models of murine heart failure. Part of our rationale for undertaking this study was our prior observations that INSL6 had roles in attenuating skeletal muscle injury.20, 22 The present study demonstrates that genetic INSL6 deficiency results in exacerbated LV systolic dysfunction in the AngII‐infusion and isoproterenol‐treatment models of heart failure in mice. In both models, cardiac fibrosis was significantly exacerbated by INSL6 deficiency, suggesting antifibrotic actions of INSL6 as a component of the mechanism that limits the progression of heart failure. Furthermore, the administration of human INSL6 protein to isoproterenol‐treated WT mice was found to significantly improve LV systolic function and reduce cardiac fibrosis. In this study, we tested the efficacy of a chemically synthesized INSL6 protein produced by methods that yield peptides joined by multiple disulfide bindings and retain oxidation‐sensitive residues.32 However, INSL6 may be subjected to additional posttranslational modifications in vivo, and additional investigations on the structure‐function relationships of recombinant INSL6 proteins will need to be defined by future studies.
AngII is a potent smooth muscle mitogen and hypertrophic agent that induces fibrosis via activation of Angiotensin II receptor type 1.44, 45 Consistent with other studies under the conditions of our assays,26 we find that low doses of AngII infusion in WT mice induced significant cardiac hypertrophy without causing significant cardiac systolic failure. Isoproterenol acts on β‐adrenergic receptors, resulting in increased heart rate and cardiac overload.25, 26 Consequently, increased myocardium oxygen demand and stress results in cardiac remodeling accompanied by myocardial necrosis, hypertrophy, and fibrosis.25, 44 In this study, we found that a moderate‐dose isoproterenol treatment resulted in LV systolic dysfunction and fibrosis without inducing cardiac hypertrophy. Notably, the patterns of fibrosis differed between these 2 pharmacological models. AngII‐induced cardiac fibrosis was focal and observed mainly in the perivascular area of coronary arteries. On the other hand, isoproterenol‐induced cardiac fibrosis was diffusely distributed throughout the LV endocardium. In both models, INSL6 deficiency exacerbated cardiac fibrosis. Correspondingly, Insl6‐knockout mice displayed increased expression of a key mediator of fibrosis, Tgfb1, and procollagen synthesis (ie, Col3a1) in both models. These data demonstrate, for the first time, the antifibrotic actions of Insl6. In comparison, the antifibrosis effect of relaxin‐2 has been well characterized.46, 47 Relaxin‐2 knockout mice display age‐dependent increases in fibrosis in the heart, lung, kidney, and reproductive systems.46 At the cellular level, relaxin‐2 administration has been shown to counteract the effects of transforming growth factor‐β or AngII on fibroblast proliferation and differentiation, reduce collagen secretion, and promote matrix metalloproteinase activities.48 Similarly, we find that recombinant INSL6 can inhibit AngII‐induced Cola1a transcript expression and suppress transcription of a reporter gene from an Smad binding element in cultured fibroblast cell lines stimulated with transforming growth factor‐β (data not shown). Given the divergence in sequence homology between INSL6 and relaxin‐2, these results suggest that antifibrotic actions may be a general property shared by the broader relaxin/INSL family members.
At baseline, Insl6‐deficient mice exhibit normal cardiac function that is no different from WT mice. These findings are in contrast with relaxin‐2–deficient mice that develop mild cardiomyopathy characterized by increased atrial weight and fibrosis accompanied by impaired LV diastolic filling with age.13 With both AngII and isoproterenol administration, Insl6‐knockout mice displayed significantly worse LV systolic function compared with WT mice, as evidenced by increased LV internal systolic dimension and reduced FS and EF. Increased or trends of increased expression of the cardiac natriuretic peptides, Nppa and Nppb, were also detected in the Insl6‐knockout mice. Relaxin‐2 serves as a vasodilator of coronary blood vessels in pregnancy and heart failure, and this is thought to be mediated by stimulating natriuretic peptide A secretion49 and inhibiting endothelin‐1 production.50 Because INSL6 deficiency and supplementation showed mixed results on Nppa transcript level in both models, these data suggest that INSL6 may act through mechanisms that differ from, as well as mechanisms that are similar to, relaxin‐2.
At the molecular level, relaxin‐2/serelaxin binds to the G‐protein–coupled receptor relaxin family peptide receptor 1 and signals via the activation of cAMP. In turn, this is reported to upregulate vascular endothelin B receptor, nitric oxide, and vascular endothelial growth factor production, resulting in decreased vascular resistance and increased cardiac output.12, 51 In contrast, INSL6, however, does not bind to relaxin family peptide receptor 1 nor activate cAMP (data not shown). Furthermore, the mechanisms of INSL6 action have received little attention, and nothing is known about how this protein confers cardiac protection. To begin to address this issue, unbiased gene profiling was performed on isoproterenol‐challenged mice that were treated with INSL6 protein or vehicle (Figure 8). This analysis revealed that INSL6 protein treatment contributes to significant transcriptomic changes in hearts that are challenged by isoproterenol infusion. Among the dysregulated genes attributable to isoproterenol challenge, 35% (n=160) genes were also altered by INSL6 protein treatment. Among those altered by both isoproterenol and INSL6 effect on isoproterenol, most of these genes (91.8%) were altered in an opposing manner. We thus focused on the signaling pathways enriched by INSL6 protein treatment on isoproterenol‐challenged heart using Ingenuity Knowledgebase. The LXR/RXR signaling pathway is one of the enriched pathways affected by INSL6 protein treatment (Figure 8D). LXRs α and β belong to the nuclear receptor superfamily of ligand‐activated transcription factors. It heterodimerizes with RXR and binds to an LXR response element in the regulatory regions of target genes.52, 53 On binding of cholesterol and oxysterol metabolites, LXR/RXR heterodimer undergoes a conformational change that transactivates target gene expression. Several lines of evidence suggest that LXR/RXR are important cardiac transcriptional regulators of LV remodeling.54 For instance, cardiac‐specific LXR α transgenic mice have been shown to be protected from AngII or transverse aortic construction–induced LV hypertrophy, cardiac function, and fibrosis.52 In WT mice, LXR agonist administration attenuated cardiac hypertrophy, systolic dysfunction, and fibrosis induced by transverse aortic construction.55
At the cellular level, LXRs have been shown to attenuate hypertrophy in neonatal cardiomyocyte via improved metabolic substrate use, downregulation of collagen synthesis, and profibrotic gene expression in cardiac fibroblasts55; and they have reduced apoptosis by inhibition of caspase‐3 protein expression.56 Analysis of the gene profiling data revealed that the expression of cholesterol 7α‐hydroxylase (Cyp7a), a target gene activated by LxRα,57 was significantly increased by 3.3‐fold in INSL6‐treated mouse hearts compared with nontreated mouse hearts subjected to isoproterenol insult. Overexpression of the Cyp7a1 in cultured mouse macrophages has been shown to enzymatically convert cholesterol to 7α‐hydroxy‐cholesterol, thus inducing cholesterol efflux.58 Cyp7a‐induced cholesterol efflux in macrophages can be further enhanced by apolipoprotein A‐I (Apoa1). Interestingly, INSL6 treatment significantly increased Apoa1 expression by 4.6‐fold as well as other members in the family, such as ApoC2, ApoC3, Apoe, and Apoh (1.5‐, 4.2‐, 1.6‐, and 2.7‐fold, respectively). In related studies, we found that INSL6 administration upregulated expression of several LXR/RXR target genes in isoproterenol‐treated heart, including Scd1 and Abca1, that control cholesterol and fatty acid homeostasis (data not shown). Thus, it is tempting to speculate that INSL6 mediates its cardioprotective actions via changes in metabolite use through regulation of LXR/RXR signaling, but further studies are needed to determine the causal role of LXR/RXR in INSL6‐mediated cardioprotection.
Conclusion
Endogenous INSL6 protects heart from adverse cardiac remodeling and fibrosis induced by isoproterenol or AngII administration to mice. Recombinant human INSL6 protein protected WT mice from isoproterenol‐induced dysfunction and fibrosis. These data suggest that INSL6 and perhaps other members of the relaxin/INSL family have therapeutic potential in the treatment of heart failure.
Sources of Funding
This work was supported by Takeda Pharmaceuticals (Walsh, Parker‐Duffen, Maruyama, Yoshida, Zhang, Wu). Maruyama was supported by HL007224, Multidisciplinary Training Grant in Cardiovascular Research. Yoshida was supported by the Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers at Tokushima University. Zhang was also supported by the State Scholarship Fund from the China Scholarship Council (No. 201408500021) and the National Natural Science Foundation of China (No. 810570212). Walsh was also supported by National Institutes of Health grants HL131006, HL129120, HL132564, and AG052160.
Disclosures
None.
Supporting information
Table S1. Transcriptome Profiling Results for Wild Type Mice Receiving Sham, Iso, or Iso+INSL6 Treatments (See Following Pages)
Acknowledgments
We thank Sam Flora for sharing the BP‐2000 Blood Pressure Analysis System. We are also grateful to Miho Sano, Jennifer Iris Reyes, and Reina Kobayashi for their assistance in mouse husbandry. We acknowledge Clinical and Translational Science Award grant UL1‐TR001430 and the Boston University Microarray and Sequencing Resource Core Facility for their assistance in microarray analysis.
(J Am Heart Assoc. 2018;7:e008441 DOI: 10.1161/JAHA.117.008441.)
References
- 1. Lu C, Lam HN, Menon RK. New members of the insulin family: regulators of metabolism, growth and now… reproduction. Pediatr Res. 2005;57:70R–73R. [DOI] [PubMed] [Google Scholar]
- 2. Wilkinson TN, Speed TP, Tregear GW, Bathgate RA. Evolution of the relaxin‐like peptide family. BMC Evol Biol. 2005;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lok S, Johnston DS, Conklin D, Lofton‐Day CE, Adams RL, Jelmberg AC, Whitmore TE, Schrader S, Griswold MD, Jaspers SR. Identification of INSL6, a new member of the insulin family that is expressed in the testis of the human and rat. Biol Reprod. 2000;62:1593–1599. [DOI] [PubMed] [Google Scholar]
- 4. Ivell R, Agoulnik AI, Anand‐Ivell R. Relaxin‐like peptides in male reproduction: a human perspective. Br J Pharmacol. 2017;174:990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gunnersen JM, Fu P, Roche PJ, Tregear GW. Expression of human relaxin genes: characterization of a novel alternatively‐spliced human relaxin mRNA species. Mol Cell Endocrinol. 1996;118:85–94. [DOI] [PubMed] [Google Scholar]
- 6. Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–480. [DOI] [PubMed] [Google Scholar]
- 7. Downing SJ, Sherwood OD. The physiological role of relaxin in the pregnant rat, III: the influence of relaxin on cervical extensibility. Endocrinology. 1985;116:1215–1220. [DOI] [PubMed] [Google Scholar]
- 8. Downing SJ, Sherwood OD. The physiological role of relaxin in the pregnant rat, II: the influence of relaxin on uterine contractile activity. Endocrinology. 1985;116:1206–1214. [DOI] [PubMed] [Google Scholar]
- 9. Downing SJ, Sherwood OD. The physiological role of relaxin in the pregnant rat, I: the influence of relaxin on parturition. Endocrinology. 1985;116:1200–1205. [DOI] [PubMed] [Google Scholar]
- 10. Winn RJ, O'Day‐Bowman MB, Sherwood OD. Hormonal control of the cervix in pregnant gilts, IV: relaxin promotes changes in the histological characteristics of the cervix that are associated with cervical softening during late pregnancy in gilts. Endocrinology. 1993;133:121–128. [DOI] [PubMed] [Google Scholar]
- 11. Dschietzig T, Richter C, Bartsch C, Laule M, Armbruster FP, Baumann G, Stangl K. The pregnancy hormone relaxin is a player in human heart failure. FASEB J. 2001;15:2187–2195. [DOI] [PubMed] [Google Scholar]
- 12. Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ. Enhanced serelaxin signalling in co‐cultures of human primary endothelial and smooth muscle cells. Br J Pharmacol. 2016;173:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du XJ, Samuel CS, Gao XM, Zhao L, Parry LJ, Tregear GW. Increased myocardial collagen and ventricular diastolic dysfunction in relaxin deficient mice: a gender‐specific phenotype. Cardiovasc Res. 2003;57:395–404. [DOI] [PubMed] [Google Scholar]
- 14. Bathgate RA, Lekgabe ED, McGuane JT, Su Y, Pham T, Ferraro T, Layfield S, Hannan RD, Thomas WG, Samuel CS, Du XJ. Adenovirus‐mediated delivery of relaxin reverses cardiac fibrosis. Mol Cell Endocrinol. 2008;280:30–38. [DOI] [PubMed] [Google Scholar]
- 15. Samuel CS, Cendrawan S, Gao XM, Ming Z, Zhao C, Kiriazis H, Xu Q, Tregear GW, Bathgate RA, Du XJ. Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest. 2011;91:675–690. [DOI] [PubMed] [Google Scholar]
- 16. Samuel CS, Hewitson TD, Zhang Y, Kelly DJ. Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology. 2008;149:3286–3293. [DOI] [PubMed] [Google Scholar]
- 17. Samuel CS, Bodaragama H, Chew JY, Widdop RE, Royce SG, Hewitson TD. Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. Hypertension. 2014;64:315–322. [DOI] [PubMed] [Google Scholar]
- 18. Parikh A, Patel D, McTiernan CF, Xiang W, Haney J, Yang L, Lin B, Kaplan AD, Bett GC, Rasmusson RL, Shroff SG, Schwartzman D, Salama G. Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ Res. 2013;113:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burnicka‐Turek O, Shirneshan K, Paprotta I, Grzmil P, Meinhardt A, Engel W, Adham IM. Inactivation of insulin‐like factor 6 disrupts the progression of spermatogenesis at late meiotic prophase. Endocrinology. 2009;150:4348–4357. [DOI] [PubMed] [Google Scholar]
- 20. Zeng L, Akasaki Y, Sato K, Ouchi N, Izumiya Y, Walsh K. Insulin‐like 6 is induced by muscle injury and functions as a regenerative factor. J Biol Chem. 2010;285:36060–36069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu CL, Satomi Y, Walsh K. RNA‐seq and metabolomic analyses of Akt1‐mediated muscle growth reveals regulation of regenerative pathways and changes in the muscle secretome. BMC Genomics. 2017;18:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeng L, Maruyama S, Nakamura K, Parker‐Duffen JL, Adham IM, Zhong X, Lee HK, Querfurth H, Walsh K. The injury‐induced myokine insulin‐like 6 is protective in experimental autoimmune myositis. Skelet Muscle. 2014;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou X, Chen X, Cai JJ, Chen LZ, Gong YS, Wang LX, Gao Z, Zhang HQ, Huang WJ, Zhou H. Relaxin inhibits cardiac fibrosis and endothelial‐mesenchymal transition via the Notch pathway. Drug Des Devel Ther. 2015;9:4599–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belge C, Hammond J, Dubois‐Deruy E, Manoury B, Hamelet J, Beauloye C, Markl A, Pouleur AC, Bertrand L, Esfahani H, Jnaoui K, Gotz KR, Nikolaev VO, Vanderper A, Herijgers P, Lobysheva I, Iaccarino G, Hilfiker‐Kleiner D, Tavernier G, Langin D, Dessy C, Balligand JL. Enhanced expression of beta3‐adrenoceptors in cardiac myocytes attenuates neurohormone‐induced hypertrophic remodeling through nitric oxide synthase. Circulation. 2014;129:451–462. [DOI] [PubMed] [Google Scholar]
- 25. Nichtova Z, Novotova M, Kralova E, Stankovicova T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen Physiol Biophys. 2012;31:141–151. [DOI] [PubMed] [Google Scholar]
- 26. Ikeda Y, Aihara K, Yoshida S, Sato T, Yagi S, Iwase T, Sumitomo Y, Ise T, Ishikawa K, Azuma H, Akaike M, Kato S, Matsumoto T. Androgen‐androgen receptor system protects against angiotensin II‐induced vascular remodeling. Endocrinology. 2009;150:2857–2864. [DOI] [PubMed] [Google Scholar]
- 27. Clark AL, Maruyama S, Sano S, Accorsi A, Girgenrath M, Walsh K, Naya FJ. miR‐410 and miR‐495 are dynamically regulated in diverse cardiomyopathies and their inhibition attenuates pathological hypertrophy. PLoS One. 2016;11:e0151515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jadhav R, Dodd T, Smith E, Bailey E, Delucia AL, Russell JC, Madison R, Potter B, Walsh K, Jo H, Rocic P. Angiotensin type I receptor blockade in conjunction with enhanced Akt activation restores coronary collateral growth in the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2011;300:H1938–H1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, Ohashi K, Kielbasa OM, Mathew M, Ewen EP, Walsh K, Gavras H, Naya FJ. Modulation of angiotensin II‐mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. 2010;106:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee HY, Chung JW, Youn SW, Kim JY, Park KW, Koo BK, Oh BH, Park YB, Chaqour B, Walsh K, Kim HS. Forkhead transcription factor FOXO3a is a negative regulator of angiogenic immediate early gene CYR61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2007;100:372–380. [DOI] [PubMed] [Google Scholar]
- 31. Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, Lecker S, Taegtmeyer H, Goldberg AL, Walsh K. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu F, Mayer JP, Zaykov AN, Zhang F, Liu F, DiMarchi RD. Chemical synthesis of human insulin‐like peptide‐6. Chemistry. 2016;22:9777–9783. [DOI] [PubMed] [Google Scholar]
- 33. Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, Asaumi Y, van den Hoff MJ, Ouchi N, Recchia FA, Walsh K. Follistatin‐like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med. 2016;8:949–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshida S, Fuster JJ, Walsh K. Adiponectin attenuates abdominal aortic aneurysm formation in hyperlipidemic mice. Atherosclerosis. 2014;235:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brouwers FP, van Gilst WH, Damman K, van den Berg MP, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van der Harst P, de Boer RA. Clinical risk stratification optimizes value of biomarkers to predict new‐onset heart failure in a community‐based cohort. Circ Heart Fail. 2014;7:723–731. [DOI] [PubMed] [Google Scholar]
- 37. Writing Group Members , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee, Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 38. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG; American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis, Thrombosis, and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council . Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M; RELAXin in Acute Heart Failure (RELAX‐AHF) Investigators . Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet. 2013;381:29–39. [DOI] [PubMed] [Google Scholar]
- 40. Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G. Relaxin for the treatment of patients with acute heart failure (Pre‐RELAX‐AHF): a multicentre, randomised, placebo‐controlled, parallel‐group, dose‐finding phase IIb study. Lancet. 2009;373:1429–1439. [DOI] [PubMed] [Google Scholar]
- 41. Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld L, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Prescott MF, Edwards C, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin T, Teerlink JR; RELAX‐AHF Investigators . Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX‐AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. [DOI] [PubMed] [Google Scholar]
- 42. Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E, Filippone S, Das A, Toldo S, Abbate A, Salloum FN. Reperfusion therapy with recombinant human relaxin‐2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS‐dependent mechanism. Cardiovasc Res. 2017;113:609–619. [DOI] [PubMed] [Google Scholar]
- 43. Zhang J, Qi YF, Geng B, Pan CS, Zhao J, Chen L, Yang J, Chang JK, Tang CS. Effect of relaxin on myocardial ischemia injury induced by isoproterenol. Peptides. 2005;26:1632–1639. [DOI] [PubMed] [Google Scholar]
- 44. Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overload and apoptosis in heart failure: implications for therapy. J Card Fail. 2000;6:1–7. [PubMed] [Google Scholar]
- 45. Billet S, Aguilar F, Baudry C, Clauser E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int. 2008;74:1379–1384. [DOI] [PubMed] [Google Scholar]
- 46. Samuel CS, Zhao C, Bathgate RA, Du XJ, Summers RJ, Amento EP, Walker LL, McBurnie M, Zhao L, Tregear GW. The relaxin gene‐knockout mouse: a model of progressive fibrosis. Ann N Y Acad Sci. 2005;1041:173–181. [DOI] [PubMed] [Google Scholar]
- 47. Samuel CS, Lekgabe ED, Mookerjee I. The effects of relaxin on extracellular matrix remodeling in health and fibrotic disease. Adv Exp Med Biol. 2007;612:88–103. [DOI] [PubMed] [Google Scholar]
- 48. Samuel CS, Unemori EN, Mookerjee I, Bathgate RA, Layfield SL, Mak J, Tregear GW, Du XJ. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. [DOI] [PubMed] [Google Scholar]
- 49. Toth M, Taskinen P, Ruskoaho H. Relaxin stimulates atrial natriuretic peptide secretion in perfused rat heart. J Endocrinol. 1996;150:487–495. [DOI] [PubMed] [Google Scholar]
- 50. Dschietzig T, Bartsch C, Richter C, Laule M, Baumann G, Stangl K. Relaxin, a pregnancy hormone, is a functional endothelin‐1 antagonist: attenuation of endothelin‐1‐mediated vasoconstriction by stimulation of endothelin type‐B receptor expression via ERK‐1/2 and nuclear factor‐kappaB. Circ Res. 2003;92:32–40. [DOI] [PubMed] [Google Scholar]
- 51. Diez J, Ruilope LM. Serelaxin for the treatment of acute heart failure: a review with a focus on end‐organ protection. Eur Heart J Cardiovasc Pharmacother. 2016;2:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cannon MV, Sillje HH, Sijbesma JW, Vreeswijk‐Baudoin I, Ciapaite J, van der Sluis B, van Deursen J, Silva GJ, de Windt LJ, Gustafsson JA, van der Harst P, van Gilst WH, de Boer RA. Cardiac LXRalpha protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med. 2015;7:1229–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–993. [DOI] [PubMed] [Google Scholar]
- 54. Cannon MV, van Gilst WH, de Boer RA. Emerging role of liver X receptors in cardiac pathophysiology and heart failure. Basic Res Cardiol. 2016;111:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cannon MV, Yu H, Candido WM, Dokter MM, Lindstedt EL, Sillje HH, van Gilst WH, de Boer RA. The liver X receptor agonist AZ876 protects against pathological cardiac hypertrophy and fibrosis without lipogenic side effects. Eur J Heart Fail. 2015;17:273–282. [DOI] [PubMed] [Google Scholar]
- 56. Lei P, Baysa A, Nebb HI, Valen G, Skomedal T, Osnes JB, Yang Z, Haugen F. Activation of liver X receptors in the heart leads to accumulation of intracellular lipids and attenuation of ischemia‐reperfusion injury. Basic Res Cardiol. 2013;108:323. [DOI] [PubMed] [Google Scholar]
- 57. Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. [DOI] [PubMed] [Google Scholar]
- 58. Moore GL, Davis RA. Expression of cholesterol‐7alpha‐hydroxylase in murine macrophages prevents cholesterol loading by acetyl‐LDL. J Lipid Res. 2002;43:629–635. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Transcriptome Profiling Results for Wild Type Mice Receiving Sham, Iso, or Iso+INSL6 Treatments (See Following Pages)