

Abstract
Background
NADPH Oxidase 5 (Nox5) is a calcium‐sensitive superoxide‐generating Nox. It is present in lower forms and higher mammals, but not in rodents. Nox5 is expressed in vascular cells, but the functional significance remains elusive. Given that contraction is controlled by calcium and reactive oxygen species, both associated with Nox5, we questioned the role of Nox5 in pro‐contractile signaling and vascular function.
Methods and Results
Transgenic mice expressing human Nox5 in a vascular smooth muscle cell–specific manner (Nox5 mice) and Rhodnius prolixus, an arthropod model that expresses Nox5 endogenoulsy, were studied. Reactive oxygen species generation was increased systemically and in the vasculature and heart in Nox5 mice. In Nox5‐expressing mice, agonist‐induced vasoconstriction was exaggerated and endothelium‐dependent vasorelaxation was impaired. Vascular structural and mechanical properties were not influenced by Nox5. Vascular contractile responses in Nox5 mice were normalized by N‐acetylcysteine and inhibitors of calcium channels, calmodulin, and endoplasmic reticulum ryanodine receptors, but not by GKT137831 (Nox1/4 inhibitor). At the cellular level, vascular changes in Nox5 mice were associated with increased vascular smooth muscle cell [Ca2+]i, increased reactive oxygen species and nitrotyrosine levels, and hyperphosphorylation of pro‐contractile signaling molecules MLC20 (myosin light chain 20) and MYPT1 (myosin phosphatase target subunit 1). Blood pressure was similar in wild‐type and Nox5 mice. Nox5 did not amplify angiotensin II effects. In R. prolixus, gastrointestinal smooth muscle contraction was blunted by Nox5 silencing, but not by VAS2870 (Nox1/2/4 inhibitor).
Conclusions
Nox5 is a pro‐contractile Nox isoform important in redox‐sensitive contraction. This involves calcium‐calmodulin and endoplasmic reticulum–regulated mechanisms. Our findings define a novel function for vascular Nox5, linking calcium and reactive oxygen species to the pro‐contractile molecular machinery in vascular smooth muscle cells.
Keywords: cell signaling, contraction, vascular biology
Subject Categories: Cell Signalling/Signal Transduction, Vascular Biology
Clinical Perspective
What Is New?
We define a biologically important role for NADPH Oxidase 5 (Nox5) as a regulator of vascular contraction.
Molecular mechanisms whereby Nox5 regulates contraction involve redox‐sensitive processes that influence calcium channels, calcium mobilization, and pro‐contractile signaling.
Nox5 is a point of cross‐talk between vascular calcium and redox signaling.
Vascular Nox5 activation also influences cardiac fibrosis.
What Are the Clinical Implications?
We identify Nox5 as a key regulator of vascular contraction and cardiac fibrosis.
Dysregulation of Nox5 may be associated with oxidative stress and amplified calcium signaling, leading to vascular hypercontractility and dysfunction in cardiovascular diseases.
Modulating Nox5 may have therapeutic potential by targeting both redox and calcium signalling pathways.
Noxs (NADPH oxidases) are a family of enzymes that function primarily to produce reactive oxygen species (ROS) by using molecular oxygen and NADPH as substrates. In phagocytic cells, ROS play a role in host‐defense responses, and in nonphagocytic cells, ROS control cell differentiation, proliferation, apoptosis, and senescence.1, 2 In the vascular system, ROS regulate vasculogenesis, endothelial function, and vascular tone.3, 4 In cardiovascular diseases, such as hypertension,5 atherosclerosis,6 and pulmonary arterial hypertension,7 Nox‐derived ROS generation is increased, leading to endothelial dysfunction and vascular injury. The mammalian Nox family comprises 7 isoforms, Nox1 to 5 and dual oxidase 1 and dual oxidase 2, of which Nox1, 2, 4, and 5 have been identified in human vessels.8, 9 Vascular Noxs have different functions in pathological conditions. Nox1 and Nox2 play a role in angiogenesis and atherosclerosis,3, 6 and Nox4 seems to be vasoprotective in ischemic and hypoxic conditions,10 but injurious in diabetes mellitus, where it promotes vascular fibrosis, diabetic nephropathy, and intravitreal neovascularization.11 Although Nox5 is expressed in mammalian vessels,12, 13, 14 there is a paucity of information on its role and biological significance in the cardiovascular system.
Unlike the other vascular Nox isoforms, Nox5 gene is absent in rodents; it generates ROS from a single gene product and does not require classical NADPH oxidase subunits (p22phox, p47phox, and p67phox) for its activation.15 Nox5 is the only Nox isoform that has been crystalized.16 Unique to Nox5 is its long N‐terminal extension that contains 4 calcium (Ca2+)‐binding EF hands, which undergoes conformational change upon Ca2+ binding.17, 18 Increased intracellular free Ca2+ concentration ([Ca2+]i) is essential for the activity of Nox5,18, 19, 20 as evidenced in cell‐culture studies where ROS are not synthesized in Ca2+‐free buffer or from cells with truncation of the N‐terminal region of Nox5.21 This is of particular significance in vascular smooth muscle cells (VSMCs), given that increased [Ca2+]i is the major trigger for vascular contraction.22, 23 Nox5 is expressed in all vascular cell types, including endothelial cells, VSMCs, and perivascular adventitial fibroblast,12, 14, 24 and plays a role in angiotensin II (Ang II)‐ and endothelin‐1 (ET‐1)‐mediated redox signaling.25 Nox5 expression is increased in human atherosclerotic and aneurysm lesions26, 27 and in kidneys from diabetic patients.28 We demonstrated that mice expressing human Nox5 in a podocyte‐specific manner exhibit podocyte injury and renal dysfunction,28 and that human Nox5 expression in mesangial cells causes renal fibrosis, nephropathy, and exacerbated atherosclerosis in diabetic mice.29 Findings from recent genome wide‐association studies of 475 000 individuals identified Nox5 as a candidate blood pressure–associated gene.30 Together, these data suggest an important (patho)physiological role for Nox5 in the cardiovascular system. However, the exact function of VSMC Nox5 and its biological significance in the vascular system are unknown.
Using a multidisciplinary approach including transgenic Nox5 mice with selective expression of Nox5 in smooth muscle cells, cultured VSMCs from Nox5‐expressing mice, and an arthropod model that endogenously expresses Nox5,31 we identify a novel role for Nox5 as an important pro‐contractile NADPH oxidase isoform that regulates vascular contraction and reactivity. We also elucidate some molecular mechanisms underlying Nox5 vascular effects. Nox5 dysregulation may contribute to vascular injury and dysfunction in pathological conditions.
Methods
Mice Expressing Human Nox5 in VSMCs
Generation of Nox5 transgenic mice was approved by the Animal Ethics Committee of the Ottawa Hospital Research Institute, University of Ottawa (Ottawa, Ontario, Canada) and carried out according to the recommendations of the Canadian Council for Animal Care. NOX5β cDNA was PCR amplified from pDONRNox5β plasmid. Purified PCR NOX5β gene‐coding region was ligated into the Tet‐responsive promoter, Pbi‐1. A 3.7‐kb TetO/NOX5β fragment was excised and the resulting band gel‐purified and provided to the University of Ottawa Core Transgenic Facility for pronuclear injection into fertilized FVB/N oocytes. Subsequent pBI‐NOX5β founders on a pure FVB/N background were identified by PCR genotyping. To generate VSMC‐specific knockout animals, pBI‐Nox5 animals were crossed with the SM22‐tTA‐FVB/N mouse strain to produce Nox5+/SM22+ and control transgenic mice (SM22+, Nox5+).
In additional studies, adult (20 weeks) WT, SM22+, Nox5+, and Nox5+/SM22+ mice were infused with Ang II (600 ng/kg/day) for 4 weeks by osmotic minipumps. At the end of treatment, tissues were snap‐frozen or fixed in formalin for analysis. Animal experiments were performed in accord with the United Kingdom Animals Scientific Procedures Act 1986 and ARRIVE (Animal Research: Reporting of In Vivo Experiments) Guidelines32 and approved by the Home Office under Project Licence No. 7009021.
Measurement of Blood Pressure
Systolic blood pressure was assessed by tail‐cuff plethysmography.33
Plasma Biochemistry and Plasma Lipid Peroxidation Products
Plasma biochemistry was determined by an automated analyzer. Plasma lipid peroxidation (malondialdehyde) was determined by quantifying thiobarbituric acid‐reactive substances by ELISA.
Myography to Assess Vascular Function and Structure
Vascular function was assessed in resistance arteries by wire myography as we previously described.34 Endothelium‐dependent relaxation was assessed in all vessels by concentration‐responses to acetylcholine (Ach) where vessels were precontracted with U46619 (thromboxane A2 analogue). Thereafter, endothelium‐independent relaxation was assessed by concentration responses to sodium nitroprusside and contractile responses mediated by U46619 evaluated in endothelium‐intact arteries. At the end of relaxation and contraction curves, ET‐1 induced contraction was assessed. Maximal contraction to KCl was also evaluated before and after addition of pharmacological inhibitors. In some experiments, arteries were preincubated with N‐acetylcysteine (ROS scavenger), GKT137831 (Nox1/Nox4 inhibitor), diltiazem (Ca2+ channels blocker), calmidazolium (calmodulin inhibitor), and dantrolene. Dantrolene is considered a potent receptor antagonist of the ryanodine receptor Ca2+ channel in the endoplasmic reticulum (ER), which decreases [Ca2+]i. Studies have demonstrated that dantrolene acts directly on the ryanodine receptor to reduce channel activation by calmodulin and decreases Ca2+ sensitivity of channel activation.
Vascular structure and mechanical properties were assessed in resistance arteries prepared as pressurized systems on a pressure myograph as we previously described.34
Cardiac Histology and Fibrosis Staining
For histochemical analysis, heart sections were stained with picrosirius red (0.1% w/v). Total collagen content (%) was measured in whole tissue under polarized light. To assess cardiomyocyte area, hematoxylin and eosin staining of heart sections was performed. Data were quantified by digital image analysis software (ImageJ; NIH, Bethesda, MD).
Mouse VSMC Culture
VSMCs from adult male WT and Nox5+SM22+ mice were studied.35 Low‐passage cells (4–7) were used. VSMCs were stimulated with U46619. In some experiments, cells were preincubated with pharmacological modulators.
Immunofluorescence
Nox5 immunofluorescence was performed in human arteries obtained from normotensive and hypertensive subjects and in aortas isolated from WT, SM22+, Nox5+, and Nox5+SM22+ mice.
Lucigenin‐Enhanced Chemiluminescence Assay
Vascular ROS generation was measured by a luminescence assay with lucigenin as the electron acceptor and NADPH as the substrate.
Measurement of Nitrotyrosine and H2O2 Levels
Nitrotyrosine, a measure of peroxynitrite (ONOO−) formation, was assessed in aortic tissue from WT, Nox5+SM22+, SM22+, and Nox5+ mice by ELISA. H2O2 was assessed in cardiac tissue from WT, Nox5+SM22+, SM22+, and Nox5+ by Amplex Red (Life Technologies).
Measurement of Superoxide Anion in Cardiac Tissue
Superoxide levels in hearts from WT, Nox5+SM22+, SM22+, and Nox5+ was measured by high‐performance liquid chromatography.
Western Blotting
Total protein was extracted from mouse VSMCs and from mesenteric arteries from WT, SM22+, Nox5+, and Nox5+SM22+ mice. Antibodies were as follows: MYPT1 (myosin phosphatase target subunit 1; Thr696); p‐MLC20 (myosin light chain 20; Thr19/Ser 19); Nox1; Nox2; Nox4; and Nox5. Antibodies to β‐actin or GAPDH were used as housekeeping controls. After incubation with secondary fluorescence‐coupled antibodies (LI‐COR Biosciences, Lincoln, NE), signals were visualized by an infrared laser scanner.
Real‐Time PCR
Total RNA was isolated from cardiac samples from WT, SM22+, Nox5+, and Nox5+SM22+ mice. Real‐time PCR was performed with specific mouse primers for Nox2 and Nox4. Data are shown as the fold change in expression of the target gene relative to the internal control gene GAPDH.36
Measurement of Intracellular Free Ca2+ Concentration ([Ca2+]i) in VSMCs
[Ca2+]i was measured in VSMCs from WT and Nox5+/SM22+ mice using the fluorescent Ca2+ indicator, Cal‐520 acetoxymethyl ester (Cal‐520/AM).
Nox5‐Expressing Arthropod Model: Rhodnius prolixus
Rhodnius prolixus was studied as an arthropod model that expresses Nox5 endogenously. Insects were fed with rabbit blood as a stimulus for gut contraction.
siRNA Downregulation of Nox5 in R. prolixus
A 457‐bp fragment from the Nox5 gene was amplified from reverse‐transcribed RNAs extracted from R. prolixus midguts using the primer pairs NOX5Ds1. For gene‐silencing experiments, adult R. prolixus were injected with dsRNA. Six days after dsRNA injection, insects were fed.
RNA Extraction, PCR, and qPCR: R. prolixus
Total RNA was extracted from anterior midguts using TRIzol and cDNA was synthesized. cDNA from anterior midguts were PCR‐amplified using the PCR master mix, and the same primers were used for qPCR. The R. prolixus EF‐1 S rRNA gene was used as an endogenous control.
VAS2870 Injection in R. prolixus
In some experiments, R. prolixus were injected with VAS2870 to inhibit Nox 1, 2, and 4 six days before feeding.
Images and Video Acquisition of Gut Contractions in Nox5‐Silenced R. prolixus
Nox5‐siRNA–injected and VAS2870‐treated insects were studied 7 days after a blood meal. Peristaltic contractions of the anterior midguts were recorded for 2 minutes by video fluorescence microscopy.
Statistical Analysis
Data are presented as mean±SEM. Statistical comparisons were made with 1‐way ANOVA followed by Dunnet's post‐hoc or 2‐tailed Student t test, when appropriate. P<0.05 was considered statistically significant.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Additional methods can be found in Data S1.
Results
Phenotypic Characterization of Nox5 Mice
The mouse genome lacks Nox5 and, accordingly, to study the biological in vivo significance of vascular Nox5, we created humanized transgenic mice that selectively express Nox5 in smooth muscle cells (Nox5+SM22+). Nox5 expression, assessed by immunofluorescence, was observed only in Nox5+SM22+ mice and not in any of the control groups (WT, SM22+, and Nox5+), confirming the selective Nox5 expression in the experimental transgenic mice (Figure 1A). Systemic oxidative stress, measured by plasma levels of lipid peroxidation (plasma malondialdehyde levels), was already increased in basal conditions in Nox5+SM22 mice (Figure 1B) and was not further enhanced by Ang II infusion.
Figure 1.
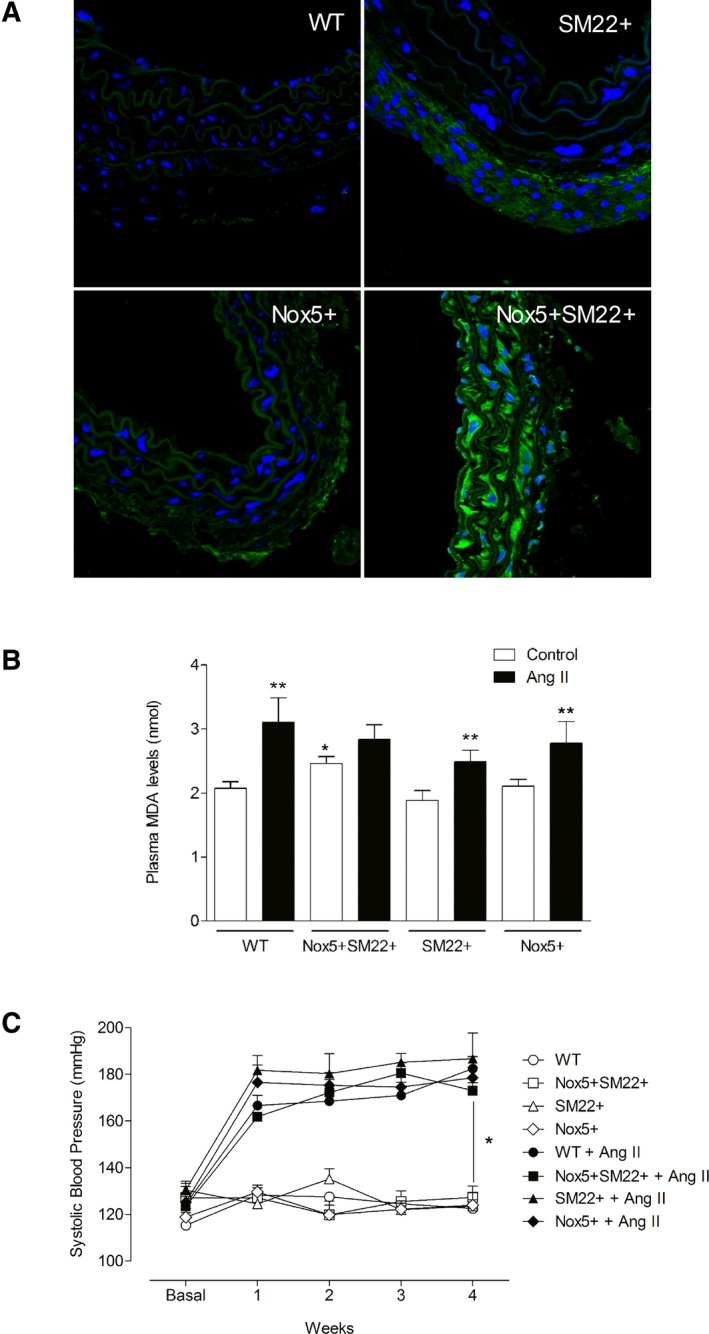
Phenotypic characterization of Nox5+ SM22+ mice. (A) Immunolocalization of Nox5 (green fluorescence) in WT, SM22+, Nox5+, and Nox5+/SM22+ mice. Blue fluorescence indicates nucleus (DAPI). B, Plasma TBARS levels in WT, SM22+, Nox5+, and Nox5+/SM22+ mice. C, Systolic blood pressure assessed by plethysmography for 4 weeks in WT, SM22+, Nox5+, and Nox5+ SM22+ mice treated or not with Ang II (600 ng/kg/day). Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs control WT group or untreated group (BP); **P<0.05 vs nontreated counterpart (TBARS). Ang II indicates angiotensin II; BP, blood pressure; DAPI, 4′,6‐diamidino‐2‐phenylindole; Nox5, NADPH Oxidase 5; TBARS, thiobarbituric acid reactive substances; WT, wild‐type.
Ang II infusion for 4 weeks increased systolic blood pressure in all groups, without differences between control and Nox5+SM22+ mice (Figure 1C). Body weight and plasma biochemistry were not significantly different between groups, although Ang II increased plasma phosphate levels in WT, SM22+, and Nox5+SM22+ mice (Table S1). Causes for the hyperphosphatemia are unclear.
Cardiac superoxide anion (Figure 2A), H2O2 (Figure S1A), and thiobarbituric acid‐reactive substances levels (Figure S1B) were increased in Nox5+SM22+. Ang II increased superoxide, H2O2, and TBARS levels only in wild‐type (WT) mice. Heart weight was only increased in control mice treated with Ang II (Figure 2B). Collagen deposition was increased in hearts from Nox5+SM22+ mice (Figure 2C and 2D). Ang II increased collagen deposition in cardiac tissue from WT, mice. Histological analysis demonstrated that cardiomyocyte area was increased in cardiac tissue from Nox5+SM22+ mice, to a similar extent to that in Ang II–infused WT mice (Figure 2E).
Figure 2.
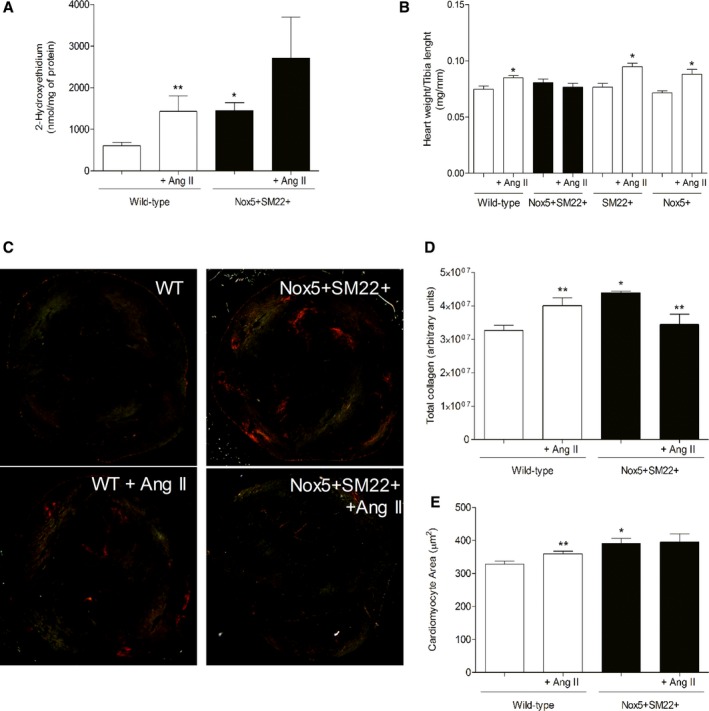
Cardiac phenotypic characterization of Nox5+ SM22+ mice. A, Superoxide anion levels assessed by HPLC in heart tissue from WT (open bars) and SM22+Nox5+ (closed bars) mice, before and after Ang II treatment. B, Heart weight in WT, SM22+, and Nox5+ mice, before and after Ang II treatment. C, Representative images of Sirius Red staining of heart tissue from WT and SM22+Nox5+ mice, treated or not with Ang II, exposed to polarized light microscopy. D, Total collagen deposition in heart tissue from WT and SM22+Nox5+ mice, before and after Ang II treatment. E, Cardiomyocyte area in heart tissue from WT and SM22+Nox5+ mice, before and after Ang II treatment. Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs control WT group; **P<0.05 vs nontreated counterpart. Ang II indicates angiotensin II; HPLC, high‐performance liquid chromatography; Nox5, NADPH Oxidase 5; WT, wild‐type.
Vascular Function in Nox5 Mice
Concentration‐response curves to the thromboxane A2 analogue, U46619 (contraction), Ach (endothelial‐dependent relaxation), and sodium nitroprusside (endothelial‐independent relaxation) were performed in all groups to assess whether expression of Nox5 in VSMCs influences vascular function. Vascular contraction induced by U46619 was increased in Nox5+SM22+ mice compared with all control groups (WT, Nox5+, and SM22+; Figure 3A). In Ang II–infused mice, contractile responses were significantly enhanced in WT and Nox5+SM22+ mice (Figure 3B). There were no significant differences in contractile responses between the control groups (WT, Nox5+, and SM22+) in basal conditions or in mice treated with Ang II (Figure S2). The importance of ROS was observed by exposure of isolated arteries to N‐acetylcysteine, an ROS scavenger, that significantly attenuated U46619‐induced contractile responses in Nox5+SM22+ mice, with only a modest effect in WT mice (Figure 3C). GKT137831, a Nox1/4 inhibitor, did not significantly alter contractile responses in WT or Nox5+SM22+ mice (Figure 3D). In previous studies, we showed that activation of the Ca2+/calmodulin pathway is important for Nox5‐dependent effects in VSMCs. Here, we found that contractile responses in WT and Nox5+SM22+ groups were significantly reduced in vessels exposed to the calmodulin inhibitor, calmidazolium (Figure 3E). Dantrolene, which inhibits ryanodine receptors and prevents Ca2+ release from the ER, attenuated contraction in Nox5+/SM22+ mice without effect in WT mice (Figure 3F).
Figure 3.
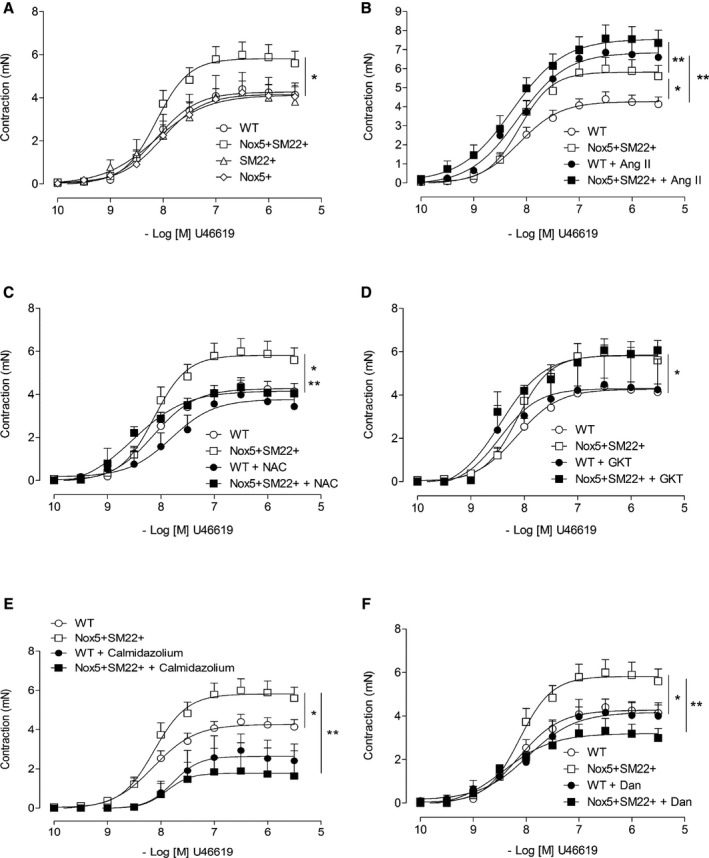
Vascular contraction is increased in Nox5+/SM22+ mice. A, Vascular contractility to U46619 assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus) and Nox5+/SM22+ (open square). B, Vascular contractility to U46619 in mesenteric arteries from WT (open circle), WT+Ang II (closed circle), Nox5+/SM22+ (open square), and Nox5+/SM22++Ang II (closed square) mice. C, Vascular contractility to U46619 in mesenteric arteries from WT (open circle) and Nox5+/SM22+ (open square) mice in the presence of N‐acetylcysteine (NAC; 10 μmol/L) (WT, closed circle; Nox5+/SM22+, closed square). Vessels were pre‐incubated with NAC for 1 hour. D, Vascular contractility to U46619 in arteries from WT (open circle) and Nox5+/SM22+ (open square) mice in the presence of GKT137831 (10 μmol/L; WT, closed circle; Nox5+/SM22+, closed square). Vessels were preincubated with GKT137831 for 1 hour. E, Vascular contractility to U46619 in mesenteric arteries from WT (open circle) and Nox5+/SM22+ (open square) mice in the presence of calmidazolium (Calmid; 1 μmol/L; WT, closed circle; Nox5+/SM22+, closed square). Vessels were preincubated with calmidazolium for 1 hour. F, Vascular contractility to U46619 in arteries from WT (open circle) and Nox5+/SM22+ (open square) mice in the presence of dantrolene (Dant; 10 μmol/L; WT, closed circle; Nox5+/SM22+, closed square). Vessels were preincubated with dantrolene for 1 hour. Results are mean±SEM of 3 to 8 mice/group. *P<0.05 vs WT; **P<0.05 vs untreated WT or Nox5+/SM22+ (open symbols). Ang II indicates angiotensin II; Nox5, NADPH Oxidase 5; WT, wild‐type.
Increased vascular contraction in Nox5+SM22+ mice was not agonist specific, but appears to be a generalized phenomenon, given that maximal responses to KCl and ET‐1 were also exaggerated in Nox5‐expressing mice compared with control groups (Figure 4). Contractile responses to KCl and ET‐1 were significantly reduced in Nox5+SM22+ vessels exposed to diltiazem, calmidozilium, and N‐acetylcysteine (Figure 4B through 4F). GKT137831 did not reduce KCl‐ or ET‐1–induced contraction (Figure 4C through 4E). Associated with exaggerated contraction, Ach‐induced vasorelaxation (endothelium‐dependent relaxation) was significantly reduced in Nox5+SM22+ mice (Figure 5A). In Ang II–infused WT and Nox5+SM22+ mice, Ach‐mediated vasorelaxation was significantly reduced, with maximal dilatory responses of only 10% to 20% (Figure 5B). Ach‐mediated responses were not different between respective WT, Nox5+, and SM22+ control groups (Figure 5C). SNP‐induced vasorelaxation was similar in all groups and was unaffected by Nox5 expression or by Ang II infusion (Figure S3A through S3C).
Figure 4.
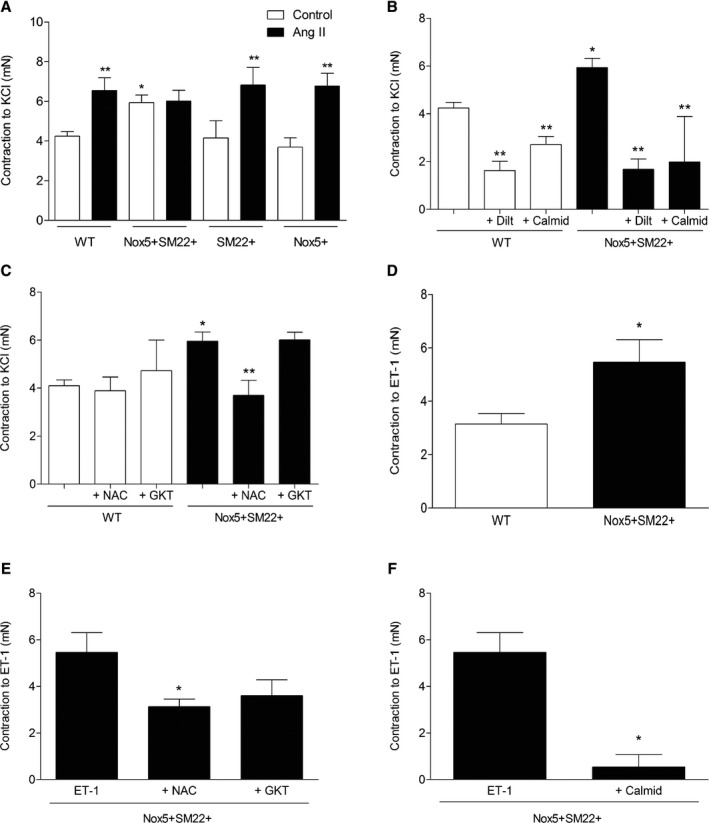
Maximum contraction to KCl and ET‐1–induced contraction are increased in arteries from Nox5+/SM22+ mice. A, Vascular contraction to a single concentration of KCl (62.5 mmol/L) in mesenteric arteries from WT, Nox5+/SM22+, SM22+, and Nox5+ mice, before and after treatment with Ang II. B, Vascular contraction to KCl in mesenteric arteries from WT and Nox5+/SM22+ mice, before and after preincubation (1 hour) with diltiazem (10 μmol/L) or calmidazolium (10 μmol/L). C, Vascular contraction to KCl in mesenteric arteries from WT and Nox5+/SM22+ mice, before and after preincubation (1 hour) with NAC (10 μmol/L) or GKT137831 (10 μmol/L). D, Vascular contraction to a single concentration of ET‐1 (0.1 mmol/L) in mesenteric arteries from WT and Nox5+/SM22+ mice. Vascular contraction to ET‐1 in mesenteric arteries from WT and Nox5+/SM22+ mice, before and after preincubation (1 hour) with NAC (10 μmol/L) or GKT137831 (10 μmol/L; E) or calmidazolium (10 μmol/L; F). Results are mean±SEM of 3 to 8 mice/group. *P<0.05 vs WT; **P<0.05 vs untreated mice. Ang II indicates angiotensin II; ET‐1, endothelin‐1; NAC, N‐acetylcysteine; Nox5, NADPH Oxidase 5; WT, wild‐type.
Figure 5.
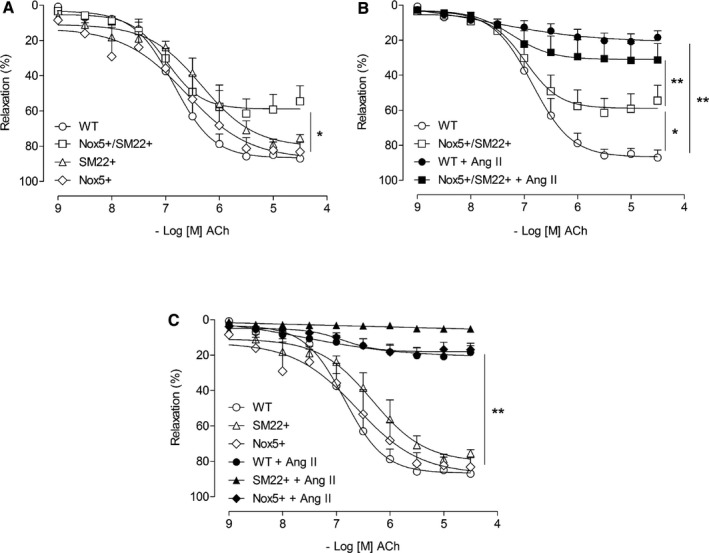
Vascular expression of Nox5 induces endothelial dysfunction. A, Vascular relaxation to ACh assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+ SM22+ (open square). B, Vascular relaxation to ACh in arteries from WT (open circle), WT+Ang II (closed circle), Nox5+/SM22+ (open square), and Nox5+/SM22++Ang II (closed square) mice. C, Vascular relaxation to ACh assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), and Nox5+ (open rhombus) before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs WT; **P<0.05 vs untreated WT or Nox5+/SM22+ (open symbols). ACh indicates acetylcholine; Ang II, angiotensin II; Nox5, NADPH Oxidase 5; WT, wild‐type.
Vascular Structure and Mechanical Properties in Nox5+SM22+ Mice
Small mesenteric arteries were also studied as pressurized systems to assess vascular structure and mechanical properties. In basal conditions, there were no structural differences between any groups as assessed by measuring cross‐sectional area (Figure 6A) and wall‐to‐lumen ratio (Figure S4A). Cross‐sectional area was increased in all Ang II–infused groups (Figure 6B and 6C), with modest effect in Nox5‐expressing mice compared with WT mice. In Ang II–infused mice, wall‐to‐lumen ratio was increased in Nox5+SM22+ mice compared with WT and vehicle‐treated Nox5+/SM22+ counterparts (Figure S4C and S4D). Vascular stiffness and distensibility, assessed by stress/strain relationships, were not significantly different between any groups (Figure S5).
Figure 6.
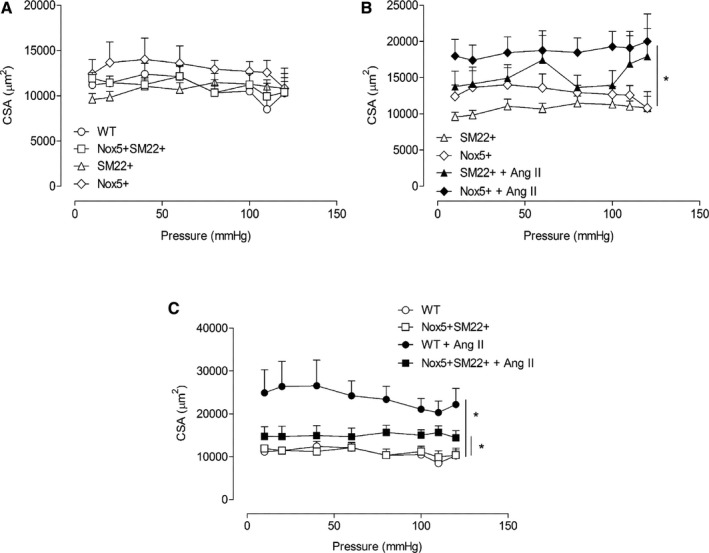
Cross‐sectional area (CSA) in resistance arteries from Nox5+/SM22+ mice. A, Vascular CSA assessed by pressure myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+/SM22+ (open square). B, Vascular CSA assessed by pressure myography in mesenteric arteries from SM22+ (open triangle) and Nox5+ (open rhombus), before and after Ang II treatment (closed symbols). C, Vascular CSA assessed by pressure myography in mesenteric arteries from WT (open circle) and Nox5+/SM22+ (open square), before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 animals per group. *P<0.05 vs untreated groups. Ang II indicates angiotensin II; Nox5, NADPH Oxidase 5; WT, wild‐type.
Molecular Mechanisms Associated With Vascular Dysfunction in Nox5 Mice
Nox5 produces that interacts with nitric oxide to form ONOO−, which binds to tyrosine residues of proteins to form nitrotyrosine. As shown in Figure 7A, vascular nitrotyrosine levels were significantly increased in vessels from Nox5+SM22+ mice. To better evaluate molecular mechanisms contributing to hypercontractility in Nox5+SM22+ mice, VSMCs cultured from mesenteric arteries from WT and Nox5+SM22+ mice were studied. Nox5+SM22+ VSMCs exhibited increased NADPH‐induced ROS production (Figure 7B), effects that were attenuated by verapamil, calmidazolium, and dantrolene (Figure 7C). Stimulation of WT and Nox5+/SM22+ VSMCs with the vasoconstrictor, U46619, induced a rapid increase in ROS production (within 5–15 minutes; Figure 7D and 7E). U46619‐induced ROS production was inhibited in Nox5+SM22+ VSMCs pretreated with verapamil and dantrolene (Figure 7F).
Figure 7.
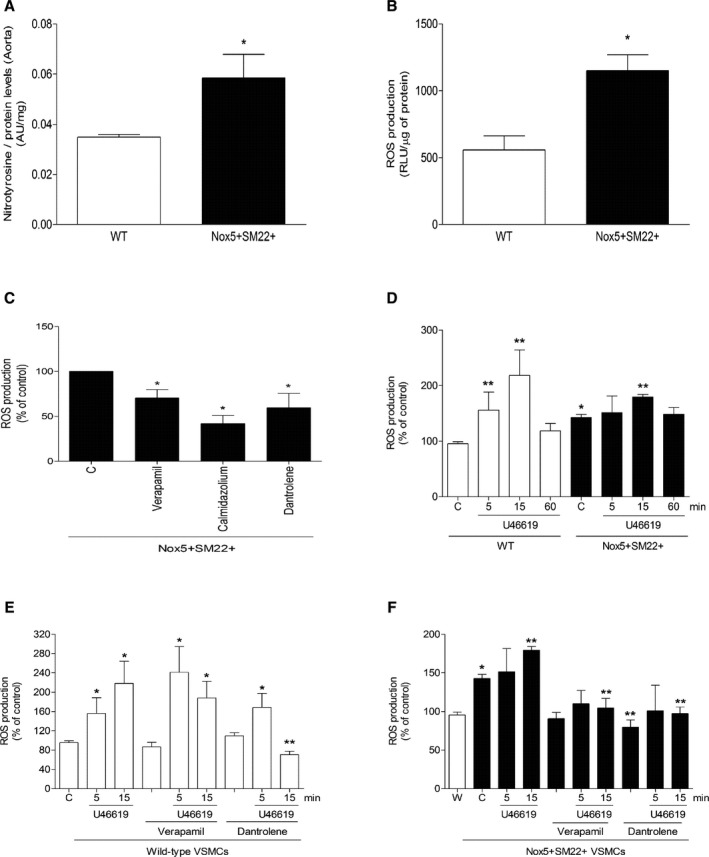
ROS production in arteries and VSMCs from Nox5+ SM22+ mice. A, Nitrotyrosine levels, a marker of ONOO−, in aorta from WT and Nox5+/SM22+ mice measured by ELISA. B, ROS generation in VSMCs from WT and Nox5+ SM22+ mice assessed by lucigenin chemiluminescence. Relative luminescence units (RLU) were corrected by protein concentration of each sample. C, Basal ROS production in VSMCs from mesenteric arteries from Nox5+ SM22+ mice after incubation with verapamil (10 μmol/L), calmidazolium (1 μmol/L), and dantrolene (10 μmol/L). D, U46619‐stimulated ROS generation in VSMCs from mesenteric arteries from WT and Nox5+ SM22+ mice. U46619‐induced ROS production in VSMCs from mesenteric arteries from WT (E) and Nox5+ SM22+ (F) after preincubation with verapamil and dantrolene. Results are mean±SEM of 3 to 6 experiments. *P<0.05 vs control WT; **P<0.05 vs control nonstimulated WT or Nox5+ SM22+. W in (F) represents basal ROS generation in WT VSMCs. Nox5 indicates NADPH Oxidase 5; ONOO−, peroxynitrite; ROS, reactive oxygen species; VSMCs, vascular smooth muscle cells; WT, wild‐type.
Key molecular processes involved in vascular contraction are increased [Ca2+]i and phosphorylation of MYPT1 and MLC20, which trigger actin‐myosin cross‐bridge formation. Ionomycin‐stimulated Ca2+ influx was significantly greater in Nox5+SM22+ VSMCs compared with WT cells (Figure 8A). These processes were associated with increased phosphorylation of the inhibitory regulatory subunit of MLCP (myosin light chain phosphatase), MYPT1 (Figure 8B through 8D). Phosphorylation of MLC20 (Thr18/Ser 19) was greater in Nox5+SM22+ VSMCs compared with WT controls (Figure 8C and 8D).
Figure 8.
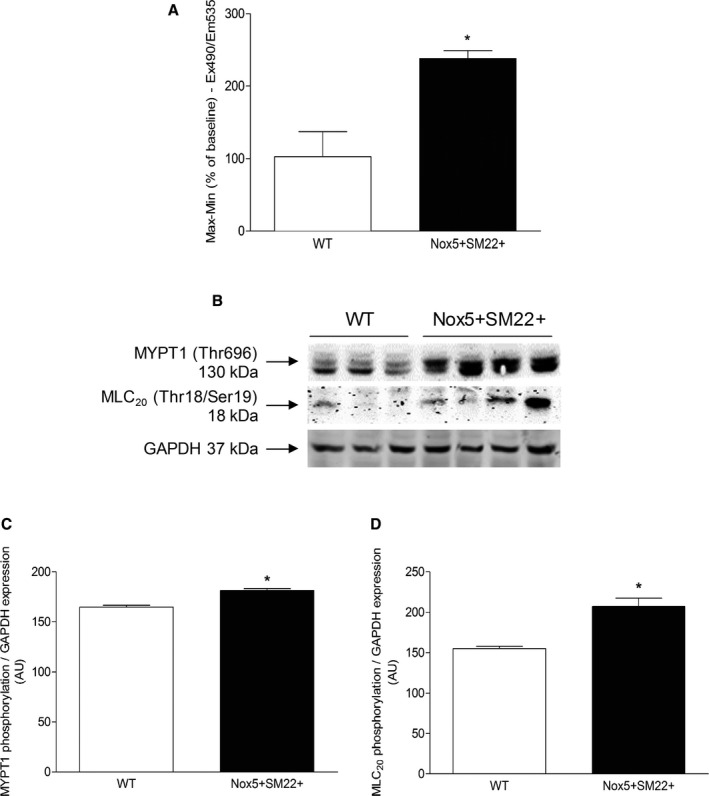
Molecular mechanisms of vascular dysfunction in Nox5+ SM22+ mice. A, Ca2+ influx induced by ionomycin in VSMCs from WT and Nox5+/SM22+ mice measured by live cell microscopy using the fluorescent probe, CAL520‐AM. B, Representative immunoblotting images of phosphorylation of MYPT1 and MLC20, corrected to total GAPDH expression. C, MYPT1 phosphorylation in VSMCs from WT and Nox5+/SM22+ mice. D, MLC20 phosphorylation in VSMCs from WT and Nox5+/SM22+ mice. Results are mean±SEM of 3 to 5 experiments. *P<0.05 vs WT. GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; MLC20, myosin light chain 20; MYPT1, myosin phosphatase target subunit 1; Nox5, NADPH Oxidase 5; VSMCs, vascular smooth muscle cells; WT, wild‐type.
Differential Expression of Vascular Nox Isoforms in Nox5+SM22+ Mice
To evaluate whether human Nox5 influences expression of endogenous Nox isoforms in vessels from Nox5+SM22+ mice, we measured protein content of Nox1, Nox2, and Nox4 in vascular tissue from WT and Nox5+SM22+ mice. As shown in Figure S6, expression of Nox2 and Nox4 was not different between WT and Nox5+SM22+ groups. However, expression of Nox1 was increased in Nox5+SM22+ mice compared with WT counterparts. In heart tissue, we did not observe any differences in mRNA levels of Nox2 (Figure S7A) and Nox4 (Figure S7B), whereas mRNA levels of Nox1 were undected.
Decreased Gut Contraction in Nox5‐Silenced R. prolixus
To further examine the role of Nox5 in in intact system that endogenously expresses functionally active Nox5, we studied R. prolixus. As shown in Figure S8A, exposure of WT R. prolixus to a feeding stimulus induced a significant increase in Nox5 expression. Silencing of Nox5 with dsRNA resulted in a significant reduction in frequency of midgut contraction (peristalsis) following feeding, suggesting an important role for Nox5 in gut muscle contraction (Figure S8B). VAS2870, a Nox1/4 inhibitor, at low (0.02 mmol/L) and high (0.1 mmol/L) concentrations, had no effect on feeding‐stimulated peristaltic contraction (Figure S8C).
Discussion
Since its discovery, the function of Nox5 and the significance of its absolute dependency on intracellular Ca2+ for its activation have been elusive. Here, we provide new insights into the pathophysiological role of Nox5, identifying it as a novel regulator of smooth muscle contraction important in vascular function. Major findings from our study demonstrate that: (1) transgenic mice expressing human Nox5 in VSMCs exhibit systemic and vascular oxidative stress and vascular hypercontractility, responses that are normalized by inhibitors of Ca2+ channels, calmodulin, and ER ryanodine receptors; (2) vascular expression of Nox5 is associated with cardiac oxidative stress and fibrosis; and (3) Nox5 does not influence Ang II pressor responses. These data define Nox5 as a pro‐contractile Nox isoform that regulates vascular contraction through molecular processes involving ROS, Ca2+, calmodulin, and the ER. In support of our findings, we demonstrated that muscle contraction in an invertebrate model that expresses Nox5 endogenously is attenuated when Nox5 is silenced, but not when Nox1 and Nox4 are inhibited. Together, our studies identify a novel function for Ca2+‐sensitive Nox5 in redox regulation of smooth muscle contraction, processes that may have significant implications in vascular (patho)physiology.
Noxs are increasingly being recognized as key sources of ROS in vascular cells, with specific Nox isoforms regulating distinct signaling pathways. For example, whereas Nox1‐ and Nox2‐derived regulate proinflammatory and mitogenic signaling pathways,37, 38 Nox4 generates mainly H2O2, an endothelium‐derived hyperpolarizing factor that controls vasodilation.39 The importance of NADPH oxidase‐derived ROS in the regulation of vascular tone was demonstrated in patients with hereditary deficiency of Nox2, who exhibit increased flow‐mediated vasodilation.40 Despite advances in Nox research, progress in Nox5 biology, especially in the cardiovascular system, in in vivo settings, has been slow because of lack of research tools and experimental models. Nox5 was originally identified in testis, ovaries, spleen, and immature lymphocytes and is heavily expressed in various cancers.17 More recently, Nox5 has been demonstrated in the vascular system. In bovine aortic endothelial cells, Nox5 is required for stromal cell–derived factor‐1α and MAPK (mitogen‐activated protein kinase) signaling, which regulate endothelial migration and angiogenesis.41 In human endothelial cells, Ang II– and ET‐1–induced redox signaling and MAPK activation are Nox5 dependent.25 In adenovirus‐mediated Nox5 overexpression in endothelial cells, phosphorylation of p38 MAPK, JAK2 (Janus kinase 2), JNK (c‐Jun N‐terminal kinase), and ERK1/2 (extracellular signal‐regulated kinases 1 and 2) was increased,12 whereas in VSMCs, Nox5 only increased ERK1/2 phosphorylation.
To investigate the pathophysiological significance of vascular smooth muscle Nox5 in vivo, we generated mice expressing human Nox5 exclusively in smooth muscle cells. These mice exhibited significant oxidative stress, both systemically and in the vasculature and heart, indicating the functional activation of ROS‐generating Nox5 in our model. Although these mice had normal blood pressure, they exhibited significant changes in vascular function as well as some evidence of cardiac changes, particularly fibrosis and cardiomyocyte hypertrophy, suggesting that Nox5‐induced ROS generation in the vasculature impacts cardiac cells, possibly through redox‐sensitive signaling pathways. The most marked phenotype in the Nox5 mice was vascular hypercontractility, which was not exacerbated by Ang II. Potential mechanisms associated with Nox5‐induced vascular changes involve ROS, Ca2+, and calmodulin, because vascular hyper‐reactivity was normalized by N‐acetylcysteine, Ca2+ channel blockers, and calmidozilium. These findings are in line with the fact that Nox5‐derived ROS production is dependent on [Ca2+]i and calmodulin.42 We also showed that ER Ca2+ mobilization may be important because dantrolene, which blocks ryanodine receptor, reduced contraction only in Nox5 mice. Previous studies in coronary arterial myocytes and pulmonary arterial smooth muscle cells demonstrated that ryanodine receptor/Ca2+ regulation involves ER‐associated Nox (2 and 4)‐derived ROS.43, 44 Our findings are supportive of this association, although the exact role of Nox5 in ER function warrants further investigation.
Nox isoforms may regulate one another, either through redox‐dependent processes that influence transcription factors and gene expression or directly through protein‐protein interactions.45, 46 In our study, Nox5 did not influence Nox2 or Nox4 expression, in the vasculature or heart, but it was associated with an increase in vascular Nox1 content. Reasons for this remain unclear. However, Nox1 upregulation did not seem to significantly influence vascular hypercontractility in Nox5 mice because GKT137831 did not inhibit exaggerated contractile responses in Nox5‐expressing mice.
Molecular processes underlying Nox5‐induced vascular dysfunction were further interrogated in VSMCs cultured from WT and Nox5 mice. In Nox5‐expressing cells, oxidative stress, [Ca2+]i and pro‐contractile signaling pathways were increased. In particular, phosphorylation of MYPT1 at Thr‐696 and phosphorylation of MLC20 at Thr18/Ser19 were increased in VSMCs from Nox5 mice. Phosphorylation of MYPT1 causes inhibition of myosin phosphatase, which leads to increased Ca2+ sensitization and increased force of vascular smooth muscle contraction.47 Hence, the increased phosphorylation of MYPTI and MLC20 in the Nox5‐expressing mice may be indicative of increased pro‐contractile signaling, which is in line with our other vascular findings.
In addition to increased contraction, Nox5‐expressing mice exhibited impaired endothelium‐dependent vasorelaxation, processes that were associated with reduced phosphorylation of endothelial nitric oxide synthase and increased vascular ONOO− content. Given that Nox5 was expressed exclusively in vascular smooth muscle in our mouse model, it is unlikely that the endothelial changes are directly attributed to Nox5. However, there may be important cross‐talk between the endothelium and vascular media through Nox5‐derived ROS, where vascular oxidative stress in Nox5 mice may promote endothelial nitric oxide synthase uncoupling, decreased endothelial nitric oxide synthase activation with reduced nitric oxide generation, increased superoxide production, and consequent ONOO− formation, processes that cause endothelial dysfunction.48
In Ang II–infused WT mice, contractile responses were increased and endothelium‐dependent vasorelaxation was impaired. These effects were not further aggravated by Nox5. Reasons for this are unclear, but may relate, at least in part, to the fact that in Nox5 mice vessels are already hypercontractile with endothelial dysfunction in the basal state before Ang II treatment and that further responses may be blunted.
Despite the critical role of Nox5 in the regulation of vascular function, it seemed to be less important in processes that control vascular structure and mechanics, given that vascular wall thickness, stiffness, and distensibility were not influenced by Nox5 in basal conditions, despite increased oxidative stress. When challenged with Ang II, arteries from WT mice exhibited significant remodeling as evidenced by an increase in cross‐sectional area. In Ang II–infused Nox5 mice, remodeling was less marked than WT counterparts. Preservation of vascular structure and mechanical integrity, and modest remodeling induced by Ang II, may explain, at least in part, why Nox5 did not significantly influence blood pressure in basal or Ang II–stimulated conditions.
During evolution, for unknown reasons, the Nox5 gene was lost in rodents, but is present and functionally active in some invertebrates and in higher mammals.17 Arthropods possess Nox5, an ortholog of human Nox5, besides dual oxidase, and some of them also possess Nox4‐art, a gene related to Nox4.31, 49 As such, we further examined the relationship between Nox5 and contraction by studying R. prolixus where midgut contraction is easily studied by videomicroscopy. Feeding induced a significant increase in Nox5 expression in R. prolixus, with associated increase in midgut contraction. However, in Nox5‐silenced R. prolixus, feeding‐stimulated contractile responses were markedly blunted, highlighting the importance of Nox5 in the regulation of muscle contraction. This was confirmed in our studies with VAS2870, a NOX1/4 inhibitor that had no effect on feeding‐stimulated peristaltic contractions. The biological importance of Nox5 in smooth muscle contraction was also shown in Drosophila melanogaster, where depletion of Nox5 (dNox) in the musculature was studied using RNA interference approaches.50 dNox‐RNAi Drosophila exhibited retention of mature eggs, impaired ovarian muscle contractions, and sterility attributed to altered agonist‐induced calcium flux and decreased muscle contraction. The muscles that control Rhodnius gut and Drosophila ovarian muscles are typical of smooth muscle and derive from mesoderm, similar to that in mammals, and hence biological processes controlling contraction may be similar between species. Together these findings strongly support the notion that Nox5 is a pro‐contractile NADPH oxidase isoform, an ancestral role that is conserved during animal evolution.
In conclusion, using a multidisciplinary approach, including Nox5 transgenic mice, cultured VSMCs from Nox5 mice, and Nox5‐expressing arthropod models, we define an important role for Nox5 in the regulation of smooth muscle contraction and vascular function. Nox5 may be a key nexus linking Ca2+ and ROS to the pro‐contractile molecular machinery in VSMCs. Upregulation of the Ca2+‐regulated Nox5‐ROS‐Ca2+ pathway may be especially important in cardiovascular diseases associated with vascular hypercontractility, such as coronary artery disease, vasospastic conditions, hypertension, and cerebral vasospasm.
Sources of Funding
This work was supported by grants from the British Heart Foundation (BHF) (RG/13/7/30099; RE/13/5/30177). Montezano was supported through a Walton Fellowship in Cardiovascular Medicine.
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. Plasma Biochemistry (mmol/L) of WT, Nox5+SM22+, SM22+, and Nox5+ Mice Before and After Treatment With Ang II. *P<0.05 vs nontreated group.
Figure S1. Cardiac H2O2 and MDA levels in Nox5+SM22+ mice. A, H2O2 levels in hearts from WT (open bars) and Nox5+SM22+ (closed bars) mice, before and after Ang II treatment, measured by Amplex Red. B, MDA levels, a marker of lipid peroxidation, in hearts from WT (open bars) and Nox5+SM22+ (closed bars) mice, before and after Ang II treatment, measured by ELISA. Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs WT group; **P<0.05 vs untreated group. Ang II indicates angiotensin II; MDA, malondialdehyde; WT, wild‐type.
Figure S2. Vascular contraction in mesenteric arteries from control mice (WT, SM22+, and Nox5+ mice). Vascular contractility to U46619 assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), and Nox5+ (open rhombus) before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. **P<0.05 vs untreated WT, SM22+, or Nox5+ (open symbols). Ang II inidcates angiotensin II; WT, wild‐type.
Figure S3. Endothelium‐independent relaxation in mesenteric arteries from Nox5+SM22+ and control mice. A, Vascular relaxation to SNP (endothelium‐independent) in arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular relaxation to SNP in arteries from WT (open circle), WT+Ang II (closed circle), Nox5+SM22+ (open square), and Nox5+SM22++Ang II (closed square) mice. C, Vascular relaxation to SNP assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), and Nox5+ (open rhombus) before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. **P<0.05 vs untreated WT, SM22+, or Nox5+ (open symbols). Ang II indicates angiotensin II; SNP, sodium nitroprusside; WT, wild‐type.
Figure S4. Wall‐to‐lumen ratio in resistance arteries Nox5+SM22+ mice. A, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from SM22+ (open triangle) and Nox5+ (open rhombus), before and after Ang II treatment (closed symbols). C, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from WT (open circle) and Nox5+SM22+ (open square), before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs untreated Nox5+SM22+. Ang II indicates angiotensin II; WT, wild‐type.
Figure S5. Vascular stiffness in Nox5+/SM22+ mice. A, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from SM22+ (open triangle) and Nox5+ (open rhombus), before and after Ang II treatment (closed symbols). C, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from WT (open circle) and Nox5+SM22+ (open square), before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. Ang II indicates angiotensin II; WT, wild‐type.
Figure S6. Nox1, Nox2, and Nox4 regulation in arteries from Nox5+SM22+ mice. A, Nox1 protein expression in mesenteric arteries from WT and Nox5+SM22+ mice assessed by immunoblotting. B, Nox2 and (C) Nox4 protein expression in mesenteric arteries from WT and Nox5+SM22+ mice assessed by immunoblotting. Results are mean±SEM of 3 experiments. *P<0.05 vs WT. Ang II indicates angiotensin II; WT, wild‐type.
Figure S7. Cardiac mRNA levels of Nox isoforms in Nox5+SM22+ and control mice. Gene levels of Nox2 (A) and Nox4 (B) in Nox5+SM22+ (closed bars) and control groups (open bars), before and after Ang II treatment. Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs WT group; **P<0.05 vs untreated group. Ang II indicates angiotensin II; WT, wild‐type.
Figure S8. Nox5 is important in midgut contraction of Rhodnius prolixus. A, Nox5 gene expression in female R. prolixus midgets before and after a bloody meal assessed by video fluorescence microscopy. Postfeeding midgut contraction (peristalsis) in female R. prolixus after Nox5 downregulation (B) and Nox1/Nox4 inhibition (VAS2870—VAS). C, Results are mean±SEM of 5 experiments. *P<0.05 vs unfed insects (A) or dsMal (B).
Acknowledgments
We thank Wendy Beattie, Laura Haddow, Jacqueline Thomson, and Carol Jenkins (University of Glasgow) for technical assistance.
(J Am Heart Assoc. 2018;7:e009388 DOI: 10.1161/JAHA.118.009388.)
The data were presented and published as an abstract at the Council on Hypertension meeting of the American Heart Association, September 14 to 17, 2017, in San Francisco, CA.
References
- 1. Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep. 2010;15:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang H, Hartnett ME. Roles of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in angiogenesis: isoform‐specific effects. Antioxidants (Basel). 2017;6:E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal. 2014;20:164–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harvey AP, Montezano AC, Hood KY, Lopes RA, Rios F, Ceravolo G, Graham D, Touyz RM. Vascular dysfunction and fibrosis in stroke‐prone spontaneously hypertensive rats: the aldosterone‐mineralocorticoid receptor‐Nox1 axis. Life Sci. 2017;179:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El‐Osta A, Andrews KL, Chin‐Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit‐Dahm KA. Nadph oxidase 1 plays a key role in diabetes mellitus‐accelerated atherosclerosis. Circulation. 2013;127:1888–1902. [DOI] [PubMed] [Google Scholar]
- 7. Hood KY, Montezano AC, Harvey AP, Nilsen M, MacLean MR, Touyz RM. Nicotinamide adenine dinucleotide phosphate oxidase‐mediated redox signaling and vascular remodeling by 16alpha‐hydroxyestrone in human pulmonary artery cells: implications in pulmonary arterial hypertension. Hypertension. 2016;68:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen F, Yin C, Dimitropoulou C, Fulton DJ. Cloning, characteristics, and functional analysis of rabbit NADPH oxidase 5. Front Physiol. 2016;7:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011;11:27–35. [DOI] [PubMed] [Google Scholar]
- 10. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110:1217–1225. [DOI] [PubMed] [Google Scholar]
- 11. Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, Burns KD, Cooper ME, Jandeleit‐Dahm K, Page P, Szyndralewiez C, Heitz F, Hebert RL, Touyz RM. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci. 2013;124:191–202. [DOI] [PubMed] [Google Scholar]
- 12. BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. Nox5 variants are functionally active in endothelial cells. Free Radic Biol Med. 2007;42:446–459. [DOI] [PubMed] [Google Scholar]
- 13. Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. [DOI] [PubMed] [Google Scholar]
- 14. Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, Barman SA, Venema RC, Stepp DW, Rudic RD, Fulton DJ. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol. 2012;302:H1919–H1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montezano AC, Burger D, Ceravolo GS, Yusuf H, Montero M, Touyz RM. Novel Nox homologues in the vasculature: focusing on Nox4 and Nox5. Clin Sci. 2011;120:131–141. [DOI] [PubMed] [Google Scholar]
- 16. Magnani F, Nenci S, Millana Fananas E, Ceccon M, Romero E, Fraaije MW, Mattevi A. Crystal structures and atomic model of NADPH oxidase. Proc Natl Acad Sci U S A. 2017;114:6764–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)‐activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem. 2001;276:37594–37601. [DOI] [PubMed] [Google Scholar]
- 18. Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (Nox5). J Biol Chem. 2004;279:18583–18591. [DOI] [PubMed] [Google Scholar]
- 19. Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. Calcium sensitization via phosphorylation. J Biol Chem. 2007;282:6494–6507. [DOI] [PubMed] [Google Scholar]
- 20. Pandey D, Gratton JP, Rafikov R, Black SM, Fulton DJ. Calcium/calmodulin‐dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol Pharmacol. 2011;80:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Chen F, Le B, Stepp DW, Fulton DJ. Impact of Nox5 polymorphisms on basal and stimulus‐dependent ROS generation. PLoS One. 2014;9:e100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ganitkevich V, Hasse V, Pfitzer G. Ca2+‐dependent and Ca2+‐independent regulation of smooth muscle contraction. J Muscle Res Cell Motil. 2002;23:47–52. [DOI] [PubMed] [Google Scholar]
- 23. Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. 2015;7:a006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jay DB, Papaharalambus CA, Seidel‐Rogol B, Dikalova AE, Lassegue B, Griendling KK. Nox5 mediates PDGF‐induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. 2008;45:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin‐1 is mediated via calcium/calmodulin‐dependent, rac‐1‐independent pathways in human endothelial cells. Circ Res. 2010;106:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium‐dependent Nox5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52:1803–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guzik B, Sagan A, Ludew D, Mrowiecki W, Chwala M, Bujak‐Gizycka B, Filip G, Grudzien G, Kapelak B, Zmudka K, Mrowiecki T, Sadowski J, Korbut R, Guzik TJ. Mechanisms of oxidative stress in human aortic aneurysms—association with clinical risk factors for atherosclerosis and disease severity. Int J Cardiol. 2013;168:2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holterman CE, Thibodeau JF, Towaij C, Gutsol A, Montezano AC, Parks RJ, Cooper ME, Touyz RM, Kennedy CR. Nephropathy and elevated BP in mice with podocyte‐specific NADPH oxidase 5 expression. J Am Soc Nephrol. 2014;25:784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jha JC, Banal C, Okabe J, Gray SP, Hettige T, Chow BSM, Thallas‐Bonke V, De Vos L, Holterman CE, Coughlan MT, Power DA, Skene A, Ekinci EI, Cooper ME, Touyz RM, Kennedy CR, Jandeleit‐Dahm K. NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes. 2017;66:2691–2703. [DOI] [PubMed] [Google Scholar]
- 30. Kraja AT, Cook JP, Warren HR, Surendran P, Liu C, Evangelou E, Manning AK, Grarup N, Drenos F, Sim X, Smith AV, Amin N, Blakemore AIF, Bork‐Jensen J, Brandslund I, Farmaki AE, Fava C, Ferreira T, Herzig KH, Giri A, Giulianini F, Grove ML, Guo X, Harris SE, Have CT, Havulinna AS, Zhang H, Jorgensen ME, Karajamaki A, Kooperberg C, Linneberg A, Little L, Liu Y, Bonnycastle LL, Lu Y, Magi R, Mahajan A, Malerba G, Marioni RE, Mei H, Menni C, Morrison AC, Padmanabhan S, Palmas W, Poveda A, Rauramaa R, Rayner NW, Riaz M, Rice K, Richard MA, Smith JA, Southam L, Stancakova A, Stirrups KE, Tragante V, Tuomi T, Tzoulaki I, Varga TV, Weiss S, Yiorkas AM, Young R, Zhang W, Barnes MR, Cabrera CP, Gao H, Boehnke M, Boerwinkle E, Chambers JC, Connell JM, Christensen CK, de Boer RA, Deary IJ, Dedoussis G, Deloukas P, Dominiczak AF, Dorr M, Joehanes R, Edwards TL, Esko T, Fornage M, Franceschini N, Franks PW, Gambaro G, Groop L, Hallmans G, Hansen T, Hayward C, Heikki O, Ingelsson E, Tuomilehto J, Jarvelin MR, Kardia SLR, Karpe F, Kooner JS, Lakka TA, Langenberg C, Lind L, Loos RJF, Laakso M, McCarthy MI, Melander O, Mohlke KL, Morris AP, Palmer CNA, Pedersen O, Polasek O, Poulter NR, Province MA, Psaty BM, Ridker PM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sever PJ, Skaaby T, Stafford JM, Starr JM, van der Harst P, van der Meer P; Understanding Society Scientific G , van Duijn CM, Vergnaud AC, Gudnason V, Wareham NJ, Wilson JG, Willer CJ, Witte DR, Zeggini E, Saleheen D, Butterworth AS, Danesh J, Asselbergs FW, Wain LV, Ehret GB, Chasman DI, Caulfield MJ, Elliott P, Lindgren CM, Levy D, Newton‐Cheh C, Munroe PB, Howson JMM; CHARGE EXOME BP, CHD Exome+, Exome BP, GoT2D:T2DGenes Consortia, The UK Biobank Cardio‐Metabolic Traits Consortium Blood Pressure Working Group. New blood pressure‐associated loci identified in meta‐analyses of 475 000 individuals. Circ Cardiovasc Genet. 2017;10:e001778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gandara ACP, Torres A, Bahia AC, Oliveira PL, Schama R. Evolutionary origin and function of NOX4‐art, an arthropod specific NADPH oxidase. BMC Evol Biol. 2017;17:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Callera GE, Antunes TT, He Y, Montezano AC, Yogi A, Savoia C, Touyz RM. c‐Src inhibition improves cardiovascular function but not remodeling or fibrosis in angiotensin II‐induced hypertension. Hypertension. 2016;68:1179–1190. [DOI] [PubMed] [Google Scholar]
- 34. Schiffrin EL, Park JB, Intengan HD, Touyz RM. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 2000;101:1653–1659. [DOI] [PubMed] [Google Scholar]
- 35. Montezano AC, Lopes RA, Neves KB, Rios F, Touyz RM. Isolation and culture of vascular smooth muscle cells from small and large vessels. Methods Mol Biol. 2017;1527:349–354. [DOI] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 37. Yogi A, Mercure C, Touyz J, Callera GE, Montezano AC, Aranha AB, Tostes RC, Reudelhuber T, Touyz RM. Renal redox‐sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II‐dependent chronic hypertension. Hypertension. 2008;51:500–506. [DOI] [PubMed] [Google Scholar]
- 38. Touyz RM, Mercure C, He Y, Javeshghani D, Yao G, Callera GE, Yogi A, Lochard N, Reudelhuber TL. Angiotensin II‐dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox‐containing NADPH oxidase. Hypertension. 2005;45:530–537. [DOI] [PubMed] [Google Scholar]
- 39. Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. [DOI] [PubMed] [Google Scholar]
- 40. Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S, Loffredo L. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120:1616–1622. [DOI] [PubMed] [Google Scholar]
- 41. Pi X, Xie L, Portbury AL, Kumar S, Lockyer P, Li X, Patterson C. Nadph oxidase‐generated reactive oxygen species are required for stromal cell‐derived factor‐1alpha‐stimulated angiogenesis. Arterioscler Thromb Vasc Biol. 2014;34:2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tirone F, Cox JA. Nadph oxidase 5 (Nox5) interacts with and is regulated by calmodulin. FEBS Lett. 2007;581:1202–1208. [DOI] [PubMed] [Google Scholar]
- 43. Zhang F, Jin S, Yi F, Xia M, Dewey WL, Li PL. Local production of O2‐ by NAD(P)H oxidase in the sarcoplasmic reticulum of coronary arterial myocytes: cADPR‐mediated Ca2+ regulation. Cell Signal. 2008;20:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee S, Paudel O, Jiang Y, Yang XR, Sham JS. CD38 mediates angiotensin II‐induced intracellular Ca(2+) release in rat pulmonary arterial smooth muscle cells. Am J Respir Cell Mol Biol. 2015;52:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jackson HM, Kawahara T, Nisimoto Y, Smith SM, Lambeth JD. Nox4 B‐loop creates an interface between the transmembrane and dehydrogenase domains. J Biol Chem. 2010;285:10281–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawahara T, Jackson HM, Smith SM, Simpson PD, Lambeth JD. Nox5 forms a functional oligomer mediated by self‐association of its dehydrogenase domain. Biochemistry. 2011;50:2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khromov A, Choudhury N, Stevenson AS, Somlyo AV, Eto M. Phosphorylation‐dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J Biol Chem. 2009;284:21569–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Montezano AC, Touyz RM. Reactive oxygen species and endothelial function—role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmac Toxicol. 2012;110:87–94. [DOI] [PubMed] [Google Scholar]
- 49. Kawahara T, Lambeth JD. Molecular evolution of Phox‐related regulatory subunits for NADPH oxidase enzymes. BMC Evol Biol. 2007;7:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ritsick DR, Edens WA, Finnerty V, Lambeth JD. Nox regulation of smooth muscle contraction. Free Radic Biol Med. 2007;43:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Plasma Biochemistry (mmol/L) of WT, Nox5+SM22+, SM22+, and Nox5+ Mice Before and After Treatment With Ang II. *P<0.05 vs nontreated group.
Figure S1. Cardiac H2O2 and MDA levels in Nox5+SM22+ mice. A, H2O2 levels in hearts from WT (open bars) and Nox5+SM22+ (closed bars) mice, before and after Ang II treatment, measured by Amplex Red. B, MDA levels, a marker of lipid peroxidation, in hearts from WT (open bars) and Nox5+SM22+ (closed bars) mice, before and after Ang II treatment, measured by ELISA. Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs WT group; **P<0.05 vs untreated group. Ang II indicates angiotensin II; MDA, malondialdehyde; WT, wild‐type.
Figure S2. Vascular contraction in mesenteric arteries from control mice (WT, SM22+, and Nox5+ mice). Vascular contractility to U46619 assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), and Nox5+ (open rhombus) before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. **P<0.05 vs untreated WT, SM22+, or Nox5+ (open symbols). Ang II inidcates angiotensin II; WT, wild‐type.
Figure S3. Endothelium‐independent relaxation in mesenteric arteries from Nox5+SM22+ and control mice. A, Vascular relaxation to SNP (endothelium‐independent) in arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular relaxation to SNP in arteries from WT (open circle), WT+Ang II (closed circle), Nox5+SM22+ (open square), and Nox5+SM22++Ang II (closed square) mice. C, Vascular relaxation to SNP assessed by wire myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), and Nox5+ (open rhombus) before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. **P<0.05 vs untreated WT, SM22+, or Nox5+ (open symbols). Ang II indicates angiotensin II; SNP, sodium nitroprusside; WT, wild‐type.
Figure S4. Wall‐to‐lumen ratio in resistance arteries Nox5+SM22+ mice. A, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from SM22+ (open triangle) and Nox5+ (open rhombus), before and after Ang II treatment (closed symbols). C, Vascular wall‐to‐lumen ratio assessed by pressure myography in mesenteric arteries from WT (open circle) and Nox5+SM22+ (open square), before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs untreated Nox5+SM22+. Ang II indicates angiotensin II; WT, wild‐type.
Figure S5. Vascular stiffness in Nox5+/SM22+ mice. A, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from WT (open circle), SM22+ (open triangle), Nox5+ (open rhombus), and Nox5+SM22+ (open square). B, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from SM22+ (open triangle) and Nox5+ (open rhombus), before and after Ang II treatment (closed symbols). C, Vascular stiffness (strain vs stress curves) assessed by pressure myography in mesenteric arteries from WT (open circle) and Nox5+SM22+ (open square), before and after Ang II treatment (closed symbols). Results are mean±SEM of 5 to 8 mice/group. Ang II indicates angiotensin II; WT, wild‐type.
Figure S6. Nox1, Nox2, and Nox4 regulation in arteries from Nox5+SM22+ mice. A, Nox1 protein expression in mesenteric arteries from WT and Nox5+SM22+ mice assessed by immunoblotting. B, Nox2 and (C) Nox4 protein expression in mesenteric arteries from WT and Nox5+SM22+ mice assessed by immunoblotting. Results are mean±SEM of 3 experiments. *P<0.05 vs WT. Ang II indicates angiotensin II; WT, wild‐type.
Figure S7. Cardiac mRNA levels of Nox isoforms in Nox5+SM22+ and control mice. Gene levels of Nox2 (A) and Nox4 (B) in Nox5+SM22+ (closed bars) and control groups (open bars), before and after Ang II treatment. Results are mean±SEM of 5 to 8 mice/group. *P<0.05 vs WT group; **P<0.05 vs untreated group. Ang II indicates angiotensin II; WT, wild‐type.
Figure S8. Nox5 is important in midgut contraction of Rhodnius prolixus. A, Nox5 gene expression in female R. prolixus midgets before and after a bloody meal assessed by video fluorescence microscopy. Postfeeding midgut contraction (peristalsis) in female R. prolixus after Nox5 downregulation (B) and Nox1/Nox4 inhibition (VAS2870—VAS). C, Results are mean±SEM of 5 experiments. *P<0.05 vs unfed insects (A) or dsMal (B).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Additional methods can be found in Data S1.