

Abstract
A new domino reaction sequence for the construction of 2-pyridone structures is reported. The reaction sequence begins with diacetyldiketopiperazine and proceeds via aldol condensation, alkene isomerization, and intramolecular Diels–Alder cycloaddition. The intermediate [2.2.2]diazabicycloalkene cycloadducts can be isolated or can engage in a base- accelerated extrusion of one lactam bridge to provide the 2-pyridone cycloreversion products. The operation leading to pyridone products can occur in one reaction vessel and proceeds at convenient temperatures.
Graphical Abstract
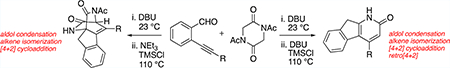
Cycloaddition/cycloreversion sequences have held the attention of chemists for the synthesis of a variety of both carbocyclic and heterocyclic aromatic compounds.1 Pyridines are one of the most prevalent aromatic heterocycles found in bioactive molecules and functional materials.2 Cascade pericyclic processes3 such as merged cycloaddition/ cycloreversion strategies,4 particularly those employing 1,2,3- and 1,2,4-triazines,5 are a valuable synthetic complement to traditional and more contemporary6 condensation methods.7 In comparison, merged cycloaddition/cycloreversion approaches for the direct synthesis of 2-pyridones are not common;8 however, Hoornaert and co-workers validated the possibility of this approach using cycloaddition of chlorinated pyrazinone intermediates.9 The cycloreversion of the intermediate [2.2.2]diazabicycloadducts derived from these chlorinated pyrazinone precursors has two primary limitations: (1) extrusion of cyanide or cyanate derivatives requires high temperatures10 (at or above 140 °C), and (2) extrusion is often nonselective and gives mixtures of both pyridine and pyridone products.9
Our preliminary efforts in this area sought to combine basepromoted aldol chemistry with a pyrazinone cycloaddition/ cycloreversion sequence in order to access highly substituted 2- pyridone structures.11 While these efforts (Scheme 1, eq 1) led to key insights, the overall success of the effort was measured. The length of the synthetic operation was not ideal and was unlikely to be adopted by the community except possibly in niche contexts. We now report a more expedient synthetic sequence for the conversion of readily available diketopiper-azine starting materials into 2-pyridone products that can be accomplished in one reaction vessel at conveniently accessible temperatures. The pyridone products obtained in this preliminary publication are embedded within a tricyclic 1-azafluorene scaffold. Such azafluorene structures and related derivatives are valuable bioactive scaffolds12 and have desirable photophysical properties13 for material applications. Additionally, only a limited number of methods have been exploited for the synthesis of 1-azafluorene structures.14,15
Scheme 1.

Synthesis of 2-Pyridone from Diketopiperazine Precursors
We planned the synthetic sequence to begin with bisacetoxyglycine anhydride (1) (Scheme 1, eq 2), which is commercially available in bench-scale quantities or can be prepared in one step (99% yield) from the corresponding cyclo-gly gly diketopiperazine (DKP).16 The availability of DKP 1 stands in contrast to the first-generation route (eq 1) where the requisite DKP lactim methyl ether starting material required four steps to prepare. Additionally, the first-generation DKP starting material needed a strong inorganic base (LiHMDS) to promote enolization. However, because bisacetoxy-DKP 1 contains an imide functional group, enolization and aldol addition are more easily accomplished. Mild bases including carbonate or amines have proven competent in this regard, and due to facile intramolecular N → O acyl transfer, elimination to condensation products is observed without effort.17
In order to engage in the desired cycloaddition process, the aldol condensation product 2 needs to undergo isomerization to the pyrazinone intermediate. Because 2 does not contain the lactim ether function, the requisite isomerization is more challenging as compared to the first-generation chemistry. Precedent for an analogous isomerization of DKP alkylidene substrates resembling 2 is restricted to a proline-derived substrate that can be isomerized only under strongly acidic conditions (refluxing HBr18 or AcCl19). We hoped to realize this isomerization under milder conditions by generating a transient lactim ether 3, which we anticipated would enable alkene isomerization to 4 and [4 + 2] cycloaddition. We anticipated that silyl-transfer reagents would be effective to initiate this isomerization and subsequent cycloaddition.
The most significant limitation of the first-generation chemistry was a direct result of the high activation barrier for cycloreversion. In order to achieve cycloreversion to the pyridone, in most cases it was necessary to activate one lactam bridge as the derived imide. Cycloreversion was performed on this intermediate using microwave heating (1 h, 300 W) at maximum temperatures near 200 °C. In this way, pyridone products could reliably be prepared from cycloadducts by a three-step sequence (lactim ether deprotection, activation, thermolysis). We predicted that this three-step sequence could be avoided with cycloadducts derived from 1 because these cycloadducts (e.g., 5) contain an imide lactam bridge already primed for extrusion.
With the project framework in place, the aldol condensation was explored with 1 and 2-ethynylbenzaldehyde (7) (Scheme 2). The desired reaction was easily accomplished, and we found Cs2CO3 was the optimal base for this transformation.20 Performing the reaction in a 1:1 mixture of DMF and toluene permitted direct precipitation of the desired condensation product 8 from the reaction medium in good yield without the need for additional purification.
Scheme 2.

Investigation of Domino Sequence
Alkylidene DKP 8 served as our substrate to evaluate the isomerization/Diels-Alder sequence, the results of which are summarized in Scheme 2. In our first attempt, we selected N,O-bis(trimethylsilyl)acetamide (BSA) as a silyl-transfer reagent and DBU as base (entry 1). Although no reaction occurred at ambient temperatures, heating at 120 °C led to consumption of starting material. We were slightly surprised that the isolated product was not the derived cycloadduct 9, but rather exclusive formation of the cycloreversion product, tricyclic pyridone 10, was observed. The unpurified reaction mixture was relatively pure and contained only 10 and trace impurities. In order to better understand the reaction, several other parameters were explored. Replication of the conditions (BSA and DBU) in refluxing toluene (entry 2) also effected the merged cycloaddition/cycloreversion sequence, and 10 was similarly produced from the reaction. A lower temperature (85 °C, entry 3) led to isolation of only cycloadduct 9; none of the cycloreversion product was obtained at this temperature. When Cs2CO3 was used as base (entry 4), only unidentified decomposition products were obtained. However, when a weaker amine base was used (NEt3, entry 5) with BSA at 120 °C in DMF, the cycloadduct 9 was cleanly produced without any cycloreversion product. When the reaction was attempted with only BSA (and no base), starting material was returned without change (entry 6). Use of a substoichiometric amount of DBU (0.2 equiv, entry 7) led to a mixture of both cycloadduct 9 and pyridone 10. Overall, entries 1–7 suggest that a strong organic base (DBU) is effective for promoting the cycloreversion at temperatures significantly below that required in the first-generation route. Entry 7 suggests that a full equivalent of DBU is needed to promote complete cycloreversion. This result is consistent with product inhibition, where the more acidic pyridone produced on cycloreversion serves to buffer the basicity of DBU.21
Use of trimethylsilyl chloride (TMSCl) as the silyl-transfer reagent led to superior results for these reactions. Products from reactions with TMSCl were cleaner, higher yielding, and did not contain acetamide (from BSA) as a byproduct. Selection of the appropriate base, either NEt3 (entry 8) or DBU (entry 9) with TMSCl at 110 °C, permitted selective formation of either cycloadduct 9 or pyridone 10 (99%, or 31% isolated yields, respectively).
In order to better comprehend the cycloreversion operation to convert cycloadduct 9 to the tricyclic pyridone 10, the reaction was explored independent from other chemical transformations such as the isomerization and cycloaddition steps. Simple heating (without base additives) at temperatures near 200 °C was required to promote some decomposition of 9. After 1 h at 200 °C, the resulting mixture contained several products, including unreacted starting cycloadduct 9 and desired pyridone 10 as well as an isomeric pyridone resulting from unselective extrusion of either lactam bridging function. Significant rate enhancement for cycloreversion was observed in the presence of strong organic bases. Similar to the reaction sequence highlighted previously, we found that conversion of 9 to pyridone 10 was efficiently promoted at lower temperatures (110 °C, refluxing toluene) in the presence of DBU (Scheme 3). We propose that DBU serves to increase the equilibrium concentration of the lactam conjugate base 11, thereby lowering the reaction threshold for cycloreversion and permitting conversion to the derived conjugate base of 10, which has greater aromatic character.
Scheme 3.

Base-Promoted Cycloreversion to 2-Pyridone
Although the unpurified product mixture was reasonably pure (ca. 85–90% as judged by 1H NMR) and mass recovery was consistently near unity, the reported isolated yield for the formation of 10 is poor (27%). We observed significant yield loss during chromatographic purification, a feature that we attribute partly to the undesirable physical properties of 10, which is poorly soluble in most solvents and difficult to manipulate. In our hands, recrystallization did not remove the trace impurities.
Anion acceleration is well documented for certain pericyclic processes,22 most notably the oxy-Cope reaction, which is reported to increase reaction rate by up to 1017 over the neutral sigmatropic process.23 Due to the electrophilic nature of the imide bridge in substrate 9, a survey of inorganic bases (such as NaH) with differently coordinating counterions (e.g., Li, Na, K) was problematic and caused unwanted nucleophilic and enolate-based decomposition. However, use of stronger organic bases,24 specifically methyl 1,5,7-triazabicyclo[4.4.0]- decene (12) and phosphazene25 base BTTP (13), were effective and permitted the cycloreversion to occur at 80 °C.26 Although the reaction threshold is lower with these stronger bases, the yield of 10 was diminished in these cases, and with 13, separation of the desired pyridone from the phosphazene base further complicated purification efforts. As such, because DBU promoted the desired cycloreversion at temperatures that are operationally easy to access (refluxing toluene), and also because DBU is both inexpensive and widely available in most laboratory environments, we chose to move forward with this base.
Armed with a greater experimental understanding of each step of the synthetic sequence, we sought to expand the reaction scope and determine conditions that would permit a “one-pot” synthesis of tricyclic pyridone products from the DKP starting material 1. To this end, we found that addition of DBU (1 equiv) to a mixture of 1 and alkynyl benzaldehyde 7 or 14–18 in DMF resulted in aldol condensation. When the consumption of starting materials was observed by TLC, the reaction was brought to reflux with additional DBU and TMSCl to promote the cycloaddition/cycloreversion steps (Scheme 4). In this way, tricyclic pyridone products 10 and 19–21 could be obtained without isolation of intermediate products (method A). These reaction conditions were not effective for all substrates. In two cases, a two-step procedure (method B), which included isolation of the intermediate cycloadduct 25 or 26 and subsequent base induced cycloreversion, was desirable for the formation of pyridone products 22 and 23. A few substrates warrant additional comment. Synthesis of 10 by the “one pot” method A provided material of higher purity as compared to material obtained by the stepwise sequence. As such, the need for chromatographic purification was obviated. Pyridone 10 could also be converted to the derived chloropyridine 24 (POCl3, 87% yield), which was soluble in nonpolar solvents and easily purified by chromatography. The alkynylbenzaldehyde substrate bearing a tert-butyl substituent at R1 was very effective in both the cycloaddition and cycloreversion operations leading to product 20 in 81% yield (method A). This result suggests that the cycloaddition step is not particularly sensitive to the steric environment at the propargylic position. Intermediate cycloadduct 25, which has aryl substitution at R1, was recalcitrant and slow to participate in cycloreversion, requiring heating for 48 h at 110 °C to obtain full conversion. In this specific example, we found that thermolysis at higher temperatures (200 °C, 1 h, microwave heating) afforded the desired product 22 more cleanly, albeit in low yield. At this point, we do not have a cogent explanation for the divergent reactivity and whether this behavior is shared among other sp2 hybridized substituents. Exclusive of 10 (which was uniquely poorly soluble), pyridone products were isolated in pure form following normal chromatographic purification.
Scheme 4.

“One Pot” Conditions and Reaction Scope
In summary, an efficient method has been developed for the construction of 2-pyridone alkaloids from diketopiperazine precursors through a domino reaction sequence featuring aldol condensation, alkene isomerization, cycloaddition, and cycloreversion. Intermediate aldol condensation or cycloaddition products can be isolated if desired, or in most cases, the synthetic sequence can be accomplished in one reaction vessel. Central to the reaction development was the discovery of a base-accelerated cycloreversion process, which permitted the final step to occur at temperatures that are operationally easy to achieve with conventional heating and common solvents. The multicomponent reaction sequence described in this paper is a fruitful area for continued development that combines anionic, base-promoted chemistry with pericyclic processes.
Supplementary Material
■ ACKNOWLEDGMENTS
The authors acknowledge support from the National Institutes of Health (R15 GM107702 to J.R.S.).
Footnotes
Notes
The authors declare no competing financial interest.
■ ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.or-glett.8b02145.
■ REFERENCES
- (1).(a) Rickborn B The retro–Diels–Alder reaction. Part I. C-C Dienophiles. Org. React. (N. Y.) 1998, 52, 1–393. [Google Scholar]; (b) Rickborn B The retro–Diels–Alder reaction. Part II. Dienophiles with one or more heteroatom. Org. React. (N. Y.) 1998, 53, 223–630. [Google Scholar]; (c) Kotha S; Banerjee S RSC Adv 2013, 3, 7642–7666. [Google Scholar]
- (2).Yet L Privileged Structures in Drug Discovery: Medicinal Chemistry and Synthesis; Wiley & Sons: New York, 2018. [Google Scholar]
- (3).(a) Allais C; Grassot JM; Rodriguez J; Constantieux T Chem. Rev 2014, 114, 10829–10868. [DOI] [PubMed] [Google Scholar]; (b) Hamzik PJ; Goutierre AS; Sakai T; Danheiser RL J. Org. Chem 2017, 82, 12975–12991. [DOI] [PubMed] [Google Scholar]; (c) Robinson JM; Sakai T; Okano K; Kitawaki T; Danheiser RL J. Am. Chem. Soc 2010, 132, 11039–11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Foster RAA; Willis MC Chem. Soc. Rev 2013, 42, 63–76. [DOI] [PubMed] [Google Scholar]; (b) van der Plas HC InAdvances in Heterocyclic Chemistry; Katritzky AR, Ed.; Elsevier Academic Press: San Diego, 2003; Vol. 74, pp 31–71. [Google Scholar]
- (5).(a) Anderson ED; Boger DL J. Am. Chem. Soc 2011, 133, 12285–12292. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Anderson ED; Boger DL Org. Lett 2011, 13, 2492–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boger DL Chem. Rev 1986, 86, 781–793. [Google Scholar]; (d) Boger DL; Panek JS; Meier MM J. Org. Chem 1982, 47, 895–897. [Google Scholar]; (e) Raw SA; Taylor RJK Recent Advances in the Chemistry of 1,2,4-Triazines In Advances in Heterocylic Chemistry; Katritzky AR, Ed.; Elsevier Academic Press: San Diego, 2010; Vol. 100, pp 75–100. [Google Scholar]; (f) Duret G; Le Fouler V; Bisseret P; Bizet V; Blanchard N Eur. J. Org. Chem 2017, 2017, 6816–6830. [Google Scholar]
- (6).(a) Selected contemporary condensation strategies for the synthesis of pyridine derivatives: Zhao MN; Ren ZH; Yu L; Wang YY; Guan ZH Org. Lett 2016, 18, 1194–1197. [DOI] [PubMed] [Google Scholar]; (b) He Z; Dobrovolsky D; Trinchera P; Yudin AK Org. Lett 2013, 15, 334– 337. [DOI] [PubMed] [Google Scholar]; (c) Chen MZ; Micalizio GC J. Am. Chem. Soc 2012, 134, 1352–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gati W; Rammah MM; Rammah MB; Couty F; Evano GJ Am. Chem. Soc 2012, 134, 9078–9081. [DOI] [PubMed] [Google Scholar]; (e) Movassaghi M; Hill MD; Ahmad OK J. Am. Chem. Soc 2007, 129, 10096. [DOI] [PubMed] [Google Scholar]
- (7).(a) Hill MD Chem. - Eur. J 2010, 16, 12052–12062. [DOI] [PubMed] [Google Scholar]; (b) Henry GD Tetrahedron 2004, 60, 6043–6061. [Google Scholar]
- (8).(a) Reviews on the synthesis of 2-pyridones: Varela JA; Saa C Chem. Rev 2003, 103, 3787–3801. [DOI] [PubMed] [Google Scholar]; (b) Comprehensive Heterocyclic Chemistry III; Katritzky AR, Ramsden CA, Scriven EFV, Taylor RJK, Eds.; Elsevier: Amsterdam, 2008; Vol. 7, pp 1–336. [Google Scholar]; (c) Tieckelmann H Pyridinols and Pyridones In Heterocyclic Compounds; Abranovitch RA, Ed.; Wiley and Sons: New York, 1974; Vol. 14, pp 597–1180. [Google Scholar]; (d) Torres M; Gil S; Parra M Curr. Org. Chem 2005, 9, 1757–1779. [Google Scholar]
- (9).(a) Tutonda M; Vanderzande D; Vekemans J; Toppet S; Hoornaert G Tetrahedron Lett 1986, 27, 2509–2512. [Google Scholar]; (b) Tutonda M; Vanderzande D; Hendrickx M; Hoornaert G Tetrahedron 1990, 46, 5715–5732. [Google Scholar]; (c) Vandenberghe SM; Buysens KJ; Meerpoel L; Loosen PK; Toppet SM; Hoornaert GJ J. Org. Chem 1996, 61, 304–308. [Google Scholar]
- (10).Chlorinated pyrazinones have also been engaged with alkynylboronates to form pyridone boronic esters at 180 °C: Delaney PM; Huang J; Macdonald SJF; Harrity JPA Org. Lett 2008, 10, 781–783. [DOI] [PubMed] [Google Scholar]
- (11).(a) Margrey KM; Hazzard AD; Scheerer JR Org. Lett 2014, 16, 904–907. [DOI] [PubMed] [Google Scholar]; (b) Leibowitz MK; Winter ES; Scheerer JR Tetrahedron Lett 2015, 56, 6069–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stauffer KJ; Williams PD; Selnick HG; Nantermet PG; Newton CL; Homnick CF; Zrada MM; Lewis SD; Lucas BJ; Krueger JA; Pietrak BL; Lyle EA; Singh R; Miller-Stein C; White RB; Wong B; Wallace AA; Sitko GR; Cook JJ; Holahan MA; Stranieri-Michener M; Leonard YM; Lynch JJ; McMasters DR; Yan YM J. Med. Chem 2005, 48, 2282–2293. [DOI] [PubMed] [Google Scholar]
- (13).Krotko DG; Fedotov KV; Tolmachev AI Dyes Pigm 2005, 65, 183–189. [Google Scholar]
- (14).Desrosiers JN; Wei XD; Gutierrez O; Savoie J; Qu B; Zeng XZ; Lee H; Grinberg N; Haddad N; Yee NK; Roschangar F; Song JHJ; Kozlowski MC; Senanayake CH Chem. Sci 2016, 7, 5581–5586 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Urbina GA Synth. Commun 1979, 9, 245–250. [Google Scholar]; (b) Mayor C; Wentrup CJ Am. Chem. Soc 1975, 97, 7467–7480. [Google Scholar]
- (16).Sivanathan S; Korber F; Tent JA; Werner S; Scherkenbeck JJ Org. Chem 2015, 80, 2554–2561. [DOI] [PubMed] [Google Scholar]
- (17).(a) Several bases are competent, including t-BuOK, Cs2CO3, NEt3, and DBU. For examples, see the respective references: Ando S; Burrows J; Koide K Org. Lett 2017, 19, 1116–1119. [DOI] [PubMed] [Google Scholar]; (b) Liao SR; Qin XC; Wang Z; Li D; Xu L; Li JS; Tu ZC; Liu YH Eur. J. Med. Chem 2016, 121, 500–509. [DOI] [PubMed] [Google Scholar]; (c) Katritzky AR; Fan WQ; Szajda M; Li QL; Caster KC J. Heterocycl. Chem 1988, 25, 591– 597. [Google Scholar]; (d) Zipfel HF; Carreira EM Org. Lett 2014, 16, 2854–2857. [DOI] [PubMed] [Google Scholar]
- (18).Jin SD; Wessig P; Liebscher J Eur. J. Org. Chem 2000, 2000, 1993–1999. [Google Scholar]
- (19).a The alkene isomerization to the metastable intermediate pyrazinone can be intercepted by cycloaddition: Jin SD; Wessig P; Liebscher J J. Org. Chem 2001, 66, 3984–3997. [DOI] [PubMed] [Google Scholar]; (b) Sanz- Cervera JF; Williams RM J. Am. Chem. Soc 2002, 124, 2556– 2559. [DOI] [PubMed] [Google Scholar]
- (20).Kelley EW; Norman SG; Scheerer JR Org. Biomol Chem 2017, 15, 8634–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).The equilibrium acidities of valerolactam and 2-pyridone in DMSO are 26.6 and 17.0: Bordwell FG Acc. Chem. Res 1988, 21, 456–463. [Google Scholar]
- (22).Chogii I; Das P; Fell JS; Scott KA; Crawford MN; Houk KN; Njardarson JT J. Am. Chem. Soc 2017, 139, 13141– 13146. [DOI] [PubMed] [Google Scholar]
- (23).(a) Evans DA; Baillargeon DJ; Nelson JV J. Am. Chem. Soc 1978, 100, 2242–2244. [Google Scholar]; (b) Evans DA; Golob AM J. Am. Chem. Soc 1975, 97, 4765–4766. [Google Scholar]
- (24).Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; Ishikawa T, Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2009. [Google Scholar]
- (25).(a) Schwesinger R; Schlemper H Angew. Chem., Int. Ed. Engl 1987, 26, 1167–1169. [Google Scholar]; (b) Schwesinger R; Schlemper H; Hasenfratz C; Willaredt J; Dambacher T; Breuer T; Ottaway C; Fletschinger M; Boele J; Fritz H; Putzas D; Rotter HW; Bordwell FG; Satish AV; Ji GZ; Peters EM; Peters K; vonSchnering HG; Walz L Liebigs Ann 1996, 1996, 1055–1081. [Google Scholar]
- (26).Phosphazene bases have been used to promote the oxy-Cope reactions: Mamdani HT; Hartley RC Tetrahedron Lett 2000, 41, 747–749. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.