

 Antidepressants are extensively used during pregnancy and associated with severe outcomes, including innate malformations, prematurity, and low birth weight, etc.
Antidepressants are extensively used during pregnancy and associated with severe outcomes, including innate malformations, prematurity, and low birth weight, etc.
Abstract
Antidepressants are extensively used during pregnancy and associated with severe outcomes, including innate malformations, prematurity, and low birth weight, etc. A recent study suggested that prenatal exposure to antidepressants may impair child neurodevelopment process. Thus, the aim of this review is to investigate the potential association between prenatal use of selective 5-HT reuptake inhibitors (SSRIs) and the risk of autism spectrum disorders (ASDs). Twelve studies related to the linkage between SSRI exposure during pregnancy and ASD in children were explored and compiled. However, there is a knowledge gap concerning the potential link between gestational exposure to antidepressants and the risk of ASDs. Despite such limitations, the available data show that some signal exists and signifies that antenatal exposure to SSRIs may increase the risk of ASDs. Thus, there is a vital need for further, large and well-designed research to definitively evaluate the existence and the magnitude of this severe risk.
1. Introduction
Antidepressants are widely used during pregnancy.1 Several studies have shown that the use of antidepressants during pregnancy is linked to adverse outcomes, including innate malformations, prematurity and low birth weight. Among several neurodevelopmental disorders, autism is an unidentified, almost certainly mixed etiology that is obvious in the toddler to preschool years.2,3 As illustrated by Kanner in 1943, three main features in individuals with autism are (i) impairments in reciprocal social interactions, (ii) an abnormal development in the use of language, and (iii) limited behavior and interests.4,5 Basic symptoms are behaviors that are commonly established in autism but are not limited to autism and may be common across the autism spectrum disorders ASDs.
Nowadays, it is estimated that prevalence of ASD has increased over time from 0.04% to1% in the United States.6,7 This pragmatic increase in the occurrence of ASD can be partially endorsed to an extrinsic aspect, including altered diagnostic criteria and their overlap with other diagnoses, better screening of the overall population (higher detection), and greater awareness of the general population.8,9 However, an increase in ASD occurrence is due to genetic and environmental factors over time.10 These factors may include de novo mutations,11 specific genes awarding susceptibility,12 higher maternal age,13 maternal diseases such as diabetes and hypertension14,15 and a maternal record of psychiatric disorder.16,17
Neuropathological alterations that have appeared in previous years in pre- and post-natal developmental modifications engage numerous areas of the brain, like the cerebellum, amygdale, and cerebral cortex, etc.18 While the causation of autism is not yet recognized, the relationship between autism and psychiatric disorders related to abnormal serotonin (5-HT) activity and the association between autism and neurological comorbidities affected by 5-HT dysregulation are interesting.
An increasing use of antidepressant treatment through pregnancy rose from 1%–6% in the 1990s to up to 7%–13% in recent years.19–22 Many psychiatric conditions and depression-like disorders are considered for treatment with selective 5-HT reuptake inhibitors (SSRIs) due to their efficiency and better protection outline. Many antidepressant treatments cross the placenta19–21 and are secreted in breast milk, and as a result raise anxiety regarding severe effects on fetal and infant exposure. In utero exposure to SSRIs, non-selective monoamine reuptake inhibitors (tricyclic antidepressants) and Norepinephrine–dopamine reuptake inhibitors contributed significantly towards the dramatic increase in the observed prevalence of autism spectrum disorders.23,24 However, the vital biologic cycle involved in the expression of 5-HT in the peripheral blood of ASD is yet to be explained. That exposure to antidepressants during late pregnancy results in poor neonatal adaptation has been reported.25–27 Directly related to concerns about a likely relationship with ASD, most studies have explained the possible effects of prenatal exposure on longer-term outcomes, such as milestone achievement, cognitive skills, and behavioral outcomes.28 Assessment of fetal exposure to SSRIs and other antidepressant medications as a possible risk factor for ASD and other neurodevelopmental conditions is complicated by the difficulty in characterizing the effects of medication exposure from the effects of the primary condition that led to the treatment.29 The presence of this dissimilarity is especially significant for the study of autism since a previous history of psychiatric disorders, including depression, has been reported to be more frequent between mothers of infants later diagnosed with autism than among mothers of unaffected children, a signifying fundamental, preexisting genetic risk.
2. Human and 5-HT impression
5-HT or 5-hydroxytryptamine (5-HT) is a monoamine neurotransmitter. Biochemically resulting from tryptophan, 5-HT is chiefly established in the gastrointestinal tract (GI tract), blood platelets, and the central nervous system (CNS) of animals, including humans.30 It is commonly thought to be a provider of feelings of well-being and happiness. Tryptophan hydroxylase (TPH) 1 and 2 are the producers of 5-HT both in human and mammalian species. Its production is most common in the brain raphe, gut, and pineal gland (where 5-HT converts to melatonin). 5-HT is mostly synthesized in the body by TPH1.31 The remaining 5-HT is produced by TPH2 neurons of the raphe nuclei and the enteric nervous system. 5-Hydroxytryptophan (5-HTP) is synthesized from Tryptophan by TPH1 and finally converted into 5-HT by the aromatic l-amino acid decarboxylase (AADC). For this reason, TPH is considered to be the rate-limiting enzyme in the biosynthesis of 5-HT. In enterochromaffin cells, up to 95% of the 5-HT in the body is found in the bowel and are synthesized by TPH1. Up to 95% of 5-HT in the body is found in the bowel, mostly produced by enterochromaffin cells and as a result, synthesized by TPH1. Approximately most of the 5-HT in the bloodstream is situated in blood platelets, obtained by the overflow 5-HT from the gut.32 Many studies indicate that in knockout mice blood platelets have no 5-HT, which suggests a lack of the plasma lemma 5-HT transporter (SERT).33
During the first month of gestation, serotonergic neurons starts producing in primates.34 Raphe neurons begin synthesizing 5-HT one day following their production and commence to develop profuse axon tracts: the caudal group projects into the spinal cord and the rostral group into the forebrain. In rodents, full maturation of the axon terminal network requires more time and is attained only after birth.35 The neurons of the brain stem also generate 5-HT mostly in the raphe nuclei, on the midline of the rhombencephalon, with a small amount in the reticular formation, but project their axons into a range of cortical and subcortical regions and innervate target tissues.36
Ascending projections from brainstem 5-HT neurons terminate in the prearranged manner in cortical, limbic, midbrain, and hindbrain regions.37 5-HT neurons modulate the activity of an extensive range of human brain circuits that elucidate the pleiotropic behavioral effects of brain 5-HT.38
2.1. Autism in relation to 5-HT
5-HT plays a vital role in the CNS and periphery to control several physiological activities.39 Sensory cortex disruption in synaptic connectivity in the brain causes a reduced or elevated brain 5-HT through the postnatal period of growth in rodents.40–42 5-HT has been confirmed to be imperative for prenatal and postnatal brain growth in human studies.43 Problem in serotonergic signaling systems has been anxious as comorbidities that occur with autism.44 In many past years, the developing brain has been commonly accepted as hyper 5-HT hypotheses while many studies sustained that hypo-5-HT may also be a cause of autism.45 There are numerous biological methods that might trigger the relationship between prenatal SSRI disclosure and the cause of neurodevelopmental disorders including ASD.46 5-HT plays a significant function in the development of the brain.47
Many studies demonstrate that 5-HT alters numerous pre- and post-natal progression and developmental processes, together with cell division, neuronal migration, cell differentiation, and synaptogenesis.42 In addition, several studies explained that 5-HT performs as a morphogen through embryonic development, manipulating the maturation of the brain.48 5-HT can be detected in the fetal brain by the fifth week of gestation and is involved in critical neurodevelopmental processes, including neurogenesis or neuronal removal, or both; neuronal differentiation; and synaptogenesis.49 Antidepressants cross the placental barrier and intrauterine exposure to serotonergic agents have been shown to promote persistent changes in the brain circuitry, decreased serotonergic reactivity, and behavioral features analogous to autism in animal models.50 Although SSRIs selectively act on the serotonergic system, almost all other antidepressants also have serotonergic activity.51
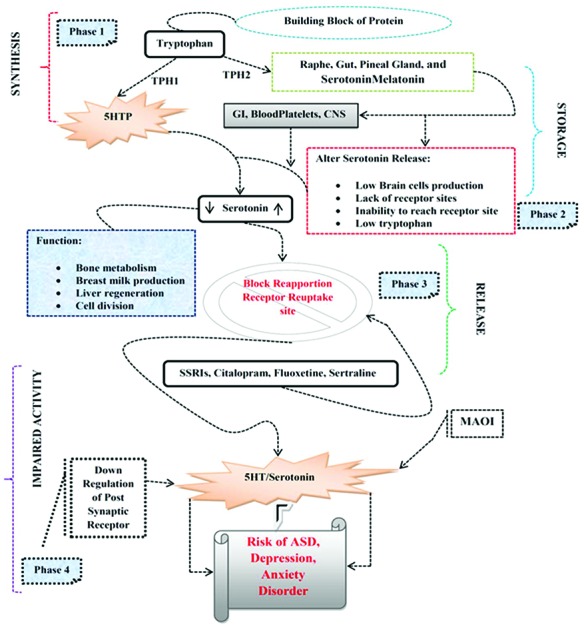
Rodent models indicated that transient inhibition of the 5-HT transporter by fluoxetine hydrochloride, an SSRI, through brain development results in behavioral deficits in later life, indicative of a decisive role for 5-HT in the maturation of the brain systems.45 An inherent feedback mechanism influences the circulating 5-HT levels and the morphological alteration of serotonergic neurons. Many studies show evidence that the serotonergic system plays a key role in ASD as SSRIs block the 5-hydroxytryptamine transporter (5-HTT), causing increased levels of 5-HT in the extracellular space; such exposure has the potential to change 5-HT signaling and innervations; this, in turn, may lead to aberrant circuitry, altered gene and/or protein expression, and altered behavior.52 Alteration of 5-HT during the development in animals can lead to anatomical and behavioral changes that resemble aspects of autism spectrum disorder (ASD).53 Embryonic SSRI exposure in rodents alters 5-HT levels and the expression of SERT and 5-HT receptors54 and it was reported that autistic individuals have elevated 5-HT levels in their blood platelets (Fig. 1).55
Fig. 1. Mechanism of action of antidepressants and risk of ASD. Phase 1: Indicates the synthesis of TPH and production of 5-HT. Phase 2: 5-HT stored in GI, blood platelets, CNS. Phase 3: Action potential causes the release of 5-HT in synapses. Phase 4: Alteration in the 5-HT release SSRIs blocks receptor reabsorption sites or enzymatic degeneration by MAOI, or down-regulation of postsynaptic receptors may increase the risk of ASD and depressive-like disorders.

3. 5-HT impact on reproduction
5-HT is remarkably the most primal of neurotransmitters seeing that an association among antidepressant use and neurodevelopment issues, 5-HT play vital role in early reproduction, and it has extensively been accepted that 5-HT causes significant problems during gestation.56 Altered 5-HT levels have a strong association with the socioemotional disturbances in offspring, and thus increase stress.57 In 1960, connected to research of the effects of antidepressants on 5-HT, Ashcroft explained that there may be a decrease in 5-HT during depression.58
The first published report about increased 5-HT levels causing autism59 was in 1961. This verdict has often been repeated since the early 1960s, and a primary relationship was made between 5-HT and birth deficits in animal studies.60 The first study that shows a connection among 5-HT reuptake inhibiting antidepressants to birth deficits appeared in the early 1970s. In 1990, the first self-governing research involving the SSRI intake to the teratogenic prospective in animal studies was published.61 These improvements resulted in different companies manufacturing novel centrally acting drugs in the 1980s, together with SSRIs, to embark on animal studies. The animal studies came across embryo lethality and gross malformations by the SSRIs, but the linked purpose was to examine the behavior of neonatal pups. Many of these studies illustrate a dose linked fetal death and other features identifiable with a teratogenic potential. All these studies were designed to give evidence of the behavioral consequences of SSRI intake during gestation but most of the evidence remains unknown with the studies unpublished and the data unapproachable. Recent studies in humans suggest that brain serotonergic systems are dysfunctional in patients with anxiety and affective disorders. Patients with panic disorder show a 4-fold increase in brain 5-HT turnover compared with healthy controls62 and this increase is positively correlated with disease severity. Likewise, patients with major depressive disorder (MDD) show an elevated brain 5-HT turnover compared with healthy controls,63 an effect that is associated with carriage of the s allele of the 5-HT transporter (slc6a4) gene.64 High 5-HT and SERT levels may indicate that these biomarkers have a role in the autism pathogenesis and support the possibility of using 5-HT and SERT to diagnose autism severity.65 Finally, published animal studies time after time explain that pups from the animals induced with antidepressants during gestation show a variety of behavioral changes representative of lower social confidence and communication defects consistent with a diagnosis of neurodevelopment spectrum disorders in humans.66,67
3.1. Fetal antidepressant exposure
Depression during gestation is widely treated by antidepressants.68 The prevalence of antidepressant medication use during pregnancy rises from 5.7%–13.3% in 2003 in the USA; 4.5% of pregnant women reported using them from 2001–2006 in Canada. Nevertheless, there is a sustained perplexity about the appropriate use of SSRIs during this decisive time period.69 Fetal psychotropic drug experience is administered by the pharmacokinetic properties of the maternal, placental, and fetal compartments. Umbilical circulation and amniotic fluid are the key routes of fetal exposure.70 TCAs (tricyclic amines) and SSRIs show that the cord blood collection at delivery attained concentrations over 50% of those seen in maternal circulation and, in some cases, are equal to maternal blood concentrations due to the paired maternal and umbilical cord. Tricyclic antidepressants and their metabolites go readily into umbilical cord serum.71 SSRIs have higher umbilical cord transfer rates in contrast with TCAs, as SSRI cord serum–maternal serum ratios are between 0.52 and 1.1 depending on the compound.72 In many cases, metabolites (e.g. norfluoxetine and desmethylvenlafaxine) are active in inhibiting the 5-HT transporter from readily transferring across the placental barrier. Likewise, antidepressants are also established in amniotic fluid,73 even though significant disparity exists among individual medications, animal exposure studies indicate that sufficient antidepressant crosses the placenta to block more than 90% of the transporter sites in the rodent brain.74
3.2. Prenatal antidepressants doses
Pregnancy can influence the metabolism and apparent clearance of centrally active medications.57 The clearance of several SSRIs has been shown to increase over the period of pregnancy.75 Correspondingly, citalopram, escitalopram, or sertraline, taken by postpartum mothers, have also been revealed in a potentially refractory metabolic state.75 If it is decided to take antidepressants during pregnancy, it is important to adjust the maternal dose of the antidepressant during pregnancy to maintain maternal health. Clinicians should be aware that the fetus is exposed to the maternal serum concentration and not the maternal dose, so dose adjustments (increase or decrease) would not considerably modify the amount of fetal central nervous exposure. During pregnancy, those women who choose to breastfeed are also related to the extent of fetal exposure. Recently, antidepressant exposure during breastfeeding has been vigorously quantified, and pregnancy exposure is magnitudes of order greater than breastfeeding exposure. Consequently, for women taking antidepressants during pregnancy, unease about breastfeeding exposure is not necessary.76 To date, twelve published studies investigated the relationship between antidepressant use during pregnancy and the risk of ASD. The outcomes of these studies are presented in Table 1.
Table 1. Characteristics of the studies investigating the relationship between antidepressant use during pregnancy and the risk of ASD.
| Author | Antidepressants | Study outcome |
| Christensen H. D. 2000 79 | Prenatal paroxetine | Behavioral abnormalities, risk of ASD |
| Bakker 2010 80 | Prenatal paroxetine | Increased risk of ASD |
| Kjaersgaard M. 2013 81 | Prenatal SSRI | Spontaneous abortion, increased risk of ASD |
| Petersen I. 2014 77 | Prenatal SSRI | Risk of ASD, prenatal depressive symptoms |
| I. Pereira 2014 82 | Sertraline (SERT) | Behavioral deficits, emotional regulation, risk of ASD |
| Glover M. E. 2015 52 | Prenatal paroxetine | Behavioral abnormalities, risk of ASD |
| Pedersen L. H. 2015 83 | Prenatal SSRI | Increased risk of ASD |
| Avitsur R. 2015 84 | Prenatal fluoxetine | Behavioral deficits, increased risk of ASD |
| Zimmerberg B. 2015 85 | Prenatal fluoxetine | Risk of ASD, social behavior deficits |
| Sprowles J. L. 2016 78 | Citalopram | Risk of ASD, behavior deficits |
| Mao Y. 2016 86 | Prenatal SSRI | Increased risk of ASD and Epilepsy |
| Zucker 2017 87 | Prenatalfluoxetine, VPA, acetaminophen | Risk of ASD, ADHD |
On this issue,77 in Table 1 proposed a relationship between prenatal selective 5-HT reuptake inhibitor (SSRI) exposure and autistic behavior in children, along with prenatal depressive indications. Though antidepressants like SSRIs will possibly be a meager indicator of the severity of underlying illnesses and it may be untimely to make such conclusions regarding the effects of treatment. Many other studies like this increase apprehension as this may enhance more anxiety and guilt among women who are going through depression in pregnancy and perhaps leave several women with no treatment77.
Glover et al. explain that 20% of pregnant and postpartum women who suffer major depression are treated with a selective 5-HT reuptake inhibitor (SSRI), but the consequences of SSRIs on their children's brain development and later emotional health are inadequately understood. SSRI treatment during gestation can extract antidepressant exclusion in newborns and increase toddlers’ anxiety and social evasion (Table 1). In rodents, prenatal SSRI exposure amplifies adult depression- and anxiety-like behavior; and some individuals are more susceptible to these effects than others. They study a rodent model of individual differences in vulnerability to prenatal SSRI exposure, exploiting selectively bred Low Responder (bLR) and High Responder (bHR) rats that were previously bred for a high versus low behavioral response to novelty. BHR/bLR females were treated with the SSRI paroxetine during gestation to examine its effects on the offspring's emotional behavior and gene expression in the developing brain. They found that bLR offspring, obviously prone to an inhibited/anxious temperament, were vulnerable to behavioral deficits related to prenatal SSRI exposure, whereas high risk-taking bHR offspring were resistant. Microarray studies exposed robust prenatal SSRI-induced gene expression alterations in the developing bLR hippocampus and amygdale (postnatal days 7–21), including transcripts concerned in neurogenesis, synaptic vesicle components, and energy metabolism. This explains that the bLR/bHR model is a useful tool to investigate the neurobiology of individual differences in vulnerability to prenatal SSRI exposure.52
Prowles et al. demonstrated that prenatal SSRI revelation has been related with an increased prevalence of autism spectrum disorder (ASD), and peripheral 5-HT is high in some ASD patients. SSRI exposure in rodents during gestation has been linked with elevated depression and anxiety-like behavior, reduced sociability, and impaired learning in the offspring, behaviors frequently observed in ASD (Table 1). They investigated that prenatal exposure to citalopram (Cit) results in persistent neurobehavioral effects in rats. Cit-exposed rats were impaired in the MWM (Morris water maze) during acquisition and probe, but not during the reversal, shift, or cued trials. Cit-exposed rats also illustrated an increased marble burying, reduced time in the center of the open-field, reduced latency to immobility in a forced swim, and elevated acoustic startle across prepulse intensities with no effects on CWM (Cincinnati water maze). The consequences are consistent with citalopram exposing several ASD-like effects. The conclusion appends to concerns about the use of SSRIs during pregnancy. Additional research on diverse types of antidepressants, dose-effect relationships, the timing of exposure periods, and mechanisms for these effects are required.78 Such findings may result in significant public health concerns and media interest, with the potential consequences of cessation of antidepressant treatment during pregnancy and its impact on both maternal and child health, and in this regard the benefit risk should be considered in terms of both mother and offspring.
4. Prospective methods
In expressions of the antidepressants effects on neurodevelopment, we have abstracted a variety of methods through which gestational SSRIs might impact on cognitive function that is supported by recent literature (Fig. 2).
Fig. 2. Different methods of adverse drug SSRI responses on neurodevelopment. SSRI intake results in abnormalities of the cardiac system and cerebral architecture, as well as cognitive problem and behavioral deficits.

• In embryogenesis, 5-HT has a major role and there is persuasive data that distressing this function results in abnormalities of many systems from cardiac through to neurological.88
• SSRIs can lead to abnormalities in cerebral architecture over a decade.45 These changes result in a decrease in exploratory behavior and reactivity.89
• SSRIs have reflective effects on sexual behavior and this can probably cause longer-term abnormalities in the social domain.90
• SSRIs can persuade a compulsive alcohol intake91 and this might be likely to have some role in the epidemiological picture.
• SSRIs also generate the experience of blunted emotion.92
• The recent analyses of ASD may include children with common cognitive problems rather than the specific disorder of a discrete communication function, a variety of treatment associated factors from SSRI withdrawal to postnatal operative interference to accurate birth defect may add to the potential of these drugs to increase ASD.
These behavioral effects are consistent with changes that will register on instruments currently used to detect communication difficulties and are likely to lead to an ASD diagnosis.93,94
SSRI effects may consequently intervene through end-stage behavioral alterations. These behavioral modifications can be continuous for months after treatment stops in adults. Such changes may be even more likely to tolerate and turn out an ASD clinical picture if the drug administered in utero. It is significant to establish the full range of effects these drugs have and there is promising but as yet unpublished literature that rates of Attention deficit hyperactivity disorder (ADHD), for example, may be amplified in children born from mothers taking SSRIs. Any method anticipated is required to be able to account for such modifications also.
5. Conclusion
Altogether, these studies are indicative of an increased risk of ASD related with antidepressant use for the period of pregnancy; the elevated rate of developmental delay is associated to SSRI use in pregnancy may also be consistent with augmented rates of diagnoses of ASD in offspring from mothers exposed to these treatments. We have highlighted twelve articles here (including organized and prospective studies) that substantiate an association between SSRI exposure through the placenta and ASDs in children.
The conclusion is supported by consistent findings in the animal studies reviewed and by the presence of many biologically plausible methods in humans. There is, moreover, a range of mechanisms through which the effects of SSRIs might lead to outcomes attracting an ASD diagnosis. To move our understanding forward will require the consideration of both epidemiological findings and physiological studies. The reason for a review of these physiological mechanisms is that the epidemiological evidence is at a point where it is now clear that a greater specificity in clinical phenotypes is needed in order to make sense of the possible links between treatment and outcomes. This will fortify the reported association that 5-HT inhibition during gestation seems to play an important role in the occurrence of ASD. However, there is a knowledge gap concerning the potential link between gestational exposure to antidepressants and the risk of autism spectrum disorders (ASD). Further research is needed to specifically assess the risk of ASD associated with antidepressant types and dosages during pregnancy. Despite such limitations, the available data show that some signal exists signifying that antenatal exposure to SSRIs may increase the risk of ASDs. Thus, there is a vital obligation for further, large, well-designed research aimed to definitively evaluate the existence and magnitude of this severe risk.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 31572575 and 31602114) and by the Fundamental Research Funds for the Central Universities (2662016PY115), as well as the long-term development plan UHK.
References
- Glover V. J. Child Psychol. Psychiatry. 2011;52(4):356–367. doi: 10.1111/j.1469-7610.2011.02371.x. [DOI] [PubMed] [Google Scholar]
- Bailey A., Phillips W., Rutter M. J. Child Psychol. Psychiatry. 1996;37(1):89–126. doi: 10.1111/j.1469-7610.1996.tb01381.x. [DOI] [PubMed] [Google Scholar]
- Amaral D. G. Brain Res. 2011;1380:3–9. doi: 10.1016/j.brainres.2010.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagnostic and statistical manual of mental disorders, Am Psychiatric Assoc, 4th edn, 1994. [Google Scholar]
- AP Association, Diagnostic and statistical manual of mental disorders Text revision, American Psychatric Assocaition, Arlington, VA, 4th edn, 2000. [Google Scholar]
- Kogan M. D., Blumberg S. J., Schieve L. A., Boyle C. A., Perrin J. M., Ghandour R. M. Pediatrics. 2009;124(5):1395–1403. doi: 10.1542/peds.2009-1522. [DOI] [PubMed] [Google Scholar]
- Baio J., Prevalence of Autism Spectrum Disorders: Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008, Morbidity and Mortality Weekly Report. Surveillance Summaries. Volume 61, Number 3. Centers for Disease Control and Prevention, 2012. [PubMed]
- Hertz-Picciotto I., Delwiche L. Epidemiology. 2009;20(1):84. doi: 10.1097/EDE.0b013e3181902d15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M., Bearman P. Int. J. Epidemiol. 2009;38(5):1224–1234. doi: 10.1093/ije/dyp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I., Croen L. A., Hansen R., Jones C. R., van de Water J., Pessah I. N. Environ. Health Perspect. 2006;114(7):1119. doi: 10.1289/ehp.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M., Iossifov I., Levy D., Wigler M. Nat. Rev. Genet. 2014;15(2):133. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- Muhle R., Trentacoste S. V., Rapin I. Pediatrics. 2004;113(5):e472–ee86. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Sandin S., Hultman C. M., Kolevzon A., Gross R., MacCabe J. H., Reichenberg A. J. Am. Acad. Child Adolesc. Psychiatry. 2012;51(5):477–486. doi: 10.1016/j.jaac.2012.02.018. [DOI] [PubMed] [Google Scholar]
- Lyall K., Pauls D. L., Spiegelman D., Ascherio A., Santangelo S. L. Autism Res. 2012;5(1):21–30. doi: 10.1002/aur.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore G. S., Kneitel A. W., Walker C. K., Gilbert W. M., Xing G. Am. J. Obstet. Gynecol. 2012;206(4):314. doi: 10.1016/j.ajog.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels J. L., Forssen U., Hultman C. M., Cnattingius S., Savitz D. A., Feychting M. Pediatrics. 2008;121(5):e1357–e1362. doi: 10.1542/peds.2007-2296. [DOI] [PubMed] [Google Scholar]
- Jokiranta E., Brown A. S., Heinimaa M., Cheslack-Postava K., Suominen A., Sourander A. Psychiatry Res. 2013;207(3):203–211. doi: 10.1016/j.psychres.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadad B. S., Hewitson L., Young K. A., German D. C. Autism Res Treat. 2013:2013. doi: 10.1155/2013/731935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra S., Born L., Sarkar M., Einarson A. Expert Opin. Drug Saf. 2005;4(2):273–284. doi: 10.1517/14740338.4.2.273. [DOI] [PubMed] [Google Scholar]
- Kendall-Tackett K., Hale T. W. J. Hum. Lact. 2010;26(2):187–195. doi: 10.1177/0890334409342071. [DOI] [PubMed] [Google Scholar]
- Alwan S., Friedman J. M. CNS Drugs. 2009;23(6):493–509. doi: 10.2165/00023210-200923060-00004. [DOI] [PubMed] [Google Scholar]
- Berle J. Ø., Steen V. M., Aamo T. O., Breilid H., Zahlsen K., Spigset O. J. Clin. Psychiatry. 2004;65(9):1228–1234. doi: 10.4088/jcp.v65n0911. [DOI] [PubMed] [Google Scholar]
- Rai D., Lee B. K., Dalman C., Golding J., Lewis G., Magnusson C. Br. Med. J. 2013;346:f2059. doi: 10.1136/bmj.f2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprowles J. L., Hufgard J. R., Gutierrez A., Bailey R. A., Jablonski S. A., Williams M. T. Int. J. Dev. Neurosci. 2017;61:92–111. doi: 10.1016/j.ijdevneu.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Yonkers K. A., Wisner K. L., Stewart D. E., Oberlander T. F., Dell D. L., Stotland N. Gen Hosp Psychiatry. 2009;31(5):403–413. doi: 10.1016/j.genhosppsych.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchkin V., Martin A. Lancet. 2005;365(9458):451–453. doi: 10.1016/S0140-6736(05)17877-5. [DOI] [PubMed] [Google Scholar]
- Sanz E. J., De-las-Cuevas C., Kiuru A., Bate A., Edwards R. Lancet. 2005;365(9458):482–487. doi: 10.1016/S0140-6736(05)17865-9. [DOI] [PubMed] [Google Scholar]
- Boukhris T., Sheehy O., Mottron L., Bérard A. JAMA Pediatr. 2016;170(2):117–124. doi: 10.1001/jamapediatrics.2015.3356. [DOI] [PubMed] [Google Scholar]
- Man K. K., Tong H. H., Wong L. Y., Chan E. W., Simonoff E., Wong I. C. Neurosci. Biobehav. Rev. 2015;49:82–89. doi: 10.1016/j.neubiorev.2014.11.020. [DOI] [PubMed] [Google Scholar]
- Gemmel M., Hazlett M., Bögi E., De Lacalle S., Hill L. A., Kokras N. Psychoneuroendocrinology. 2017;84:159–171. doi: 10.1016/j.psyneuen.2017.07.480. [DOI] [PubMed] [Google Scholar]
- Gershon M. D. J. Clin. Gastroenterol. 2005;39(5):S184–SS93. doi: 10.1097/01.mcg.0000156403.37240.30. [DOI] [PubMed] [Google Scholar]
- Chambers C. D., Hernandez-Diaz S., Van Marter L. J., Werler M. M., Louik C., Jones K. L. N. Engl. J. Med. 2006;354(6):579–587. doi: 10.1056/NEJMoa052744. [DOI] [PubMed] [Google Scholar]
- Chen J. J., Li Z., Pan H., Murphy D. L., Tamir H., Koepsell H. J. Neurosci. 2001;21(16):6348–6361. doi: 10.1523/JNEUROSCI.21-16-06348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P., Rakic P. Dev. Brain Res. 1982;4(1):35–57. doi: 10.1016/0165-3806(82)90095-5. [DOI] [PubMed] [Google Scholar]
- Lidov H. G., Molliver M. E. Brain Res. Bull. 1982;8(4):389–430. doi: 10.1016/0361-9230(82)90077-6. [DOI] [PubMed] [Google Scholar]
- Benes F. M., Taylor J. B., Cunningham M. C. Cereb. Cortex. 2000;10(10):1014–1027. doi: 10.1093/cercor/10.10.1014. [DOI] [PubMed] [Google Scholar]
- Berger M., Gray J. A., Roth B. L. Annu. Rev. Med. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B. L., The serotonin receptors: from molecular pharmacology to human therapeutics, Springer Science & Business Media, 2008. [Google Scholar]
- Shah R., Courtiol E., Castellanos F. X., Teixeira C. M. Front. Behav. Neurosci. 2018;12:114. doi: 10.3389/fnbeh.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J. B., Iny L. J., Meaney M. J. Dev. Brain Res. 1990;55(2):231–235. doi: 10.1016/0165-3806(90)90204-c. [DOI] [PubMed] [Google Scholar]
- Meaney M., Diorio J., Francis D., LaRocque S., O'DONNELL D., Smythe J. Ann. N. Y. Acad. Sci. 1994;746(1):260–274. doi: 10.1111/j.1749-6632.1994.tb39243.x. [DOI] [PubMed] [Google Scholar]
- Gaspar P., Cases O., Maroteaux L. Nat. Rev. Neurosci. 2003;4(12):1002. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia P. M., Azmitia E. C. Neurosci. Lett. 1986;67(3):307–312. doi: 10.1016/0304-3940(86)90327-7. [DOI] [PubMed] [Google Scholar]
- Rose'Meyer R. Mol. Autism. 2013;4(1):37. doi: 10.1186/2040-2392-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge M. S., Zhou M., Lira A., Hen R., Gingrich J. A. Science. 2004;306(5697):879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Healy D., Le Noury J., Mangin D. Int. J. Risk Saf. Med. 2016;28(3):125–141. doi: 10.3233/JRS-160726. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia P. M. Brain Res. Bull. 2001;56(5):479–485. doi: 10.1016/s0361-9230(01)00615-3. [DOI] [PubMed] [Google Scholar]
- Sodhi M., Sanders-Bush E. Int. Rev. Neurobiol. 2004;59(6):111–174. doi: 10.1016/S0074-7742(04)59006-2. [DOI] [PubMed] [Google Scholar]
- Torjesen I. BMJ [Br. Med. J.] 2014:349. [Google Scholar]
- Zahra A., Jiang J., Chen Y., Long C., Yang L. Exp. Neurol. 2018;307:145–154. doi: 10.1016/j.expneurol.2018.06.003. [DOI] [PubMed] [Google Scholar]
- King B. H. J. Am. Med. Assoc. 2017;317(15):1568–1569. doi: 10.1001/jama.2016.20614. [DOI] [PubMed] [Google Scholar]
- Glover M. E., Pugh P. C., Jackson N. L., Cohen J. L., Fant A. D., Akil H. Neuroscience. 2015;284:775–797. doi: 10.1016/j.neuroscience.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitoglou M., Ververi A., Antoniadis A., Zafeiriou D. I. Pediatr. Neurol. 2010;42(5):309–314. doi: 10.1016/j.pediatrneurol.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Cabrera-vera T. and Battaglia G., Prenatal Exposure to Fluoxetine (prozac) Produces Site-specific and Age-dependent Alterations in Brain Serotonin Transporters in Rat Progeny: Evidence From Autoradiographic Studies, Year Book of Psychiatry and Applied Mental Health, 2000, vol. 20001, pp. 391–392. [PubMed] [Google Scholar]
- Hanley H. G., Stahl S. M., Freedman D. X. Arch. Gen. Psychiatry. 1977;34(5):521–531. doi: 10.1001/archpsyc.1977.01770170031002. [DOI] [PubMed] [Google Scholar]
- Buznikov G. Pharmacol. Ther. 1984;25(1):23–59. doi: 10.1016/0163-7258(84)90023-8. [DOI] [PubMed] [Google Scholar]
- Ornoy A., Koren G. Expert Opin. Drug Metab. Toxicol. 2018;14(3):247–259. doi: 10.1080/17425255.2018.1430139. [DOI] [PubMed] [Google Scholar]
- Ashcroft G., Sharman D. Nature. 1960;186(4730):1050–1051. doi: 10.1038/1861050a0. [DOI] [PubMed] [Google Scholar]
- Schain R. J., Freedman D. X. J. Pediatr. 1961;58(3):315–320. doi: 10.1016/s0022-3476(61)80261-8. [DOI] [PubMed] [Google Scholar]
- Poulson E., Robson J., Sullivan F. Science. 1963;141(3582):717–718. doi: 10.1126/science.141.3582.717. [DOI] [PubMed] [Google Scholar]
- Shuey D. L., Yavarone M., Sadler T. W. and Lauder J. M., Serotonin and morphogenesis in the cultured mouse embryo. Molecular Aspects of Development and Aging of the Nervous System, Springer, 1990, pp. 205–215. [DOI] [PubMed] [Google Scholar]
- Esler M., Lambert E., Alvarenga M., Socratous F., Richards J., Esler M. Stress. 2007;10(3):295–304. doi: 10.1080/10253890701300904. [DOI] [PubMed] [Google Scholar]
- Barton D. A., Esler M. D., Dawood T., Lambert E. A., Haikerwal D., Brenchley C. Arch. Gen. Psychiatry. 2008;65(1):38–46. doi: 10.1001/archgenpsychiatry.2007.11. [DOI] [PubMed] [Google Scholar]
- Hale M. W., Shekhar A., Lowry C. A. Cell. Mol. Neurobiol. 2012;32(5):695–708. doi: 10.1007/s10571-012-9827-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulamir H. A., Abdul-Rasheed O. F., Abdulghani E. A. Saudi Med. J. 2018;39(5):487–494. doi: 10.15537/smj.2018.5.21751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciag D., Simpson K. L., Coppinger D., Lu Y., Wang Y., Lin R. C. Neuropsychopharmacology. 2006;31(1):47. doi: 10.1038/sj.npp.1300823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinast K., Peeters D., Kolk S. M., Schubert D., Homberg J. R. Front. Cell. Neurosci. 2013;7:72. doi: 10.3389/fncel.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard S. J., Weston-Green K., Newell K. A. Neurosci. Biobehav. Rev. 2017;80:743–765. doi: 10.1016/j.neubiorev.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Cooper W. O., Willy M. E., Pont S. J., Ray W. A. Am. J. Obstet. Gynecol. 2007;196(6):544. doi: 10.1016/j.ajog.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Roullet F. I., Lai J. K., Foster J. A. Neurotoxicol. Teratol. 2013;36:47–56. doi: 10.1016/j.ntt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Loughhead A. M., Stowe Z. N., Newport D. J., Ritchie J. C., DeVane C. L., Owens M. J. Biol. Psychiatry. 2006;59(3):287–290. doi: 10.1016/j.biopsych.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Hendrick V., Stowe Z. N., Altshuler L. L., Hwang S., Lee E., Haynes D. Am. J. Psychiatry. 2003;160(5):993–996. doi: 10.1176/appi.ajp.160.5.993. [DOI] [PubMed] [Google Scholar]
- Rampono J., Simmer K., Ilett K., Hackett L., Doherty D., Elliot R. Pharmacopsychiatry. 2009;42(03):95–100. doi: 10.1055/s-0028-1103296. [DOI] [PubMed] [Google Scholar]
- Capello C. F., Bourke C. H., Ritchie J. C., Stowe Z. N., Newport D. J., Nemeroff A. J. Pharmacol. Exp. Ther. 2011;339(1):275–285. doi: 10.1124/jpet.111.183855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit D. K., Perel J. M., Helsel J. C., Wisner K. L. J. Clin. Psychiatry. 2008;69(4):652. doi: 10.4088/jcp.v69n0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S., Stowe Z. N. Best Pract. Res. Clin. Obstet. Gynaecol. 2014;28(1):71–83. doi: 10.1016/j.bpobgyn.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Petersen I., Evans S., Nazareth I. Br. J. Psychiatry. 2014;205(2):105–106. doi: 10.1192/bjp.bp.113.141721. [DOI] [PubMed] [Google Scholar]
- Sprowles J. L., Hufgard J. R., Gutierrez A., Bailey R. A., Jablonski S. A., Williams M. T. Int. J. Dev. Neurosci. 2016;54:39–52. doi: 10.1016/j.ijdevneu.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Christensen H. D., Rayburn W. F., Gonzalez C. L. Neurotoxicol. Teratol. 2000;22(5):733–739. doi: 10.1016/s0892-0362(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Bakker M. K., Kerstjens-Frederikse W. S., Buys C. H., de Walle H. E., de Jong-van den Berg L. Birth Defects Res., Part A. 2010;88(2):94–100. doi: 10.1002/bdra.20641. [DOI] [PubMed] [Google Scholar]
- Kjaersgaard M. I. S., Parner E. T., Vestergaard M., Sørensen M. J., Olsen J., Christensen J. PLoS One. 2013;8(8):e72095. doi: 10.1371/journal.pone.0072095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Figueiredo I., Sancho C., Carro J., Castellano O., López D. E. Front. Behav. Neurosci. 2014;8:260. doi: 10.3389/fnbeh.2014.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen L. H. Pediatr. Drugs. 2015;17(6):443–448. doi: 10.1007/s40272-015-0141-5. [DOI] [PubMed] [Google Scholar]
- Avitsur R., Levy S., Grinshpahet R., Goren N., Hirsh O., Zalko A. J. Neuroimmunol. 2015;284:49–56. doi: 10.1016/j.jneuroim.2015.05.006. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B., Germeyan S. C. Dev. Psychobiol. 2015;57(2):141–152. doi: 10.1002/dev.21264. [DOI] [PubMed] [Google Scholar]
- Mao Y., Pedersen L. H., Christensen J., Vestergaard M., Zhou W., Olsen J. Pharmacoepidemiol. Drug Saf. 2016;25(11):1320–1330. doi: 10.1002/pds.4072. [DOI] [PubMed] [Google Scholar]
- Zucker I. Neurosci. Biobehav. Rev. 2017:107–121. doi: 10.1016/j.neubiorev.2017.03.005. [DOI] [PubMed] [Google Scholar]
- Alwan S., Reefhuis J., Rasmussen S. A., Olney R. S., Friedman J. M. N. Engl. J. Med. 2007;356(26):2684–2692. doi: 10.1056/NEJMoa066584. [DOI] [PubMed] [Google Scholar]
- Aragon M., Ayala M., Marin M., Avilés A., Damian-Matsumura P., Domínguez R. Reproduction. 2005;129(6):717–727. doi: 10.1530/rep.1.00598. [DOI] [PubMed] [Google Scholar]
- Maciag D., Coppinger D., Paul I. A. Brain Res. 2006;1125(1):171–175. doi: 10.1016/j.brainres.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Sari Y., Zhou F. C. Dev. Brain Res. 2004;150(2):151–161. doi: 10.1016/j.devbrainres.2003.02.001. [DOI] [PubMed] [Google Scholar]
- Tranter R., Bell D., Gutting P., Harmer C., Healy D., Anderson I. M. J. Affective Disord. 2009;118(1):87–93. doi: 10.1016/j.jad.2009.01.028. [DOI] [PubMed] [Google Scholar]
- Durso G. R., Luttrell A., Way B. M. Psychol. Sci. 2015;26(6):750–758. doi: 10.1177/0956797615570366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H., Hidaka N., Kawagoe C., Odagiri K., Watanabe Y., Ikeda T. Neurosci. Res. 2007;59(2):145–151. doi: 10.1016/j.neures.2007.06.1465. [DOI] [PubMed] [Google Scholar]