

Abstract
A completely regioselective and highly stereoselective palladium‐catalyzed intramolecular hydroarylation of arenesulfonyl ynamines to benzothiazoles was developed. The presence of an electron‐withdrawing group on the triple bond of the sulfonyl ynamine was crucial for the success of the reaction and our mechanistic studies suggest an alkyne‐directed 5‐exo‐dig cyclization pathway. The products easily underwent photoinduced rearrangement to 3‐amino‐1‐benzothiophene‐1,1‐diones (up to 35 % yields after two steps).
Keywords: Ynamines, Homogeneous catalysis, Palladium, C–H activation, Synthetic methods
Introduction
Sulfonyl ynamines have recently emerged as a privileged class of ynamides.1 They are stable compounds, readily prepared2 and easy to handle. In addition, the polarized alkyne system of sulfonyl ynamines shows excellent reactivity in a wide variety of reactions featuring π‐acid catalysis,3 metal‐catalyzed cyclizations,4 cycloadditions5 and rearrangements6 for the synthesis of complex nitrogen‐containing heterocycles and natural products. With the idea of extending the chemistry of these ynamines, we envisioned that arenesulfonyl ynamines 1 could be used as simple precursors for a single step, atom‐efficient synthesis of 1,2‐benzothiazole‐1,1‐diones, key elements of diverse biologically active compounds and useful reagents in organic synthesis (Scheme 1).7
Scheme 1.
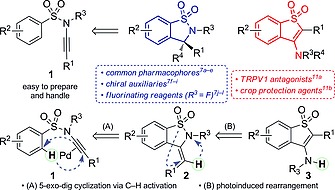
Syntheses and applications of 1,2‐benzothiazole‐1,1‐diones and 3‐amino‐1‐benzothiophene‐1,1‐diones.
Considering that compounds 1 react in the presence of alkynophilic transition metals usually through π‐activation of the triple bond, we anticipated that initial alkyne coordination might direct an aromatic ortho‐C–H activation followed by a stereocontrolled intramolecular hydroarylation (pathway A, Scheme 1), similar to that of N‐alkynyl indoles developed by Park et al.8 or alkynyl aryl ethers developed by Hiyama et al.9 Moreover, excitation of the obtained benzothiazolediones 2 with light could trigger a [1,3]sigmatropic rearrangement of the sulfonyl group by S–N bond cleavage10 followed by colligation to 3‐amino‐1‐benzothiophene‐1,1‐diones 3 (pathway B, Scheme 1).11 In particular, our interest in these compounds was awakened because they are potential crop protecting agents, and therefore beneficial for the ECHONET program we are part of.12 To the best of our knowledge, alkyne‐directed 5‐exo‐dig cyclizations of arenesulfonyl ynamines to 1,2‐benzothiazole‐1,1‐diones and its following rearrangement to 3‐amino‐1‐benzothiophene‐1,1‐diones have not been yet explored.
Results and Discussion
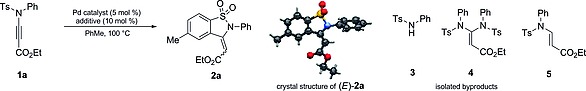
We initially investigated the behavior of sulfonyl ynamine 1a in the presence of alkynophilic transition metals and Brønsted acids (Table 1; for detailed information on the reaction optimization, see the Supporting Information). The reaction took place only with palladium‐based catalysts. Thus, sulfonyl ynamine 1a was converted to products (E/Z)‐2a, 3, 4 and 5 in the presence of Pd(OAc)2 (5 mol‐%) in toluene at 100 °C for 18 h (Table 1, entry 1). The (E)‐exocyclic alkylidene product 2a was isolated in 21 % yield after purification of the crude mixture by neutral silica‐gel column chromatography.
Table 1.
Reaction optimization
|
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ratio of products [%]a | |||||||||
| Entry | Catalyst | Additive | 1a | 2a | 3 | 4 | 5 | E/Z‐ratioa | Yield (E)‐2a [%]b |
| 1 | Pd(OAc)2 | 27 | 35 | 33 | 3 | 2 | 82:18 | 21 | |
| 2 | Pd(PPh3)4 | 30 | 10 | 58 | 2 | 0 | 90:10 | 7 | |
| 3 | Pd(PPh3)4 | HOAc | 43 | 23 | 17 | 3 | 14 | 92:8 | 14 |
| 4 | Pd(OAc)2 | Zn | 23 | 37 | 38 | 2 | 0 | 85:15 | 20 |
| 5 | Pd(OAc)2 | NaOAcc | 28 | 14 | 58 | 0 | 0 | 85:15 | 8 |
| 6 | Pd(OAc)2/PCy3 | 13 | 48 | 36 | 0 | 3 | 90:10 | 27 | |
| 7 | Pd(OAc)2/P(p‐tol)3 | 10 | 51 | 35 | 0 | 4 | 98:2 | 37 | |
Reaction conditions: 1a (1.0 mmol), Pd catalyst (5 mol‐%), phosphine ligand (10 mol‐%), additive (10 mol‐%), PhMe (10.0 mL), 100 °C for 18 h. [a] Based on the 1H NMR spectrum of the crude.
Isolated yield.
NaOAc (2.0 equiv.).
The structure of (E)‐2a was unambiguously determined by NMR spectroscopy and X‐ray crystallography. This reaction also showed degradation of sulfonyl ynamine 1a under the reaction conditions forming N‐phenyl sulfonamide 3, which was the main byproduct of the ynamide cyclization. Compound 3 reacted further with 1a to form Michael adduct 4, which was successfully isolated after purification. Furthermore, sulfonyl ynamine 1a underwent partial hydrogenation of the triple bond to yield sulfonyl enamine 5 in a trace amount.13 The reaction also took place in the presence of palladium(0), albeit in a much lower conversion compared to Pd(OAc)2 (entries 2 and 3). The use of Pd(OAc)2 and Zn, a catalytic system developed by Hiyama et al. for the cyclization of alkynyl aryl ethers,9 had no influence on the yield of 2a (entry 4). These initial studies showed that formation of benzothiazoledione 2a took place without any further additives (e.g., NaOAc, entry 5). Finally, addition of phosphine ligands promoted the cyclization. Bulky ligands led in general to higher yields and better ratios of the stereoisomers (entries 6 and 7).
Although the yields are relatively low, this unprecedented intramolecular hydroarylation reaction is completely regioselective and highly stereoselective: (E)‐exocyclic 1,2‐benzothiazole‐1,1‐dione 2a is the major isomer with an E/Z‐ratio of up to 98:2, confirmed by NOE studies in all cases.
With these results in hand, we focused our attention on the scope and limitations of the reaction varying the substituents on the alkyne terminus (R1), on the aromatic ring of the sulfonamide group (R2) and on the nitrogen (R3) for the hydroarylation of sulfonyl ynamines 1a–k (Scheme 2). Notably, the presence of an electron‐withdrawing group on the triple bond, e.g., ester or ketone, was essential for the success of the hydroarylation. Thus, sulfonyl ynamines with a ketone showed similar reactivity to sulfonyl ynamine 1a proceeding with exclusive regioselectivity and excellent stereoselectivity (2b–2c). In stark contrast, when the reaction was performed with sulfonyl ynamines 1d–1f (R = H, SiMe3, Ph) no cyclization was detected and only decomposition to 3 was observed. Changing the conditions (solvent, temperature, Pd catalyst, addition of base) did not lead to products 2d–2f either.
Scheme 2.
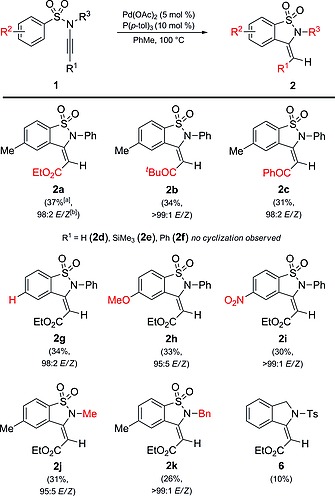
Scope of sulfonyl ynamines for intramolecular hydroarylation. Reaction conditions: 1 (1.0 mmol), Pd(OAc)2 (5 mol‐%), P(p‐tol)3 (10 mol‐%), PhMe (10.0 mL), 100 °C for 18 h. [a] Isolated yield. [b] Based on the 1H NMR spectrum of the crude mixture.
Next, a series of electronically different sulfonyl ynamines were employed for this transformation. To our delight, we observed no significant difference in the reaction efficiency when changing the substituents at the aryl group of the sulfonamide. 1,2‐Benzothiazole‐1,1‐diones (2g–i) were obtained in comparable yields and also with complete regioselectivity and with excellent stereoselectivity. Finally, sulfonyl ynamines 1j and 1k were subjected to the reaction conditions to compare the reactivity of compounds with different substitution on the nitrogen. Compound 1j formed product 2j in 31 % yield and compound 1k resulted in the formation of a mixture of 2k (>99:1 E/Z ratio) and 6 in 26 and 10 % yield, respectively.14
For a better understanding of the reaction mechanism, we investigated the reaction pathway with DFT calculations (Figure 1). The most logical mechanistic scenario for the reaction of sulfonyl ynamine 1l and Pd(OAc)2 would involve coordination of the PdII species to the substrate, as in 7, followed by an alkyne‐directed concerted deprotonation‐metalation sequence (CDM).15 Thus, calculations indicate transition state TS1 in which one of the acetate groups is acting both as a κ2 ligand and as a base. The proton abstraction presents the highest activation barrier within the catalytic cycle (22.6 kcal/mol), and therefore should be considered as the rate‐determining step of the reaction. Additionally, ortho‐C–H activation in TS1 proceeds much slower for the alkyne–H and alkyne–SiMe3 substructures (24.5 and 24.6 kcal/mol, for 1d and 1e, respectively) compared to the corresponding alkyne–CO2Me substructure 1l. The difference between these barrier values is >2.5 kcal/mol corresponding to more than 100 times slower reaction rate, which is in agreement with the experimental results. Consequently, the intermediate 8 performs a carbopalladation to the CC triple bond through TS2, leading to intermediate 9. Also, this step was predicted to be a few kcal/mol higher for sulfonyl ynamines 1d and 1e (16.0 and 14.2 kcal/mol respectively) than for sulfonyl ynamine 1l (11.2 kcal/mol). Further protonation of the “push‐pull” alkenyl–PdII species 9 with acetic acid forms an enol intermediate 10, with almost free rotation around the exocyclic C–C bond. We found that the enol structure 10 lies 12.9 kcal/mol higher in energy than palladium species 9, confirming the feasibility of this or related isomerization processes. The final protodemetalation with acetic acid renders the major product (E)‐2a and recovers the active PdII‐acetate species.
Figure 1.
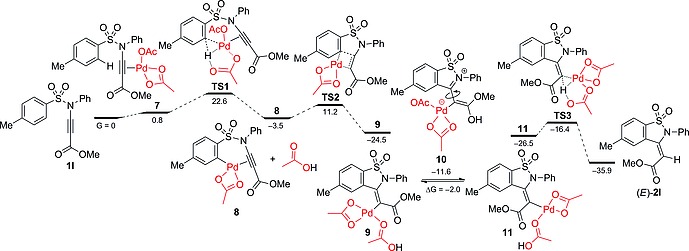
Computed pathway for cyclization of 1l. Free energies (298 K) with respect to starting materials are shown in kcal/mol.
Our DFT calculations further reveal that the (E)‐isomer is the most stable isomer [2.5 kcal/mol Lower in energy than the (Z)‐isomer, Scheme 3]. To validate this result, (E)‐2a and (Z)‐2a were subjected independently to the same experimental conditions. We observed the formation of an 85:15 E/Z mixture of isomers after 18 h of reaction time. These results suggest that the cyclization of 1a might proceed in an exceptional stereoselective manner (as proposed in Scheme 2), however the major product further undergoes partial isomerization under the reaction conditions.
Scheme 3.
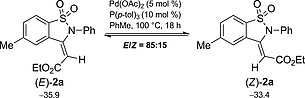
Equilibration between (E)‐ and (Z)‐isomers of 2a. Free energies (298 K) are shown in kcal/mol.
Finally, 1,2‐benzothiazole‐1,1‐diones 2 underwent a previously unexplored photoinduced rearrangement to 3‐amino‐1‐benzothiophene‐1,1‐diones 12 while being irradiated with UV light (300 nm; Scheme 4).
Scheme 4.
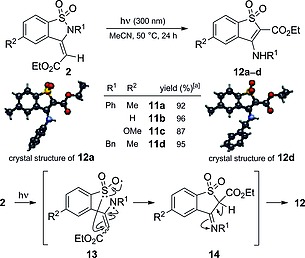
Photochemical rearrangement of 1,2‐benzothiazole‐1,1‐diones 2 to 3‐amino‐1‐benzothiophene‐1,1‐diones 12. Reaction conditions: 2 (0.1 mmol), CH3CN (10.0 mL), hν (300 nm), 24 h. [a] Isolated yield.
Thus, when either stereoisomer of 2a or the mixture of both was exposed to irradiation at 50 °C for 24 h, a complete photochemical conversion to 3‐amino‐1‐benzothiophene‐1,1‐dione 12a was observed. Furthermore, to show the scope of this transformation, several 1,2‐benzothiazoledione derivatives were subjected to the same reaction conditions. The corresponding products 12b–12d were obtained in excellent yields in all cases. The structure of the products was confirmed by selNOE spectroscopy and X‐ray crystallography of 12a and 12d. We also propose a plausible mechanism on the basis of previously reported photoinduced cleavage of sulfonamides.7 This [1,3]sigmatropic rearrangement entails a homolytic cleavage of the sulfonamide S–N bond of 1,2‐benzothiazoledione 2, followed by recombination of the resulting sulfinate radical with the C‐terminus of the enaminyl radical (13). Subsequent tautomerization of the formed imine 14 results in 3‐amino‐1‐benzothiophene‐1,1‐dione 12.
Conclusions
In summary, we have demonstrated a completely regioselective and highly stereoselective intramolecular hydroarylation of sulfonyl ynamines. This method opens an easy access to benzothiazole heterocycles, valuable scaffolds in medicinal and organic synthesis. Our mechanistic studies suggest an alkyne‐directed 5‐exo‐dig intramolecular cyclization pathway, where the presence of an electron‐withdrawing group at the triple bond was key for the success of the reaction. Moreover, we have discovered the first example of photoinduced rearrangement of 1,2‐benzothiazole‐1,1‐diones to form 3‐amino‐1‐benzothiophene‐1,1‐dione derivatives in excellent yields. Optimization studies that broaden the synthetic scope and applications of the reaction are currently under investigation.
Experimental Section
General Information: Reagents were obtained from commercial suppliers and were used without purification. Standard syringe techniques were applied for the transfer of dry solvents and air‐ or moisture‐sensitive reagents. All inert reactions were carried out under a nitrogen atmosphere using flame‐dried flasks. If stated, reactions were performed in Biotage Initiator+ Microwave Synthesizer under a nitrogen atmosphere. 1H and 13C NMR spectra were recorded at 298 K on a Varian Inova 400 MHz or Bruker 500 MHz spectrometer in the solvent indicated. Chemical shifts are given in parts per million (ppm) with respect to tetramethylsilane (δ =0.00 ppm) as internal standard for 1H NMR; and CDCl3 (δ =77.16 ppm) as internal standard for 13C NMR spectroscopy. Coupling constants are reported as J values in Hertz (Hz). 1H NMR spectroscopic data are reported as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, quint = quintet, t = triplet and combination of them), coupling constants (Hz) and integration. All NMR signals were assigned on the basis of 1H NMR, 13C NMR, gCOSY, gHSQC, gHMBS and NOESY experiments. Mass spectra were recorded on a JEOL AccuTOF CS JMST100CS (ESI) mass spectrometer. Automatic flash column chromatography was performed using Biotage Isolera Spektra One, using SNAP cartridges (Biotage, 30–100 µm, 60 Å), 10–50 g. Analysis by TLC was conducted on Silica gel F254 (Merck KGaA) plates with detection by UV absorption (254 nm) where applicable, and by dipping into a solution of aqueous KMnO4/Na2CO3/NaOH solution followed by charring at ca. 150 °C. IR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer.
General Procedure for the Synthesis of Sulfonamides: Sulfonamides 3 and S1–S5 were prepared by the literature procedure reported by Murphy et al.16 The sulfonyl chloride (11 mmol, 1.1 equiv.) was added to a solution of the amine (10 mmol, 1 equiv.) and pyridine (950 mg, 12 mmol, 1.2 equiv.) in CH2Cl2 (30 mL) portion‐wise with stirring. The reaction mixture was then stirred at 23° for 12 h before evaporation of CH2Cl2 and quenching with an aqueous NaOH solution (2 n, 100 mL). The aqueous solution was rinsed with diethyl ether (2 × 50 mL) then acidified with concentrated HCl and extracted with CH2Cl2 (3 × 50 mL). The combined organic washings were dried with sodium sulfate and concentrated in vacuo. The obtained crude product was used directly in the next step without further purification.
N‐Phenylbenzenesulfonamide (S1): Sulfonamide S1 was prepared from aniline (0.9 g, 10 mmol) and benzenesulfonyl chloride (1.9 g, 11 mmol) according to the general procedure and obtained in 90 % yield as a white solid. 1H NMR [400 MHz, CDCl3]: δ = 6.58 (br. s, 1 H): δ = 7.02–7.09 (m, 2 H), 7.09–7.18 (m, 1 H), 7.20–7.29 (m, 2 H), 7.40–7.48 (m, 2 H), 7.50–7.58 (m, 1 H), 7.72–7.79 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 121.6, 125.4, 127.3, 129.3, 129.7, 133.0, 136.5, 139.0 ppm. These data were in accordance to those reported in the literature.17
4‐Methyl‐N‐phenylbenzenesulfonamide (3): Sulfonamide 3 was prepared from aniline (0.9 g, 10 mmol) and 4‐methylbenzenesulfonyl chloride (2.1 g, 11 mmol) according to the general procedure and obtained in 92 % yield as a white solid. 1H NMR [400 MHz, CDCl3]: δ = 2.35 (s, 3 H), 6.61 (br. s, 1 H), 7.02–7.10 (m, 2 H), 7.12–7.25 (m, 5 H), 7.62–7.68 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.6, 121.5, 125.1, 127.5, 129.2, 129.6, 136.0, 136.4, 143.9 ppm. These data were in accordance to those reported in the literature.17
4‐Methoxy‐N‐phenylbenzenesulfonamide (S2): Sulfonamide S2 was prepared from aniline (0.9 g, 10 mmol) and 4‐methoxybenzenesulfonyl chloride (2.3 g, 11 mmol) according to the general procedure and obtained in 81 % yield as a white solid. 1H NMR [400 MHz, CDCl3]: δ = 3.82 (s, 3 H), 6.70 (br. s, 1 H), 6.81–6.96 (m, 2 H), 7.03–7.15 (m, 3 H), 7.18–7.31 (m, 2 H), 7.59–7.78 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 55.7, 114.3, 121.6, 125.2, 129.4, 129.5, 130.7, 136.9, 163.2 ppm. These data were in accordance to those reported in the literature.17
4‐Nitro‐N‐phenylbenzenesulfonamide (S3): Sulfonamide S3 was prepared from aniline (0.9 g, 10 mmol) and 4‐nitrobenzenesulfonyl chloride (2.4 g, 11 mmol) according to the general procedure and obtained in 94 % yield as a light yellow solid. 1H NMR [400 MHz, CDCl3]: δ = 6.02 (br. s, 1 H), 7.07–7.32 (m, 5 H), 7.89–7.93 (m, 2 H), 8.22–8.30 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 122.4, 124.3, 126.6, 128.7, 129.9, 135.2, 144.5, 150.3 ppm. These data were in accordance to those reported in the literature.18
N‐Benzyl‐4‐methylbenzenesulfonamide (S4): Sulfonamide S4 was prepared from benzylamine (1.07 g, 10 mmol) and 4‐methylbenzenesulfonyl chloride (2.1 g, 11 mmol) according to the general procedure and obtained in 91 % yield as a white solid. 1H NMR [400 MHz, CDCl3]: δ = 2.32 (s, 3 H), 4.02 (d, J = 6.5 Hz, 2 H), 6.74 (br. t, J = 6.5 Hz, 1 H), 7.05–7.21 (m, 5 H), 7.27–7.31 (m, 2 H), 7.64–7.71 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 48.3, 128.3, 128.4, 129.1, 129.5, 130.9, 139.1, 139.2, 144.3 ppm. These data were in accordance to those reported in the literature.19
N,4‐Dimethylbenzenesulfonamide (S5): Sulfonamide S5 was prepared from methylamine hydrochloride (0.68 g, 10 mmol) and 4‐methylbenzenesulfonyl chloride (2.1 g, 11 mmol) according to the general procedure and obtained in 89 % yield as brown oil. 1H NMR [400 MHz, CDCl3]: δ = 2.42 (s, 3 H), 2.62 (d, J = 5.5 Hz, 3 H), 4.45 (br. s, 1 H), 7.29–7.33 (m, 2 H), 7.70–7.76 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.2, 29.5, 127.3, 129.8, 135.7, 143.3 ppm. These data were in accordance to those reported in the literature.20
General Procedure for the Synthesis of 1,2‐Dichlorovinyl Sulfonamides: 1,2‐Dichlorovinyl sulfonamides S6–S11 were prepared by the literature procedure reported by Anderson et al.21 The sulfonamide (8 mmol, 1.0 equiv.) was added dropwise at 0 °C to a suspension of sodium hydride (670 mg, 60 % dispersion in mineral oil, 16.8 mmol, 2.1 equiv.) in DMF (30 mL) and the reaction mixture was warmed to 23° in 2 h. Trichloroethene (800 µL, 8.8 mmol, 1.1 equiv.) was slowly added to this solution, which was after stirred at 50 °C for 16 h. After cooling to 23°, the reaction mixture was quenched with water (300 mL) and extracted with AcOEt (3 × 50 mL). The combined organic washings were dried with sodium sulfate, concentrated in vacuo and the crude residue was purified by column chromatography as indicated. The E‐configuration of the obtained products including the new compounds was assigned based on the 1H NMR spectra and X‐ray crystallography data known from the literature.21
N‐[(E)‐1,2‐Dichlorovinyl]‐N‐phenylbenzenesulfonamide (S6): Column chromatography (heptane/AcOEt, 40:1 → 10:1) afforded product S6 as a yellow oil (72 %). R F (silica gel, heptane/AcOEt, 10:1): 0.34 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 6.46 (s, 1 H), 7.30–7.39 (m, 5 H), 7.43–7.50 (m, 2 H), 7.58–7.63 (m, 1 H), 7.75–7.80 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 120.7, 128.6, 128.78, 128.79, 129.2, 129.4, 130.6, 133.6, 137.6, 138.5 ppm. FTIR: ν̃ = 811, 1089, 1169, 1363, 1489, 2934, 3086 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C14H13Cl2NO2S 327.9966, found 327.9954.
N‐[(E)‐1,2‐Dichlorovinyl]‐4‐methyl‐N‐phenylbenzenesulfonamide (S7): Column chromatography (heptane/AcOEt, 40:1 → 10:1) afforded the product S7 as a white solid (91 %). R F (silica gel, heptane/AcOEt, 1:1): 0.70 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.41 (s, 3 H), 6.46 (s, 1 H), 7.19–7.27 (m, 2 H), 7.30–7.39 (m, 5 H), 7.61–7.69 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 120.5, 128.6, 128.8, 129.1, 129.3, 129.4, 130.7, 135.6, 137.7, 144.6 ppm. These data were in accordance to those reported in the literature.22
N‐[(E)‐1,2‐Dichlorovinyl]‐4‐methoxy‐N‐phenylbenzenesulfonamide (S8): Column chromatography (heptane/AcOEt, 40:1 → 10:1) afforded the product S8 as a yellow oil (87 %). R F (silica gel, heptane/AcOEt, 7:1): 0.23 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 3.86 (s, 3 H), 6.45 (s, 1 H), 6.86–6.98 (m, 2 H), 7.29–7.41 (m, 5 H), 7.65–7.73 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 55.6, 113.9, 120.4, 128.7, 129.0, 129.3, 130.0, 130.7, 130.9, 137.8, 163.6 ppm. FTIR: ν̃ = 809, 1089, 1160, 1261, 1361, 1489, 1594, 2945, 3085 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C15H15Cl2NO3S 358.0071, found 358.0092.
N‐[(E)‐1,2‐Dichlorovinyl]‐4‐nitro‐N‐phenylbenzenesulfonamide (S9): Column chromatography (heptane/AcOEt, 20:1 → 2:1) afforded the product S9 as a white solid (82 %). R F (silica gel, heptane/AcOEt, 10:1): 0.11 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 6.51 (s, 1 H), 7.31–7.49 (m, 5 H), 7.81–7.99 (m, 2 H), 8.21–8.40 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 121.3, 124.0, 128.7, 129.72, 129.77, 129.83, 129.84, 137.0, 144.1, 150.6 ppm. FTIR: ν̃ = 685, 738, 1094, 1171, 1348, 1530, 3082, 3102 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C14H12Cl2N2O4S 372.9817, found 372.9834.
N‐Benzyl‐N‐[(E)‐1,2‐dichlorovinyl]‐4‐methylbenzenesulfonamide (S10): Column chromatography (heptane/AcOEt, 40:1 → 4:1) afforded the product S10 as a white solid (73 %). R F (silica gel, heptane/AcOEt, 10:1): 0.23 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.47 (s, 3 H), 3.72–4.81 (br. s, 2 H), 6.27 (s, 1 H), 7.28–7.34 (m, 5 H), 7.32–7.37 (m, 2 H), 7.80–7.86 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 52.0, 121.7, 128.23, 128.28, 129.3, 129.4, 129.8, 133.5, 135.3, 144.7 ppm. These data were in accordance to those reported in the literature.21
N‐[(E)‐1,2‐Dichlorovinyl]‐N,4‐dimethylbenzenesulfonamide (S11): Column chromatography (heptane/ AcOEt 40:1 → 10:1) afforded the product S11 as brown oil (64 %). R F (silica gel, heptane/AcOEt, 10:1): 0.13 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.45 (s, 3 H), 2.94 (s, 3 H), 6.39 (s, 1 H), 7.31–7.37 (m, 2 H), 7.79–7.85 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 37.9, 121.4, 127.8, 129.2, 129.5, 132.4, 143.2 ppm. FTIR: ν̃ = 693, 722, 1084, 1160, 1333, 1498, 2986, 3012 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C10H13Cl2NO2S 279.9966, found 279.9983.
General Procedure A for the Syntheses of Sulfonyl Ynamines: Sulfonyl Ynamines 1a, 1d, 1e, 1g, 1h, 1i, 1j and 1k were prepared by the literature procedure reported by Davies et al.23 n‐Butyllithium (3.8 mL, 1.6 m in THF, 6 mmol, 2.1 equiv.) was slowly added to a stirred solution of 1,2‐dichlorovinyl sulfonamide (5 mmol, 1.0 equiv.) in THF (30 mL) under an argon atmosphere at –78 °C. After stirring for 1 h, the lithium acetylide was treated with the corresponding electrophile and stirred for 1 h at –78 °C. The mixture was warmed to 23 °C and stirred for 2–3 h. The reaction mixture was quenched with brine (100 mL) and extracted with Et2O (2 × 50 mL). The combined organic washings were dried with sodium sulfate, concentrated in vacuo and the crude residue was purified by column chromatography as indicated.
General Procedure B for the Syntheses of Sulfonyl Ynamines: Sulfonyl Ynamines 1b and 1c were prepared by the literature procedure reported by Wolf et al.24 Sulfonyl ynamine 1d (1.0 g, 3.7 mmol, 1.0 equiv.), CuI (70 mg, 0.37 mmol, 0.1 equiv.) and N,N‐diisopropylethylamine (1.5 mL, 7.4 mmol, 2.0 equiv.) were dissolved in chloroform (20 mL) under a nitrogen atmosphere. After 30 min, the acyl chloride (5.6 mmol, 1.5 equiv.) was added, and the mixture was stirred until completion as determined by TLC. Solvent was removed in vacuo and the crude residue was purified by column chromatography as indicated.
General Procedure C for the Syntheses of Sulfonyl Ynamines: Sulfonyl Ynamine 1f was prepared by the literature procedure reported by Hsung et al.25 Sulfonyl ynamine 1d (1.0 g, 3.7 mmol, 1.0 equiv.), iodobenzene (830 mg, 4.1 mmol, 1.1 equiv.) and Pd(PPh3)4 (214 mg, 0.185 mmol, 0.05 equiv.) were dissolved in Et3N/toluene mixture (2:1, 36 mL) under a nitrogen atmosphere. The solution was stirred at 23 °C for 10 min, and CuI (11 mg, 0.06 mmol, 0.015 equiv.) was then added. After heating the reaction mixture at 60 °C for 12 h, the mixture was diluted with AcOEt, filtered through a diatomaceous earth pad, and concentrated in vacuo. The resulting crude residue was purified by silica gel flash column chromatography as indicated.
Ethyl 3‐(4‐Methyl‐N‐phenylbenzenesulfonamido)propanoate (1a): Sulfonyl ynamine 1a was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S7 (1.7 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (heptane/AcOEt, 40:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1a as a white solid (82 %). R F (silica gel, heptane/AcOEt, 10:1): 0.11 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.31 (t, J = 7.1 Hz, 3 H), 2.46 (s, 3 H), 4.24 (q, J = 7.1 Hz, 2 H), 7.16–7.23 (m, 2 H), 7.29–7.34 (m, 2 H), 7.34–7.38 (m, 3 H), 7.58–7.67 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 21.8, 61.7, 66.6, 82.2, 126.5, 128.4, 129.3, 129.6, 130.0, 133.1, 137.5, 145.9, 154.8 ppm. These data were in accordance to those reported in the literature.23
N‐(4,4‐Dimethyl‐3‐oxopent‐1‐yn‐1‐yl)‐4‐methyl‐N‐phenylbenzenesulfon‐amide (1b): Sulfonyl ynamine 1b was prepared according to general procedure B. The reaction with pivaloyl chloride (690 µL, 6.6 mmol) was performed at 30 °C for 18 h. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1b as a light yellow oil (79 %). R F (silica gel, toluene): 0.32 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.20 (s, 9 H), 2.40 (s, 3 H), 7.16–7.20 (m, 2 H), 7.24–7.28 (m, 2 H), 7.30–7.34 (m, 3 H), 7.53–7.59 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.6, 26.3, 44.5, 73.6, 89.1, 126.3, 128.3, 129.1, 129.3, 129.7, 133.2, 127.4, 145.6, 193.2 ppm. These data were in accordance to those reported in the literature.24
4‐Methyl‐N‐(3‐oxo‐3‐phenylprop‐1‐yn‐1‐yl)‐N‐phenylbenzenesulfonamide (1c): Sulfonyl ynamine 1c was prepared according to general procedure B. The reaction with benzoyl chloride (767 µL, 6.6 mmol) was performed at 30 °C for 24 h. Column chromatography (heptane/AcOEt, 20:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1c as a light yellow oil (86 %). R F (silica gel, heptane/AcOEt, 10:1): 0.33 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.42 (s, 3 H), 7.25–7.29 (m, 4 H), 7.36–7.41 (m, 3 H), 7.47–7.55 (m, 2 H), 7.60–7.65 (m, 3 H), 8.14–8.26 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 74.9, 90.2, 126.4, 128.2, 128.7, 129.2, 129.2, 129.5, 129.9, 132.9, 133.5, 136.9, 137.1, 145.9, 176.8 ppm. These data were in accordance to those reported in the literature.24
N‐Ethynyl‐4‐methyl‐N‐phenylbenzenesulfonamide (1d): Sulfonyl ynamine 1d was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S7 (1.7 g, 5.0 mmol). The lithium acetylide was treated with water (10 mL) and the resulting mixture was stirred at 23 °C for 2 h. Column chromatography (heptane/AcOEt, 20:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1d as a light yellow oil (81 %). R F (silica gel, heptane/AcOEt, 10:1): 0.18 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.44 (s, 3 H), 2.83 (s, 1 H), 7.23–7.34 (m, 7 H), 7.56–7.61 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 59.1, 75.8, 126.2, 128.2, 128.3, 129.0, 129.4, 132.7, 138.4, 145.0 ppm. These data were in accordance to those reported in the literature.26
4‐Methyl‐N‐phenyl‐N‐[(trimethylsilyl)ethynyl]benzenesulfonamide (1e): Sulfonyl ynamine 1e was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S7 (1.7 g, 5.0 mmol). The lithium acetylide was treated with trimethylsilyl chloride (952 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (heptane/AcOEt, 20:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1e as a white solid (52 %). RF (silica gel, heptane/AcOEt, 10:1): 0.27 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 0.17 (s, 9 H), 2.45 (s, 3 H), 7.21–7.25 (m, 2 H), 7.26–7.30 (m, 2 H), 7.30–7.36 (m, 3 H), 7.54–7.60 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 0.16, 21.9, 73.4, 93.2, 126.3, 128.3, 128.6, 129.2, 129.5, 133.0, 138.7, 145.1 ppm. FTIR: ν̃ = 752, 834, 902, 1056, 1168, 1372, 1476, 1592, 2137, 2161, 2887, 3034 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H23NO2SSi 344.1141, found 344.1134.
4‐Methyl‐N‐phenyl‐N‐(phenylethynyl)benzenesulfonamide (1f): Sulfonyl ynamine 1f was prepared according to general procedure C. Column chromatography (heptane/AcOEt, 20:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1f as a light yellow solid (80 %). R F (silica gel, heptane/AcOEt, 10:1): 0.27 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.45 (s, 3 H), 7.27–7.36 (m, 9 H), 7.36–7.42 (m, 3 H), 7.61–7.65 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 21.7, 70.6, 83.2, 122.7, 126.5, 126.3, 128.1, 128.32, 128.34, 129.0, 129.5, 131.4, 132.9, 139.0, 145.1 ppm. These data were in accordance to those reported in the literature.27
Ethyl 3‐(N‐Phenylbenzenesulfonamido)propanoate (1g): Sulfonyl ynamine 1g was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S6 (1.6 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1g as a white solid (74 %). R F (silica gel, toluene): 0.35 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.31 (t, J = 7.1 Hz, 3 H), 4.24 (q, J = 7.1 Hz, 2 H), 7.14–7.22 (m, 2 H), 7.30–7.41 (m, 3 H), 7.49–7.58 (m, 2 H), 7.64–7.73 (m, 1 H), 7.74–7.80 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 61.8, 66.5, 81.9, 126.6, 128.3, 129.27, 129.29, 129.5, 134.6, 135.8, 137.0, 154.0 ppm. FTIR: ν̃ = 685, 726, 1088, 1123, 1204, 1372, 1702, 2218, 2982 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C17H17NO4S 330.0800, found 330.0812.
Ethyl 3‐(4‐Methoxy‐N‐phenylbenzenesulfonamido)propanoate (1h): Sulfonyl ynamine 1h was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S8 (1.8 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1h as colorless oil (84 %). R F (silica gel, toluene): 0.25 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.30 (t, J = 7.1 Hz, 3 H), 3.88 (s, 3 H), 4.23 (q, J = 7.1 Hz, 2 H), 6.91–7.03 (m, 2 H), 7.15–7.24 (m, 2 H), 7.3–7.40 (m, 3 H), 7.64–7.73 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 55.8, 61.7, 66.6, 82.5, 114.4, 126.4, 127.2, 129.2, 129.5, 130.6, 137.2, 154.1, 164.4 ppm. FTIR: ν̃ = 689, 1087, 1123, 1371, 1496, 1593, 1702, 2216, 2981 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO5S 360.0906, found 360.0922.
Ethyl 3‐(4‐Nitro‐N‐phenylbenzenesulfonamido)propanoate (1i) and Ethyl 3,3‐Bis(4‐nitro‐N‐phenyl‐benzenesulfonamido)acrylate (15): Sulfonyl ynamine 1i and compound 15 were prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S9 (1.9 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 3 h. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1i (42 %) and product 15 (41 %) as white solids. Product 15 was a common side product during the course of the reaction.28 Compound 1i: RF (silica gel, toluene): 0.33 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.32 (t, J = 7.1 Hz, 3 H), 4.25 (q, J = 7.1 Hz, 2 H), 7.17–7.23 (m, 2 H), 7.36–7.46 (m, 3 H), 7.92–8.00 (m, 2 H), 8.33–8.42 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.1, 62.0, 66.7, 80.4, 124.5, 126.5, 129.7, 129.81, 129.84, 136.5, 140.9, 150.7, 153.6 ppm. FTIR: ν̃ = 690, 740, 854, 1086, 1126, 1184, 1206, 1348, 1533, 1593, 1706, 2223, 2925, 3107 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C17H16N2O6S 375.0651, found 375.0659. Compound 15: RF (silica gel, toluene): 0.07 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.41 (t, J = 7.1 Hz, 3 H), 4.33 (q, J = 7.1 Hz, 2 H), 6.14 (s, 1 H), 7.11–7.16 (m, 2 H), 7.20–7.23 (m, 2 H), 7.24–7.27 (m, 2 H), 7.33–7.38 (m, 2 H), 7.40–7.45 (m, 2 H), 7.45–7.50 (m, 3 H), 7.52–7.57 (m, 1 H), 7.93–8.00 (m, 2 H), 8.09–8.18 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.1, 61.6, 110.0, 117.5, 123.4, 124.1, 129.4, 129.6, 129.7, 129.93, 129.97, 129.99, 130.0, 136.0, 136.3, 139.9, 143.4, 144.2, 149.9, 150.4, 163.9 ppm. FTIR: ν̃ = 694, 740, 855, 1086, 1139, 1172, 1202, 1350, 1531, 1720, 3106 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C29H26N4O10S2 653.1012, found 653.1034.
Ethyl 3‐(N,4‐Dimethylbenzenesulfonamido)propanoate (1j): Sulfonyl ynamine 1j was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S11 (1.4 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (heptane/AcOEt, 20:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1j as a white solid (71 %). R F (silica gel, heptane/AcOEt, 1:1): 0.6 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.31 (t, J = 7.1 Hz, 3 H), 2.47 (s, 3 H), 3.17 (s, 3 H), 4.23 (q, J = 7.1 Hz, 2 H), 7.35–7.44 (m, 2 H), 7.78–7.92 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 21.8, 38.5, 61.6, 66.0, 83.5, 128.0, 130.2, 133.0, 145.8, 154.1 ppm. These data were in accordance to those reported in the literature.29
Ethyl 3‐(N‐Benzyl‐4‐methylbenzenesulfonamido)propanoate (1k): Sulfonyl ynamine 1k was prepared according to general procedure A using 1,2‐dichlorovinyl sulfonamide S10 (1.8 g, 5.0 mmol). The lithium acetylide was treated with freshly distilled ethyl chloroformate (714 µL, 7.5 mmol) at –78 °C for 15 min and at 23 °C for 2 h. Column chromatography (heptane/AcOEt, 40:1 → 10:1; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 1k as a white solid (87 %). R F (silica gel, heptane/AcOEt, 10:1): 0.17 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.28 (t, J = 7.1 Hz, 3 H), 2.44 (s, 3 H), 4.18 (q, J = 7.1 Hz, 2 H), 4.62 (s, 2 H), 7.24–7.33 (m, 7 H), 7.68–7.75 (m, 2 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 21.6, 55.3, 61.4, 68.1, 82.5, 127.6, 128.5, 128.6, 129.3, 129.7, 133.5, 134.1, 145.4, 153.9 ppm. These data were in accordance to those reported in the literature.22
Products of Palladium‐Catalyzed Intramolecular Hydroarylation of Sulfonyl Ynamine 1a: Sulfonyl ynamine 1a (343 mg, 1.0 mmol, 1.0 equiv.) was dissolved in toluene (10 mL) under a nitrogen atmosphere in a Biotage microwave vial (10.0–20.0 mL) equipped with a magnetic stirring bar. Pd(OAc)2 (11 mg, 0.05 mmol, 0.05 equiv.) and tri(p‐tolyl)phosphine (30 mg, 0.1 mmol, 0.1 equiv.) were added at 23 °C. The vial was covered with a Teflon septum and secured via a crimped aluminum cap. The reaction was irradiated in a Biotage Initiator microwave at 100 °C for 18 h (30 second pre‐stir, Fixed Hold Time On, Low absorbance level). The reaction mixture was quenched with brine (50 mL) and extracted with AcOEt (2 × 20 mL). The resulting organic washings were dried with sodium sulfate, concentrated in vacuo and the crude residue was purified by column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography).
Ethyl (E)‐2‐[5‐Methyl‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2a]: R F (silica gel, toluene): 0.33 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.26 (t, J = 7.1 Hz, 3 H), 2.57 (s, 3 H), 4.18 (q, J = 7.1 Hz, 2 H), 5.11 (s, 1 H), 7.47–7.51 (m, 2 H), 7.54–7.62 (m, 4 H), 7.82 (d, J = 7.9 Hz, 1 H), 9.23 (quint, J = 0.8 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 22.2, 60.5, 97.0, 120.9, 127.4, 129.8, 130.36, 130.39, 130.43, 130.5, 131.0, 133.1, 144.8, 146.2, 166.1 ppm. FTIR: ν̃ = 714, 902, 1089, 1174, 1302, 1512, 1635, 1737, 2992 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO4S 344.0957, found 344.0966.
Ethyl (Z)‐2‐[5‐Methyl‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(Z)‐2a]: R F (silica gel, toluene): 0.28 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.01 (t, J = 7.1 Hz, 3 H), 2.54 (s, 3 H), 3.66 (q, J = 7.1 Hz, 2 H), 5.83 (s, 1 H), 7.36–7.49 (m, 5 H), 7.55 (m, J = 8.0, 1.2, 0.6 Hz, 1 H), 7.62 (quint, J = 0.6 Hz, 1 H), 7.79 (d, J = 8.0 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 13.9, 22.0, 60.4, 93.0, 121.5, 122.0, 126.9, 128.6, 128.9, 129.5, 130.2, 133.1, 134.9, 140.2, 144.9, 164.0 ppm. FTIR: ν̃ = 693, 907, 1082, 1145, 1266, 1327, 1494, 1635, 1708, 3029 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO4S 344.0957, found 344.0943.
4‐Methyl‐N‐Phenylbenzenesulfonamide (3): The spectroscopic data were identical to those reported above.
Ethyl 3,3‐Bis(4‐methyl‐N‐phenylbenzenesulfonamido)acrylate (4): R F (silica gel, toluene): 0.06 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.39 (t, J = 7.1 Hz, 3 H), 2.30 (s, 3 H), 2.38 (s, 3 H), 4.33 (q, J = 7.1 Hz, 2 H), 6.09 (s, 1 H), 6.86–6.94 (m, 4 H), 7.06–7.12 (m, 2 H), 7.12–7.19 (m, 6 H), 7.26–7.33 (m, 2 H), 7.33–7.40 (m, 3 H), 7.42–7.48 (m, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.1, 21.5, 21.6, 61.1, 115.5, 127.3, 128.18, 128.23, 128.93, 128.97, 129.0, 129.4, 129.5, 130.0, 130.2, 135.1, 136.0, 136.8, 137.1, 140.6, 143.5, 144.3, 164.7 ppm. FTIR: ν̃ = 695, 814, 1087, 1139, 1167, 1362, 1489, 1531, 1719, 2981 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C31H32N2O6S2 591.1624, found 591.1638.
Ethyl (E)‐3‐(4‐Methyl‐N‐phenylbenzenesulfonamido)acrylate (5): R F (silica gel, toluene): 0.12 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.24 (t, J = 7.1 Hz, 3 H), 2.44 (s, 3 H), 4.14 (q, J = 7.1 Hz, 2 H), 4.62 (d, J = 13.8 Hz, 1 H), 6.88–6.97 (m, 2 H), 7.27–7.32 (m, 2 H), 7.35–7.45 (m, 3 H), 7.53–7.60 (m, 2 H), 8.37 (d, J = 13.7 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.3, 21.7, 60.1, 100.0, 121.7, 127.8, 129.7, 129.86, 129.89, 134.8, 135.1, 143.9, 144.9, 167.1 ppm. FTIR: ν̃ = 727, 906, 1089, 1170, 1368, 1624, 1737, 2245, 2981 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H21NO4S 346.1113, found 346.1131.
General Procedure for the Intramolecular Hydroarylation of Sulfonyl Ynamines 1a–c and 1g–k: Sulfonyl ynamine 1 (1.0 mmol, 1.0 equiv.) was dissolved in toluene (10 mL) under a nitrogen atmosphere in a Biotage microwave vial (10.0–20.0 mL) equipped with a magnetic stirring bar. Pd(OAc)2 (11 mg, 0.05 mmol, 0.05 equiv.) and tri(p‐tolyl)phosphine (30 mg, 0.1 mmol, 0.1 equiv.) were added at 23 °C. The vial was covered with a Teflon septum and secured via a crimped aluminum cap. The reaction was irradiated in a Biotage Initiator microwave at 100 °C for 18 h (30 second pre‐stir, Fixed Hold Time On, Low absorbance level). The reaction mixture was quenched with brine (50 mL) and extracted with AcOEt (2 × 20 mL). The resulting organic washings were dried with sodium sulfate, concentrated in vacuo and the crude residue was purified by column chromatography as indicated.
Ethyl (E)‐2‐[5‐Methyl‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2a]: (E)‐1,2‐Benzothiazoledione 2a was prepared according to general procedure using sulfonyl ynamine 1a (343 mg, 1.0 mmol). Ratio E/Z = 98:2 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded product (E)‐2a as a white solid (127 mg, 37 %). The spectroscopic data were identical to those reported above.
(E)‐3,3‐Dimethyl‐1‐[5‐methyl‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]butan‐2‐one [(E)‐2b]: (E)‐1,2‐Benzothiazoledione 2b was prepared according to general procedure using sulfonyl ynamine 1b (355 mg, 1.0 mmol). Ratio E/Z >, 99:1 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2b as a white solid (121 mg, 34 %). R F (silica gel, toluene): 0.28 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.05 (s, 9 H), 2.57 (s, 3 H), 5.71 (s, 1 H), 7.48–7.53 (m, 2 H), 7.54–7.65 (m, 4 H), 7.82 (d, J = 7.9 Hz, 1 H), 9.10 (quint, J = 0.6 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 22.3, 26.8, 44.7, 101.5, 121.0, 127.8, 129.4, 130.4, 130.5, 130.6, 130.8, 130.9, 133.4, 145.1, 145.6, 204.3 ppm. FTIR: ν̃ = 695, 966, 1078, 1183, 1322, 1566, 1673, 2965 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C20H23NO3S 356.1320, found 356.1343.
(E)‐2‐[5‐methyl‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]‐1‐phenylethan‐1‐one [(E)‐2c]: (E)‐1,2‐Benzothiazoledione 2c was prepared according to general procedure using sulfonyl ynamine 1c (375 mg, 1.0 mmol). Ratio E/Z = 98:2 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2c as a white solid (116 mg, 31 %). R F (silica gel, toluene): 0.31 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 2.57 (s, 3 H), 6.09 (s, 1 H), 7.37–7.45 (m, 2 H), 7.48–7.55 (m, 1 H), 7.56–7.66 (m, 6 H), 7.72–7.79 (m, 2 H), 7.85 (d, J = 8.0 Hz, 1 H), 9.00 (quint, J = 0.5 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 22.2, 102.4, 121.1, 127.4, 127.7, 128.2, 128.6, 129.1, 130.5, 130.6, 130.7, 130.9, 132.8, 133.6, 139.4, 145.1, 146.4, 189.7 ppm. FTIR: ν̃ = 696, 959, 1185, 1323, 1565, 1653, 1724, 2922, 3064 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C22H19NO3S 376.1007, found 376.1015.
Ethyl (E)‐2‐[1,1‐Dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2g]: (E)‐1,2‐Benzothiazoledione 2g was prepared according to general procedure using sulfonyl ynamine 1g (330 mg, 1.0 mmol). Ratio E/Z = 98:2 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2g as a white solid (112 mg, 34 %). R F (silica gel, toluene): 0.31 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.28 (t, J = 7.1 Hz, 3 H), 4.20 (q, J = 7.1 Hz, 2 H), 5.16 (s, 1 H), 7.46–7.56 (m, 2 H), 7.57–7.67 (m, 3 H), 7.76–7.85 (m, 2 H), 7.92–8.02 (m, 1 H), 9.44 (m, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.4, 60.7, 97.6, 121.3, 127.4, 129.9, 130.5, 130.6, 130.7, 131.2, 132.5, 133.3, 133.9, 146.2, 166.3 ppm. FTIR: ν̃ = 692, 910, 1045, 1099, 1157, 1186, 1324, 1619, 1708, 2925 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C17H17NO4S 330.0800, found 330.0827.
Ethyl (E)‐2‐[5‐Methoxy‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2h]: (E)‐1,2‐Benzothiazoledione 2h was prepared according to general procedure using sulfonyl ynamine 1h (360 mg, 1.0 mmol). Ratio E/Z = 95:5 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2h as a white solid (119 mg, 33 %). R F (silica gel, toluene): 0.17 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.25 (t, J = 7.1 Hz, 3 H), 3.98 (s, 3 H), 4.16 (q, J = 7.1 Hz, 2 H), 5.12 (s, 1 H), 7.25 (dd, J = 8.7, 2.2 Hz, 1 H), 7.47–7.52 (m, 2 H), 7.53–7.64 (m, 3 H), 7.83 (d, J = 8.6 Hz, 1 H), 9.14 (d, J = 2.3 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.3, 56.1, 60.5, 97.2, 113.4, 119.7, 122.4, 125.1, 129.6, 130.43, 130.49, 130.53, 131.0, 146.3, 164.0, 166.2 ppm. FTIR: ν̃ = 1103, 1183, 1252, 1307, 1473, 1583, 1621, 1707, 2988, 3118 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO5S 360.0906, found 360.0919.
Ethyl (E)‐2‐[5‐Nitro‐1,1‐dioxo‐2‐phenyl‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2i]: (E)‐1,2‐Benzothiazoledione 2i was prepared according to general procedure using sulfonyl ynamine 1i (375 mg, 1.0 mmol). Ratio E/Z >, 99:1 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2i as a white solid (113 mg, 30 %). R F (silica gel, toluene): 0.28 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.28 (t, J = 7.1 Hz, 3 H), 4.24 (q, J = 7.1 Hz, 2 H), 5.27 (s, 1 H), 7.46–7.56 (m, 2 H), 7.58–7.69 (m, 3 H), 8.12 (d, J = 8.5 Hz, 1 H), 8.61 (dd, J = 8.5, 1.9 Hz, 1 H), 10.43 (d, J = 1.9 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.2, 61.2, 99.9, 122.5, 125.6, 127.3, 129.1, 129.6, 130.7, 130.9, 131.1, 137.6, 143.8, 151.2, 165.6 ppm. FTIR: ν̃ = 694, 740, 1095, 1113, 1186, 1341, 1537, 1706, 2926, 3111 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C17H16N2O6S 375.0651, found 375.0667.
Ethyl (E)‐2‐[2,5‐Dimethyl‐1,1‐dioxo‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2j]: (E)‐1,2‐Benzothiazoledione 2j was prepared according to general procedure using sulfonyl ynamine 1j (281 mg, 1.0 mmol). Ratio E/Z = 95:5 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2j as colorless oil (87 mg, 31 %). R F (silica gel, toluene): 0.23 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.35 (t, J = 7.1 Hz, 3 H), 2.54 (s, 3 H), 3.15 (s, 3 H), 4.25 (q, J = 7.1 Hz, 2 H), 5.33 (s, 1 H), 7.50 (d, J = 7.9 Hz, 1 H), 7.76 (d, J = 7.9 Hz, 1 H), 9.17 (s, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.3, 22.2, 26.5, 60.5, 95.0, 120.8, 127.9, 129.8, 130.5, 132.7, 144.8, 144.9, 166.0 ppm. FTIR: ν̃ = 701, 823, 1039, 1137, 1159, 1315, 1619, 1708, 2982 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C13H17NO4S 282.0800, found 282.0819.
Ethyl (E)‐2‐[2‐Benzyl‐5‐methyl‐1,1‐dioxo‐1,2‐benzothiazol‐3(2H)‐ylidene]acetate [(E)‐2k] and Ethyl (E)‐2‐[2‐(4‐Methylbenzenesulfonyl)isoindolin‐1‐ylidene]acetate (6): (E)‐1,2‐Benzothiazoledione 2k and compound 6 were prepared according to general procedure using sulfonyl ynamine 1k (357 mg, 1.0 mmol). Ratio E/Z >, 99:1 was determined by 1H NMR spectroscopy. Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product (E)‐2k as a white solid (93 mg, 26 %) and product 6 as a colorless oil (36 mg, 10 %). Compound (E)‐2k: RF (silica gel, toluene): 0.30 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.26 (t, J = 7.1 Hz, 3 H), 2.53 (s, 3 H), 4.15 (q, J = 7.1 Hz, 2 H), 4.84 (s, 2 H), 5.28 (s, 1 H), 7.27–7.33 (m, 1 H), 7.34–7.40 (m, 2 H), 7.40–7.44 (m, 2 H), 7.52 (dd, J = 7.9, 0.6 Hz, 1 H), 7.81 (d, J = 7.9 Hz, 1 H), 9.13 (quint, J = 0.6 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.4, 22.4, 44.5, 60.6, 96.5, 121.0, 127.2, 127.8, 128.1, 129.1, 130.1, 130.4, 132.9, 134.1, 143.7, 145.1, 166.0 ppm. FTIR: ν̃ = 670, 732, 831, 1136, 1168, 1311, 1626, 1706, 2986, 3098 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C19H21NO4S 358.1113, found 358.1118. Compound 6: RF (silica gel, toluene): 0.21 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.32 (t, J = 7.1 Hz, 3 H), 2.40 (s, 3 H), 4.19 (q, J = 7.1 Hz, 2 H), 5.02 (s, 2 H), 6.43 (s, 1 H), 7.28–7.39 (m, 4 H), 7.41–7.47 (m, 1 H), 7.76–7.83 (m, 2 H), 9.13 (d, J = 8.1 Hz, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.6, 21.8, 55.4, 60.1, 96.7, 121.9, 127.7, 128.4, 128.9, 130.1, 131.3, 133.0, 134.6, 138.1, 145.1, 152.0, 167.0 ppm. FTIR: ν̃ = 667, 800, 1089, 1165, 1344, 1620, 1705, 2922 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C19H21NO4S 358.1113, found 358.1132.
Intramolecular Hydroarylation of 1d, 1e and 1f: Sulfonyl ynamines 1d, 1e or 1f were submitted to the reaction conditions according to general procedure. 1H NMR spectroscopy of the crude product indicated partial degradation of the starting material to 4‐methyl‐N‐phenylbenzenesulfonamide 3 and additional unidentified products. The formation of products 2d, 2e or 2f was not observed.
Study of Equilibration Between E‐ and Z‐Isomers of 2a: (E)‐1,2‐Benzothiazole‐1,1‐dione 2a or (Z)‐1,2‐Benzothiazole‐1,1‐dione 2a (69 mg, 0.2 mmol, 1.0 equiv.) were independently dissolved in toluene (2 mL) under a nitrogen atmosphere in a Biotage microwave vial (2.0–5.0 mL) equipped with a magnetic stirring bar. Pd(OAc)2 (2.2 mg, 0.01 mmol, 0.05 equiv.) and tri(p‐tolyl)phosphine (6.1 mg, 0.02 mmol, 0.1 equiv.) were added at 23 °C. The vial was covered with a Teflon septum and secured via a crimped aluminum cap. The reaction was irradiated in a Biotage Initiator microwave at 100 °C for 18 h (30 second pre‐stir, Fixed Hold Time On, Low absorbance level). The reaction mixture was quenched with brine (50 mL) and extracted with AcOEt (2 × 20 mL). The combined organic washings were dried with sodium sulfate, filtered off and concentrated in vacuo. 1H NMR spectroscopy of the crude product indicated a mixture of isomers with ratio E/Z = 85:15. Additionally, partial decomposition of the starting material was observed.
General Procedure for Photochemical Rearrangement of 1,2‐Benzothiazole‐1,1‐diones: To 25 mL quartz flask was added 1,2‐benzothiazole‐1,1‐dione 2a, 2g, 2h or 2k (0.1–1.0 mmol, 1.0 equiv.) in 10 mL of deoxygenated MeCN with stirring. This flask was irradiated in a Rayonet RMR‐600 photochemical reactor, using eight lamps of 300 nm of wavelength for 24 h with internal temperature of 50 °C. After cooling to 23 °C, the solvent was removed in vacuo and the crude residue was purified by column chromatography as indicated.
Ethyl 5‐Methyl‐1,1‐dioxo‐3‐(phenylamino)‐1‐benzothiophene‐2‐carboxylate (12a): 3‐Amino‐1‐benzothiophene‐1,1‐dione 12a was prepared according to general procedure using 1,2‐benzothiazole‐1,1‐dione 2a (343 mg, 1.0 mmol). Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 12a as a white solid (316 mg, 92 %). R F (silica gel, toluene): 0.22 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.45 (t, J = 7.1 Hz, 3 H), 2.12 (s, 3 H), 4.43 (q, J = 7.1 Hz, 2 H), 6.40 (quint, J = 0.5 Hz, 1 H), 7.30–7.34 (m, 2 H), 7.39 (ddd, J = 7.8, 1.3, 0.7 Hz, 1 H), 7.44–7.54 (m, 3 H), 7.71 (d, J = 7.8 Hz, 1 H), 10.43 (br. s, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.4, 21.7, 61.1, 100.1, 121.2, 125.9, 126.5, 127.0, 128.6, 129.8, 133.6, 137.4, 138.2, 142.8, 155.0, 164.5 ppm. FTIR: ν̃ = 704, 730, 1027, 1122, 1160, 1247, 1285, 1569, 1657, 2854, 2925 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO4S 344.0957, found 344.0963.
Ethyl 1,1‐Dioxo‐3‐(phenylamino)‐1‐benzothiophene‐2‐carboxylate (12b): 3‐Amino‐1‐benzothiophene‐1,1‐dione 12b was prepared according to general procedure using 1,2‐benzothiazoledione 2g (33 mg, 0.1 mmol). Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 12b as a white solid (31 mg, 96 %). R F (silica gel, toluene): 0.24 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.45 (t, J = 7.1 Hz, 3 H), 4.44 (q, J = 7.1 Hz, 2 H), 6.66 (d, J = 8.0 Hz, 1 H), 7.23–7.28 (m, 1 H), 7.29–7.36 (m, 2 H), 7.43–7.54 (m, 3 H), 7.58–7.62 (m, 1 H), 7.83 (d, J = 7.6 Hz, 1 H), 10.44 (br. s, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.4, 61.3, 99.9, 121.6, 125.7, 125.9, 126.9, 128.7, 130.0, 132.1, 133.2, 137.4, 141.0, 154.8, 164.6 ppm. FTIR: ν̃ = 702, 763, 1161, 1246, 1284, 1434, 1561, 1610, 1657, 2854, 2925 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C17H17NO4S 330.0800, found 330.0813.
Ethyl 5‐Methoxy‐1,1‐dioxo‐3‐(phenylamino)‐1‐benzothiophenecarboxylate (12c): 3‐Amino‐1‐benzothiophene‐1,1‐dione 12c was prepared according to general procedure using 1,2‐benzothiazole‐1,1‐dione 2h (36 mg, 0.1 mmol). Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 12c as a white solid (31 mg, 87 %). R F (silica gel, toluene): 0.20 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.45 (t, J = 7.1 Hz, 3 H), 3.48 (s, 3 H), 4.43 (q, J = 7.1 Hz, 2 H), 6.13 (d, J = 2.2 Hz, 2 H), 7.04 (dd, J = 8.5, 2.2 Hz, 1 H), 7.32–7.38 (m, 2 H), 7.44–7.53 (m, 3 H), 7.72 (d, J = 8.5 Hz, 1 H), 10.41 (br. s, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.4, 55.4, 61.2, 100.7, 111.6, 118.3, 122.8, 127.2, 127.8, 128.8, 130.0, 132.7, 137.4, 154.4, 162.3, 164.5 ppm. FTIR: ν̃ = 703, 729, 910, 1021, 1162, 1245, 1567, 1655, 2926 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C18H19NO5S 360.0906, found 330.0927.
Ethyl 3‐(Benzylamino)‐5‐methyl‐1,1‐dioxo‐1‐benzothiophene‐2‐carboxylate (12d): 3‐Amino‐1‐benzothiophene‐1,1‐dione 12d was prepared according to general procedure using 1,2‐benzothiazole‐1,1‐dione 2k (36 mg, 0.1 mmol). Column chromatography (toluene; silica gel was washed with 1 % Et3N in heptane before being used for column chromatography) afforded the product 12d as a white solid (34 mg, 95 %). R F (silica gel, toluene): 0.25 (UV, KMnO4 solution). 1H NMR [400 MHz, CDCl3]: δ = 1.40 (t, J = 7.1 Hz, 3 H), 2.40 (s, 3 H), 4.36 (q, J = 7.1 Hz, 2 H), 5.02 (d, J = 5.9 Hz, 2 H), 7.33–7.40 (m, 3 H), 7.40–7.46 (m, 2 H), 7.49 (d, J = 7.8 Hz, 1 H), 7.59 (s, 1 H), 7.77 (d, J = 7.8 Hz, 1 H), 9.44 (br. s, 1 H) ppm. 13C NMR [101 MHz, CDCl3]: δ = 14.5, 22.2, 50.0, 61.0, 100.1, 121.2, 125.9, 126.5, 127.0, 128.6, 129.8, 133.6, 137.4, 138.2, 142.8, 154.9, 164.5 ppm. FTIR: ν̃ = 735, 786, 1141, 1216, 1268, 1577, 1659, 1738, 2925, 3254 cm–1. HRMS (ESI‐TOF) m/z: [M + H]+ calcd. for C19H21NO4S 358.1113, found 358.1133.
https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/ejoc.201801062 1828629 (for 2a), 1828630 (for 12a), and 1828631 (for 12d) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Supporting information
Supporting Information
Acknowledgements
Financial support for this research was provided by the FP7Marie Curie Actions of the European Commission via the ITN ECHONET (MCITN‐2012‐316379). Enrique Gómez‐Bengoa and Lia Sotorríos thank MINECO (CTQ2016‐78083‐P) and IZO‐SGI SGIker of UPV‐EHU for human and technical support. Huub van Amerongen is kindly acknowledged for the preparation of the starting materials.
Contributor Information
Enrique Gómez‐Bengoa, Email: enrique.gomez@ehu.es.
Floris P. J. T. Rutjes, Email: floris.rutjes@ru.nl.
References
- 1.a) Dekorver K. A., Li H., Lohse A. G., Hayashi R., Lu Z., Zhang Y. and Hsung R. P., Chem. Rev., 2010, 110, 5064–5106; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Evano G., Coste A. and Jouvin K., Angew. Chem. Int. Ed., 2010, 49, 2840; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2010, 122, 2902–2921 [Google Scholar]; Evano G., Coste A. and Jouvin K., Angew. Chem. Int. Ed., 2010, 49, 2840–2859; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2010, 122, 2902; [Google Scholar]; c) Lu T. and Hsung R. P., ARKIVOC, 2014, 1, 127–141; [PMC free article] [PubMed] [Google Scholar]; d) Wang X.‐N., Yeom H.‐S., Fang L.‐C., He S., Ma Z.‐X., Kedrowski B. L. and Hsung R. P., Acc. Chem. Res., 2014, 47, 560–578; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cook A. M. and Wolf C., Tetrahedron Lett., 2015, 56, 2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang Y., Hsung R. P., Tracey M. R., Kurtz K. C. M. and Vera E. L., Org. Lett., 2004, 6, 1151–1154. [DOI] [PubMed] [Google Scholar]
- 3.a) Wezeman T., Zhong S., Nieger M. and Bräse S., Angew. Chem. Int. Ed., 2016, 55, 3823; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 3888–3892 [Google Scholar]; Wezeman T., Zhong S., Nieger M. and Bräse S., Angew. Chem. Int. Ed., 2016, 55, 3823–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 3888; [Google Scholar]; b) Evano G., Lecomte M., Thilmany P. and Theunissen C., Synthesis, 2017, 49, 3183–3214. [Google Scholar]
- 4.a) Greenaway R. L., Campbell C. D., Holton O. T., Russell C. A. and Anderson E. A., Chem. Eur. J., 2011, 17, 14366–14370; [DOI] [PubMed] [Google Scholar]; b) Garcia P., Harrak Y., Diab L., Cordier P., Ollivier C., Gandon V., Malacria M., Fensterbank L. and Aubert C., Org. Lett., 2011, 13, 2952–2955; [DOI] [PubMed] [Google Scholar]; c) Rettenmeier E., Schuster A. M., Rudolph M., Rominger F., Gade C. A. and Hashmi A. S. K., Angew. Chem. Int. Ed., 2013, 52, 5880; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 5993–5997 [Google Scholar]; Rettenmeier E., Schuster A. M., Rudolph M., Rominger F., Gade C. A. and Hashmi A. S. K., Angew. Chem. Int. Ed., 2013, 52, 5880–5884; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 5993; [Google Scholar]; d) Siva Reddy A., Leela Siva Kumari A., Saha S. and Kumara Swamya K. C., Adv. Synth. Catal., 2016, 358, 1625–1638. [Google Scholar]
- 5.a) Dateer R. B., Shaibu B. S. and Liu R.‐S., Angew. Chem. Int. Ed., 2012, 51, 113; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 117–121 [Google Scholar]; Dateer R. B., Shaibu B. S. and Liu R.‐S., Angew. Chem. Int. Ed., 2012, 51, 113–117; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 117; [Google Scholar]; b) Karad S. N., Bhunia S. and Liu R.‐S., Angew. Chem. Int. Ed., 2012, 51, 8722; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 8852–8856 [Google Scholar]; Karad S. N., Bhunia S. and Liu R.‐S., Angew. Chem. Int. Ed., 2012, 51, 8722–8726; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 8852; [Google Scholar]; c) Karad S. N. and Liu R.‐S., Angew. Chem. Int. Ed., 2014, 53, 9072; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 9218–9222 [Google Scholar]; Karad S. N. and Liu R.‐S., Angew. Chem. Int. Ed., 2014, 53, 9072–9076; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 9218; [Google Scholar]; d) Shu C., Wang Y.‐H., Zhou B., Li X.‐L., Ping Y.‐F., Lu X. and Ye L.‐W., J. Am. Chem. Soc., 2015, 137, 9567–9570. [DOI] [PubMed] [Google Scholar]
- 6.a) Bendikov M., Duong H. M., Bolanos E. and Wudl F., Org. Lett., 2005, 7, 783–786; [DOI] [PubMed] [Google Scholar]; b) Gomes F., Fadel A. and Rabasso N., J. Org. Chem., 2012, 77, 5439–5444; [DOI] [PubMed] [Google Scholar]; c) Brioche J., Meyer C. and Cossy J., Org. Lett., 2013, 15, 1626–1629; [DOI] [PubMed] [Google Scholar]; d) Li D.‐Y., Wei Y. and Shi M., Eur. J. Org. Chem., 2015, 4108–4113. [Google Scholar]
- 7.a) Wrobel J., Dietrich A., Woolson S. A., Millen J., McCaleb M., Harrison M. C., Hohman T. C., Sredy J. and Sullivan D., J. Med. Chem., 1992, 35, 4613–4627; [DOI] [PubMed] [Google Scholar]; b) D. C. Baker, B. Jiang, U. S. Patent 6,353,112, 2002;; c) J. Mao, D. C. Baker, U. S. Patent 6,458,962, 2002;; d) J. L. Castro Pineiro, J. C. Hannam, T. Harrison, A. Madin, M. P. Ridgill, U. S. Patent 7,371,771, 2008;; e) Zhang S., Li L., Hu Y., Zha Z., Wang Z. and Loh T. P., Org. Lett., 2015, 17, 1050–1053; [DOI] [PubMed] [Google Scholar]; f) Oppolzer W., Wills M., Kelly M. J., Signer M. and Blagg J., Tetrahedron Lett., 1990, 31, 5015–5018; [Google Scholar]; g) Oppolzer W., Rodriguez I., Starkemann C. and Walther E., Tetrahedron Lett., 1990, 31, 5019–5022; [Google Scholar]; h) Oppolzer W., Kingma A. J. and Pillai S. K., Tetrahedron Lett., 1991, 32, 4893–4896; [Google Scholar]; i) Ahn K. H., Kim S. and Ham C., Tetrahedron Lett., 1998, 39, 6321–6322; [Google Scholar]; j) Differding E. and Bersier P. M., Tetrahedron, 1992, 48, 1595–1604; [Google Scholar]; k) Lal G. S., Pez G. P. and Syvret R. G., Chem. Rev., 1996, 96, 1737–1755; [DOI] [PubMed] [Google Scholar]; l) Takeuchi Y., Suzuki T., Satoh A., Shiragami T. and Shibata N., J. Org. Chem., 1999, 64, 5708–5711. [DOI] [PubMed] [Google Scholar]
- 8. Alam K., Hong S. W., Oh K. H. and Park J. K., Angew. Chem. Int. Ed., 2017, 56, 13387; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 13572–13576 [Google Scholar]; Alam K., Hong S. W., Oh K. H. and Park J. K., Angew. Chem. Int. Ed., 2017, 56, 13387–13391; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 13572. [Google Scholar]
- 9.a) Minami Y., Yamada K. and Hiyama T., Angew. Chem. Int. Ed., 2013, 52, 10611; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 10805–10809 [Google Scholar]; Minami Y., Yamada K. and Hiyama T., Angew. Chem. Int. Ed., 2013, 52, 10611–10615; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 10805; [Google Scholar]; b) Minami Y., Kanda M. and Hiyama T., Chem. Lett., 2014, 43, 181–183; [Google Scholar]; c) Minami Y., Kanda M., Sakai M. and Hiyama T., Tetrahedron, 2015, 71, 4522–4534; [Google Scholar]; d) Minami Y., Kodama T. and Hiyama T., Angew. Chem. Int. Ed., 2015, 54, 11813; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2015, 127, 11979–11982 [Google Scholar]; Minami Y., Kodama T. and Hiyama T., Angew. Chem. Int. Ed., 2015, 54, 11813–11816; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2015, 127, 11979; [Google Scholar]; e) Minami Y., Noguchi Y., Yamada K. and Hiyama T., Chem. Lett., 2016, 45, 1210–1212; [Google Scholar]; f) Minami Y., Sakai M., Anami T. and Hiyama T., Angew. Chem. Int. Ed., 2016, 55, 8701; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 8843–8847 [Google Scholar]; Minami Y., Sakai M., Anami T. and Hiyama T., Angew. Chem. Int. Ed., 2016, 55, 8701–8705; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 8843; [Google Scholar]; g) Minami Y. and Hiyama T., Acc. Chem. Res., 2016, 49, 67–77. [DOI] [PubMed] [Google Scholar]
- 10.a) Stacey F. W., Sauer J. C. and Mckusic B. C., J. Am. Chem. Soc., 1959, 81, 987–992; [Google Scholar]; b) W. Hertler, U. S. Patent 3,814,604, 1974;; c) Paquette L. A., Dura R. D. and Modolo I., J. Org. Chem., 2009, 74, 1982–1987. [DOI] [PubMed] [Google Scholar]
- 11.a) D. J. Kyle, L. Tafesse, U. S. Patent US 8,546,388, 2013;; b) R. Fischer, O. Kretschik, T. Schenke, R.‐I. Schenkel, J. Wiedemann, C. Erdelen, P. Losel, M. W. Drewes, D. Feucht, W. Andersch, U. S. Patent 6,670,385, 2003
- 12.ECHONET ITN Home Page, https://sites.google.com/a/sheffield.ac.uk/echonet-itn/ (accessed July 02, 2018).
- 13. Siva Reddy A. and Kumara Swamya K. C., Angew. Chem. Int. Ed., 2017, 56, 6984–6988; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 7088–7092 [Google Scholar]; Reddy A. Siva and Kumara Swamya K. C., Angew. Chem. Int. Ed., 2017, 56, 6984–6988; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 7088. [Google Scholar]
- 14.Product 6 is presumably formed by aromatic ortho‐C–H activation of the benzyl group followed by intramolecular hydroarylation.
- 15.For an overview on CMD sequence see: Lapointe D. and Fagnou K., Chem. Lett. 2010, 39, 1118–1126, and references therein. [Google Scholar]
- 16. O'Sullivan* S., Doni E., Tuttle T. and Murphy J. A., Angew. Chem. Int. Ed., 2014, 53, 474; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 484–488 [Google Scholar]; O′Sullivan S., Doni E., Tuttle T. and Murphy J. A., Angew. Chem. Int. Ed., 2014, 53, 474–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 484. [Google Scholar]
- 17. Zhang W., Xie J., Rao B. and Luo M., J. Org. Chem., 2015, 80, 3504–3511. [DOI] [PubMed] [Google Scholar]
- 18. Laha J. K., Jethava K. P. and Dayal N., J. Org. Chem., 2014, 79, 8010–8019. [DOI] [PubMed] [Google Scholar]
- 19. Heffernan S. J., Beddoes J. M., Mahon M. F., Hennessy A. J. and Carbery D. R., Chem. Commun., 2013, 49, 2314–2316. [DOI] [PubMed] [Google Scholar]
- 20. Li F., Xie J., Shan H., Sun C. and Chen L., RSC Adv., 2012, 2, 8645–8652. [Google Scholar]
- 21. Mansfield S. J., Campbell C. D., Jones M. W. and Anderson E. A., Chem. Commun., 2015, 51, 3316–3319. [DOI] [PubMed] [Google Scholar]
- 22. Geary L. M. and Hultin P. G., Org. Lett., 2009, 11, 5478–5481. [DOI] [PubMed] [Google Scholar]
- 23. Gillie A. D., Jannapu Reddy R. and Davies P. W., Adv. Synth. Catal., 2016, 358, 226–239. [Google Scholar]
- 24. Zhang P., Cook A. M., Liu Y. and Wolf C., J. Org. Chem., 2014, 79, 4167–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tracey M. R., Zhang Y., Frederick M. O., Mulder J. A. and Hsung R. P., Org. Lett., 2004, 6, 2209–2212. [DOI] [PubMed] [Google Scholar]
- 26. Brückner D., Tetrahedron, 2006, 62, 3809–3814. [Google Scholar]
- 27. Davies P. W., Cremonesi A. and Martin N., Chem. Commun., 2011, 47, 379–381. [DOI] [PubMed] [Google Scholar]
- 28.This side product is a result of addition of 4‐nitro‐N‐phenylbenzene‐sulfonamide 3 to 1i formed during the reaction.
- 29. Chen P., Song C.‐X., Wang W.‐S., Yua X.‐L. and Tang Y., RSC Adv., 2016, 6, 80055–80058. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information