

Abstract
Solid tumors, including gliomas, still represent a challenge to clinicians and first line treatments often fail, calling for new paradigms in cancer therapy. Novel strategies to overcome tumor resistance are mainly represented by multi‐targeted approaches, and cell vector‐based therapy is one of the most promising treatment modalities under development. Here, we show that mouse bone marrow‐derived mesenchymal stromal cells (MSCs), when primed with low‐dose irradiation (irMSCs), undergo changes in their immunogenic and angiogenic capacity and acquire anti‐tumoral properties in a mouse model of glioblastoma (GBM). Following grafting in GL261 glioblastoma, irMSCs migrate extensively and selectively within the tumor and infiltrate predominantly the peri‐vascular niche, leading to rejection of established tumors and cure in 29% of animals. The therapeutic radiation dose window is narrow, with effects seen between 2 and 15 Gy, peaking at 5 Gy. A single low‐dose radiation decreases MSCs inherent immune suppressive properties in vitro as well as shapes their immune regulatory ability in vivo. Intra‐tumorally grafted irMSCs stimulate the immune system and decrease immune suppression. Additionally, irMSCs enhance peri‐tumoral reactive astrocytosis and display anti‐angiogenic properties. Hence, the present study provides strong evidence for a therapeutic potential of low‐dose irMSCs in cancer as well as giving new insight into MSC biology and applications.
Keywords: glioma, MSC, immune modulation, angiogenesis, TGFβ
Short abstract
What's new?
Median survival for glioblastoma (GBM) patients is little more than one year, and long‐term survivors are rare. Hence, newer, more effective therapies for GBM are needed. Here, the authors explored the antitumoral properties of low‐dose irradiated mesenchymal stromal cells (irMSCs). Low‐dose irradiation was found to induce a phenotypic switch in MSCs, limiting their immune suppressive function. In a GBM mouse model, intra‐tumoral grafting of irMSCs affected immune response and tumor angiogenesis, resulting in increased survival of tumor‐bearing mice. The findings highlight the potential of cellular therapy in cancer and show that low‐dose irradiation can be used to effectively manipulate MSC phenotype.
Introduction
The ability of cancer cells to develop resistance to treatment is one of the major causes of therapy failure in oncology.1 Recent efforts aiming at targeting multiple independent pathways in cancer cells and their microenvironment2 have highlighted cell‐based therapeutic approaches as a valuable alternative. Mesenchymal stromal cells (MSCs) are multipotent cells that can be isolated from various tissues3 and whose safety has already been assessed in several clinical trials.4, 5 To date, the therapeutic use of MSCs in cancer is based on their inherent tumor‐homing ability that has been exploited to deliver cytotoxic substances selectively to neoplastic tissue.6 Glioblastoma (GBM) is the most common aggressive brain tumor in adults and is characterized by an invasive growth pattern, vivid angiogenesis and immune suppression7 and we and others have previously used GBM as a model of highly aggressive solid cancer.8 MSCs, engineered to express pro‐inflammatory cytokines such as IL2,9 IL7,10 and IFNβ,11 have been shown to exert immune‐dependent anti‐tumor effects in models of GBM as well as other types of solid cancer.12 Further, the same approach has been used to deliver anti‐angiogenic factors in GBM models.13 MSCs possess immune regulatory capacity14 and ample evidence exists that MSCs exert suppressive effects on both the innate and the adaptive immune system in various experimental systems, including neoplasia.15, 16 However, the ability of MSCs to modulate the immune system can be dynamically regulated by the environment and particularly by inflammation.17 In this respect, the behavior of MSCs resembles microglia and macrophage polarization with differentiation mainly into a pro‐inflammatory phenotype upon TLR4 stimulation and an anti‐inflammatory phenotype upon TLR3 signaling.18 Detailed information regarding the molecular mechanisms underlying the immune phenotypic switch of MSCs is lacking and the potential of MSCs as immune modulators in cancer has not been thoroughly explored. The concept of context‐dependent MSC phenotype is further corroborated by the dual effect of MSCs on angiogenesis, with several studies reporting both pro‐ and anti‐angiogenic effects of MSCs in cancer.19
Recently, it was shown that macrophages can acquire an immune stimulatory phenotype when subjected to low‐dose irradiation.20 Due to the similarities between macrophages and MSC pro‐/anti‐inflammatory behavior, the present study was undertaken to determine whether low‐dose irradiation can drive bone marrow‐derived MSCs into an immune stimulatory state, with a further focus on the anti‐angiogenic and anti‐tumor potential of irradiated MSCs (irMSCs) in an in vivo model of GBM.
Herein, we identify a narrow therapeutic window of low‐dose irradiation in which MSCs acquire anti‐tumoral properties and cure tumor‐bearing mice, opening up a new avenue of potential MSC‐based therapy in cancer.
Materials and Methods
Primary cells and cell line cultures
GL261 mouse glioma cell line of C57Bl/6 origin was a kind gift of Dr. Géza Sáfrány, Hungary. The cell line was maintained in R10 medium consisting of RPMI 1640 medium supplemented with 1 mM sodium pyruvate, 10 mM HEPES, 50 µg/ml gentamicin (Thermo Fisher Scientific Inc., Waltham, MA) and 10% FBS (Biochrom AB, Berlin, Germany). To establish primary cultures of mouse bone marrow‐derived mesenchymal stromal cells, femurs were collected from 6–8 week old female mice and bone marrow flushed out of the marrow cavity. Cells were maintained in MesenCult medium supplemented with MesenPure (STEMCELL Technologies SARL, Grenoble, France) and 1% antibiotic antimycotic solution (Sigma‐Aldrich, Stockholm, Sweden). After 3 days non‐adherent cells were removed by changing the medium and the remaining adherent cells were sub‐cultured. The isolated MSCs were identified based on cell surface markers expression (CD44+, CD29+, SCA‐1+, CD34−, and CD117−) and the ability to differentiate in adipocytes and osteoblasts.21 For adipocyte differentiation, 1 × 104 non irradiated MSCs (niMSCs) or 5 Gy irMSCs at passage 3 (p3) were seeded in triplicates in 24 well plates and the next day treated with induction medium (α‐MEM (Thermo Fisher Scientific Inc., Waltham, MA) supplemented with 10% FBS, 10−6 M dexamethasone, 0.5 µM IBMX, 10 ng/ml bovine pancreas insulin (Sigma‐Aldrich, Stockholm, Sweden). The medium was changed three times per week for a total of 14 days and the differentiation was assessed by Oil red O staining. For staining of lipid droplets, cells were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature (RT), washed with dH20, incubated 5 min with 60% isopropanol (Sigma‐Aldrich, Stockholm, Sweden) followed by 15 min incubation with Oil red O solution (Sigma‐Aldrich, Stockholm, Sweden). After 5 min incubation with 60% isopropanol the samples were rinsed with tap water and analyzed. For osteoblast differentiation, 1 × 104 ni/5 Gy irMSCs at p3 were seeded in triplicates in 24‐well plates and the next day treated with induction medium (α‐MEM supplemented with 10% FBS, 10−7 M dexamethasone, 10 mM β‐glycerol phosphate and 50 µM ascorbate‐2‐phosphate (Sigma‐Aldrich, Stockholm, Sweden)). The medium was changed 3 times per week for a total of 21 days and the differentiation was assessed by Alizarin Red staining. For staining of calcium deposits, cells were fixed with 4% PFA for 30 min at RT, washed with dH20 followed by 10 min incubation with 40 mM Alizarin Red solution (Sigma‐Aldrich, Stockholm, Sweden) and analyzed. In vitro cell viability was assessed by plating 1 × 104 niMSCs or 2/5/10/15/20 Gy irMSCs in 96 well plates and proliferation analyzed at 24 and 48 hr by Presto Blue assay (Thermo Fisher Scientific Inc., Waltham, MA) according to the manufacturer's instructions. All cell lines were kept in culture no longer than 6 weeks and MSCs were never used beyond p9. Radiation experiments were performed by using a 137Cs γ‐emitting irradiator (Gamma Cell 40, MSD Nordion, Canada). The same batch of cells was divided into groups (ni/irMSCs) where the irMSC group was subjected to irradiation, and directly used for in vitro or in vivo assays.
Ethics and animal procedures
All animal procedures were performed according to the practices of the Swedish Board of Animal Research and approved by the Committee of Animal Ethics in Lund‐Malmö, Sweden. Female C57Bl/6 mice were purchased from Taconic (Taconic Biosciences Inc., Hudson NY) and maintained under specific pathogen‐free conditions at the In Vivo Department, Lund University, Sweden. Brain tumors were induced at Day 0 by injecting 1 × 104 GL261 cells intra‐cerebrally (i.c.) into the right striatum (2.75 mm ventral of the skull bone) of anaesthetized mice (Isoflurane, Forene, Abbott Scandinavia AB, Solna, Sweden). The head of the mouse was fixed in a stereotactic frame (David Kopf Instruments, Tujunga CA), all animals received subcutaneous local anaesthesia (2.5 mg/ml Marcain adrenalin, Astra Zeneca AB, Solna, Sweden) and cells were injected using a Hamilton syringe (Hamilton Company, Switzerland). For survival study, 1 × 105 niMSCs or 2/5/10/15/20 Gy irMSCs (p7–9) were grafted intra‐tumorally (i.t.) at days 7 and 17 following tumor inoculation. The animals were euthanized either at the end of the experiment or when early signs of neurologic illness appeared (Fig. 1 a). The presence of brain tumors was macroscopically assessed by coronal transections and visual inspection following the removal of the brain from the skull. The migratory potential of MSCs was assessed by injecting 1 × 105 niMSCs‐GFP or 5 Gy irMSCs‐GFP (p7–9) into tumor‐bearing mice at days 7 and 17. At Day 22, the animals were cardially perfused with saline solution followed by 4% PFA (Fig. 4 d). In a parallel experiment, to investigate the mechanisms of tumor rejection, niMSCs or 5 Gy irMSCs (p7–9) were transplanted i.t., peripheral blood collected at days 14/21 and brains snap‐frozen at Day 22 (Fig. 2 a). Both GL261 and MSCs were injected in 5 µl of R0 medium, consisting of RPMI 1640 medium supplemented with 1 mM sodium pyruvate and 10 mM HEPES.
Figure 1.
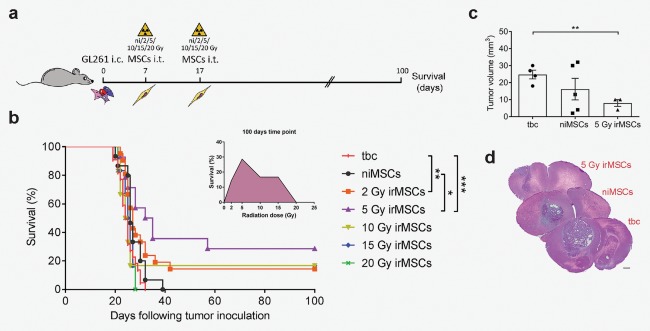
irMSCs increase the survival of GBM‐bearing mice. (a) Outline of the survival study. (b) Survival curves of mice subjected to tumor grafting (GL261), followed by intra‐tumoral niMSCs or 2/5/10/15/20 Gy irMSCs implantation. Animals injected with only GL261 cells were used as control and referred to as tumor‐bearing control (tbc). The animals were followed up to 100 days and data are from 1 to 3 independent experiments with the tbc and niMSC groups included in each experiment (tbc/niMSCs/2/5/10/15/20 Gy irMSCs, n = 22/15/21/14/6/6/6, respectively). Primary MSCs were harvested from bone marrow in 1–2 independent experiments. (c) Analysis of tumor volume and (d) representative H&E stainings; scale bar 500 µm. Data are from one experiment (n = 3–5) and are presented as mean ± SEM. Survival over time is estimated by Kaplan‐Meier analysis and curves are compared by log rank test. Comparisons between groups are performed by one‐way ANOVA, followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 4.
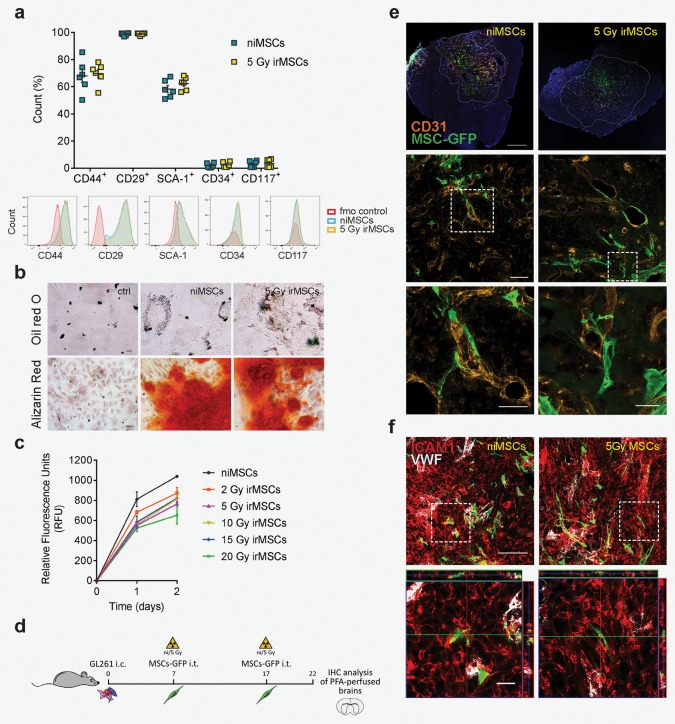
MSC basic characteristics are not affected by low‐dose irradiation in vitro or in vivo. (a) Flow cytometry analysis of MSC surface markers expression at 24 hr post‐irradiation (5 Gy); data from two independent experiments are shown as mean ± SEM (n = 6). (b) Differentiation potential as assessed by immunofluorescence (IF) for adipocyte (Oil red O) and osteoblast (Alizarin red) marker expression in niMSCs and 5 Gy irMSCs; scale bar 20 and 50 µm, respectively. (c) Proliferation capacity of MSCs after various radiation doses assessed at 24 and 48 hr by Presto Blue assay; data from two independent experiments are presented as mean ± SEM. (from d to f) Evaluation of irMSC in vivo tumor‐tropism. (d) ni/5 Gy irMSCs‐GFP were transplanted i.t. at days 7 and 17, after GL261 mouse GBM inoculation and animals were sacrificed by transcardiac perfusion at Day 22. (e) Representative images showing in vivo tumor‐ and peri‐vascular tropism of MSCs (CD31+ orange, GFP+ green, dashed lines marking tumor edges); scale bars from top to bottom: 500, 100, 50 (high magnification, left panel), and 25 (high magnification, right panel) µm. (f) Representative images of MSC (GFP+ green)‐tumor (ICAM1+ red) interaction in vivo; von Willebrand factor (VWF white) expression distinguishes endothelial cells from tumor cells; scale bar 100 µm and 20 µm for low and high magnification, respectively.
Figure 2.
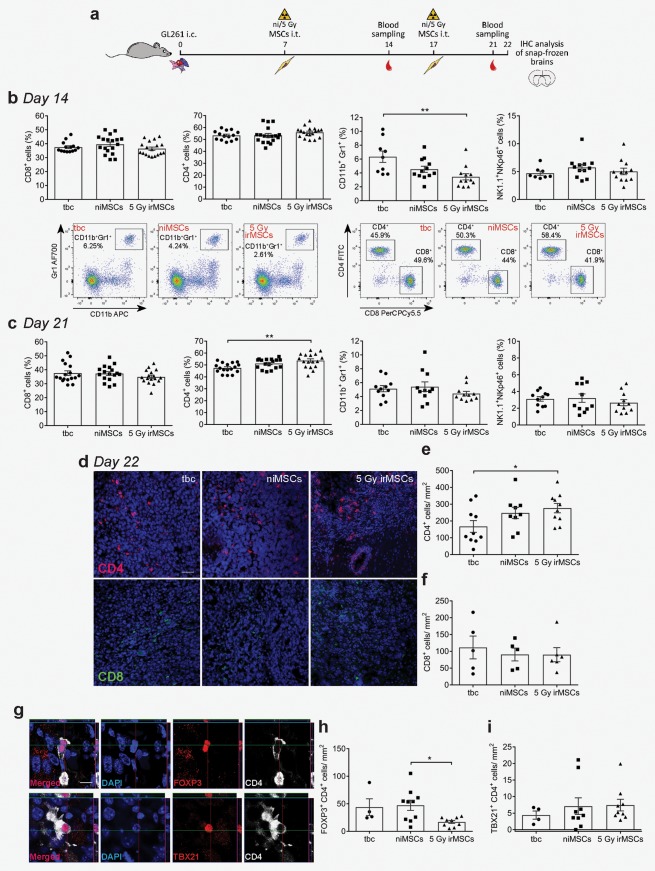
Stimulatory effect of irMSCs on the immune system in vivo. (a) Outline of the study. (b and c) Peripheral immune response analyses at days 14 (b) and 21 (c) post‐tumor inoculation. Percentages of CD4+ or CD8+ T cells (out of CD3+), NK1.1+/NKp46+ cells and CD11b+/Gr1+ cells (out of total leukocyte population) were analyzed by flow cytometry (representative plots for MDSCs [left plots] and CD4+/CD8+ T cells [right plots]). Data are from 2 to 3 independent experiments (n = 8–18, all experimental groups assessed in parallel in each independent experiment) and are presented as mean ± SEM. (from d to i) In vivo tumor‐infiltrating immune cells assessed by immunohistochemistry analysis. Representative images of intra‐tumoral CD4+ (d, red) and CD8+ (d, green) cell infiltration, quantified in (e) and (f), respectively. Data are from 1 and 2 independent experiments for CD8 and CD4 cell count, respectively (n = 5–10, all experimental groups assessed in parallel in each independent experiment) and are presented as mean ± SEM; (from g to i) FOXP3 and TBX21 analysis together with representative images of positive cells; data are from 2 independent experiments (n = 8–9), except the tbc (one experiment, n = 4) and are presented as mean ± SEM; scale bar for (d) and (g) is 50 and 10 µm, respectively. Comparisons between groups are performed by one‐way ANOVA, followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01.
Flow cytometry
Peripheral blood was collected at days 14 and 21 from vena saphena and mixed with heparin (LEO Pharma AB, Malmö, Sweden). Red blood cell depletion was performed by osmotic shock with water, then quickly neutralized with NaCl to a final concentration of 0.9%. The samples were then incubated with 10 µg/ml of CD16/CD32 (Mouse BD Fc Block, clone 2.4G2) for 5 min at RT followed by 30 min incubation at 4°C with primary monoclonal antibodies (1 µg/ml; Table 1). Cells were fixed and permeabilized using BD Pharmingen Transcription Factor Buffer Set (BD Biosciences, San Jose, CA) following the manufacturer's instructions. Antigen expression was detected using BD LSRII (BD Biosciences, San Jose, CA) and data analyzed with FlowJo software (TreeStar Inc., Ashland, OR). Fluorescence minus one (FMO) controls were used to determine positive and negative staining gates. Leukocytes were determined by forward and side scatter profile (FSC/SSC), CD4+ and CD8+ T cells percentages were obtained out of singlets and CD3+/NK1.1− cells; NKp46+ cells were gated on CD3−NK1.1+ cells and CD11b+Gr1+ were gated on singlets. To assess the expression of cell surface markers 3 × 105 ni/5 Gy irMSCs were seeded in 24 well plates and after 24 hr, collected and stained as described above.
Table 1.
List of primary antibodies used for flow cytometry and immunofluorescence
| Antibody | Fluorochrome | Clone | Brand |
|---|---|---|---|
| CD44 | APC | IM7 | BD Pharmingen |
| CD29 | APC‐Cy7 | HM β1‐1 | BioLegend |
| Ly‐6A/E | FITC | E13–161.7 | BD Pharmingen |
| CD34 | Brilliant Violet421 | RAM34 | BD Pharmingen |
| CD117 | PE‐Cy7 | 2B8 | BD Pharmingen |
| CD11b | APC‐Cy7 | M1/70 | BD Pharmingen |
| CD11b | PE‐Cy5 | M1/70 | BioLegend |
| Gr‐1 | Alexa Fluor700 | RB6–8C5 | BD Pharmingen |
| CD3ɛ | APC | 145‐2C11 | BD Pharmingen |
| CD3ɛ | APC‐eFluor780 | 145‐2C11 | eBioscience |
| CD3 | Pacific Blue | 17A2 | BioLegend |
| CD4 | FITC | GK1.5 | BD Pharmingen |
| CD4 | PE | GK1.5 | BD Pharmingen |
| CD4 | PE‐Cy7 | RM4–5 | BioLegend |
| CD8α | APC‐Cy7 | 53‐6.7 | BioLegend |
| CD8α | PerCP‐Cy5.5 | 53‐6.7 | BD Pharmingen |
| CD8α | Pacific Blue | 53‐6.7 | BD Pharmingen |
| NK1.1 | Brilliant Violet605 | PK136 | BD Pharmingen |
| NKp46 | Brilliant Violet421 | 29A1.4 | BD Pharmingen |
| Rat anti‐mouse CD4 | H129.19 | BD Pharmingen | |
| Rat anti‐mouse CD8 | 53‐6.7 | BD Pharmingen | |
| Rabbit anti‐mouse CD31 | Abcam | ||
| Rat anti‐mouse CD31 | MEC 13.3 | BD Pharmingen | |
| Chicken anti‐mouse GFAP | Chemicon | ||
| Rabbit anti‐mouse Vimentin | EPR3776 | Abcam | |
| Chicken anti‐GFP | Abcam | ||
| Rat anti‐mouse ICAM1 | YN1/1.7.4 | Abcam | |
| Rat anti‐mouse Von Willebrand Factor | Abcam | ||
| Rabbit anti‐mouse TBX21 | Invitrogen | ||
| Rabbit anti‐mouse FOXP3 | Novus Biologicals |
Immunohistochemistry and stereology
At Day 22, animals were sacrificed and brains dissected and snap frozen. For CD4, CD8, GFAP/Vimentin and CD31 stainings 6 µm thick sections were cut using a Microm HM 560 cryostat (Thermo Fisher Scientific Inc., Waltham, MA) and directly mounted onto glass slides. Frozen sections were fixed in acetone for 10 min, permeabilized for 5 min with PBS‐0.25% Triton X‐100 (referred to as T‐PBS; Sigma‐Aldrich, Stockholm, Sweden) and blocked for 20 min with 5% goat and/or donkey serum (Jackson ImmunoResearch Inc., West Grove, PA). The samples were incubated for 2.5 hr with primary antibodies (see Table 1), followed by 60 min incubation with suitable secondary antibodies. The slides were mounted wet using Pro‐Long Gold anti‐fading reagent (Thermo Fisher Scientific Inc., Waltham, MA). All the staining procedures were performed at RT and T‐PBS was used as diluent in all the steps. For the migration study, PFA‐perfused brains were post‐fixed overnight (ON) at 4°C in 4% PFA and then transferred to a 30% sucrose solution. The brains were cut into 30 µm thick coronal sections with a Leica SM200 R microtome (Leica Biosystems Nussloch GmbH, Nussloch, Germany) and stored at −20°C in anti‐freeze solution (30% ethylene glycol and 30% glycerol in 0.012 M NaH2PO4·H20 and 0.031 M Na2HPO4·2H2O (Sigma‐Aldrich, Stockholm, Sweden). The sections were washed with KPBS and then blocked for 60 min with KPBS‐0.25% Triton X‐100 (referred to as T‐KPBS) in 5% goat and/or donkey serum. After ON incubation with primary antibodies (see Table 1), the sections were washed and incubated for 2 hr with suitable secondary antibodies. Following a few washing steps in T‐KPBS and KPBS, the sections were mounted in dH2O onto glass slides and were coverslipped with PVA‐DABCO solution (Sigma‐Aldrich, Stockholm, Sweden). All the staining procedures were performed at RT and T‐KPBS plus serum was used as diluent in all the steps. Cell nuclei were counterstained with either DAPI or Hoechst 33342 (Thermo Fisher Scientific Inc., Waltham, MA). In all experiments primary antibodies were omitted in the negative controls. Cell quantification was conducted on an epifluorescence microscope equipped with Visiopharm stereology software (Visiopharm, Hoersholm, Denmark) and was carried out at 40× magnification, except for the TBX21/FOXP3 staining where the tumor‐infiltrating CD4+ cells were counted at 20× magnification. The tumor area, as determined by nuclear staining, was manually drawn, except for astrocyte counts where the whole hemisphere was analyzed. Twenty percent of the drawn area was randomly sampled by the software and cell counting carried out manually. CD4+, CD8+, FOXP3+, and TBX21+ cells, visualized by fluorescent staining, were manually counted within the tumor area. For CD31 staining of vessels, the luminal area within the tumor was measured and the number of tumor vessels counted and compared between groups. For reactive astrocytes, the GFAP+/Vimentin+ cells were counted and related to the hemisphere area. Images were taken using the Zeiss LSM 780 confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany).
Hematoxylin and eosin staining was performed on PFA‐perfused tissue and images taken using VS120 Virtual Slide Microscope (Olympus Sverige AB, Solna, Sweden).
Gene expression analysis
Total RNA was extracted from MSCs at p6 using the phenol/chloroform method.22 Any traces of DNA were removed using the TURBO DNA‐free Kit (Thermo Fisher Scientific Inc., Waltham, MA) according to the manufacturer's instructions. Single stranded cDNA was obtained with the qScript cDNA Synthesis Kit (Quanta BioSciences Inc., Gaithersburg, MD) and a total of 5 ng/µl was used to perform TaqMan Gene Expression Assays (Thermo Fisher Scientific Inc., Waltham, MA) for the following genes: Gapdh (Mm99999915_g1), Arg1 (Mm00475988_m1), Vegfa (Mm00437306_m1), Tgfβ1 (Mm01178820_m1) and Tnfα (Mm00443258_m1). qPCR was carried out on a Bio‐Rad iQ5 thermocycler (Bio‐Rad Laboratories Inc., Solna, Sweden) according to the manufacturer's instructions.
Cytokine production
For cytokine production, 1 × 105 niMSCs or 2/5/10/15/20 Gy irMSCs at p5 were seeded in 24 well plates and supernatants collected at 8 hr, centrifuged to remove cell debris and immediately stored at −80°C. For mTGFβ1 analysis of GL261 cells, ni/5 Gy irMSCs were cultured for 8 hr and supernatants from technical replicates were collected, mixed and split in equal amounts either for culturing GL261 cells (12 hr) or in empty wells as control (to estimate mTGFβ1 secreted by ni/5 Gy irMSCs). GL261 cells grown in MesenCult medium were used as control. The following mouse factors were analyzed using ELISA kits according to the manufacturer's instructions: TGFβ1, VEGFA, CCL2, and IL6 (R&D Systems Inc., Minneapolis, MN).
T cell activation assay
Mouse spleens were mashed and passed through a 40 µm cell strainer to obtain single cell suspensions. Red cells were lysed with ammonium chloride solution (STEMCELL Technologies SARL, Grenoble, France). CD3 enrichment was performed with MACS cell separation columns (Miltenyi Biotec Norden AB, Lund, Sweden) after labeling with biotin anti‐mouse CD3ɛ (clone 145‐2C11, BioLegend, San Diego, CA) and anti‐biotin microbeads ultra‐pure (Miltenyi Biotec Norden AB, Lund, Sweden). Enriched CD3+ cells were labeled with CellTrace CFSE Cell Proliferation Kit (Thermo Fisher Scientific Inc., Waltham, MA) according to the manufacturer's instructions. 1.25 × 104 ni/irMSCs (p6) were seeded in a 24 well plate and after 24 hr, 4 × 105 freshly isolated CD3+ T cells were added to the cultures (1:32 ratio); single T cell cultures were used as control. After 22 hr, 4 × 105 Dynabeads Mouse T‐Activator CD3/CD28 (Thermo Fisher Scientific Inc., Waltham, MA) were added to the cultures and the cells analyzed 72 hr later using BD LSRII (BD Biosciences, San Jose, CA; Fig. 5 a). Data were analyzed with FlowJo software (TreeStar Inc., Ashland, OR) and FMO controls were used to determine positive and negative staining gates.
Figure 5.
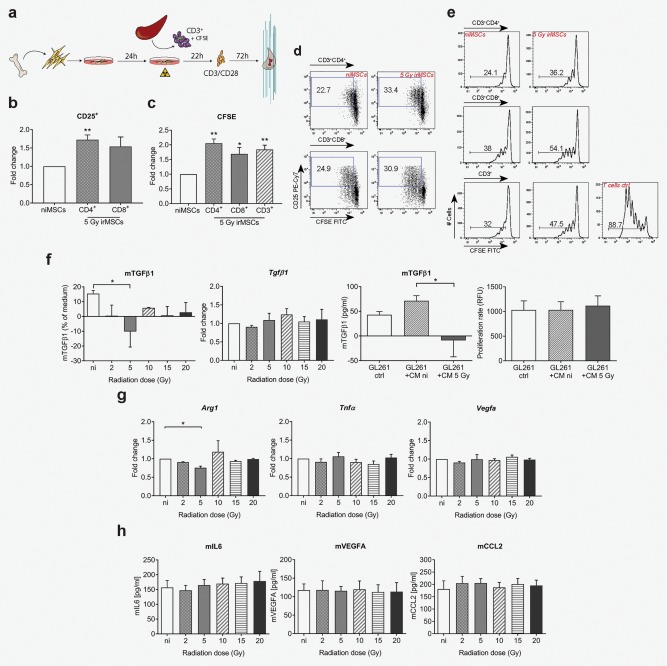
MSC inherent immune suppressive features are reduced after low‐dose irradiation and affect immune suppressive properties of tumor cells in vitro. (from a to e) T cell activation assay. ni/irMSCs were co‐cultured with splenic CD3+ T cells (1:32 ratio) and after 22 hr, cultures were supplemented with CD3/CD28 mouse T‐activator beads and analyzed 72 hr later by flow cytometry (see outline in a). (b) Fold change expression of CD25 in the CD3+CD4+ and CD3+CD8+ T cell population and representative plots (d). (c) Cell proliferation was followed in CD3+CD4+, CD3+CD8+ and total CD3+ T cell population using CFSE and expressed as fold change; representative histograms are shown in (e). Data are from one experiment with three biological replicates (n = 3) and are presented as mean ± SEM. Comparisons between groups are performed by two‐tailed Student's t test. (f) TGFβ1 analyses at 8 hr post‐irradiation; protein (first panel) and gene expression (second panel) analyses of TGFβ1 in ni/irMSCs; TGFβ1 protein analysis (third panel) and proliferation (forth panel) of GL261 cells cultured for 12 hr in ni/irMSC conditioned medium (8 hr). (g) Gene expression analysis of MSCs at 8 hr post‐irradiation; ‐fold change expression of Arg1, Vegfa and Tnfα compared to Gapdh. (h) Factors released by MSCs at 8 hr post‐irradiation, as assessed by ELISA. Data shown are from 2 to 5 biological replicates, with technical replicates, and are presented as mean ± SEM. Comparisons between groups are performed by one‐way ANOVA, followed by Tukey's multiple comparisons test. *p < 0.05, **p < 0.01.
Lentiviral production and cell transduction
pLenti PGK GFP Puro construct was a kind gift from Zaal Kokaia, Lund University, Sweden. The viral particles were produced in HEK293T cells by calcium phosphate precipitation, using 3rd generation LV packaging plasmids (kindly provided by Henrik Ahlenius, Lund University, Sweden). MSCs at 70–80% of confluence were transduced ON using protamine sulfate (8 µg/ml) (Sigma‐Aldrich, Stockholm, Sweden). Twenty‐four hours after changing medium, the cells were subjected to 3 days of puromycin selection (Thermo Fisher Scientific Inc., Waltham, MA).
Statistics. Statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla CA). Kaplan‐Meier survival curves were compared using a log rank test. Comparisons between groups were performed by two‐tailed Student's t test or by one‐way ANOVA, followed by Tukey's multiple comparisons test. p < 0.05 was considered statistically significant.
Results
Low‐dose irradiated MSCs exert anti‐tumoral effects in a mouse GBM model
To assess the anti‐tumoral properties of irMSCs, non‐irradiated or irMSCs (2, 5, 10, 15, and 20 Gy) were transplanted intra‐tumorally at days 7 and 17 into tumor‐bearing mice and the survival rate was assessed at Day 100, post GL261 grafting (Fig. 1 a). Various radiation doses resulted in differential effects on survival, with the best outcome achieved after intra‐tumoral implantation of MSCs irradiated with 5 Gy. In this group, 29% of the animals were cured (Fig. 1 b). Other radiation doses were also effective, although to a lesser degree, with 17% survival seen after grafting of 10 or 15 Gy irMSCs and 14% after 2 Gy. No difference in survival was seen when niMSCs or 20 Gy irMSCs were compared to tumor‐bearing mice (Fig. 1 b). In the surviving animals, we could neither detect tumors nor signs of illness, indicative of tumor regression. Indeed, earlier analysis of the brains (Day 22) proved that the mean tumor volume was reduced of approximately 67% in animals treated with 5 Gy irMSCs compared to the control group (Fig. 1 c). In light of these results, we chose to carry on with the 5 Gy radiation dose and investigate the mechanism of action.
Irradiated mesenchymal stromal cells modulate the anti‐tumor immunity in vivo
To investigate the mechanism of action of anti‐tumoral irMSCs, we analyzed both systemic and local immune responses. For the systemic immune response, peripheral blood was collected at days 14 and 21 and the relative proportion of immune cells analyzed by flow cytometry (Fig. 2 a). CD8+ T cells are considered amongst the main players in anti‐tumor immunity, including glioma,23 and the degree of infiltration correlated to patient survival.24 In contrast, our time course analyses did not show any effect on the CD8+ T cells but rather on the CD4+ T cell population. At 3 weeks post‐grafting of irMSCs, the CD4+ T cells accounted for 54% of the total T lymphocyte population (CD3+) compared to 47% in the control group (Fig. 2 c) and no difference was seen in the number of systemic CD8+ T or NK cells (Fig. 2 b–2 c). Interestingly, myeloid‐derived suppressor cells (MDSCs), defined as CD11b+/Gr1+ 25 and regarded as important components of cancer immune evasion,26 decreased in the systemic circulation of 5 Gy irMSC mice, dropping from 6.0% of the total leukocyte population in control animals to 2.9% in 5 Gy irMSC group (Fig. 2 b).
We next determined the presence of T lymphocytes within the tumor at Day 22 by immunohistochemistry. In agreement with previous reports, we could confirm the presence of both CD4+ and CD8+ cells in the tumor but not in the surrounding normal brain. Mirroring the findings in the peripheral blood, the intra‐tumoral infiltration of T cells mainly constituted of CD4+ cells, with a 1.7‐fold increase of cells/mm2 after treatment with 5 Gy irMSCs compared to control animals (Fig. 2 d and 2 e) whereas the intra‐tumoral infiltration of CD8+ cells was not affected by grafting of irMSCs (Fig. 2 d and 2 f). CD4+ T cells can differentiate into different subsets characterized by diverse immune modulatory functions and therefore differentially contributing to the immunologic environment.27 The FOXP3+ regulatory T cell (Treg) subset is known for promoting tumor progression via T cell suppression.28 Within the irMSC‐treated group the density of FOXP3+ T cells decreased to 36% and 39% compared to the niMSC and the tumor‐bearing control (tbc) group, respectively whilst we could not detect any changes in the TH1 subset (TBX21+; Fig. 2 g–2 i).
Finally, we confirmed the presence of a glial scar surrounding the tumors as reported elsewhere.29, 30 Quantification of reactive astrocytes showed a three‐fold increase in the number of GFAP+/Vimentin+ cells at the tumor border after grafting of irMSCs, as compared to tumor‐bearing control (Fig. 3 a and 3 b). The clear separation between tumor and reactive astrocytes seen in both tumor‐bearing control and niMSC groups was replaced by a diffuse tumor border and a more prominent intra‐tumoral infiltration of reactive astrocytes in the irMSC group (Fig. 3 a, white arrowheads).
Figure 3.
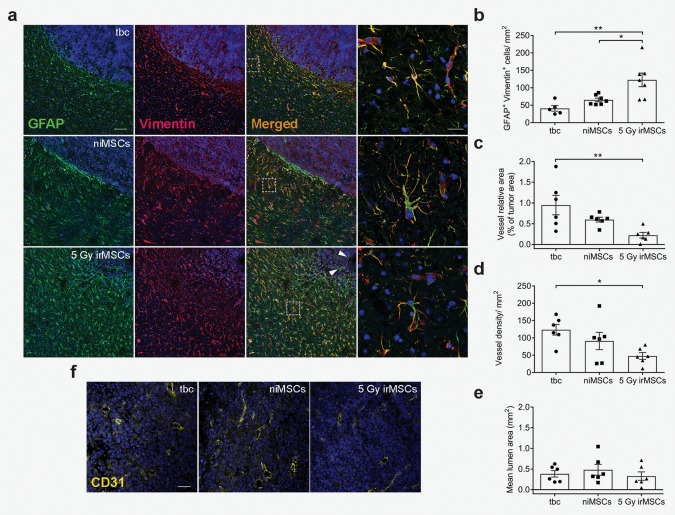
Tumor microenvironment is affected by irMSCs. (a) Representative images of reactive astrocytes (GFAP+ green, Vimentin+ red) at the tumor border, double positive cells quantified in (b); scale bar 100 µm and 20 µm for low and high (inserts) magnification images, respectively. Data are from two independent experiments (n = 5–7, all experimental groups assessed in parallel in each independent experiment) and are presented as mean ± SEM. (f) Representative images showing effect of 5 Gy irMSCs on tumor vasculature (CD31+, yellow), quantified in (c), (d), and (e); scale bar 50 µm. Data are from two independent experiments (n = 6, all experimental groups assessed in parallel in each independent experiment) and are presented as mean ± SEM. Comparisons between groups are performed by one‐way ANOVA, followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01.
Taken together, these results indicate that i.t. grafted irMSCs decrease the general immune suppression occurring in cancer, favoring the anti‐tumor action of the immune system both at the systemic and local level.
Irradiated mesenchymal stromal cells decrease the number of tumor microvessels
We next wanted to assess the effect of irMSCs on tumor vasculature. The GL261 mouse glioma model is characterized by an aberrant tumor vasculature, mimicking human GBM and therefore making it suitable for studies on glioma angiogenesis. We first analyzed the vessel coverage within the tumor by immunohistochemistry and found a marked decrease in the tumor area occupied by vessels after intra‐tumoral grafting of 5 Gy irMSCs (Fig. 3 c–3 f). Specifically, the proportion of area covered by the vessels decreased by 4.3‐fold in the 5 Gy irMSC‐treated tumors compared to the control group (Fig. 3 c). A more detailed analysis of the vasculature indicated that this effect was mainly due to a decreased number of intra‐tumoral vessels (2.6‐fold compared to the control; Fig. 3 d) rather than a difference in their size (Fig. 3 e).
Altogether, we demonstrate that irMSCs not only affect the immune system but also the tumor vasculature, displaying anti‐angiogenic properties in our tumor model of GBM.
Mesenchymal stromal cells retain their phenotypic and tumor‐tropic properties after irradiation
It has been previously reported that MSCs, when transplanted i.t., migrate within the tumor area and its extensions without infiltrating the normal brain tissue.31 Here, we investigated the effect of irradiation on MSC basic characteristics and tumor‐tropism. We first analyzed the expression of MSC surface markers by flow cytometry. As shown in Fig. 4 a, both niMSCs and 5 Gy irMSCs were positive for CD44, CD29, SCA‐1 and negative for the hematopoietic markers CD34 and CD117. Further, irMSCs maintained their differentiation potential and were able to differentiate into adipocytes and osteoblasts (Fig. 4 b).
We next assessed whether the radiation treatment would affect the proliferative ability of MSCs in vitro and confirmed that MSCs subjected to various radiation doses did not alter their growth capacity (Fig. 4 c). In order to evaluate the tumor‐tropic ability of irMSCs, we transplanted GFP‐labeled ni/5 Gy irMSCs in tumor‐bearing mice at days 7 and 17 and collected the brains at Day 22 post‐grafting (Fig. 4 d). As shown in Figure 4 e (first panel), low‐dose irradiation did not affect the tumor‐tropism of MSCs and the transplanted cells migrated within the tumor area and its extensions and interact with tumor cells (Fig. 4 f) without infiltrating the normal brain tissue. Furthermore, irradiation did not influence MSC morphology in vivo and in both experimental groups the cells were mainly localized within the tumor peri‐vascular niche (Fig. 4 e, high magnification).
Taken together, these results show that low‐dose irradiation does not alter inherent MSC properties and the irradiated cells meet the standards for their designation as well as preserve their tumor‐tropic ability.
Irradiation decreases mesenchymal stromal cells inherent immune suppressive properties in vitro
MSCs have been shown to possess an immune suppressive phenotype in their native state, therefore supporting cancer immune escape mechanisms.32 To ascertain whether irradiation could affect the immune regulatory properties of MSCs, we analyzed their effect on T cells by measuring proliferation by CFSE and upregulation of CD25 surface expression33 (Fig. 5 a). As shown in Figure 5 e, co‐culturing of splenic CD3+ T cells with niMSCs inhibits T cell activation and proliferation in vitro. Low‐dose irradiation alters this immune suppressive function and following co‐culture with irMSCs, both CD3+CD4+ T cells and CD3+CD8+ T cells upregulate the activation marker CD25 of 1.7‐ and 1.5‐fold, respectively (Fig. 5 b and 5 d). In accordance, the T cell proliferation increases 2‐ and 1.7‐fold for CD4+ and CD8+ T cells, respectively upon co‐culture with irMSCs (Fig. 5 c and 5 e).
Next, we analyzed MSC secretome, primarily focusing on the release of immune modulatory/angiogenic molecules. Interestingly, we found a reduction in the amount of immune suppressant active TGFβ1, specifically in the 5 Gy irMSCs (Fig. 5 f, first panel), with levels being even lower than in the control medium. The percentage difference of active TGFβ1 compared to control medium spanned from +15% in niMSCs to −10% found upon low‐dose irradiation. This led us to speculate that soluble factor(s) interfering with TGFβ1 post‐transcriptional regulation may be released by irMSCs, a hypothesis further reinforced by gene expression analysis showing no changes in the expression level of Tgfβ1 after irradiation (Fig. 5 f, second panel). To address this question, we cultured GL261 glioma cells in niMSCs or 5 Gy irMSC conditioned medium (CM). Surprisingly, we found that glioma cells grown in 5 Gy CM had a reduced amount of active TGFβ1 (Fig. 5 f, third panel), partially confirming the hypothesis of an interference with the TGFβ1 pathway by irMSCs. Further, to rule out the possibility that this effect was due to a reduced proliferation of the tumor cells, we tested their proliferation rate upon culturing in ni/5 Gy irMSCs CM and found no change compared to control (Fig. 5 f, forth panel).
Lastly, we assessed the effect of irradiation on other important immune modulatory molecules by both gene expression analysis and ELISA. As shown in Fig. 5 g, we found a decrease in the expression of the anti‐inflammatory marker Arg1, specifically in the 5 Gy irradiated group, whereas the expression or secretion of other factors was not affected by irradiation (Fig. 5 g and 5 h), even upon priming with TNFα and/or IFNγ (data not shown).
Taken together, these results demonstrate that low‐dose irradiation alters MSC immune phenotype, limiting the cells’ immune suppressive properties in vitro.
Discussion
In our study, we show that, by a single low‐dose irradiation, the immune suppressive phenotype of bone marrow‐derived MSCs can be reverted and the cells acquire an anti‐angiogenic and anti‐tumoral profile. Low‐dose irradiated MSCs grafted orthotopically in the GL261 mouse glioma model increased survival and led to rejection of established tumors in a significant number of animals. The immune suppressive properties of MSCs are altered upon irradiation both in vivo and in vitro. The immunologic milieu undergoes a substantial rearrangement, characterized by a more favorable immune response and a general decline in both local and systemic immune suppression. The tumor microenvironment changes its prospect with regard to peri‐tumoral reactive astrocytosis and angiogenesis.
In vitro, the effect of irradiation is mainly represented by a marked downregulation of active TGFβ1 in irMSCs as well as in tumor cells subjected to irMSC‐conditioned medium. TGFβ1 inhibits lymphocyte function by influencing both T cell proliferation and differentiation34 and it has been correlated to poor clinical outcome in cancer patients.35, 36, 37 Indeed, when we co‐cultured irMSCs with T cells we could detect a decrease in the immune suppressive properties of MSCs. In accordance with our results, downregulation of TGFβ1 is seen after TLR4 priming of MSCs and the resulting conversion into pro‐inflammatory MSCs.18
Downregulation of arginase 1 may further contribute to the acquisition of a less immune suppressive phenotype by irMSCs and therefore favor the anti‐tumor immunologic response. Expression of this enzyme is seen in immune suppressive macrophages, whereas immune stimulatory macrophages lack arginase 1 expression.38
Tumor treatment induces changes in immune cell composition and our kinetic analysis of the in vivo immune response revealed alternate waves of immune response. This phenomenon may reflect a general decline in tumor‐induced immune suppression, where the expansion of MDSCs, a population of immature myeloid cells typically induced in cancer,39 is compromised. This would create a favorable environment for the immune system to bustle about and perform its anti‐tumor functions. Moreover, the increased lymphocyte infiltration may also benefit from the normalization of tumor vasculature induced by irMSCs that facilitates immune cell extravasation.40 The effect on the CD4+ population appears to be quite specific in that no changes were detected when analyzing CD8+ or NK cell populations. Recently, CD4+ cells have been shown to acquire cytotoxic properties in tumor settings, offering a plausible explanation for our findings.41, 42
Analysis of tumor tissue revealed an increased quantity of reactive astrocytes at the tumor border upon treatment with irMSCs. Reactive astrocytes form a border of reactive gliosis surrounding the tumor where they interact with microglia and infiltrating monocytes and in this way affect tumor immune response.29 Moreover, glioma‐associated astrocytes have been shown to upregulate MHCII,30 further supporting an active role for reactive astrocytes in cancer immunity. Recently, it was reported that microvesicles are released from reactive astrocytes in response to inflammatory brain damage and trigger the recruitment of peripheral immune cells into the brain lesion region.43 In light of this, a more pronounced peri‐tumoral activation of astrocytes, as shown here, may support immune cell trans‐migration into the glioma by means of increased brain‐to‐periphery microvesicle communication.
Bone marrow‐derived mesenchymal stromal cells are radio‐resistant. Even when subjected to radiation doses lethal for hematopoietic cells, MSCs survive mainly due to cell cycle arrest and activation of DNA repair mechanisms.44 Low‐dose irradiation does not lead to random cellular changes due to non‐selective DNA damage45 and it has been demonstrated that MSCs retain their defining stem cell characteristics after exposure to ionizing radiation.46 In accordance, we show that MSCs retained their ability to differentiate as well as the expression of typical MSC surface markers after low‐dose irradiation. Furthermore, the proliferative capacity of MSCs was not affected by irradiation, indicating that the anti‐tumor effects seen in vivo are not secondary to an unspecific immune response triggered by dying MSCs. This concept was further strengthened by the finding that MSCs subjected to higher doses than 5 Gy did not elicit the same favorable effect on animal survival.
Importantly, irradiated cells retained the ability to selectively and efficiently infiltrate the growing tumor, localizing mainly within the tumor peri‐vascular niche.31 Indeed, MSCs have been shown to promote angiogenesis in different types of tumors by secreting a panel of cytokines and pro‐angiogenic molecules.47, 48 In contrast, here we show that the aberrant angiogenesis characteristic of GBM49 was considerably reduced after irMSC treatment. The tumor area covered by vessels significantly decreased after intra‐tumoral irMSC grafting. A possible mechanism for the observed reduction in tumor microvessel formation is the marked irMSC‐induced decrease of active TGFβ1. Solid evidence for a crucial role of TGFβ1 in tumor‐, including GBM, angiogenesis exists and TGFβ1 blockade is a potential therapeutic approach under development.50, 51
Targeting only one aspect of tumor formation has been shown to favor the development, or improvement, of cancer cell resistance mechanisms. These self‐defense mechanisms may be overcome by acting on multiple hallmarks of cancer simultaneously, and in this way, limiting the ability of malignant cells to circumvent the therapy. In this respect, low‐dose irradiated mesenchymal stromal cells present great potential in as much as they may attack glioma through both immune system‐dependent and anti‐angiogenic mechanisms.
In summary, the present series of experiments shows that upon low‐dose irradiation, MSCs reduce their innate immune suppressive features and acquire anti‐angiogenic and anti‐tumoral properties. After intra‐tumoral grafting of irMSCs, this phenotypic switch ultimately leads to cure in ∼30% of animals challenged with brain tumors. The therapeutic range of radiation on MSCs is narrow, with effects on animal survival seen between 2 and 15 Gy and a peak at 5 Gy. The findings thus provide proof‐of‐principle for the use of low‐dose irradiated MSCs as a therapeutic tool in GBM. However, further investigations will be necessary to define the molecular mechanisms governing the characteristics of irMSCs, allowing for a better understanding and control of the potential therapeutic properties of these cells.
Author Contributions
FRS designed and performed the experiments, analyzed the data and wrote the article. SE designed and performed the experiments. SV and TAK designed and performed the T cell activation experiment and analyzed the data. JB designed the experiments and wrote the article. All the authors critically revised the article.
Acknowledgments
This work was supported by a donation from Viveca Jeppsson, ALF grants from the Medical Faculty at Lund University and funds from Region Skåne. The authors thank Drs. Mattias Belting, Daniel Bexell, Fredrik Ivars and Henrik Ahlenius for fruitful discussion and critical reviewing of the article. We also thank Dr. Tania Ramos Moreno for help with animals’ perfusion, Klaus Sejdini for MSC differentiation, Dr. Alya Zriwil for valuable comments on the article and assistance with flow cytometry analysis. We are grateful to Dr. Zaal Kokaia for sharing laboratory equipment, the StemTherapy Imaging Core Facility and FACS Core Facility at the Lund Stem Cell Center for excellent technical assistance. The authors have declared that no conflict of interest exists.
References
- 1. Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. [DOI] [PubMed] [Google Scholar]
- 2. Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33:407–20. [DOI] [PubMed] [Google Scholar]
- 3. Keating A. Mesenchymal stromal cells. Curr Opin Hematol. 2006;13:419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trounson A, McDonald C. Stem cell therapies in clinical trials: Progress and challenges. Cell Stem Cell. 2015;17:11–22. [DOI] [PubMed] [Google Scholar]
- 5. National Institutes of Health . ClinicalTrials.gov, Updated March 13, 2017. Accessed August 28, 2017. [Google Scholar]
- 6. Sage EK, Thakrar RM, Janes SM. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy. 2016;18:1435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vega EA, Graner MW, Sampson JH. Combating immunosuppression in glioma. Future Oncol. 2008;4:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenting K, Verhaak R, Ter Laan M, et al. Glioma: experimental models and reality. Acta Neuropathol. 2017;133:263–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakamura K, Ito Y, Kawano Y, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–64. [DOI] [PubMed] [Google Scholar]
- 10. Gunnarsson S, Bexell D, Svensson A, et al. Intratumoral IL‐7 delivery by mesenchymal stromal cells potentiates IFNgamma‐transduced tumor cell immunotherapy of experimental glioma. J Neuroimmunol. 2010;218:140–4. [DOI] [PubMed] [Google Scholar]
- 11. Nakamizo A, Marini F, Amano T, et al. Human bone marrow‐derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. [DOI] [PubMed] [Google Scholar]
- 12. Studeny M, Marini FC, Champlin RE, et al. Bone marrow‐derived mesenchymal stem cells as vehicles for interferon‐beta delivery into tumors. Cancer Res. 2002;62:3603–8. [PubMed] [Google Scholar]
- 13. Wang Q, Zhang Z, Ding T, et al. Mesenchymal stem cells overexpressing PEDF decrease the angiogenesis of gliomas. Biosci Rep. 2013;33:e00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma S, Xie N, Li W, et al. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014;21:216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poggi A, Giuliani M. Mesenchymal stromal cells can regulate the immune response in the tumor microenvironment. Vaccines (Basel). 2016;4:41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi Y, Du L, Lin L, et al. Tumour‐associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16:35–52. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, Chen X, Cao W, et al. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–16. [DOI] [PubMed] [Google Scholar]
- 18. Waterman RS, Tomchuck SL, Henkle SL, et al. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro‐inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One. 2010;5:e10088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marfy‐Smith SJ, Clarkin CE. Are mesenchymal stem cells so bloody great after all? Stem Cells Transl Med. 2017;6:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klug F, Prakash H, Huber PE, et al. Low‐dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602. [DOI] [PubMed] [Google Scholar]
- 21. Zhu H, Guo ZK, Jiang XX, et al. A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat Protoc. 2010;5:550–60. [DOI] [PubMed] [Google Scholar]
- 22. Rio DC, Ares M, Jr. , Hannon GJ, et al. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc. 2010;2010:pdb.prot5439 pdb prot5439. [DOI] [PubMed] [Google Scholar]
- 23. Strojby S, Eberstal S, Svensson A, et al. Intratumorally implanted mesenchymal stromal cells potentiate peripheral immunotherapy against malignant rat gliomas. J Neuroimmunol. 2014;274:240–3. [DOI] [PubMed] [Google Scholar]
- 24. Kmiecik J, Poli A, Brons NH, et al. Elevated CD3+ and CD8+ tumor‐infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83. [DOI] [PubMed] [Google Scholar]
- 25. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid‐derived suppressor cell nomenclature and characterization standards. Nat Comms. 2016;7:12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khaled YS, Ammori BJ, Elkord E. Myeloid‐derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. 2013;91:493–502. [DOI] [PubMed] [Google Scholar]
- 27. Kim HJ, Cantor H. CD4 T‐cell subsets and tumor immunity: the helpful and the not‐so‐helpful. Cancer Immunol Res. 2014;2:91–8. [DOI] [PubMed] [Google Scholar]
- 28. Sonabend AM, Rolle CE, Lesniak MS. The role of regulatory T cells in malignant glioma. Anticancer Res. 2008;28:1143–50. [PubMed] [Google Scholar]
- 29. Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34. [DOI] [PubMed] [Google Scholar]
- 30. Katz AM, Amankulor NM, Pitter K, et al. Astrocyte‐specific expression patterns associated with the PDGF‐induced glioma microenvironment. PLoS One. 2012;7:e32453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bexell D, Gunnarsson S, Tormin A, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte‐like migratory vehicles in experimental gliomas. Mol Ther. 2009;17:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669–82. [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y, Day A, Haykal S, et al. Mesenchymal stromal cells augment CD4+ and CD8+ T‐cell proliferation through a CCL2 pathway. Cytotherapy. 2013;15:1195–207. [DOI] [PubMed] [Google Scholar]
- 34. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta‐induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8. [DOI] [PubMed] [Google Scholar]
- 36. Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor‐beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–70. [DOI] [PubMed] [Google Scholar]
- 37. Jiang L, Zhou J, Zhong D, et al. Overexpression of SMC4 activates TGFbeta/Smad signaling and promotes aggressive phenotype in glioma cells. Oncogenesis. 2017;6:e301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas AC, Mattila JT. Of mice and men”: arginine metabolism in macrophages. Front Immunol. 2014;5:479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ganss R, Ryschich E, Klar E, et al. Combination of T‐cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–70. [PubMed] [Google Scholar]
- 41. Quezada SA, Simpson TR, Peggs KS, et al. Tumor‐reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez‐Diez A, Joncker NT, Choi K, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dickens AM, Tovar‐y‐Romo LB, Yoo S‐W, et al. Astrocyte‐shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. 2017;10:eaai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sugrue T, Lowndes NF, Ceredig R. Mesenchymal stromal cells: radio‐resistant members of the bone marrow. Immunol Cell Biol. 2013;91:5–11. [DOI] [PubMed] [Google Scholar]
- 45. Islam MS, Stemig ME, Takahashi Y, et al. Radiation response of mesenchymal stem cells derived from bone marrow and human pluripotent stem cells. J Radiat Res. 2015;56:269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nicolay NH, Sommer E, Lopez R, et al. Mesenchymal stem cells retain their defining stem cell characteristics after exposure to ionizing radiation. Int J Radiat Oncol Biol Phys. 2013;87:1171–8. [DOI] [PubMed] [Google Scholar]
- 47. Liu Y, Han ZP, Zhang SS, et al. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem. 2011;286:25007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coffelt SB, Marini FC, Watson K, et al. The pro‐inflammatory peptide LL‐37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci Usa U S A. 2009;106:3806–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jain RK, di Tomaso E, Duda DG, et al. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–22. [DOI] [PubMed] [Google Scholar]
- 50. Han J, Alvarez‐Breckenridge CA, Wang QE, et al. TGF‐beta signaling and its targeting for glioma treatment. Am J Cancer Res. 2015;5:945–55. [PMC free article] [PubMed] [Google Scholar]
- 51. Neuzillet C, Tijeras‐Raballand A, Cohen R, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. [DOI] [PubMed] [Google Scholar]